

Abstract
The literature on the pharmacokinetics of vancomycin in patients undergoing extracorporeal membrane oxygenation (ECMO) therapy is sparse. A population pharmacokinetic (PK) model for vancomycin in ECMO patients was developed using a nonlinear mixed effects modeling on the concentration–time profiles of 14 ECMO patients who received intravenous vancomycin. Model selection was based on log‐likelihood criterion, goodness of fit plots, and scientific plausibility. Identification of covariates was done using a full covariate model approach. The pharmacokinetics of vancomycin was adequately described with a two‐compartment model. Parameters included clearance of 2.83 L/hr, limited central volume of distribution 24.2 L, and low residual variability 0.67%. Findings from the analysis suggest that standard dosing recommendations for vancomycin in non‐ECMO patients are adequate to achieve therapeutic trough concentrations in ECMO patients. This further shows that ECMO minimally affects the PK of vancomycin in adults including in higher‐weight patients.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Vancomycin is a commonly used agent to prevent and treat suspected MRSA infections in the hospital. ECMO therapy has the potential to complicate the pursuit of therapeutic drug concentrations by altering the pharmacokinetics of vancomycin. Previous studies have investigated whether ECMO can affect vancomycin pharmacokinetics with differing results.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addresses the question of the ability of ECMO therapy to alter the vancomycin pharmacokinetic profile and how to dose patients who are administered vancomycin while on ECMO therapy.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This study produces a robust model for vancomycin in adult patients receiving ECMO therapy. This model describes higher‐weight patients, resulting in a unique covariate selection.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS
☑ This study suggests that ECMO causes minimal difference in pharmacokinetics and recommends a potential dosing strategy. Standard vancomycin dosing approaches should produce therapeutic drug concentrations in heavier patients treated with ECMO.
Extracorporeal membrane oxygenation (ECMO) is a cardiopulmonary support procedure in which patient blood is drained via cannulas through an external circuit to be oxygenated, warmed, and returned to the patient.1, 2 There are two types of ECMO: venovenous (VV) ECMO, which provides support for the lungs, and venoarterial (VA) ECMO, which provides support for both the heart and lungs.3, 4 VV ECMO is used to treat severe but potentially reversible respiratory failure, while VA ECMO is primarily used for treating severe cardiac or cardiorespiratory failure.
Patients requiring ECMO are critically ill, with varying diagnoses, age, body size, and degrees of end‐organ dysfunction.5 Patients treated with ECMO are at increased risk of infection.6 Vancomycin is a bactericidal, glycopeptide antibiotic used in patients receiving ECMO for procedural prophylaxis or treatment of infections caused by Gram‐positive organisms, particularly methicillin‐resistant Staphylococcus aureus (MRSA). The effectiveness of vancomycin therapy is dependent on achieving and maintaining optimal plasma concentrations. In the critically ill, a trough concentration between 15–20 mg/L is recommended by the Infectious Disease Society of America guidelines for the treatment of MRSA infections.7
Patients undergoing ECMO therapy are treated with multiple medications. The continuous pathophysiological changes often make it difficult to attain adequate therapeutic concentration.8 Multiple organ dysfunction or failure significantly alters the PK of drugs, including volume of distribution and clearance.9 In addition, previous studies have shown that ECMO therapy results in an increase in vancomycin volume of distribution and a decrease in clearance, most dramatically in infants, with an increase in volume of distribution of 75% and a decrease in clearance of approximately 40%10. Similar results are seen in children and adults, with an increase of volume and decrease in clearance of approximately 40% and 35%, respectively.11 There are several biochemical properties of drugs that may influence sequestration by the components of the ECMO circuitry, including lipophilicity, molecular size, and plasma protein binding.11, 12 The added surface area of the tubing and membrane, in addition to these drug properties, has revealed varying pharmacokinetic differences among ECMO patients compared to other patient populations.13
While the use of ECMO in adult patients continues to increase, the literature describing the use of vancomycin for adult patients on ECMO is still limited, especially in overweight and obese patients. Optimized antibacterial therapy necessitates a clear understanding of vancomycin disposition during ECMO therapy. The objective of this study was to determine the PK profile of vancomycin in a population of high bodyweight, adult ECMO patients and to provide a quantitative framework to determine the appropriate dosing regimen for these patients.
METHODS
Patient eligibility
This open‐labeled single‐center, prospective, observational study was approved by the Internal Review Board at Thomas Jefferson University Hospital. Patients ≥18 years of age on VA or VV ECMO and receiving vancomycin were eligible for this study. Patients who received vancomycin within 48 hours of study entry, who had a documented vancomycin allergy, or whose blood cultures with identified vancomycin‐resistant Gram‐positive bacteria necessitating a more appropriate antibiotic were excluded from this study. Patients were recruited after informed consent was obtained from the patient's representative.
ECMO apparatus
The ECMO system was comprised of a ROTAFLOW centrifugal pump and CARDIOHELP system (Maquet, Rastatt, Germany) in configuration with the poly‐methyl‐pentene (PMP) QUADROX‐D diffusion membrane hollow‐fiber oxygenator (Maquet), and Fem‐Flex II femoral arterial or Femtrak femoral venous cannulas (Edwards Lifescience, Irvine, CA). The ECMO circuit was primed with ∼600 mL of normal saline. Blood pumps can generate up to 5–6 L/min depending on cannula size.
Clinical protocol
Subjects received vancomycin per standard of care dosing regimens. In addition to routine trough level monitoring, patients enrolled in this study required five additional blood samplings after the first vancomycin infusion. Blood samples were collected 30, 60, 120, 240, and 360 minutes after vancomycin infusion.
Vancomycin drug concentrations were analyzed with a Roche Cobas C501 instrument (Roche Diagnostics, Indianapolis, IN) using a glucose‐6‐phosphate dehydrogenase‐based enzyme immunoassay method. This assay is based on a homogenous enzyme immunoassay technique used for the quantitative analysis of vancomycin in human serum or plasma.14 The lower detection limit of this assay is 1.7 μg/mL (1.2 μmol/L). The measuring range of the assay is 1.7–80 μg/mL (1.2–55.2 μmol/L). Assay precision was determined using human serum/plasma samples and controls and in a modified NCCLS EP5‐T2 protocol. No significant crossreactivity or interference was noted with additional drugs tested in this assay.
Population PK analysis
The population PK data were analyzed using nonlinear mixed‐effects modeling implemented in NONMEN 7.3 (Icon Development Solutions, Hanover, MD) and a G77 FORTRAN compiler. Xpose (xpose.sourceforge.net), PsN (psn.sourceforge.net), and R (R‐project, www.rproject.org, v. 3.2.2) were used for the exploratory analysis and postprocessing of NONMEM output. Prior to modeling, observed vancomycin concentrations were plotted against time (i.e., time since the last dose) and stratified by key study design elements such as dose. These exploratory graphs were used to inform the selection of a starting structural model. Base model building started with one‐ and two‐compartment models with and without bodyweight allometric scaling. The model was refined by testing intersubject variability (ISV) on each PK parameter. ISV was modeled using exponential functions. The apparent percent coefficient of variation (%CV) for ISV was computed as the square root of EXP(OMEGA)−1 × 100% with OMEGA describing the variance term. Additive, proportional, and combined error models were all attempted to describe residual variability, which is a composite measure of assay error, dose/sample time collection errors, model misspecification, and any other unexplained variability within a subject. In each of the pivotal models, selection of competing models was based on successful minimization and completion of covariance ($COV) steps in NONMEM, reductions in NONMEM objective function value OFV (Δ OFV of 6.63, P < 0.01 for 1 degree of freedom) for hierarchical models, precision and plausibility of parameter estimates, and a variety of goodness‐of‐fit plots. Additionally, the condition number of the correlation matrix of the parameter estimates, i.e., the ratio of the largest to smallest eigenvalues was required to be less than 1,000.
Identification of covariates was performed using a full covariate model as described previously.15 This method was selected instead of the stepwise covariate selection approach in order to avoid selection bias, which is a risk in small datasets.16 Initial covariates were selected by choosing covariate effects that were biologically plausible, grounded in prior knowledge, and reflected clinical interest. This included age, bodyweight, gender, serum creatinine, creatinine clearance, and usage of renal replacement therapy. The available covariates were then reduced by eliminating correlated covariates. All covariate pairs with an absolute correlation coefficient greater than 0.3 were considered for removal with the more biologically plausible covariate retained in the analysis. The remaining covariates were included using a linear covariate‐parameter relationship in the model and reparameterized as necessary to achieve a stable model.
A nonparametric bootstrap method was used to evaluate the stability of the final model.17 One thousand samples were created by resampling from the original dataset. No stratification was considered during the sampling of the dataset. The model was run on each sample, and results from the 2.5th and 97.5th percentiles of converged runs from the 1,000 bootstrap dataset were used to estimate the 95% confidence interval (CI). This was then compared to the estimates provided by the original sample. A visual predictive check (VPC) was used to assess the ability of the model to predict the data. One thousand Monte Carlo simulations of the final model were performed.18 The median and 95% CI of the simulated data was then compared to the observed data.
Model predictive checks (PCs) were also used to evaluate the final model.18 Three test statistics of clinical importance were chosen: clearance, area under the curve (AUC), and trough concentration. Clearance and AUC were calculated using a noncompartmental method. Trough concentration was defined as the last concentration reported for each patient. The distributions of the test statistics were calculated from the observed data. The median was calculated as a measure of central tendency, and the 5th and 95th percentiles of the test statistics were calculated as bounds around the median. The final population PK model, including final fixed and random effect parameters, was used to simulate 1,000 replicates of the observed dataset, and test statistics were computed from each of the simulated datasets. The predictive distributions of each test statistic from the simulated datasets were compared with the median value of the corresponding observed test statistic. The model was considered predictive if the median test statistic from the observed data fell within the 95% CI of the test statistics of the simulated data.
All covariates obtained from the covariate analysis were included in the final model. Fixed effect as well as parameter precision obtained from the final model was sampled 5,000 times using a parametric bootstrap method.19 The obtained samples were used to derive the distribution of change in the value relative to reference (typical value) of each covariate obtained from the covariate analysis. To be clinically relevant, the absolute change in the value relative to reference (typical value) should be more than 20%. For continuous covariates, the changes were calculated on the extreme values (i.e., the 10th and 90th percentiles of the covariate distributions).
The results of the model were used to simulate potential dosing strategies for the median patient. One thousand Monte Carlo simulations were performed using a patient of the median weight (WT) and creatinine clearance (CRCL) of the observed values. Several doses (1–2 g) and dose schedules (1–3 times daily) were used for the simulation. A dose strategy was selected based on the goal of reaching a trough between 15 and 20 mg/L by steady state after the third dose had been administered.
RESULTS
A total of 14 patients were recruited for this trial, resulting in 65 vancomycin concentrations for analysis. The demographic factors of these patients are presented in Table 1. A two‐compartment model was found to adequately describe the concentration–time profile of vancomycin. The residual intraindividual variability was accounted for using a proportional error model. Removal of the interindividual variability terms for intercompartmental clearance (Q) and peripheral volume of distribution (V2) did not increase the objective value function significantly. From the final base model, the typical value for total clearance (CL) was estimated to be 2.83 L/hr (%RSE of 34%), central volume of distribution (V1) was estimated to be 24.2 L (%RSE of 15%), the intercompartmental clearance was 11.2 L/hr (%RSE of 15%), and the typical value for peripheral volume of distribution was 32.3 L (%RSE of 12%). The proportional residual variability was 0.0067 (%RSE of 47%). The final base model described the observed serum vancomycin concentration data adequately, as no serious systematic trends were seen in the residual diagnostic plots. The condition number (calculated as the ratio of the largest eigenvalue to the smallest eigenvalue) for the base model was 345, indicating the adequacy of model parameterization. ETA shrinkages for final base model were 6.9% for V1 and 9.6% for CL. Epsilon shrinkage was 18%. Values for ETA shrinkage and epsilon shrinkage are within the range of values where empirical Bayes estimates (EBE)‐based diagnostics are generally still a valid metric to identify potential covariates.20
Table 1.
Demographic factors
| Mean (SD) | |
|---|---|
| N | 14 |
| Female | 21% |
| Age (yr) | 47 (16) |
| Range: 19–72 | |
| Weight (kg) | 95 (27) |
| CrCl (mL/min) | 84 (37) |
| Renal impairment | 50% |
| Mild | 28.6% |
| Moderate | 14.3% |
| Severe | 7.15% |
| Renal replacement | 0% |
| ECMO type | |
| VA | 86% |
| VV | 14% |
Renal impairment definition based on creatinine clearance calculated using the Cockcroft‐Gault equation: mild (60–89 mL/min), moderate (30–59 mL/min), and severe (15–29 mL/min).
Following covariate reduction due to heavy correlation between covariates, the covariate effects of CRCL on CL, bodyweight on V1, and bodyweight on V2 were included in the model. All covariate‐parameter relationships were described using a linear approach centered around the median covariate. After covariate addition, improvements were attempted to improve the covariate model by adjusting the OMEGA structure including perturbing the initial estimates of ETA and attempting to estimate an OMEGA block instead of an OMEGA diagonal. However, this did not improve the model either. Therefore, the full covariate model was considered to be the final covariate model.
The goodness‐of‐fit plots (Figure 1) revealed no systemic bias. In general, there was good agreement between the predicted and observed vancomycin concentrations for the subjects in this analysis. However, the model for one patient resulted in significant underprediction of vancomycin concentrations. The VPC is shown in Figure 2 a. This shows that the model was successful in predicting the concentrations seen in the samples, with the exception of the aforementioned patient. The goodness‐of‐fit is further demonstrated in the individual plots of observed concentration, individual prediction, and population prediction in Supplementary Figure 1.
Figure 1.
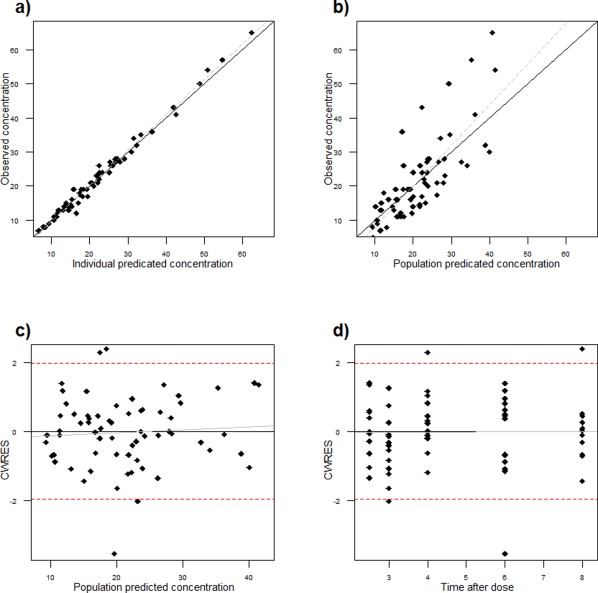
Goodness‐of‐fit plots. (a) Individual prediction vs. observed. (b) Population prediction vs. observed. (c) Conditional weighted residuals (CWRES) vs. population prediction. (d) CWRES vs. time.
Figure 2.
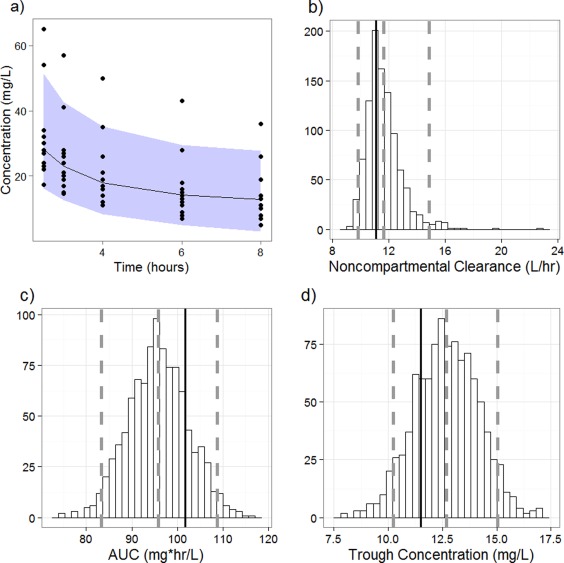
Predictive checks. (a) Visual predictive check. The black circles represent the observed vancomycin concentrations. The black line represents the median concentrations from the simulations. The gray shaded area represents the 2.5th and 97.5th percentile of the simulated data points to denote a 95% CI. The five uppermost points represent concentrations from one patient who was considered an outlier in this analysis. b–d: Clinical predictive check. The histogram represents posterior predictive distribution of noncompartmental clearance (b), AUC (c), and trough concentration (d) from the 1,000 simulated datasets. The black line represents the mean of the observed data. The gray lines represent the median and 2.5th and 97.5th percentiles of the simulated datasets.
Table 2 presents parameter estimates of the final covariate models along with the corresponding 95% CI from the bootstrap resampling technique. The PK parameter estimates from the final covariate model are close to the respective mean values from the bootstrap runs, and the 95% CIs are reasonably small. However, there is some bias between the parameter estimate and the mean value of the bootstrap. This is likely due to small sample size and sample heterogeneity.
Table 2.
Parameter estimates
| Parameter | Estimate | Relative standard error | Intersubject variability | Mean bootstrap | 95% CI |
|---|---|---|---|---|---|
| V1 (L) | 24.2 | 14.5% | 34% | 20.8 | [11.9,27.8] |
| CL (L/hr) | 2.83 | 33.5% | 77% | 3.31 | [1.72,5.49] |
| V2 (L) | 32.3 | 11.8% | — | 29.3 | [20.7,40.7] |
| Q (L/hr) | 11.2 | 15% | — | 11.9 | [9.04,15.2] |
| CLCRCL | 0.0154 | 21.3% | — | 0.0125 | [0.002,0.024] |
| V1WT | 0.00638 | 98% | 0.00816 | [0.001,0.018] | |
| V2WT | 0.0169 | 14.6% | — | 0.0135 | [0.001, 0.020] |
| Proportional error (σ2) | 0.0067 | 46.9% | — | — | — |
This table reflects the parameter estimates of the final model as produced by NONMEM output. The mean bootstrap represents the mean of the successful bootstrap runs. The 95% CI represents the values at the 2.75th and 97.5th percentile of the successful bootstrap runs. V1, central volume of distribution; V2, peripheral volume of distribution; Q, intercompartmental clearance; CL, total clearance; CLCRCL, change in CL per unit change in CRCL (mL/min); V1WT, change in V1 per unit change in WT (kg); V2WT, Change in V2 per unit change in WT (kg).
Figure 2 b–d presents the comparisons obtained from the PC using noncompartmental CL, AUC, and trough concentration as the test statistics. Overall, the observed and simulated datasets appear to match with no major systemic bias. The median observed mean of the test statistics are within the 95% CI of the simulated means, and the medians of the simulated and observed datasets appear in concordance with the simulated means. The variability of the simulated data is small and physiologically plausible relative to the observed data. Taken together, this suggests that the model predicts the selected clinically relevant parameters very well.
Figure 3 presents the analysis of clinical relevance of the final covariates selected. The shaded region represents the 20% difference from the typical value. The extreme values of CRCL and WT resulted in clinically significant changes to the clearance and peripheral volume parameters, respectively. The effects of the extreme values of WT on central volume showed trends towards clinical significance but appear to lack statistical significance, as the 95% CI for each crosses the line demarking the null value. This suggests a potentially meaningful relationship that was unable to be fully described. The effect of CRCL on CL is further significant, as CL directly affects the AUC. In this sample, the 10th and 90th percentiles of CRCL result in a roughly 60% increase and decrease in the AUC from the median value, respectively.
Figure 3.
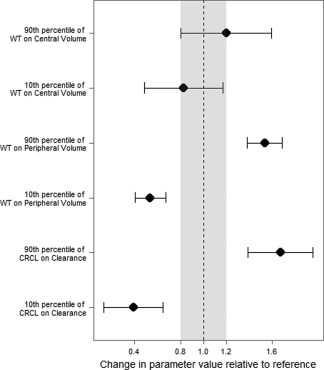
Clinical relevance of covariates. This graph represents the calculation of covariate effects on relevant PK parameters. The shaded region represents a 20% difference from the typical value, a clinical equivalence range. The bars represent the 95% CI of parameter changes relative to each extreme of the covariate.
After finalizing and demonstrating the viability of the PK model, it was used to simulate the vancomycin concentration–time profile for the patient of median bodyweight and creatinine clearance in the study. Figure 4 demonstrates the results of the dosing simulation; 1,000 mg given twice daily and 2,000 mg given daily were both able to adequately reach the selected trough region of 15–20 mg/L during steady‐state concentration. This suggests that either dose regimen of vancomycin will show therapeutic efficacy in ECMO patients with a bodyweight close to 95 kg and with a CRCL of 85 mL/min, while higher or more frequent doses would increase the possibility of supratherapeutic troughs. Given the focus on patients of high bodyweight, the dose simulation was performed in patients above median bodyweight; 3,000 mg daily in 2–3 divided doses was found to reach adequate trough concentrations in patients between 95 and 149 kg.
Figure 4.
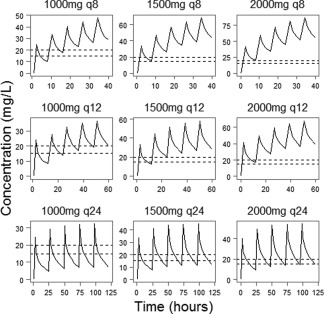
Dose simulation. This figure shows the result of the dose simulation for the patient of median weight (95 kg) and creatinine clearance (84 ml/min). In all, 1,000 Monte Carlo simulations were performed using the median patient at different dosage regimens. The dashed lines represent the target trough concentration. The straight curve represents the median prediction of vancomycin concentration. A dosing strategy was considered successful if it produced a trough in the target range in between the third and fifth administration of vancomycin.
DISCUSSION
Accurately describing the concentration–time profile of vancomycin is of high clinical interest. Despite extensive reporting, an optimal therapeutic drug monitoring approach for vancomycin has not been established.21, 22 This is even more of an issue for special populations, such as patients treated with ECMO. Vancomycin primarily exhibits time‐dependent killing properties; an AUC/MIC (minimum inhibitory concentration) ratio of 400 is associated with successful bacterial eradication.23 In turn, the trough concentration is a convenient predictor of the AUC/MIC ratio and thus a useful tool to guide therapeutic decisions. In critically ill patients with suspected serious infections, the target trough concentration is between 15–20 mg/L as recommended by the Infectious Disease Society of America guidelines for the treatment of MRSA infections.7 Targeting vancomycin troughs between 15–20 mg/L increases the probability of achieving an AUC/MIC target of 400, which has been advocated as a target to achieve clinical effectiveness with vancomycin.24 However, high vancomycin concentrations have been linked to higher risk of adverse events such as ototoxicity and nephrotoxicity.25, 26
A population pharmacokinetic (PopPK) model was developed to adequately describe vancomycin disposition in critically ill patients. This model identified CRCL and bodyweight as clinically relevant factors impacting CL, V1, and V2, respectively. This finding agrees with the generally accepted knowledge for vancomycin and the dosing strategies employed in most hospitals, where the vancomycin‐dosing regimen is selected according to bodyweight and creatinine clearance. The covariate of bodyweight on V2 appeared to be more clinically relevant than the covariate of bodyweight on V1. The patients in the study skewed toward a higher weight, which may better be reflected by increases in adipose tissue, which is linked more closely to V2 than V1. Furthermore, there were limited samples drawn during the distribution phase (1.5–2.5 hours) of vancomycin, which may have made it difficult to fully characterize the distribution phase and V1. Regardless, characterizing the trough concentration is the most clinically relevant aspect of vancomycin pharmacokinetics, and it occurs well after the distribution phase has ended. Thus, any imprecision in the calculation of the central volume parameter may not be clinically significant as the volume at steady state, the combination of V1 and V2, is well described. In addition, the results from the predictive checks indicate that the model is well equipped to predict clinically relevant parameters.
The current PopPK model was developed based on a limited sample of critically ill patients. While the illness would be the main factor necessitating ECMO therapy, it raises the possibility that the illness may have affected the patient in ways that are difficult to predict. This may partially explain the predominate outlier in our analysis. This female patient differed significantly from the others on the basis of youth (22 years) and had an extremely low body size (54 kg), which may explain why the current model built on older, higher‐weight patients underestimated this patient's vancomycin concentrations. The variability due to illness may also have led to the moderate ISV seen in the model for CL. However, based on a large body of published investigation, vancomycin is notorious for a relatively wide PK variability, with a CL ISV ranging between 16 and 45%, volume of distribution at steady state (Vss) ISV between 13 and 48%, and a residual variability between 7 and 40%.27, 28 In this study, the ISV on V2 was not estimated and the residual variability was very low, increasing the possibility that more of the ISV was explained by CL. As such, the level of intersubject error seen in the current investigation is not atypical. The remaining intraindividual error was small in the final model, suggesting low variability once the variability between patients was calculated.
The current study is comparable to other analyses of vancomycin PK. (Table 3). Two‐compartment models have often been used to describe the distribution of vancomycin, although some studies utilize a one‐compartment model. Because Q is high with respect to CL, the distribution phase is relatively fast. Therefore, a one‐compartment model may appear to fit the data without much sampling in the distribution phase. Our study also reports similar results to other PK studies in adults receiving vancomycin and ECMO therapy.11, 29, 30 These studies generally report either no or limited differences in patients receiving ECMO compared to non‐ECMO patients. The studies reporting differences state a trend toward increased Vss and decreased CL. This difference appears to be more pronounced in the pediatric population, with special regard to neonates.
Table 3.
Comparison to literature sources
| Study | Population | Analysis method | Scaled? | Vss | CL |
|---|---|---|---|---|---|
| Adane et al.31 | Non‐ECMO | PopPK | Yes | 48.2 | 4.3 |
| Lim et al.32 | Non‐ECMO | PopPK | Yes | 81.4 | 4.0 |
| Donadello et al.29 | Non‐ECMO | NCA | No | 92.3 | 2.3 |
| Donadello et al.29 | ECMO | NCA | No | 99.3 | 2.4 |
| Wu et al.30 | Non‐ECMO | NCA | No | 76.6 ± 29.3 | 7.2 ± 4.3 |
| Wu et al.30 | ECMO | NCA | No | 79.4 ± 22.7 | 5.9 ± 3.5 |
| Mulla & Pooboni11 | ECMO | PopPK | Yes | 69.0 ± 26.5 | 3.8 ± 1.9 |
| Present | ECMO | PopPK | Yes | 56.5 ± 10.1 | 2.8 ± 1.1 |
This table includes parameter estimates of vancomycin PK in both ECMO and non‐ECMO patient populations. If the PK parameters were linked to weight and creatinine clearance, the final parameter was scaled to the current study population. Standard deviations were included if provided by the study. Volume at steady state (Vss) was calculated as the sum of the central volume and the peripheral volume if a two‐compartment model was selected or Vss was equal to the central volume if a one‐compartment model was selected. NCA, noncompartmental analysis; PopPK, population pharmacokinetic.
While our study did not include a control cohort of patients receiving vancomycin who were not receiving ECMO, it can be compared to literature studies performed with ECMO/non‐ECMO cohorts or non‐ECMO patients entirely. Table 3 shows parameter estimates for vancomycin scaled to the median weight and CRCL of patients in the current study when available. The Adane et al. and Lim et al. studies were performed in non‐ECMO patients with high and normal weights, respectively.31, 32 However, their estimate of volume at steady state is markedly different, although covariate differences in the respective study populations were considered in the estimate. Likewise, there appears to be a similar decrease in the volume parameter when comparing the present study to other studies performed in ECMO patients. This emphasizes the necessity of studying a higher‐weight population and guided the comparison of the present study to a non‐ECMO study. Our results suggest a trend towards decreased CL and increased Vss relative to the analysis by Adane et al.,31 which had a similar population of higher‐weight patients. However, the magnitude of the difference in PK parameters is relatively small, with high overlap of CIs, thus suggesting that the observed difference may not be clinically significant.
Despite these limitations, this model is a useful tool to characterize vancomycin PK models in ECMO. While other models have been created for vancomycin in ECMO patients, there are potential differences due to the tubing and membranes involved in the ECMO apparatus. The enrollment of higher‐weight patients, the use of a two‐compartment model, and the covariates found also differentiate this analysis from other studies. The simulation that was performed suggested a dose of 1,000 mg every 12 hours or 2,000 mg every day for the median patient in the study to reach the specified therapeutic trough of 15–20 mg/L. The simulated dose appears to concord well with standard vancomycin dosing regimens, which suggest a dose of 10–15 mg/kg twice daily or 20–30 mg/kg per day.33
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
National Institutes of Health Postdoctoral training grant no. T32GM008562 for postdoctoral training in Clinical Pharmacology supported Dr. Moore and Dr. Healy. Dr. Ahamadi is an employee of Merck. We greatly appreciate the help from all the nursing professionals in the Surgical Cardiovascular Intensive Critical Care Unit at Thomas Jefferson University Hospital. We also thank Dr. Lance Wollenberg (Array BioPharma, Boulder, CO), Dr. Marc Gastonguay (Metrum Institute, Tariffville, CT), and Dr. Ryan Vargo (Merck & Co, Upper Gwynedd, PA) for technical advice.
Conflict of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the article apart from those disclosed.
Author Contributions
J.N.M., J.R.H., M.A., and W.K.K. wrote the article; B.N.T., M.M.P., and N.C.C. designed the study; B.N.T., L.S., and M.M.P. performed the research; J.N.M. and M.A. analyzed the data.
References
- 1. Bartlett, R.H. & Gattinoni, L. Current status of extracorporeal life support (ECMO) for cardiopulmonary failure. Minerva Anestesiol. 76, 534–540 (2010). [PubMed] [Google Scholar]
- 2. MacLaren, G. , Combes, A. & Bartlett, R.H. Contemporary extracorporeal membrane oxygenation for adult respiratory failure: life support in the new era. Intensive Care Med. 38, 210–220 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Sidebotham, D. , McGeorge, A. , McGuinness, S. , Edwards, M. , Willcox, T. & Beca, J. Extracorporeal membrane oxygenation for treating severe cardiac and respiratory disease in adults: Part 1. Overview of extracorporeal membrane oxygenation. J. Cardiothorac. Vasc. Anesth. 23, 886–892 (2009) [DOI] [PubMed] [Google Scholar]
- 4. Sidebotham, D. , McGeorge, A. , McGuinness, S. , Edwards, M. , Willcox, T. & Beca, J. Extracorporeal membrane oxygenation for treating severe cardiac and respiratory failure in adults: Part 2. Technical considerations. J. Cardiothorac. Vasc. Anesth. 24, 164–172 (2010). [DOI] [PubMed] [Google Scholar]
- 5. Allen, S. , Holena, D. , McCunn, M. , Kohl, B. & Sarani, B. A review of the fundamental principles and evidence base in the use of extracorporeal membrane oxygenation (ECMO) in critically ill adult patients. J. Intensive Care Med. 26, 13–26 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Aubron, C. et al Infections acquired by adults who receive extracorporeal membrane oxygenation: risk factors and outcome. Infect. Control Hosp. Epidemiol. 34, 24–30 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Liu, C. et al Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin‐resistant Staphylococcus aureus infections in adults and children: executive summary. Clin. Infect. Dis. 52, 285–292 (2011). [DOI] [PubMed] [Google Scholar]
- 8. Shekar, K. et al Can physicochemical properties of antimicrobials be used to predict their pharmacokinetics during extracorporeal membrane oxygenation? Illustrative data from ovine models. Crit. Care. 19, 437 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ulldemolins, M. , Roberts, J.A. , Lipman, J. & Rello, J. Antibiotic dosing in multiple organ dysfunction syndrome. Chest 139, 1210–1220 (2011). [DOI] [PubMed] [Google Scholar]
- 10. Amaker, R.D. , DiPiro, J.T. , Bhatia, J. Pharmacokinetics of vancomycin in critically ill infants underdoing extracorporeal membrane oxygenation. Antimicrob. Agents Chemother. 40, 1139–1142 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mulla, H. & Pooboni, S. Population pharmacokinetics of vancomycin in patients receiving extracorporeal membrane oxygenation. Br. J. Clin. Pharmacol. 60, 265–275 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buck, M.L. Pharmacokinetic changes during extracorporeal membrane oxygenation: implications for drug therapy of neonates. Clin. Pharmacokinet. 42, 403–417 (2003). [DOI] [PubMed] [Google Scholar]
- 13. Shekar, K. et al Sequestration of drugs in the circuit may lead to therapeutic failure during extracorporeal membrane oxygenation. Crit. Care 16, R194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsu, P. , Ernst, R. & Levy, M. EMIT 2000 tobramycin and vancomycin assays [abstract]. Clin. Chem. 46 (suppl. 6) (2000). [Google Scholar]
- 15. Gastonguay, M.R. A full model estimation approach for covariate effects: Inference based on clinical importance and estimation precision. AAPS J. 6, Abstract W4354 (2004). [Google Scholar]
- 16. Ribbing, J. & Jonsson, E.N. Power, selection bias and predictive performance of the Population Pharmacokinetic Covariate Model. J. Pharmacokinet . Pharmacodyn. 31, 109–134 (2004). [DOI] [PubMed] [Google Scholar]
- 17. Baverel, P.G. , Savic, R.M. & Karlsson, M.O. Two bootstrapping routines for obtaining imprecision estimates for nonparametric parameter distributions in nonlinear mixed effects models. J. Pharmacokinet. Pharmacodyn. 38, 63–82 (2011). [DOI] [PubMed] [Google Scholar]
- 18. Yano, Y. , Beal, S.L. & Sheiner, L.B. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J. Pharmacokinet. Pharmacodyn. 28, 171–192 (2001). [DOI] [PubMed] [Google Scholar]
- 19. Gastonguay M.R. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies. PAGE Abstracts of the Annual Meeting of the Population Approach Group in Europe, Abstract 2229 (2011).
- 20. Savic, R.M. & Karlsson, M.O. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 11, 558–569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rybak, M. et al Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health‐System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health Syst. Pharm. 66, 82–98 (2009). [DOI] [PubMed] [Google Scholar]
- 22. Ye, Z.K. , Li, C. & Zhai, S.D. Guidelines for therapeutic drug monitoring of vancomycin: a systematic review. PLoS One 9, e99044 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moise‐Broder, P.A. , Forrest, A. , Birmingham, M.C. & Schentag, J.J. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin. Pharmacokinet. 43, 925–942 (2004). [DOI] [PubMed] [Google Scholar]
- 24. Mohr, J.F. & Murray, B.E. Point: Vancomycin is not obsolete for the treatment of infection caused by methicillin‐resistant Staphylococcus aureus. Clin. Infect. Dis. 44, 1536–1542 (2007). [DOI] [PubMed] [Google Scholar]
- 25. Bosso, J.A. et al Relationship between vancomycin trough concentrations and nephrotoxicity: a prospective multicenter trial. Antimicrob. Agents Chemother. 55, 5475–5479 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elyasi, S. , Khalili, H. , Dashti‐Khavidaki, S. & Mohammadpour, A. Vancomycin‐induced nephrotoxicity: mechanism, incidence, risk factors and special populations. A literature review. Eur. J. Clin. Pharmacol. 68, 1243–1255 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Marsot, A. , Boulamery, A. , Bruguerolle, B. & Simon, N. Vancomycin: a review of population pharmacokinetic analyses. Clin. Pharmacokinet. 51, 1–13 (2012). [DOI] [PubMed] [Google Scholar]
- 28. Murphy, J.E. , Gillespie, D.E. & Bateman, C.V. Predictability of vancomycin trough concentrations using seven approaches for estimating pharmacokinetic parameters. Am . J. Health Syst. Pharm. 63, 2365–2370 (2006). [DOI] [PubMed] [Google Scholar]
- 29. Donadello, K. et al Vancomycin population pharmacokinetics during extracorporeal membrane oxygenation therapy: a matched cohort study. Crit. Care 18, 632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu, C.C. , Shen, L.J. , Hsu, L.F. , Ko, W.J. & Wu, F.L. Pharmacokinetics of vancomycin in adults receiving extracorporeal membrane oxygenation. J. Formos. Med. Assoc. 115, 560–570 (2015). [DOI] [PubMed] [Google Scholar]
- 31. Adane, E.D. , Herald, M. & Koura, F. Pharmacokinetics of vancomycin in extremely obese patients with suspected or confirmed Staphylococcus aureus infections. Pharmacotherapy. 35, 127–139 (2015). [DOI] [PubMed] [Google Scholar]
- 32. Lim, H.S. , Chong, Y.P. , Noh, Y.H. , Jung, J.A. & Kim, Y.S. Exploration of optimal dosing regimens of vancomycin in patients infected with methicillin‐resistant Staphylococcus aureus by modeling and simulation. J. Clin. Pharm. Ther. 39, 196–203 (2014). [DOI] [PubMed] [Google Scholar]
- 33. Morrill, H.J. , Caffrey, A.R. , Noh, E. & LaPlante, K.L. Vancomycin dosing considerations in a real‐world cohort of obese and extremely obese patients. Pharmacotherapy 35, 869–875 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information