

Abstract
Microbial pathogens have developed several mechanisms to modulate and interfere with host cell cycle progression. In this study, we analyzed the effect of the human pathogen Neisseria meningitidis on the cell cycle of epithelial cells. Two pathogenic isolates, as well as two carrier isolates, were tested for their ability to adhere to and invade into the epithelial cell lines Detroit 562 and NP69 and to modulate the cell cycle. We found that all isolates adhered equally well to both Detroit 562 and NP69 cells, whereas the carrier isolates were significantly less invasive. Using propidium iodide staining and 5-ethynyl-2′-deoxyuridine pulse-labeling, we provide evidence that meningococcal infection arrested cells in the G1 phase of the cell cycle at 24 h postinfection. In parallel, a significant decrease of cells in the S phase was observed. Interestingly, G1-phase arrest was only induced after infection with live bacteria but not with heat-killed bacteria. By Western blotting we demonstrate that bacterial infection resulted in a decreased protein level of the cell cycle regulator cyclin D1, whereas cyclin E expression levels were increased. Furthermore, N. meningitidis infection induced an accumulation of the cyclin-dependent kinase inhibitor (CKI) p21WAF1/CIP1 that was accompanied by a redistribution of this CKI to the cell nucleus, as shown by immunofluorescence analysis. Moreover, the p27CIP1 CKI was redistributed and showed punctate foci in infected cells. In summary, we present data that N. meningitidis can interfere with the processes of host cell cycle regulation.
INTRODUCTION
Recent studies have shown that many bacteria produce and secrete compounds, e.g., toxins and effectors, that interfere with the host cell cycle. These factors are summarized as “cyclomodulins” and have been proposed to be a new class of virulence-associated factors (1, 2). The cell cycle is a series of events that describe the growth, DNA replication, distribution of the duplicated chromosomes to daughter cells and division of a cell. It is divided into four phases: M phase (mitosis), G1 (the period between mitosis and the initiation of nuclear DNA replication), S (the period of nuclear DNA replication), and G2 (the period between the completion of nuclear DNA replication and mitosis). Cells in G1 phase can enter a resting state called G0, which represents nongrowing and nonproliferating cells.
The progression from one cell cycle phase to another occurs in an orderly fashion and is regulated by different cellular proteins: key regulatory proteins are the cyclin-dependent kinases (CDKs), a family of serine/threonine protein kinases, that are activated at specific points of the cell cycle (3). CDKs form complexes with different cyclins that are required at different phases of the cell cycle. Three D type cyclins—cyclin D1, cyclin D2, and cyclin D3—bind to CDK4 and to CDK6. CDK-cyclin D complexes are essential for entry in G1 (4). Another G1 cyclin is cyclin E, which associates with CDK2 to regulate progression from G1 into S phase (5). Downstream targets of CDK-cyclin complexes include the retinoblastoma protein (pRB) and E2F transcription factors. CDK activity can be counteracted by cell cycle inhibitory proteins, called CDK inhibitors (CKI), which bind to CDK alone or to the CDK-cyclin complex and regulate CDK activity. CKIs are classified into two groups, the INK4 and Cip/Kip families. INK4 family members bind only to CDK4/6 and inhibit their activities, whereas Cip/Kip family members (including p21WAF1/CIP1, p27CIP1, and p57CIP2) can inhibit the activities of G1 CDK-cyclin complexes and, to a lesser extent, the CDK1-cyclin B complex (6, 7).
During coevolution with their hosts, bacteria have established multiple mechanisms that allow them to interfere with cell proliferation. During the last decade, a growing family of bacterial effectors and toxins has been described that interferes with the host cell cycle (1, 2, 8, 9). The cytolethal distending toxin of Escherichia coli was the first bacterial toxin described to act as a “cyclomodulin” and has been shown to cause growth arrest at the G2/M phase (10). Further candidates are the cycle inhibiting factors (Cifs) produced by enteropathogenic and enterohemorrhagic E. coli (EPEC and EHEC), that trigger an irreversible cell cycle arrest at G2 with complete inhibition of mitosis by inhibition of the CDK1-cyclin B complex, whose activation is necessary for the cell cycle G2/M transition (11). Other than G2 arrest, Cif also induces G1 cell cycle arrest in a process that involves the stabilization of the CKIs p21WAF1/CIP1 and p27CIP1 (12). Whereas these bacterial “cyclomodulins” induce cell cycle arrest, other bacterial toxins can also induce DNA replication and cell proliferation (1). These include the Pasteurella multocida toxin PMT (13), which upregulates cyclins D and E and p21WAF1/CIP1; the cytotoxic necrotizing factors from E. coli (14); the dermonecrotic toxin from Bartonella spp. (14); and CagA from Helicobacter pylori (15). Finally, alteration of cell cycle progression has also been observed during pathogen-plant interaction (16).
Neisseria meningitidis, the meningococcus, is a Gram-negative organism and an obligate human pathogen. Meningococci are often found in the nasopharynx as asymptomatic colonizers and carriage rates vary between 10 and 35% for healthy adults (17–19). In populations with individuals in close contact, such as university students or military recruits, carriage rates of ca. 70% have been found (20). For colonization of the human nasopharynx, the microorganism must adhere to the mucosal surface, utilize locally available nutrients, and evade the human immune system. N. meningitidis expresses a range of structures and molecules that facilitate adhesion and invasion, including the type IV pili, the outer membrane proteins Opa and Opc, and a number of newly identified minor adhesion or adhesion-like proteins (21–28). Compared to the carriage rate, meningococcal disease is a rare event, and disease rates vary in different geographic regions of the world. The mechanisms that drive the colonization state of the organism into a disease state are still not entirely clear. It seems that a combination of bacterial virulence factors and host susceptibility, including age, smoking, prior viral infection, and genetic polymorphisms (29, 30), may facilitate meningococcal disease.
We have recently shown that N. meningitidis can interfere with the cell cycle of brain endothelial cells (31), inducing an accumulation of infected cells in the S phase, which is triggered by the major adhesins/invasins of N. meningitidis, the Opa proteins and the Opc protein (31). Mechanistically, cell cycle arrest could be correlated to an increased expression of the CKI p21WAF1/CIP1 (31). In the present study, we analyzed the effect of N. meningitidis on the cell cycle of epithelial cells. We show that infection with N. meningitidis caused a G1 cell cycle arrest, which correlated with an accumulation of CKI p21WAF1/CIP1 protein levels and a redistribution of this CKI into the nucleus. Furthermore, meningococcal infection resulted in a decreased cyclin D1 and increased cyclin E protein amount. In addition to our previous study, we extended our analyses and included two pathogenic and two genetically related carrier isolates to decipher whether disease and carrier isolates differ in their ability to interfere with the host cell cycle of epithelial cells.
MATERIALS AND METHODS
Bacterial strains, mutants, and culture conditions.
Neisseria meningitidis strain MC58, a serogroup B isolate (United Kingdom, 1983) of the sequence type 74 (ST-74) (ST-32 clonal complex [cc]) was kindly provided by E. R. Moxon. The nonencapsulated mutant MC58 siaD has previously been described (32). N. meningitidis serogroup C strain 8013/clone12 (ST-18cc, ST-177) was kindly provided by M. Taha (33). N. meningitidis strain α4 (ST-18cc, ST-19) and strain α711 (ST-32cc, ST-32) were isolated during the Bavarian meningococcal carriage study (19) (the strains and mutants are summarized in Table 1).
TABLE 1.
N. meningitidis strains used in this studya
| Strain | Serogroup | ST | cc | Epidemiology | Source | Country | Yr | Reference | Genome accession no. |
|---|---|---|---|---|---|---|---|---|---|
| MC58 | B | 74 | 32 | Invasive | IMD | United Kingdom | 1983 | 73 | AE002098 |
| 8013/clone12 | C | 177 | 18 | Invasive | IMD | France | 1989 | 74 | FM999788 |
| α4 | B | 19 | 18 | Carriage | Carrier | Germany | 1999 | 19 | |
| α711 | B | 32 | 32 | Carriage | Carrier | Germany | 2000 | 19 |
ST, sequence type; cc, clonal complex; IMD, invasive meningococcal disease.
All meningococcal isolates were routinely grown on commercially acquired Columbia blood agar plates (bioMérieux) at 37°C in a humidified 5% CO2 incubator and were cultured in proteose-peptone medium (PPM) supplemented with 1× Kellogg's supplements I and II, 0.01 M MgCl2, and 0.005 M NaHCO3 (PPM+). Meningococcal supernatants were prepared as follows: Dulbecco modified Eagle medium (DMEM) with 1× Kellogg's supplements I and II was inoculated with meningococcal strain MC58, 8013/12, α4, or α711, followed by incubation for 2 h at 37°C at 200 rpm. The optical density at 600 nm (OD600) was measured, and a second culture was inoculated at an OD600 of 0.05, followed by incubation overnight at 37°C with 200-rpm shaking. The following day, the culture was centrifuged at 4,000 × g for 20 min at 4°C, and the supernatant was sterilized through a 0.2-μm-pore size filter. The supernatants were transferred to a 20-ml VivaSpin column (molecular weight cutoff, 10,000; Sartorius) and centrifuged at 4,000 × g for 20 min at 20°C. Concentrated proteins were removed from the concentrator pocket and analyzed by SDS-PAGE as described below. The supernatants were stored at −20°C until use.
Epithelial cell lines.
The cell line Detroit 562 (human pharyngeal cells; ATCC CCL-138) was maintained in DMEM with GlutaMAX (Life Technologies) supplemented with 10% fetal calf serum (FCS; Life Technologies), 4 mM l-glutamine (Life Technologies), 1 mM sodium pyruvate (Life Technologies), and 1× nonessential amino acids (GE Healthcare Europe GmbH, Freiburg, Germany) at 37°C and 5% CO2. Nasopharyngeal epithelial cell line NP69 (NP69 cells harboring SV40T) was kindly provided by George Sai Wah Tsao (University of Hong Kong) (34). Keratin profiles and studies of the response to TGFβ1 treatment have shown that NP69 cells retain the characteristics of primary nasopharyngeal cells. Cells were maintained in serum-free keratinocyte medium (KSFM), supplemented with human recombinant epidermal growth factor (5 ng/ml) and bovine pituitary extract (50 μg/ml) (Life Technologies). The cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
Reagents and antibodies.
The following antibodies and reagents were used in this study: anti-β-actin (catalog no. 4967) and anti-p21WAF1/CIP1 (clone 12D1; catalog no. 2947) (both from Cell Signaling); anti-cyclin E (clone HE12; sc-247; Santa Cruz Biotechnologies) and anti-cyclin D1 (clone EP272Y; 04-221; Merck) for Western blotting; and anti-p21WAF1/CIP1 (clone 12D1; 2947; Cell Signaling) and anti-p27CIP1 (clone G173-534 [RUO]; 554069; BD Biosciences) for immunofluorescence analyses. Cy3-conjugated rabbit anti-mouse (315-166-047) was from Dianova, Alexa 488-conjugated goat anti-rabbit (A-11008) and DAPI (4′,6′-diamidino-2-phenylindole; D1306) were purchased from Life Technologies, and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG/IgM (115-035-044) and HRP-goat anti-rabbit IgG (111-035-006) were from Dianova.
Gentamicin protection assay.
Gentamicin protection assays were conducted as described previously (32). Briefly, 5 × 104 Detroit 562 cells and 1 × 105 NP69 cells per well were seeded in 24-well plates. Cells were infected with bacteria at a multiplicity of infection (MOI) of 30 for the indicated time points. Infections were carried out in DMEM–10% FCS for Detroit 562 cells and supplemented KSFM for NP69 cells. The medium was then replaced with DMEM or supplemented KSFM containing 200 μg/ml gentamicin. After 2 h of incubation in gentamicin-containing medium, the cells were lysed by the addition of 1% saponin in DMEM or supplemented KSFM for 15 min. Suitable dilutions were plated in duplicate on blood agar plates to determine the numbers of recovered viable bacteria.
Real-time PCR.
Total RNA was extracted using the RNeasy minikit (Qiagen), including an on-column DNase digestion step using an RNase-free DNase set (Qiagen). The quality of extracted RNA was assessed by the Agilent 2100 Bioanalyzer (Agilent Technologies) using an RNA Nano-Chip for eukaryotic RNA according to the manufacturer's protocol. Only RNA samples with an RNA integrity number that was ≥9 were used for further analysis (35). First-strand cDNA was synthesized from 1.0 μg of total RNA using SuperScript II reverse transcriptase (Thermo Scientific) and a QIAquick PCR purification kit (Qiagen). The following primers were used for RT-PCR: forward cyclin D (5′-CGG TAG TAG GAC AGG AGG TT-3′) and reverse cyclin D (5′-CTG TGC CAC AGA TGT GAA GT-3′); forward cyclin E (5′-AGC GGT AAG AAG CAG AGC AG-3′) and reverse cyclin E (5′-CGC TGC AAC AGA CAG AAG AG-3′); and forward human GAPDH (5′-GCA CCG TCA AGG CTG AGA AC-3′) and reverse human GAPDH (5′-ATG GTG GTG AAG ACG CCA GT-3′). Transcribed cDNA and primers were mixed with RT2 SYBR green ROX qPCR Mastermix (Qiagen) and RNase-free water (Qiagen), and 25 μl was loaded into each well of a 96-well plate. Quantitative real-time PCR was performed on a StepOnePlus real-time system (Applied Biosystems). For PCR, the mixture was denatured at 95°C for 3 min, and the target genes were amplified by 35 cycles of reaction: cyclin D1 (95°C for 30 s, 61°C for 30 s, and 72°C for 1 min), cyclin E (95°C for 30 s, 61°C for 30 s, and 72°C for 1 min), or GAPDH (95°C for 30 s, 61°C for 30 s, and 72°C for 1 min). Three independent experiments were performed for infected and uninfected cells at each time point. The gene expression values were normalized against GAPDH and were calculated according to the comparative ΔCT method (36). We used a cutoff of ≥1.5-fold change and a P value of ≤0.01.
Flow cytometry assay.
Cell cycle was monitored by propidium iodide (PI) staining, followed by flow cytometry as previously described (31). Briefly, Detroit 562 or NP69 cells were seeded in six-well plates at 2 × 105 cells per well for 24 h and allowed to grow to 50 to 60% confluence, since tight confluence can induce a cell cycle arrest. Detroit 562 cells or NP69 cells were infected with bacteria in DMEM–10% FCS or supplemented KSFM, respectively, at an MOI of 100, and the bacteria were allowed to adhere for 6 h at 37°C and 5% CO2. Subsequently, the cells were washed with 1× PBS, and the medium was replaced with DMEM–10% FCS or supplemented KSFM containing 200 μg/ml gentamicin to prevent bacterial overgrowth. The same infection conditions were used for the EdU cell proliferation assay, the immunofluorescence studies, and the Western blot analyses. In some experiments, cells were exposed to heat-inactivated bacteria or meningococcal supernatants.
Treated cells were harvested by trypsinization, washed twice with 1 ml of 1× PBS using centrifugation, and then fixed by adding 70% ethanol dropwise at 4°C. Next, the cells were incubated for at least 60 min at 4°C, centrifuged, and washed once with 1× PBS, followed by lysis in 0.05% Triton X-100 (Roth), treatment with 100 μg/ml RNase A (Roche Diagnostic) for 30 min at 37°C, and staining with PI (100 μg/ml; Sigma) for 30 min at 37°C, protected from light. The cells were subsequently analyzed on a FACSCalibur flow cytometer (BD Biosciences), and BD CellQuest Pro software (BD Biosciences) was used for data acquisition. To determine the proportion of cells at each stage of the cell cycle, the cell cycle curve-fitting option of the software package WEASEL v3.1 (Walter and Eliza Hall Institute of Medical Research) was used.
EdU cell proliferation assay.
The quantification of 5-ethynyl-2′-deoxyuridine (EdU) incorporation into replicating DNA was performed using the Click-iT EdU flow cytometry assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions with the following modifications. A total of 2 × 105 cells per well were plated in six-well plates and grown for 24 h at 37°C. The cells were infected with bacteria (MOI of 100) as described above. After 22 h of infection, the cells were labeled with 20 μM EdU, followed by incubation for another 2 h. Afterward, the cells were fixed and permeabilized as described above, and click labeled with Alexa Fluor 488 azide (Thermo Fisher Scientific) for 30 min in the presence of copper(II) sulfate and a reducing agent to afford the copper-catalyzed click reaction (37). In addition, the cells were stained with 1 μl of FxCycle Far Red (Invitrogen) for 30 min at 4°C. EdU and FxCycle Far Red measurements were performed using a BD FACSCalibur flow cytometer. Alexa Fluor 488 signals were measured on FL1 with a 530/30-nm band-pass filter, and FxCycle Far Red on FL3 was measured with a 670-nm long-pass filter. Sample measurements were performed with BD CellQuest Pro software.
Immunofluorescence assay.
The procedure for immunofluorescence was performed as described previously (31). Briefly, Detroit 562 cells were grown on glass coverslips, infected as described above, washed with 1× PBS, and fixed with 3.7% formaldehyde for 20 min. After washing, the cells were permeabilized with 0.1% Triton X-100 (Roth) for 15 min and blocked with blocking buffer (BB; PBS, 1% FCS, and 2% bovine serum albumin) for at least 45 min at 4°C. Monoclonal anti-p21WAF1/CIP1 IgG antibody was used at a 1:400 dilution in BB, and monoclonal anti-p27 IgG antibody was used at a 1:50 dilution in BB. A goat anti-rabbit Alexa Fluor 488-labeled secondary antibody and a rabbit anti-mouse Cy3-labeled secondary antibody were used at a 1:200 dilution. Nuclei were stained with DAPI at a 1:10,000 dilution in PBS, and the cells were examined by fluorescence microscopy using a Zeiss Axio Imager Z1 fluorescence microscope at a ×40 or a ×63 magnification. Images were processed with ImageJ software and documented using Adobe Photoshop CS.
Western blot analysis.
The levels of cyclins and cell cycle regulating proteins were examined by using cell extracts prepared as described recently (31). All primary antibodies (p21WAF1/CIP1, cyclin D1, and cyclin E) were used at 1:1,000 in TBST (0.1% Tween). Western blots are shown for one of three independent experiments. Band analysis for each protein was quantified using ImageJ software. The area under the curve (AUC) of the specific signal was corrected for the AUC of the β-actin loading control before comparison for changes (recorded as the n-fold change) in a two-step normalization method. First, the relative densities were calculated by dividing the intensity of the protein band by the intensity of the corresponding β-actin band. Second, all of the relative band intensities were adjusted to the band intensity obtained from uninfected control cells. SDS-PAGE and Western blotting for pilin, Opa, and Opc expression was conducted as previously described (32) using the SM1 monoclonal antibody specifically recognizing class I pilin types (38), the 4B12/C11 pan-Opa antibody, and the Opc-specific monoclonal antibody B306 (39).
Statistical analysis.
Statistical differences between groups were calculated using the Student unpaired t test (two tailed). P values of ≤0.05 were considered statistically significant.
RESULTS
Adherence to and invasion of the nasopharyngeal epithelial cell lines Detroit 562 and NP69 by invasive and carriage N. meningitidis isolates.
We have previously shown that N. meningitidis is able to interfere with the host cell cycle of human brain microvascular endothelial cells after infection (31). To investigate whether N. meningitidis infection might also result in an alteration of the cell cycle of nasopharyngeal cells, two cell lines were implemented, the pharyngeal carcinoma epithelial cell line Detroit 562 and NP69 cells. NP69 cells were established from primary nonmalignant nasopharyngeal epithelial cells and were transduced with the SV40T antigen (34). Keratin profiles and response to TGFβ1 treatment have shown that NP69 cells retained characteristics of primary nasopharyngeal cells. First, adhesion and invasion properties of two disease and two carriage meningococcal isolates were quantified by using gentamicin protection assays as described previously (32, 40). Detroit 562 and NP69 cells were infected with the serogroup B disease strain MC58, a corresponding isogenic unencapsulated mutant strain MC58 siaD, the serogroup C disease isolate 8013/clone12, and the two serogroup B carrier isolates α711 and α4 (Table 1). N. meningitidis MC58 was chosen as a prototype strain belonging to the ST-32 clonal complex (cc), whereas strain α711 was chosen as a carrier isolate related to MC58 from the ST-32 cc (6). Furthermore, N. meningitidis isolate 8013/clone 12 (also known as clone 12 or 2C43) was included as a prototype ST-18 cc disease strain, while isolate α4 was chosen as a carrier isolate related to 8013/clone12. The invasion and adherence kinetics were estimated at 2, 4, and 6 h postinfection (p.i.), and the cells were infected at an MOI of 30 (Fig. 1).
FIG 1.

Carrier isolates and strain 8013/clone12 are less invasive than meningococcal disease isolates in epithelial host cells. Adhesion to and invasion of N. meningitidis into Detroit 562 and NP69 cells were assessed over a 6 h infection period. (A and B) Adherence to (A) and invasion of (B) both cell lines of serogroup B strain MC58 (■) and unencapsulated strain MC58 siaD (□) were determined by gentamicin protection assays at the time intervals indicated. Both cell lines were infected at an MOI of 30. (C and D) Adherence to (C) and invasion of (D) serogroup C disease isolate N. meningitidis 8013/clone12 and two serogroup B carrier isolates α711 and α4 (MOI 30) were determined by gentamicin protection assays at 6 h p.i. and are represented as relative data compared to MC58. The data show mean values ± the standard deviation (SD) of three independent experiments conducted in duplicate. *, P < 0.05.
Meningococci adhered very effectively to both Detroit 562 and NP69 cells, reaching a maximum of about 1 × 107 adherent bacteria per cell monolayer for strain MC58 at 6 h p.i. (Fig. 1A). In addition to adherence, the meningococcal wild-type strain MC58 elicited a time-dependent increase of bacterial uptake ranging from 3.2 × 102 at 2 h p.i. up to 1.54 × 103 at 6 h p.i. for Detroit 562 cells and from 1.53 × 103 at 2 h p.i. up to 2.5 × 104 at 6 h p.i. for NP69 cells. The unencapsulated strain MC58 siaD showed a significantly higher rate of invasion into Detroit 562 cells (about 7.9 × 103 at 2 h p.i. and 1.9 × 104 at 6 h p.i.) than its capsulated parent strain according to previously published data (Fig. 1) (32). Interestingly, the two carriage isolates and isolate 8013/clone12 were significantly less invasive than N. meningitidis MC58 into both Detroit 562 and NP69 cells. Invasion rates of about 25.6% invasion for 8013/clone12, 42.5% for α711%, and 9.8% for α4 were observed compared to MC58 at 6 h p.i. for Detroit 562 cells and 26.7% invasion for 8013/clone12, 28.0% for α711, and 55.5% for α4 using NP69 cells (Fig. 1D). The meningococcal isolates used in the present study were characterized for the expression of some of the major adhesins/invasins and phenotypes by Western blotting (see Fig. S1 in the supplemental material). This analysis showed that all four strains expressed class I pilin. (there was no change in the migration or reactivity of pilin from MC58, 8013/clone12, α711, and α4, respectively). However, 8013/clone12 did not express any Opa proteins, as analyzed by Western blotting and 8013/clone12, α711, and α4 did not react with the Opc-specific monoclonal antibody B306 either due to a deletion of the opc gene (8013/clone12 and α4 [data not shown]) or as a result of transcriptionally regulated phase variation (α711) (see Fig. S1B in the supplemental material), which is mediated by a variable polycytidine stretch in the promoter region of the gene (41).
N. meningitidis infection causes an accumulation of epithelial cells in the G1 phase of the cell cycle.
To investigate the effects of pathogenic and carrier N. meningitidis isolates on the host cell cycle of human epithelial cells, asynchronous cultures of Detroit 562 and NP69 cells were either left uninfected or were infected with the disease isolates MC58 and 8013/clone12 or the two carrier isolates α711 and α4 using an MOI of 100 for 24 h (see Materials and Methods), and the DNA content of infected cells was determined by propidium iodide (PI) staining and subsequent flow cytometry analyses. The cells were infected over a maximum time period equal to the doubling time of the cell, which was estimated as 24 h for Detroit 562 cells and 30 h for NP69 cells. The cells were harvested at 2, 4, 8, 12, and 24 h p.i. or at 3, 6, 12, 24, and 30 h p.i., respectively, for PI staining.
Unfortunately, NP69 cells were not suited for PI staining when infected with N. meningitidis at time points later than 12 h p.i. in our hands. A significant fraction of colonies of clumped up cells was observed and resulted in an increase of super G2 fraction, which interfered with our cell cycle curve-fitting analysis. We therefore focused in the following assays on Detroit 562 cells. As a consequence of meningococcal infection, Detroit 562 cells displayed a significant accumulation of cells in the G1 phase at 24 h p.i. in response to the pathogenic isolates MC58 and 8013/clone12 to a level of about 47.4% for MC58 and 47.0% for 8013/clone12 compared to 35.4 and 35.9% in control cells (Fig. 2). Interestingly, carrier isolates also induced an accumulation of cells in the G1 phase of about 40.7% for isolate α711 and 41.6% for isolate α4, respectively, compared to about 34.0% in control cells (Fig. 2). In parallel, a significant reduction of cells in the S phase was detected at 24 h p.i. (35.9% for MC58, 30.1% for 8013/clone12, 34.4% for α711, and 33.4% for α4, compared to about 41% of the cells in S phase for uninfected control cells) (Fig. 2). These results are indicative of an arrest at the G1 phase of epithelial cells in response to both pathogenic and carrier isolates.
FIG 2.

Both pathogenic N. meningitidis isolates and carrier strains cause an accumulation of epithelial cells in the G1 phase. Detroit cells either were left uninfected (control cells) or were infected with N. meningitidis strain MC58, 8013/clone12, α711, or α4 at an MOI of 100. Cells were collected at the time points indicated for propidium iodide (PI) staining and subsequent flow cytometry analysis of the cell cycle. In the left panel, representative histograms (24 h p.i.) for uninfected control cells and cells infected with the different meningococcal isolates are shown. The G1 and G2/M phases are indicated. In the right panel, the histograms were analyzed with the software package WEASEL v3.1, and the percentages of the cells in the G1, S, and G2/M phases are shown. The results are presented as the means and the SD from three independent experiments. The statistical significance was determined for comparison between uninfected control cells and N. meningitidis-infected cells at each time point. *, P < 0.05.
To verify that meningococcal infection arrested Detroit 562 cells in G1 phase and reduced the number of cells in S phase, we next used MC58 and carrier isolate α711 and used an improved cell proliferation assay based upon the incorporation of 5-ethynyl-2′-deoxyuridine (EdU) for the direct measurement of cells in the S phase. In this approach, EdU was added, incorporated into replicating DNA and measured after treatment with the Alexa Fluor 488 Click-iT kit to label newly synthesized DNA during arrest. FxCycle Far Red was used to quantify nuclear DNA. In comparison to uninfected control cells, we found a significantly smaller amount of incorporated EdU in Detroit 562 at 24 h p.i. when cells were infected with MC58 (42.2%) or α711 (40.9%) compared to control cells (51.1%) (Fig. 3A), indicating that N. meningitidis-infected Detroit 562 cells remained mostly in the G1 phase. Moreover, the percentages of cells in each stage of the cell cycle was determined by FxCycle Far Red staining, which revealed, that the reduction of EdU-positive cells during bacterial infection was due to the sequestration of cells in the G1 phase. The cell cycle distribution pattern obtained by this technique was in line with the data obtained by PI staining and proved that meningococcal infection caused an arrest of host cells in the G1 cell cycle phase (Fig. 3B).
FIG 3.
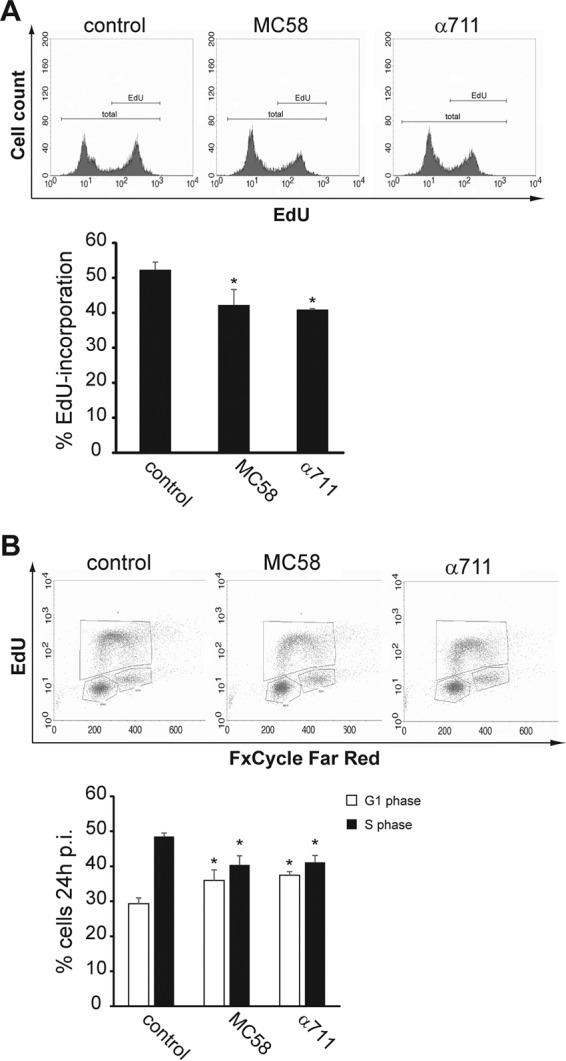
Meningococcal infection decreases the number of Detroit 562 cells in the S phase. Detroit 562 cells were either left uninfected or were infected with MC58 or α711 (MOI of 100) for a 24-h time period. Cell proliferation analysis was performed using a Click-iT EdU Alexa Fluor 488 flow cytometry assay kit and FxCycle Far Red stain. During the final 2 h of infection, Detroit 562 cells were treated with 20 μM EdU, harvested, and then analyzed by flow cytometry. EdU was stained with Alexa Fluor 488 azide according to the manufacturer's protocol, followed by staining with 1 μM FxCycle Far Red stain for DNA content analysis. The cells were then analyzed by flow cytometry using either 488-nm excitation for the EdU Alexa Fluor 488 dye or 633-nm excitation for the FxCycle Far Red stain. (A, upper panel) The results of a representative experiment are shown. The complete population (total) and the EdU-positive (EdU) populations are indicated. (Lower panel) The results of a statistical analyses of the percentages of the cell population incorporating EdU in uninfected control cells, MC58-infected cells, or α711-infected cells from three independent experiments are shown. The data are shown as means ± the standard deviations (*, P < 0.05). (B, upper panel) The results of a representative experiment are shown, and cells in the G1, S, and G2/M phases are gated in boxes. (Lower panel) The results of a statistical analyses of the percentages of the cell population in the S phase in uninfected control cells, MC58-infected cells, or α711-infected cells from three independent experiments are shown. The data are shown as means ± the standard deviations (*, P < 0.05).
Live but not heat-killed meningococci induce an accumulation of epithelial cells in the G1 phase of the cell cycle.
We next examined whether Detroit 562 cells were also arrested in G1 phase after treatment with heat-killed bacteria. Detroit 562 cells were exposed to both live and heat-killed meningococcal isolates MC58, 8013/clone12, α711, and α4 for 24 h and cellular DNA content was measured by PI staining as described above. Although live bacteria induced an accumulation of cells in the G1 phase as demonstrated earlier (Fig. 4), heat-killed (hk) bacteria of all N. meningitidis isolates had no effect on the cell cycle distribution (29.1% for hk MC58, 28.2% for hk 8013/clone12, 29.1% for hk α711, and 29.3% for hk α4, compared to 27.2% of the cells in the G1 phase for uninfected control cells) (Fig. 4). These findings suggested that the alteration of the cell cycle might be attributed to heat-labile bacterial proteins or might be due to less internalized bacteria that are able to initiate various signaling cascades.
FIG 4.

G1-phase arrest is induced by viable bacteria but not by heat-killed bacteria. Detroit 562 cells either were infected with viable N. meningitidis strain MC58, 8013/clone12, α711, or α4 (MOI 100) or were exposed to heat-killed (hk) bacteria. Uninfected cells served as a control. Cells were harvested for PI staining at 24 h p.i. and analyzed by flow cytometry. (A) The results of representative experiments are shown. (B) The results of statistical analyses of the percentages of the cell population in the G1, S, and G2/M phases are shown. The data are shown as means ± the SD obtained from three independent experiments (*, P < 0.05).
Meningococcal supernatants do not account for G1-phase arrest of the cell cycle in Detroit 562 cells.
To explore the effects of the soluble factors, including outer membrane vesicles, secreted by N. meningitidis on cell cycle alteration, Detroit 562 cells were next treated with bacterial supernatants of the different meningococcal isolates. Bacterial supernatants were collected from overnight cultures, followed by protein concentration (see Materials and Methods). Detroit 562 cells were exposed to viable N. meningitidis MC58 or 25- and 100-μl portions of meningococcal supernatants, correlating to 28.3 and 113.0 μg of protein, respectively. Although live bacteria induced an accumulation of cells in the G1 phase as demonstrated before, meningococcal supernatants of all four N. meningitidis isolates had no effect on the cell cycle distribution (Fig. 5). A similar amount of cells in G1 phase, S phase, and G2/M phase was observed when cells were either left untreated (control cells) or treated with bacterial supernatants of MC58, 8013/clone12, α711, or α4 (Fig. 5). These data suggest that it seems likely that continuous bacterial presence is necessary.
FIG 5.

Meningococcal supernatants do not have any effect on the cell cycle distribution in epithelial cells. N. meningitidis strain MC58, 8013/clone 12, α711, or α4 was grown to the exponential phase and harvested by centrifugation, and supernatants were collected, filter sterilized, and transferred to VivaSpin columns for protein concentration. Detroit 562 cells either were left uninfected or were infected with viable MC58 (as a positive control) or were exposed to 25- or 100-μl portions of concentrated meningococcal supernatants. Cells were harvested for PI staining at 24 h p.i. and analyzed by flow cytometry. (A) The results of representative experiments are shown. (B) The graphs show the statistical analyses of the percentages of the cell population in the G1, S, and G2/M phases. The data are shown as means ± the SD obtained from three independent experiments (*, P < 0.05).
N. meningitidis infection affects cyclin D1 and cyclin E levels in the epithelial cell line Detroit 562.
Alterations of the cell cycle are predicted to result in changes in the abundance of the major cyclins, which, when complexed with a specific cyclin-dependent kinase (CDK), are one of the main drivers of cell cycle progression. Since the appropriate temporal activation of cyclin D/cdk4/6 and cyclin E/cdk2 is required for progression through the G1 to the S phase (42–44), we investigated whether the observed accumulation of cells in the G1 phase was reflected by a change in the abundance of these cyclins by analyzing protein expression levels by immunoblot analysis. Asynchronously growing Detroit 562 cells were infected with N. meningitidis at an MOI of 100 for a 24-h time period, cell lysates were collected and tested for cyclin D1 and cyclin E protein levels. After chemiluminescence band analysis, each protein signal was normalized to β-actin, and the changes were calculated (n-fold) between N. meningitidis-infected cells and uninfected control cells. The data indicated that the abundance of cyclin D1 significantly changed in Detroit 562 cells infected with carrier strains: cells infected with isolates α711 and α4 showed relative decreases by 0.4 and 0.2, respectively, in cyclin D1 expression at 24 h p.i. (P < 0.05), whereas cells infected with disease isolates 8013/12 and MC58 showed no significant changes in cyclin D1 levels (Fig. 6A). In contrast to cyclin D1 protein levels, N. meningitidis infection induced an increase in the levels of cyclin E at 24 h p.i. in response to all tested isolates: infected cells revealed 2.5-fold (MC58), 3.1-fold (8013/clone12), 2.7-fold (α711), and 1.8-fold (α4) increases, respectively, compared to uninfected control cells (Fig. 6A).
FIG 6.

Pathogenic N. meningitidis strains and carrier isolates influence cyclin D1 and cyclin E protein levels. (A) Detroit 562 cells were either left uninfected (control) or infected with N. meningitidis MC58, 8013/clone 12, α711, or α4 (MOI 100) for 24 h. Cell lysates of control and N. meningitidis-infected cells were collected and analyzed for cyclin D1 and cyclin E protein expression by immunoblot analysis. β-Actin was used to normalize protein loading. The figure shows a representative Western blot with lanes from different areas of the same blot. (B) Bar diagram showing the fold increase in the normalized mRNA expression of cyclin D1 and cyclin E in Detroit 562 cells infected with MC58 or α711 in comparison to uninfected control cells. Real-time PCR data were analyzed according to the comparative ΔCT method by first normalizing the sample values (cyclin D1 and cyclin E) to the reference gene values (GAPDH) in infected and uninfected control cells, respectively, and then calculating the relative change in expression (as a fold increase) in infected compared to uninfected cells. Experiments were performed three times, and error bars represent the standard errors of the mean (ns, not significant).
Accumulation of cyclin E and loss of cyclin D1 expression are not controlled at transcriptional level.
To address the mechanism by which N. meningitidis induced cyclin E accumulation, we examined the levels of cyclin E mRNA at 24 h after infection of Detroit 562 cells with MC58 and carrier isolate α711. In parallel, we determined the levels of cyclin D1 mRNA in response to the two isolates (Fig. 6B). No differences in the cyclin E and cyclin D1 mRNA levels were observed between the cells exposed to meningococcal isolates or uninfected control cells. This result confirmed that N. meningitidis does not induce the synthesis of cyclin E at a transcriptional level. Increases in cyclin E protein levels are believed to be mainly regulated transcriptionally, although other control mechanisms have been described, including mRNA stabilization or prevention of protein degradation. In particular, transcription factor E2F-1 stabilizes cyclin E protein by preventing its ubiquitination, preserving the ability of the cyclin to bind and activate its kinase moiety (45).
N. meningitidis infection results in an accumulation of the CKI p21WAF1/CIP1.
The activities of cyclins and cyclin-CDK complexes are regulated by various mechanisms, including inhibition by CDK inhibitors (CKIs). Among them, the CKIs p21WAF1/CIP1 and p27CIP1, both members of the kinase inhibitor proteins (KIPs), inhibit G1-specific CDK complexes and negatively regulate cellular proliferation (46, 47). When complexed with their respective cyclin binding partners, p21WAF1/CIP1 and p27CIP1 are able to block the kinase activity of CDKs (48). To analyze whether the observed cell cycle arrest in Detroit 562 cells treated with different meningococcal isolates was in correlation with altered activity of the CKI p21WAF1/CIP1 or p27CIP1, asynchronously growing Detroit 562 cells were infected with both the pathogenic and the carrier isolates at an MOI of 100 for a 24-h time period, and cell lysates were subjected to immunoblot analysis to determine the abundance of p21WAF1/CIP1 and p27CIP1. As shown in Fig. 7A, an increase of p21WAF1/CIP1 total protein levels was observed in the infected cells in response to all N. meningitidis strains, reaching significantly higher values for MC58 and α711 (3.2- and 2.9-fold increases relative to the uninfected control, respectively). Unfortunately, we failed to detect p27CIP1 in whole-cell lysates from infected Detroit 562 cells due to technical issues (data not shown).
FIG 7.

Pathogenic N. meningitidis strains and carrier isolates increase p21WAF1/CIP1 protein levels in Detroit 562 cells and induce redistribution of p21WAF1/CIP1 and p27CIP1. (A) Cells were either left uninfected (control) or infected with N. meningitidis MC58, 8013/clone12, α711, or α4 (MOI 100) for a 24-h time period and were analyzed for p21WAF1/CIP1 protein expression by Western blot analysis. Band intensities were quantified by densitometric analysis as described for cyclin D1 and cyclin E using ImageJ and normalized to β-actin. The figure shows a representative Western blot with lanes from different areas of the same blot. (B and C) Subcellular distribution of p21WAF1/CIP1 (B) and p27CIP1 (C) in N. meningitidis-infected Detroit 562 cells assessed by immunofluorescence microscopy. Colors in panel B: green, p21WAF1/CIP1 protein stained with specific antibody and secondary antibody conjugated with Alexa Fluor 488; blue, nuclei stained with DAPI; light green, colocalization. Colors in panel C: red, p27CIP1 stained with specific antibody and secondary antibody conjugated with Cy3; blue, nuclei stained with DAPI.
N. meningitidis infection results in a subcellular redistribution of p21WAF1/CIP1 and p27CIP1.
We next investigated the subcellular localization of p21WAF1/CIP1 and p27CIP1 in response to N. meningitidis infection by immunofluorescence microscopy. In infected Detroit 562 cells, strong nuclear accumulation of p21WAF1/CIP1 was observed: 4/100 cells were positive for p21WAF1/CIP1 nuclear staining in control cells versus 16/100 cells in MC58-infected cells at 24 h p.i. (Fig. 7B). These experiments indicated that in Detroit 562 cells, upon meningococcal infection, p21WAF1/CIP1 accumulated in nuclei. Such an accumulation is in agreement with the function of p21WAF1/CIP1 in the regulation of the cell cycle. Of note, much less profound accumulation of p21WAF1/CIP1 was seen in the cytoplasm (Fig. 7B). Similar to p21WAF1/CIP1, p27CIP1 needs to be transported into the nucleus to exert its inhibitory action. In noninfected cells, p27CIP1 was visible inside the nucleus; however, in infected cells, staining revealed punctate p27CIP1 foci in cells exposed to meningococci (Fig. 7C).
DISCUSSION
Like many viruses that perturb the cell cycle machinery to adapt their own replication, bacteria can hijack checkpoints of the cell cycle to establish infection (1, 2, 8, 9). During microbial pathogenesis, the particular function to induce cell cycle arrest might represent a strategy to prevent maturation and exfoliation of epithelial cells to support prolonged bacterial persistence and survival in the epithelial cells. The concept that cell cycle modulation might favor bacterial colonization was demonstrated for the gut invasive pathogen Shigella (49) and for Helicobacter pylori that has adapted to the inhospitable conditions found at the gastric mucosal surface (50). The epithelium of the upper respiratory tract is classified as pseudostratified columnar epithelium; the epithelial cells closest to the basal lamina also undergo continuous mitosis, and their progeny replace the surface cells. Therefore, interference with cell cycle progression might be an advantage for a bacterial pathogen that adheres to the epithelium of the upper respiratory tract to prolong its colonization in this particular niche. In the present study, we therefore investigated the effects of pathogenic N. meningitidis strains, as well as carrier isolates, on the cell cycle of the epithelial host cell. We demonstrate that both disease and carrier isolates can induce a G1 arrest in the epithelial cell line Detroit 562 at 24 h p.i., which was paralleled by a significant decrease of cells in the S phase. We show that G1 arrest was only induced after infection with live bacteria but not after infection with heat-killed bacteria. Bacterial infection with three of the tested meningococcal isolates resulted in a decreased protein level of cyclin D1, whereas cyclin E expression levels were increased in response to all meningococcal isolates. Furthermore, N. meningitidis infection induced an accumulation of the cyclin-dependent kinase inhibitor (CKI) p21WAF1/CIP1 that was accompanied by a redistribution to the cell nucleus. In addition, the p27CIP1 CKI was redistributed and showed punctate foci in infected cells.
In order to implement a suitable model for investigation of cell cycle alteration during meningococcal infection, we chose Detroit 562 cells, a pharyngeal epithelial carcinoma cell line, and NP69 cells, a nasopharyngeal epithelial cell line harboring SV40T (34). Detroit 562 cells are commonly used for N. meningitidis host cell interaction studies and exhibit high levels of bacterial adhesion (51–53). This cell line has been shown to express high levels of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), a major Opa receptor, after exposure to cytokines or when infected with Opa-expressing N. meningitidis (52). NP69 cells were established from primary nonmalignant nasopharyngeal epithelial cells and may represent a model of premalignant nasopharyngeal epithelial cells (34, 54). The cell line was used to study Epstein-Barr virus interactions with the host cell (54, 55).
Both cell lines demonstrated equivalent data of bacterial adhesion when infected with the pathogenic meningococcal strain MC58. Moreover, the meningococcal wild-type strain MC58 elicited a time-dependent increase of uptake. In addition, the isogenic unencapsulated mutant showed a significantly higher rate of invasion compared to the wild-type strain due to the inhibitory effect of the serogroup B capsule polysaccharide of strain MC58 to bacterial invasion (22, 23, 32, 51, 56). We observed that meningococcal disease strain 8013/clone12 and carrier isolate α711 effectively adhered to Detroit 562 cells, whereas they were poorly invasive for both epithelial cell lines. In addition, low invasion rates were detected for carrier isolate α4, most probably due to low adhesion rates. For colonization of the human nasopharynx, the microorganism must adhere to the mucosal surface, utilize locally available nutrients, and evade the human immune system. For effective adhesion and invasion, N. meningitidis produces a number of structures and molecules, including the type IV pili, the outer membrane proteins Opa and Opc, and a number of newly identified minor adhesion or adhesion-like proteins (21–28). For our study, we chose N. meningitidis MC58 as a prototype ST-32 cc strain and strain α711 as a genetically related carrier isolate from the ST-32 cc. We also included N. meningitidis isolate 8013/clone 12 from the ST-18 cc (also known as clone 12 or 2C43), which has been used to study the interaction between N. meningitidis and brain endothelial cells (57–60). Isolate α4 was chosen as a genetically related carrier isolate to 8013/clone 12. To decipher whether the poor invasive bacterial phenotype of the two carrier isolates and isolate 8013/clone 12 was due to differential expression of known adhesins or invasins, we characterized the tested isolates for expression of some adhesins/invasins, including the pili, Opc, and Opa. All isolates showed reactivity with the pilin antiserum and three expressed Opa proteins. However, all less invasive isolates did not express the Opc protein, either due to a deletion of the opc gene (8013/clone12 and α4) or as a result of transcriptionally regulated phase variation (α711), which is mediated by a variable polycytidine stretch in the promoter region of the gene (41). The contribution of Opc to mediate efficient uptake by epithelial and endothelial cells has been shown in previous studies (22, 23) and seems to be assignable for Detroit 562 and NP69 cells.
To establish colonization, bacteria have to counteract epithelial cell turnover. The nasal epithelium, as well as the mucosa of the extranasal upper respiratory tract, is in a steady state of cell renewal in which cell production balances cell loss (61). Epithelial cell proliferation is controlled either intracellularly via cell cycle regulatory factors, including cyclins, cyclin-dependent kinases (CDKs), cyclin-dependent kinase inhibitors (CKIs), or retinoblastoma protein (pRb), or by extracellular factors (growth and differentiation factors). During the last decade, it became evident that bacteria have virulence mechanisms that target the cell cycle. To determine whether N. meningitidis is also able to impact the host cell cycle of epithelial cells, we determined the DNA profiles of PI-stained infected Detroit 562 cells. These data demonstrated that infected cells showed a significant accumulation of cells in the 2n DNA peak at 24 h p.i., suggesting an arrest or block of infected cells in the G1 phase. In parallel, PI staining data could be verified by analyzing cells that were allowed to incorporate EdU and were simultaneously stained for DNA content. Moreover, we could show that only viable bacteria are capable to arrest epithelial cells in G1 phase, whereas heat-killed bacteria had no effect. This suggested that the accumulation of cells in G1 phase is most likely induced by a heat-labile bacterial factor or factors. Likewise, accumulation of cells in G1 phase might be a result of a lower number of internalized bacteria that would be able to initiate various signaling cascades.
However, treatment with bacterial supernatants did not alter cell cycle distribution. Therefore, it seems likely that a continuous bacterial presence is necessary. Interestingly, N. meningitidis differently impacts the cell cycle depending on the cell type. Whereas in epithelial cells, meningococci induce a G1 cell cycle arrest, we recently showed that N. meningitidis induces an accumulation of cells in the S phase of brain endothelial cells. Moreover, cell cycle alterations in brain endothelial cells could be assigned to the outer membrane proteins (Opa and the Opc protein) of N. meningitidis (31).
Progression through the cell cycle is driven by the activities of CDKs, in association with their regulatory subunits, the cyclins (43, 44). In particular, type D and E cyclins mediate G1- to S-phase cell cycle progression through the activation of specific CDKs that phosphorylate pRb, thus inactivating it and releasing the transcription factor E2F from its inhibition. E2F then activates a number of growth-promoting genes, which drive the cell cycle into the S phase. Whereas D-type cyclins associate with cdk4 and cdk6, cyclin E associates with cdk2. Our studies showed that cyclin D1 protein levels were partially decreased in response to meningococcal isolates, whereas the cyclin D1 mRNA levels were not changed in infected cells. Since the levels of cyclins are regulated at the level of transcription or by targeted degradation via the ubiquitin pathway (62), our data suggest that the decrease in the cyclin D1 protein levels during meningococcal infection was most likely due to proteasomal degradation. In addition, our data showed that cyclin E protein levels increased at 24 h p.i. in response to all meningococcal strains, whereas the mRNA levels in infected cells were comparable to those in uninfected cells, suggesting that increased cyclin E protein levels were not due to increase of gene transcription. Whether inhibition of cyclin E ubiquitination and proteasomal degradation occurs during meningococcal infection is questionable and remains to be elucidated.
Our data furthermore clearly indicated a significant increase of p21WAF1/CIP1 protein levels. p21WAF1/CIP1 is the archetypal mammalian CKI and is tightly controlled by the tumor suppressor protein p53 through which this protein mediates the p53-dependent cell cycle G1-phase arrest in response to a variety of stress stimuli. In addition, it can be induced by p53-independent mechanisms. p21WAF1/CIP1 can bind to cyclinD-cdk4 or cyclinD-cdk6 complexes and prevents them from phosphorylating pRb. Moreover, cyclinE-cdk2 complexes can be negatively regulated by the CKI p21WAF1/CIP1. Similar to p21WAF1/CIP1, p27CIP1 can inhibit cyclin-CDK complexes in G1 phase, and it is functional in the nucleus. Unfortunately, we failed to detect p27CIP1 expression by immunoblotting in infected Detroit 562 cells due to technical issues. However, we could detect significant increase of punctate foci of p27CIP1 in infected cells by immunofluorescence analysis. Such an accumulation as observed during meningococcal infection is in line with the function of p27CIP1 in the regulation of the cell cycle. p27CIP1 expression is regulated at different levels, i.e., transcriptional, translational, and posttranslational mechanisms, including ubiquitin-proteasome-induced degradation, as well as protein phosphorylation. Taken together, we would assume that the downstream mechanisms of N. meningitidis-induced G1 cell cycle arrest might involve p21WAF1/CIP1 recruitment to cyclinD-cdk4/6 complexes and/or cyclin E-cdk2 complexes, thereby inhibiting CDK activity and thus the phosphorylation of pRb (Fig. 8). However, future studies have to clarify the exact mechanism by which an accumulation of p21WAF1/CIP1 results in G1 arrest in epithelial cells in response to meningococcal infection.
FIG 8.

Proposed model of G1 cell cycle arrest in epithelial cells during meningococcal infection. Infection of pharyngeal epithelial cells with both invasive meningococcal disease isolates and apathogenic carriage isolates results in increased protein levels of the CKI p21WAF/CIP1 and nuclear redistribution of p21WAF/CIP1 and p27CIP1. Increased p21WAF/CIP1 levels may be required for inhibition of the G1-phase complexes cyclin D-cdk4/6 and/or cyclin E-cdk2 by one or both of the CKIs, resulting in subsequent sequestration of cells in the G1 phase. G1-phase, gap phase 1; S-phase, DNA synthesis; G2-phase, gap phase 2; M-phase, mitosis; CDK, cyclin-dependent kinase.
During the past decade, several bacterial effectors have been shown to interfere with p21WAF1/CIP1 expression, such as the cyclomodulin cytolethal distending toxin from E. coli (63, 64), toxin A from Clostridium difficile (65), or the CagA effector from Helicobacter pylori (66–68). Interestingly, the cycle inhibiting factor (Cif) produced by EPEC and EHEC bacteria differs from the toxins mentioned above in that Cif-induced accumulation of p21WAF1/CIP1 does not involve transcription mechanisms but acts on a pathway controlling protein stability (69).
N. meningitidis can asymptomatically colonize the upper respiratory tract of healthy carriers for months or years (70). The asymptomatic carriage of N. meningitidis is of concern since it is the mechanism by which the reservoir of endemic meningococci is maintained within the population. Adaptation to this niche not only requires mechanisms to effectively adhere to and invade the host cells but also includes mechanisms that help to counteract epithelial cell turnover. In this study, we now provide evidence that both disease and carrier isolates of N. meningitidis can interfere with cell cycle progression by sequestration of targeted host cells in G1. The cell cycle arrest induced by N. meningitidis may represent a strategy to prevent maturation and exfoliation of epithelial cells to support prolonged bacterial persistence and survival in the epithelial cells of the upper respiratory tract. However, further investigations are needed to monitor the attachment of meningococci to epithelial cells that have been arrested in the G1 phase. Interestingly, the related species N. gonorrhoeae has also been shown to modulate the host cell cycle (71, 72). In line with our findings, N. gonorrhoeae also arrests cells in the early G1 phase of the cell cycle after 24 h of infection, as shown for unsynchronized HeLa cells (71). In a follow-up study, the authors could show that the accumulation of infected cells in G1 was a result of a delay in the G2-to-M-phase progression and the sequestration of cells in G2 (72). Mechanistically, the sequestration of cells in G2 was a result of increased expression of p21WAF1/CIP1 and p27CIP1. Relevant to our study, neither supernatants from infected cell cultures nor bacterial supernatants had any effect on the cell cycle. Although research in the last decade has shown that pathogenic bacteria can interfere with host cell cycle progression (1, 2, 8), one of the open questions remained whether commensal bacteria also produce “cyclomodulins” and whether commensal bacteria can interfere with epithelial cell turnover. Here, we now provide evidence that carrier isolates of the species Neisseria can also impact on the host cell cycle. Future studies are necessary to identify the molecular components of the meningococci responsible for eliciting cell cycle arrest in epithelial cells. Since both the rate of cell proliferation and cell migration contribute to the turnover time of the respiratory epithelium, future studies will also rely on more complex cell culture models, for example, the established three-dimensional organotypic tissues of the human mucosa, to achieve a full understanding of the interference and control of the cell cycle during bacterial pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to George Sai Wah Tsao (University of Hong Kong) for generously providing the NP69 cell line used in this study.
This study was supported by German Research Foundation (DFG) grant SCHU 2394/3-1. Wilhelm F. Oosthuysen holds a postdoctoral research fellowship from the National Research Foundation of South Africa.
The authors have no conflicts of interest that are directly relevant to the content of this article.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00296-16.
REFERENCES
- 1.Nougayrede JP, Taieb F, De Rycke J, Oswald E. 2005. Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol 13:103–110. doi: 10.1016/j.tim.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Oswald E, Nougayrede JP, Taieb F, Sugai M. 2005. Bacterial toxins that modulate host cell-cycle progression. Curr Opin Microbiol 8:83–91. doi: 10.1016/j.mib.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Vermeulen K, Van Bockstaele DR, Berneman ZN. 2003. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherr CJ. 1994. G1 phase progression: cycling on cue. Cell 79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 5.Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. 1995. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol 15:2612–2624. doi: 10.1128/MCB.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherr CJ, Roberts JM. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 7.Hengst L, Reed SI. 1998. Inhibitors of the Cip/Kip family. Curr Top Microbiol Immunol 227:25–41. [DOI] [PubMed] [Google Scholar]
- 8.Tran Van Nhieu G, Arbibe L. 2009. Genetic reprogramming of host cells by bacterial pathogens. F1000 Biol Rep 1:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim M, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Sasakawa C. 2010. Bacterial interactions with the host epithelium. Cell Host Microbe 8:20–35. doi: 10.1016/j.chom.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Peres SY, Marches O, Daigle F, Nougayrede JP, Herault F, Tasca C, De Rycke J, Oswald E. 1997. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol Microbiol 24:1095–1107. doi: 10.1046/j.1365-2958.1997.4181785.x. [DOI] [PubMed] [Google Scholar]
- 11.Marches O, Ledger TN, Boury M, Ohara M, Tu X, Goffaux F, Mainil J, Rosenshine I, Sugai M, De Rycke J, Oswald E. 2003. Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol Microbiol 50:1553–1567. doi: 10.1046/j.1365-2958.2003.03821.x. [DOI] [PubMed] [Google Scholar]
- 12.Samba-Louaka A, Taieb F, Nougayrede JP, Oswald E. 2009. Cif type III effector protein: a smart hijacker of the host cell cycle. Future Microbiol 4:867–877. doi: 10.2217/fmb.09.60. [DOI] [PubMed] [Google Scholar]
- 13.Lax AJ, Pullinger GD, Baldwin MR, Harmey D, Grigoriadis AE, Lakey JH. 2004. The Pasteurella multocida toxin interacts with signaling pathways to perturb cell growth and differentiation. Int J Med Microbiol 293:505–512. doi: 10.1078/1438-4221-00287. [DOI] [PubMed] [Google Scholar]
- 14.Horiguchi Y. 2001. Escherichia coli cytotoxic necrotizing factors and Bordetella dermonecrotic toxin: the dermonecrosis-inducing toxins activating Rho small GTPases. Toxicon 39:1619–1627. doi: 10.1016/S0041-0101(01)00149-0. [DOI] [PubMed] [Google Scholar]
- 15.Chang YJ, Wu MS, Lin JT, Pestell RG, Blaser MJ, Chen CC. 2006. Mechanisms for Helicobacter pylori CagA-induced cyclin D1 expression that affect cell cycle. Cell Microbiol 8:1740–1752. doi: 10.1111/j.1462-5822.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- 16.Bao Z, Hua J. 2015. Linking the cell cycle with innate immunity in Arabidopsis. Mol Plant 8:980–982. doi: 10.1016/j.molp.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Cartwright KA, Stuart JM, Jones DM, Noah ND. 1987. The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol Infect 99:591–601. doi: 10.1017/S0950268800066449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orr HJ, Gray SJ, Macdonald M, Stuart JM. 2003. Saliva and meningococcal transmission. Emerg Infect Dis 9:1314–1315. doi: 10.3201/eid0910.030344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claus H, Maiden MC, Wilson DJ, McCarthy ND, Jolley KA, Urwin R, Hessler F, Frosch M, Vogel U. 2005. Genetic analysis of meningococci carried by children and young adults. J Infect Dis 191:1263–1271. doi: 10.1086/428590. [DOI] [PubMed] [Google Scholar]
- 20.Caugant DA, Hoiby EA, Rosenqvist E, Froholm LO, Selander RK. 1992. Transmission of Neisseria meningitidis among asymptomatic military recruits and antibody analysis. Epidemiol Infect 109:241–253. doi: 10.1017/S0950268800050196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Virji M. 2009. Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat Rev Microbiol 7:274–286. doi: 10.1038/nrmicro2097. [DOI] [PubMed] [Google Scholar]
- 22.Virji M, Makepeace K, Ferguson DJ, Achtman M, Moxon ER. 1993. Meningococcal Opa and Opc proteins: their role in colonization and invasion of human epithelial and endothelial cells. Mol Microbiol 10:499–510. doi: 10.1111/j.1365-2958.1993.tb00922.x. [DOI] [PubMed] [Google Scholar]
- 23.Virji M, Makepeace K, Ferguson DJ, Achtman M, Sarkari J, Moxon ER. 1992. Expression of the Opc protein correlates with invasion of epithelial and endothelial cells by Neisseria meningitidis. Mol Microbiol 6:2785–2795. doi: 10.1111/j.1365-2958.1992.tb01458.x. [DOI] [PubMed] [Google Scholar]
- 24.Virji M, Kayhty H, Ferguson DJ, Alexandrescu C, Heckels JE, Moxon ER. 1991. The role of pili in the interactions of pathogenic Neisseria with cultured human endothelial cells. Mol Microbiol 5:1831–1841. doi: 10.1111/j.1365-2958.1991.tb00807.x. [DOI] [PubMed] [Google Scholar]
- 25.Virji M, Makepeace K, Ferguson DJ, Watt SM. 1996. Carcinoembryonic antigens (CD66) on epithelial cells and neutrophils are receptors for Opa proteins of pathogenic neisseriae. Mol Microbiol 22:941–950. doi: 10.1046/j.1365-2958.1996.01551.x. [DOI] [PubMed] [Google Scholar]
- 26.Nassif X. 1999. Interaction mechanisms of encapsulated meningococci with eucaryotic cells: what does this tell us about the crossing of the blood-brain barrier by Neisseria meningitidis? Curr Opin Microbiol 2:71–77. doi: 10.1016/S1369-5274(99)80012-5. [DOI] [PubMed] [Google Scholar]
- 27.Nassif X, Beretti JL, Lowy J, Stenberg P, O'Gaora P, Pfeifer J, Normark S, So M. 1994. Roles of pilin and PilC in adhesion of Neisseria meningitidis to human epithelial and endothelial cells. Proc Natl Acad Sci U S A 91:3769–3773. doi: 10.1073/pnas.91.9.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheuerpflug I, Rudel T, Ryll R, Pandit J, Meyer TF. 1999. Roles of PilC and PilE proteins in pilus-mediated adherence of Neisseria gonorrhoeae and Neisseria meningitidis to human erythrocytes and endothelial and epithelial cells. Infect Immun 67:834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill DJ, Griffiths NJ, Borodina E, Virji M. 2010. Cellular and molecular biology of Neisseria meningitidis colonization and invasive disease. Clin Sci 118:547–564. doi: 10.1042/CS20090513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emonts M, Hazelzet JA, de Groot R, Hermans PW. 2003. Host genetic determinants of Neisseria meningitidis infections. Lancet Infect Dis 3:565–577. doi: 10.1016/S1473-3099(03)00740-0. [DOI] [PubMed] [Google Scholar]
- 31.Oosthuysen WF, Mueller T, Dittrich MT, Schubert-Unkmeir A. 2016. Neisseria meningitidis causes cell cycle arrest of human brain microvascular endothelial cells at S phase via p21 and cyclin G2. Cell Microbiol 18:46–65. doi: 10.1111/cmi.12482. [DOI] [PubMed] [Google Scholar]
- 32.Unkmeir A, Latsch K, Dietrich G, Wintermeyer E, Schinke B, Schwender S, Kim KS, Eigenthaler M, Frosch M. 2002. Fibronectin mediates Opc-dependent internalization of Neisseria meningitidis in human brain microvascular endothelial cells. Mol Microbiol 46:933–946. doi: 10.1046/j.1365-2958.2002.03222.x. [DOI] [PubMed] [Google Scholar]
- 33.Nassif X, Lowy J, Stenberg P, O'Gaora P, Ganji A, So M. 1993. Antigenic variation of pilin regulates adhesion of Neisseria meningitidis to human epithelial cells. Mol Microbiol 8:719–725. doi: 10.1111/j.1365-2958.1993.tb01615.x. [DOI] [PubMed] [Google Scholar]
- 34.Tsao SW, Wang X, Liu Y, Cheung YC, Feng H, Zheng Z, Wong N, Yuen PW, Lo AK, Wong YC, Huang DP. 2002. Establishment of two immortalized nasopharyngeal epithelial cell lines using SV40 large T and HPV16E6/E7 viral oncogenes. Biochim Biophys Acta 1590:150–158. doi: 10.1016/S0167-4889(02)00208-2. [DOI] [PubMed] [Google Scholar]
- 35.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. 2006. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salic A, Mitchison TJ. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Virji M, Heckels JE. 1983. Antigenic cross-reactivity of Neisseria pili: investigations with type- and species-specific monoclonal antibodies. J Gen Microbiol 129:2761–2768. [DOI] [PubMed] [Google Scholar]
- 39.Merker P, Tommassen J, Kusecek B, Virji M, Sesardic D, Achtman M. 1997. Two-dimensional structure of the Opc invasin from Neisseria meningitidis. Mol Microbiol 23:281–293. doi: 10.1046/j.1365-2958.1997.2051567.x. [DOI] [PubMed] [Google Scholar]
- 40.Simonis A, Hebling S, Gulbins E, Schneider-Schaulies S, Schubert-Unkmeir A. 2014. Differential activation of acid sphingomyelinase and ceramide release determines invasiveness of Neisseria meningitidis into brain endothelial cells. PLoS Pathog 10:e1004160. doi: 10.1371/journal.ppat.1004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarkari J, Pandit N, Moxon ER, Achtman M. 1994. Variable expression of the Opc outer membrane protein in Neisseria meningitidis is caused by size variation of a promoter containing polycytidine. Mol Microbiol 13:207–217. doi: 10.1111/j.1365-2958.1994.tb00416.x. [DOI] [PubMed] [Google Scholar]
- 42.Hwang HC, Clurman BE. 2005. Cyclin E in normal and neoplastic cell cycles. Oncogene 24:2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- 43.Coleman ML, Marshall CJ, Olson MF. 2004. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat Rev Mol Cell Biol 5:355–366. doi: 10.1038/nrm1365. [DOI] [PubMed] [Google Scholar]
- 44.Sherr CJ. 2000. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res 60:3689–3695. [PubMed] [Google Scholar]
- 45.Pajalunga D, Crescenzi M. 2004. Regulation of cyclin E protein levels through E2F-mediated inhibition of degradation. Cell Cycle 3:1572–1578. doi: 10.4161/cc.3.12.1279. [DOI] [PubMed] [Google Scholar]
- 46.Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E, Fox MP, Wei N. 1995. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell 6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toyoshima H, Hunter T. 1994. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 48.Abukhdeir AM, Park BH. 2008. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med 10:e19. doi: 10.1017/S1462399408000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwai H, Kim M, Yoshikawa Y, Ashida H, Ogawa M, Fujita Y, Muller D, Kirikae T, Jackson PK, Kotani S, Sasakawa C. 2007. A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 130:611–623. doi: 10.1016/j.cell.2007.06.043. [DOI] [PubMed] [Google Scholar]
- 50.Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, Fujita Y, Nagamatsu K, Ishijima N, Koyasu S, Haas R, Sasakawa C. 2007. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2:250–263. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 51.Bartley SN, Tzeng YL, Heel K, Lee CW, Mowlaboccus S, Seemann T, Lu W, Lin YH, Ryan CS, Peacock C, Stephens DS, Davies JK, Kahler CM. 2013. Attachment and invasion of Neisseria meningitidis to host cells is related to surface hydrophobicity, bacterial cell size and capsule. PLoS One 8:e55798. doi: 10.1371/journal.pone.0055798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bradley CJ, Griffiths NJ, Rowe HA, Heyderman RS, Virji M. 2005. Critical determinants of the interactions of capsule-expressing Neisseria meningitidis with host cells: the role of receptor density in increased cellular targeting via the outer membrane Opa proteins. Cell Microbiol 7:1490–1503. doi: 10.1111/j.1462-5822.2005.00572.x. [DOI] [PubMed] [Google Scholar]
- 53.Quattroni P, Li Y, Lucchesi D, Lucas S, Hood DW, Herrmann M, Gabius HJ, Tang CM, Exley RM. 2012. Galectin-3 binds Neisseria meningitidis and increases interaction with phagocytic cells. Cell Microbiol 14:1657–1675. doi: 10.1111/j.1462-5822.2012.01838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lo AK, Liu Y, Wang XH, Huang DP, Yuen PW, Wong YC, Tsao GS. 2003. Alterations of biologic properties and gene expression in nasopharyngeal epithelial cells by the Epstein-Barr virus-encoded latent membrane protein 1. Lab Invest 83:697–709. doi: 10.1097/01.LAB.0000067480.44925.10. [DOI] [PubMed] [Google Scholar]
- 55.Lo AK, Lo KW, Tsao SW, Wong HL, Hui JW, To KF, Hayward DS, Chui YL, Lau YL, Takada K, Huang DP. 2006. Epstein-Barr virus infection alters cellular signal cascades in human nasopharyngeal epithelial cells. Neoplasia 8:173–180. doi: 10.1593/neo.05625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Vries FP, van Der Ende A, van Putten JP, Dankert J. 1996. Invasion of primary nasopharyngeal epithelial cells by Neisseria meningitidis is controlled by phase variation of multiple surface antigens. Infect Immun 64:2998–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lambotin M, Hoffmann I, Laran-Chich MP, Nassif X, Couraud PO, Bourdoulous S. 2005. Invasion of endothelial cells by Neisseria meningitidis requires cortactin recruitment by a phosphoinositide-3-kinase/Rac1 signalling pathway triggered by the lipo-oligosaccharide. J Cell Sci 118:3805–3816. doi: 10.1242/jcs.02514. [DOI] [PubMed] [Google Scholar]
- 58.Coureuil M, Lecuyer H, Scott MG, Boularan C, Enslen H, Soyer M, Mikaty G, Bourdoulous S, Nassif X, Marullo S. 2010. Meningococcus hijacks a β2-adrenoceptor/beta-arrestin pathway to cross brain microvasculature endothelium. Cell 143:1149–1160. doi: 10.1016/j.cell.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 59.Coureuil M, Mikaty G, Miller F, Lecuyer H, Bernard C, Bourdoulous S, Dumenil G, Mege RM, Weksler BB, Romero IA, Couraud PO, Nassif X. 2009. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science 325:83–87. doi: 10.1126/science.1173196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bernard SC, Simpson N, Join-Lambert O, Federici C, Laran-Chich MP, Maissa N, Bouzinba-Segard H, Morand PC, Chretien F, Taouji S, Chevet E, Janel S, Lafont F, Coureuil M, Segura A, Niedergang F, Marullo S, Couraud PO, Nassif X, Bourdoulous S. 2014. Pathogenic Neisseria meningitidis utilizes CD147 for vascular colonization. Nat Med 20:725–731. doi: 10.1038/nm.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fabrikant JI, Cherry J. 1970. The kinetics of cellular proliferation in normal and malignant tissues. X. Cell proliferation in the nose and adjoining cavities. Ann Otol Rhinol Laryngol 79:572–578. [DOI] [PubMed] [Google Scholar]
- 62.Sherr CJ. 1993. Mammalian G1 cyclins. Cell 73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 63.Ge Z, Schauer DB, Fox JG. 2008. In vivo virulence properties of bacterial cytolethal-distending toxin. Cell Microbiol 10:1599–1607. doi: 10.1111/j.1462-5822.2008.01173.x. [DOI] [PubMed] [Google Scholar]
- 64.Smith JL, Bayles DO. 2006. The contribution of cytolethal distending toxin to bacterial pathogenesis. Crit Rev Microbiol 32:227–248. doi: 10.1080/10408410601023557. [DOI] [PubMed] [Google Scholar]
- 65.Kim H, Kokkotou E, Na X, Rhee SH, Moyer MP, Pothoulakis C, Lamont JT. 2005. Clostridium difficile toxin A-induced colonocyte apoptosis involves p53-dependent p21(WAF1/CIP1) induction via p38 mitogen-activated protein kinase. Gastroenterology 129:1875–1888. doi: 10.1053/j.gastro.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 66.Sommi P, Savio M, Stivala LA, Scotti C, Mignosi P, Prosperi E, Vannini V, Solcia E. 2002. Helicobacter pylori releases a factor(s) inhibiting cell cycle progression of human gastric cell lines by affecting cyclin E/cdk2 kinase activity and Rb protein phosphorylation through enhanced p27(KIP1) protein expression. Exp Cell Res 281:128–139. doi: 10.1006/excr.2002.5629. [DOI] [PubMed] [Google Scholar]
- 67.Ahmed A, Smoot D, Littleton G, Tackey R, Walters CS, Kashanchi F, Allen CR, Ashktorab H. 2000. Helicobacter pylori inhibits gastric cell cycle progression. Microbes Infect 2:1159–1169. doi: 10.1016/S1286-4579(00)01270-3. [DOI] [PubMed] [Google Scholar]
- 68.Yokoyama K, Higashi H, Ishikawa S, Fujii Y, Kondo S, Kato H, Azuma T, Wada A, Hirayama T, Aburatani H, Hatakeyama M. 2005. Functional antagonism between Helicobacter pylori CagA and vacuolating toxin VacA in control of the NFAT signaling pathway in gastric epithelial cells. Proc Natl Acad Sci U S A 102:9661–9666. doi: 10.1073/pnas.0502529102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Samba-Louaka A, Nougayrede JP, Watrin C, Jubelin G, Oswald E, Taieb F. 2008. Bacterial cyclomodulin Cif blocks the host cell cycle by stabilizing the cyclin-dependent kinase inhibitors p21 and p27. Cell Microbiol 10:2496–2508. doi: 10.1111/j.1462-5822.2008.01224.x. [DOI] [PubMed] [Google Scholar]
- 70.Yazdankhah SP, Caugant DA. 2004. Neisseria meningitidis: an overview of the carriage state. J Med Microbiol 53:821–832. doi: 10.1099/jmm.0.45529-0. [DOI] [PubMed] [Google Scholar]
- 71.Jones A, Jonsson A-B, Aro H. 2007. Neisseria gonorrhoeae infection causes a G1 arrest in human epithelial cells. FASEB J 21:345–355. doi: 10.1096/fj.06-6675com. [DOI] [PubMed] [Google Scholar]
- 72.Vielfort K, Soderholm N, Weyler L, Vare D, Lofmark S, Aro H. 2013. Neisseria gonorrhoeae infection causes DNA damage and affects the expression of p21, p27 and p53 in non-tumor epithelial cells. J Cell Sci 126:339–347. doi: 10.1242/jcs.117721. [DOI] [PubMed] [Google Scholar]
- 73.McGuinness BT, Clarke IN, Lambden PR, Barlow AK, Heckels JE, Poolman JT, Jones DM. 1991. Point mutation in meningococcal porA gene associated with increased endemic disease. Lancet 337:514–517. doi: 10.1016/0140-6736(91)91297-8. [DOI] [PubMed] [Google Scholar]
- 74.Rusniok C, Vallenet D, Floquet S, Ewles H, Mouze-Soulama C, Brown D, Lajus A, Buchrieser C, Medigue C, Glaser P, Pelicic V. 2009. NeMeSys: a biological resource for narrowing the gap between sequence and function in the human pathogen Neisseria meningitidis. Genome Biol 10:R110. doi: 10.1186/gb-2009-10-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.