

Summary
This report illustrates the value of whole genome sequencing (WGS) in elucidating the genetic cause of disease in patients with primary immunodeficiency (PID). As sequencing costs decline, we predict that utilization of next generation sequencing (NGS) in the clinical setting will increase.
Keywords: Next generation sequencing (NGS), Whole genome sequencing (WGS), Primary immunodeficiency (PID), Severe combined immunodeficiency (SCID), Combined immunodeficiency (CID), Chronic granulomatous disease (CGD), Hyper IgM syndrome (HIGM)
To the Editor
Next-generation sequencing (NGS), including whole-exome and whole-genome sequencing (WES and WGS, respectively), has been successful at identifying causes of Mendelian diseases, even when the condition is seen in a single patient.1,2,3 Here, we report our findings from WGS in six patients with primary immunodeficiency (PID) from five families in whom the molecular defect was unknown.
Patients 1 and 2 were full sisters with a history of recurrent infections, including: tuberculous lymphadenitis, granulomas, and pneumonias. They had a similarly affected brother. Both patients had an absent rhodamine based respiratory burst, confirming the diagnosis of chronic granulomatous disease (CGD). The parents are distant relatives. Genetic testing was performed at a CLIA-certified commercial laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen only examined mutations in exon 2, which harbors the 2GT deletion that causes most reported cases of NCF1-related CGD.4 WGS revealed a homozygous 579G>A substitution causing a premature stop codon (Trp193X) in NCF1 that had previously been reported as causal for CGD.5
Patient 3 was a boy who developed Pneumocystis jiroveci pneumonia during the first year of life. There was no family history of PID. Immune evaluation demonstrated absent serum IgG and IgA. He had normal numbers of B, T and natural killer (NK) cells by flow cytometry and had normal T cell proliferative responses to mitogens and antigens. However, the patient lacked any detectable expression of CD40 ligand (CD154) on T cells after stimulation with ionomycin and phorbol myristate acetate, consistent with a diagnosis of X-linked hyper IgM syndrome (HIGM1). CD40 ligand gene (CD40LG) sequencing was performed at a CLIA-certified laboratory and no mutations were found, somewhat conflicting with the CD40L expression results. WGS revealed a novel G>T nucleotide substitution in the CD40LG on the X-chromosome resulting in a premature stop codon at Glu-230 (Glu230X). Nonsense mutations in neighboring codons (G227X and Q232X) have been previously reported as causes of HIGM1.6,7 Based on the mutation’s absence in control subjects and in Exome Variant Server (EVS, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA [URL: http://evs.gs.washington.edu/EVS/]), its predicted functional consequence, and the described disease causing mutations in neighboring codons, this mutation was judged the likely cause of this patient’s immune defect.
Patient 4 was a girl who presented in infancy with a history of failure to thrive (FTT) and recurrent infections. Flow cytometry showed that she had no T or B cells, but had an elevated percentage of NK cells. T cell proliferation studies showed no responses to mitogens. A diagnosis of SCID was made. RAG1 and RAG2 genes were sequenced and no mutations were identified. WGS revealed a homozygous 82kb deletion spanning exons 1–4 in DCLRE1C (see Figure E1A in the Online Repository).
Patient 5 was a girl who presented in infancy with FTT and recurrent infections. Immune studies showed absent T cells and B cells and no T cell proliferative responses to mitogens. All of her lymphocytes were NK cells. These findings led to a diagnosis of SCID. Gene sequencing was performed at a CLIA-certified commercial laboratory for RAG1, RAG2 and DCLRE1C, which are known to be associated with NK phenotype SCID8,9 and the results were negative. WGS revealed a novel heterozygous single nucleotide variant (SNV) observed at an essential splice site in DCLRE1C. This splice site mutation (c.362+2T>A) is absent in controls and in EVS, and is predicted to destroy the canonical splice donor site in intron 5. A splice site variant at the neighboring nucleotide position (c.362+1G>T) is associated with SCID.9 Further investigation revealed an 82kb deletion involving exons 1–4 in DCLRE1C (see Figure E1B in the Online Repository). The combined presence of this partial deletion and the c.362+2T>A (IVS5+2T>A) splice site mutation is the likely cause of SCID in this patient.
Patient 6 was a girl whose brother had combined immunodeficiency with a predominance of NK cells. She was investigated immunologically at an early age and had normal T lymphocyte proliferative responses to mitogens, antigens and allogeneic cells. She did well clinically until around 7 years of age, when she became profoundly lymphopenic and developed very low T-cell function. WGS showed a homozygous essential splice site mutation (c.109+1 G>T) (IVS1+1 G>T) in DCLRE1C that was absent in controls. This SNV is extremely rare (MAF=0.00023 in EVS). This splice site mutation is predicted to destroy the canonical splice donor site in intron 1. However, given the rarity of this variant and the lack of consanguinity in the family, homozygosity was considered unlikely. Deletion screening showed that the patient also had the same 82kb hemizygous deletion involving exons 1–4 of DCLRE1C observed in patient 5 (see Figure E1C in the Online Repository), which had resulted in miscalling of the SNV as homozygous.
The difference in disease severity between patients 5 and 6 is interesting, as both patients are compound heterozygotes for similar deletion but have different essential splice site mutations. Patient 5 had no T cell function, while patient 6 had normal T-cell function until around 7 years of age. To explain the difference in phenotypic severity, we constructed minigenes for each of the two newly identified DCLRE1C variants (IVS1+1 G>T and IVS5+2 T>A) (see Figure E2 in online repository). A decrease in the WT DCLRE1C isoform was observed in the IVS1+1 G>T variant construct (Figure 1A). Follow-up quantitative RT-PCR of the wild type and mutant minigene transcript revealed that the variant produced only 16% of the wild type transcript (Figure 1B). The IVS5+2 T>A variant construct resulted in a novel isoform in which exon 5 was skipped (Figure 1C and 1D). Both novel isoforms are predicted to result in abnormal gene and/or protein expression and/or function. We examined the mRNA and protein expression of the full-length WT-DCLRE1C cDNA and the IVS5+2 T>A isoform, in which exon 5 is skipped, using FLAG-tagged constructs (Figure 2A). While both isoforms showed similar expression at the mRNA level, only the wild type isoform showed a detectable level of protein (Figure 2B and 2C).
Figure 1.
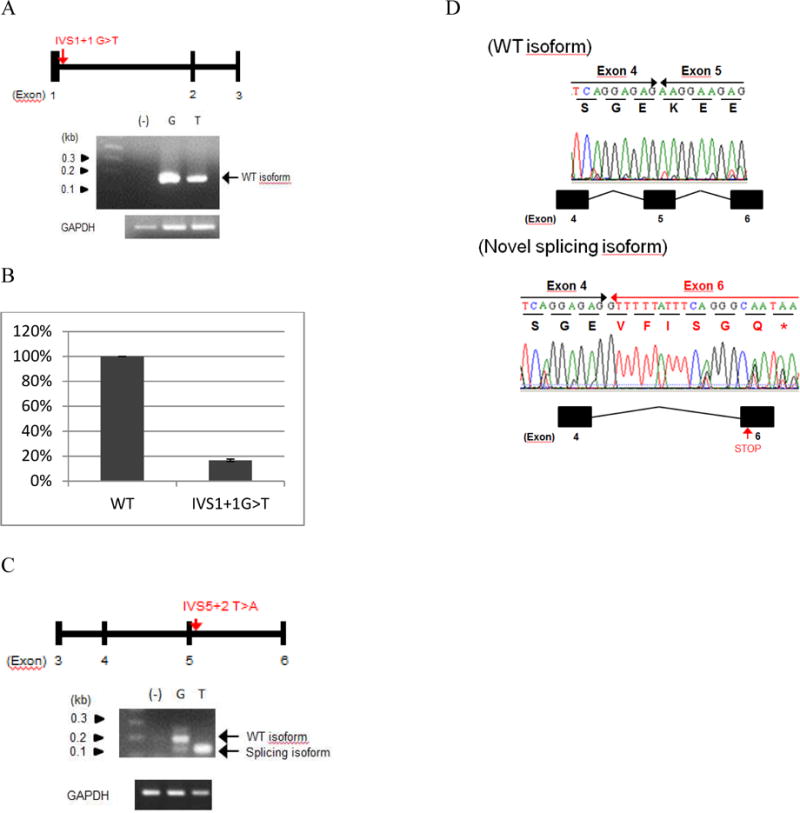
Semi-quantitative PCR and quantitative real-time PCR results of Minigene expression. A) Minigene 1 (IVS1+1G>T) mRNA expression, B) quantitative Taqman results showing only 16% expression of the wild type mRNA in Minigene-1, C) Minigene-5 (IVS5+2T>A) mRNA expression, and D) Sanger sequencing confirmation of IVS5+2T>A resulting in exon 5 skipping.
Figure 2.
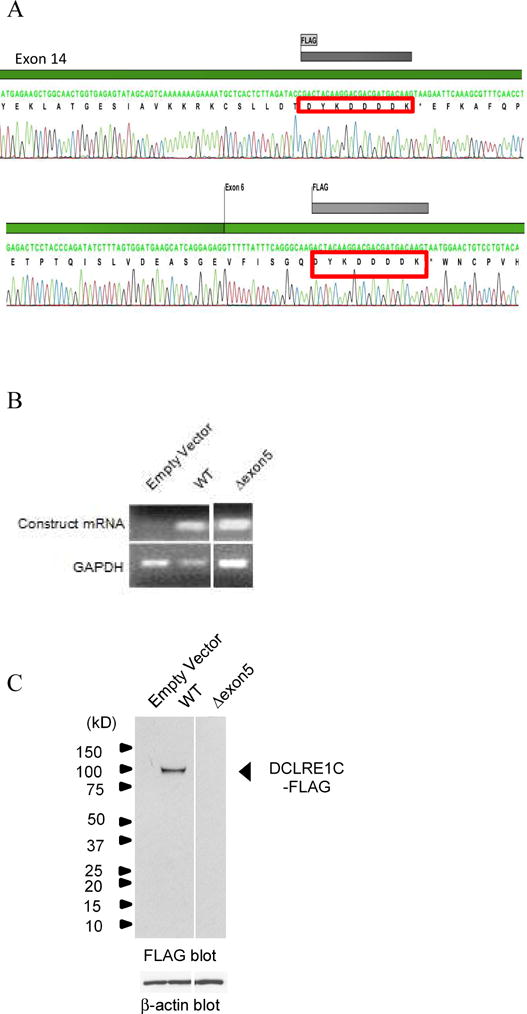
DCLRE1C protein assays. A) FLAG-tag insertion in exon 14 of the wild-type DCLRE1C construct and exon 6 of the Δexon5 construct, B) semi-quantitative PCR showing presence of Δexon5 mRNA expression, C) immunoblotting showing absence of DCLRE1C protein expression in mutant Δexon5 isoform.
Hence, the IVS1+1 G>T variant found in patient 6 produced some of the canonical wild type isoform, suggesting that this mutation decreases the splicing efficiency. The IVS5+2 T>A variant from patient 5 did not produce any wild type isoform, but did produce another isoform. This result is consistent with a model in which patient 6 produced enough wild type Artemis protein to delay the onset of the patient’s combined immunodeficiency, resulting in a less severe phenotype.
The use of NGS is accepted for investigating undiagnosed genetic conditions. Here, we show the value of WGS in PID patients without identified causal mutations.
Supplementary Material
Acknowledgments
We would like to acknowledge the following individuals for the contributions of control samples: Dr. William B. Gallentine, Dr. Erin L. Heinzen, Dr. Aatif M. Husain, Ms. Kristen N Linney, Dr. Rodney A. Radtke, Dr. Saurabh R. Sinha, and Ms. Nicole M. Walley; Dr. Joseph McEvoy, Dr. Anna Need, Mr. Jordan Silver, Ms. Marlyne Silver; Dr. Elizabeth Cirulli; Dr. Julie Hoover-Fong, Dr. Nara L. Sobreira and Dr. David Valle; Dr. Robert Brown; Dr. Ruth Ottman; Dr. Deborah Levy; K. Welsh-Bomer, C. Hulette, J. Burke; Mr. David H. Murdock and the MURDOCK community registry and biorepository; Dr. Vandana Shashi and Ms. Kelly Schoch; Dr. Demetre Daskalakis.
The collection of control samples and the production of sequence data were funded in part by NIAID Grant UO1AIO67854 (Center for HIV/AIDS Vaccine Immunology (“CHAVI”)).
The authors would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
This work was supported by NIH grants (including T32 training grants) and a Senior Fellowship Award from the Clinical Immunology Society/Grifols to TM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, et al. Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011 Mar;13(3):255–62. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 2.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009 Nov 10;106(45):19096–101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Need AC, Shashi V, Hitomi Y, Schoch K, Shianna KV, McDonald MT, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012 Jun;49(6):353–61. doi: 10.1136/jmedgenet-2012-100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noack D, Rae J, Cross AR, Ellis BA, Newburger PE, Curnutte JT, et al. Autosomal recessive chronic granulomatous disease caused by defects in NCF-1, the gene encoding the phagocyte p47-phox: mutations not arising in the NCF-1 pseudogenes. Blood. 2001 Jan 1;97(1):305–11. doi: 10.1182/blood.v97.1.305. [DOI] [PubMed] [Google Scholar]
- 5.Roos D, de Boer M, Koker MY, Dekker J, Singh-Gupta V, Ahlin A, et al. Chronic granulomatous disease caused by mutations other than the common GT deletion in NCF1, the gene encoding the p47phox component of the phagocyte NADPH oxidase. Hum Mutat. 2006 Dec;27(12):1218–29. doi: 10.1002/humu.20413. [DOI] [PubMed] [Google Scholar]
- 6.Lin Q, Rohrer J, Allen RC, Larche M, Greene JM, Shigeoka AO, et al. A single strand conformation polymorphism study of CD40 ligand. Efficient mutation analysis and carrier detection for X-linked hyper IgM syndrome. J Clin Invest. 1996 Jan 1;97(1):196–201. doi: 10.1172/JCI118389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rangel-Santos A, Wakim VL, Jacob CM, Pastorino AC, Cunha JM, Collanieri AC, et al. Molecular characterization of patients with X-linked Hyper-IgM syndrome: description of two novel CD40L mutations. Scand J Immunol. 2009 Feb;69(2):169–73. doi: 10.1111/j.1365-3083.2008.02198.x. [DOI] [PubMed] [Google Scholar]
- 8.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Science. 1996 Oct 4;274(5284):97–9. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- 9.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001 Apr 20;105(2):177–86. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.