

Abstract
Motivation: Determining the methylation state of regions with high copy numbers is challenging for second-generation sequencing, because the read length is insufficient to map reads uniquely, especially when repetitive regions are long and nearly identical to each other. Single-molecule real-time (SMRT) sequencing is a promising method for observing such regions, because it is not vulnerable to GC bias, it produces long read lengths, and its kinetic information is sensitive to DNA modifications.
Results: We propose a novel linear-time algorithm that combines the kinetic information for neighboring CpG sites and increases the confidence in identifying the methylation states of those sites. Using a practical read coverage of ∼30-fold from an inbred strain medaka (Oryzias latipes), we observed that both the sensitivity and precision of our method on individual CpG sites were ∼93.7%. We also observed a high correlation coefficient (R = 0.884) between our method and bisulfite sequencing, and for 92.0% of CpG sites, methylation levels ranging over [0,1] were in concordance within an acceptable difference 0.25. Using this method, we characterized the landscape of the methylation status of repetitive elements, such as LINEs, in the human genome, thereby revealing the strong correlation between CpG density and hypomethylation and detecting hypomethylation hot spots of LTRs and LINEs. We uncovered the methylation states for nearly identical active transposons, two novel LINE insertions of identity ∼99% and length 6050 base pairs (bp) in the human genome, and 16 Tol2 elements of identity >99.8% and length 4682 bp in the medaka genome.
Availability and Implementation: AgIn (Aggregate on Intervals) is available at: https://github.com/hacone/AgIn
Contact: ysuzuki@cb.k.u-tokyo.ac.jp or moris@cb.k.u-tokyo.ac.jp
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
There has been a great deal of interest in identification of genome-wide epigenetic DNA modifications in recent years, because DNA modifications play an essential role in cellular and developmental processes (Anway et al., 2005; Miller, 2010; Molaro et al., 2011; Schmitz et al., 2011; Smith et al., 2012; Weaver et al., 2004; Zemach et al., 2010). Some of human transposable elements (TEs), such as long interspersed nuclear elements (LINE), transpose actively within somatic cells along differentiation of neural tissues and are partly regulated by DNA methylation (Muotri et al., 2005, 2010). Each family of human TEs exhibits a variety of methylation statuses in different tissue types, which was found by looking at the mixture of methylation information on the consensus sequence of TEs in the same family (Xie et al., 2013). Many human diseases are also associated with DNA methylation state of TEs. In particular, unmethylation of repetitive elements (REs), such as LINE-1 (L1) elements, has been related to some cancers (Ross et al., 2010; Wilson et al., 2007). Although only a few L1 elements exhibit activity in the human genome (Beck et al., 2010), it has been reported in various cancer genomes (Goodier, 2014; Lee et al., 2012), and importantly, transposition is correlated with unmethylation in the promoter region of L1 elements (Tubio et al., 2014). Therefore, it is essential to develop an experimental framework that can characterize the methylation state of REs in a genome-wide manner.
The advent of second-generation sequencing technology has increased the efficiency of the generation of precise genome-wide methylation maps at a single-base resolution using bisulfite treatment (Cokus et al., 2008; Lister et al., 2008, 2009; Harris et al., 2010; Meissner et al., 2008); however, these sequencing-based technologies have difficulty in characterizing the methylation status of CpGs in regions that are highly similar to other regions. Bisulfite-treated short reads from these regions often fail to map uniquely to their original positions; instead, they are likely to be aligned ambiguously to multiple genomic positions. Especially, the younger and more active transposons retain higher fidelity and are therefore difficult to address using short reads.
The PacBio RS II sequencing system uses DNA polymerases to perform single-molecule real-time (SMRT) sequencing (Eid et al., 2009; Korlach et al., 2008), and is able to sequence reads of an average length of >10 kb. It is also able to sequence genomic regions with extremely high GC content. A striking example is the sequencing of a >2-kb region with GC content of 100% (Loomis et al., 2012), indicating that SMRT sequencing is less vulnerable to sequence composition bias than first/second-generation sequencing is.
SMRT sequencing of bisulfite-treated DNA fragments may allow identification of DNA methylation; however, this approach is unlikely to process long, highly identical repeats because bisulfite treatment breaks DNA into fragments of <1500 bp (Miura et al., 2012; Yang et al., 2015). Instead, we explored another advantage of SMRT sequencing to detect DNA modifications directly.
2 Approach
In SMRT sequencing, we observe the base sequence in a single DNA molecule as the time course of the fluorescence pulses which reflect the incorporation processes of nucleotides. From this time course information, we define the inter-pulse duration (IPD), the time interval separating the two pulses of consecutive bases. Importantly, the IPD of the same genomic position varies and has a significant and predictable response to the presence of DNA modifications and damages (Flusberg et al., 2010).
Since the IPD tends to be perturbed systematically when DNA modifications are present, SMRT sequencing has been used to detect 5-hydroxymethylcytosine (Flusberg et al., 2010), N4-methylcytosine (Clark et al., 2012), N6-methylademine (Fang et al., 2012; Feng et al., 2013; Flusberg et al., 2010; Greer et al., 2015) and damaged DNA bases (Clark et al., 2011) in bacteria and mitochondria. Though the sequence motifs with modifications can be detected with very low coverage (Beckmann et al., 2014), estimation of 5-methylcytosine (5-mC) residues using low-coverage reads is challenging. It requires extensive coverage (∼500×) at each position to clarify the base-wise 5-mC state and therefore becomes costly (Fang et al., 2012; Flusberg et al., 2010; Schadt et al., 2012). Clark et al. (2013) attempted to improve the detection of microbial 5-mC in the Escherichia coli and Bacillus halodurans genomes using Tet1-mediated oxidation to convert 5-mC into 5caC in SMRT reads of ∼150× coverage per DNA strand. Therefore, kinetic information from low-coverage SMRT reads at a single CpG site is not reliable for predicting the methylation status.
In this study, we exploited the facts that unmethylated CpG dinucleotides are rare (∼10%) in vertebrates and generally do not exist in isolation but often range over long hypomethylated regions (Bock et al., 2008; Eckhardt et al., 2006; Gifford et al., 2013; Nautiyal et al., 2010; Qu et al., 2012; Shoemaker et al., 2010; Xie et al., 2013). Su et al. (2012) reported that the average length of hypomethylated regions in five human cell types is ∼2 kb. Thus, estimating regions with unmethylated CpG sites is informative in most cases. Moreover, integrating kinetic information over many CpG sites in a long region can increase the confidence in detecting methylation when the status of those sites is correlated. Therefore, it shows promise for predicting the methylation status in a block using low-coverage SMRT reads. In the rest of this article, we examine the feasibility of the approach and present a novel computational algorithm that integrates SMRT sequencing kinetic data and determines the methylation status of CpG sites.
3 Methods
3.1 Outline of our method AgIn
Figure 1A shows a schematic representation of the basic concept of our method. To eliminate the context-dependent fluctuation of the IPD values, we calculated the IPD ratio (IPDR) on each genomic position as previously described (Flusberg et al., 2010). This normalization is essential to compare the IPD values from different genomic positions with various sequence contexts. Then, we defined the IPDR profile of a CpG site as an array of IPDR measurements of 21 bp surrounding the CpG site because these neighboring positions have proven to be effective in predicting 5-hydroxymethylcytosine, N4-methylcytosine and N6-methylademine in bacteria genomes in previous studies (Clark et al., 2011, 2012; Fang et al., 2012; Flusberg et al., 2010). With low coverage, the IPDR profiles at individual CpG sites are noisy and insufficient for determining whether each CpG site is methylated or not. However, if we could somehow identify the boundaries of hypomethylated/hypermethylated regions and take the average of the IPDR profiles for the CpGs within each region, then it would allow better prediction of the methylation state of each region from its average IPDR profile, which has less noise than the profile of a single CpG site. Averaging the IPDR profiles is also expected to alleviate the possible confounding effect from other types of modifications found in DNA. An example of our prediction for the human genome is shown in Figure 1B. Our method was able to estimate hypomethylation of long duplicated regions while the bisulfite sequencing provided little information. Supplementary Figure S1C illustrates another example in which both methods were consistent in showing hypomethylation in the gene promoters.
Fig. 1.

Outline of our integration method. (A) The top three distributions show the typical inter-pulse duration ratio (IPDR) profiles within 10 bp of the CpG sites in the raw data. The IPDR profiles of individual CpG sites were treated as points in the 21-dimensional feature space. Red-colored unmethylated CpGs and blue-colored methylated CpGs are often difficult to separate using a hyperplane. Therefore, initially, we had little knowledge about the methylation status of each CpG site from the raw data, as illustrated by the question marks at the CpG sites. Our algorithm predicts the boundary of unmethylated and methylated CpG sites. The average IPDR profiles of the two regions, which have clearly distinct IPDR profiles, are shown below the two regions separated by the boundary (see the detailed IPDR profiles in Supplementary Fig. S1B). Red circles and blue boxes represent unmethylated and methylated CpGs, respectively, predicted by our algorithm (annotated as ‘predicted regions’) and were observed by bisulfite sequencing (labeled ‘answer’). In the feature space, red and blue disks represent the IPDR profiles of predicted regions. (B) Comparison of our prediction with the available human genome methylome data. From top to bottom, black bars indicate hypomethylated regions predicted from SMRT sequencing data using our method. Yellow and black bars show the methylation level and read coverage obtained from public bisulfite sequencing data, respectively, and blue boxes show hypomethylated regions predicted from the bisulfite data. Green bars below indicate the alignability of short (100-bp) reads. The bottom row shows repeat masker tracks. Both methods are consistent in showing hypomethylation on the three blue-colored regions. No read counts of the bisulfite data are available in long duplicated regions where the alignability is quite low, but our method can estimate hypomethylation in these regions
3.2 Estimating the methylation status at each CpG site
Suppose that the focal genome has n CpG sites. We denote the genomic position of C of the ith CpG site by . For example, if the C of the second CpG site is at the 10th genomic position, .” ‘.’ Our goal is to predict the methylation status, unmethylated or methylated, at pi using information of the read coverage and the IPDRs at positions surrounding pi. 21 neighboring positions are denoted by for in the plus strand. For example, the positions 5 bases upstream and downstream of pi are and , respectively.
We used the SMRT Analysis pipeline to process raw kinetic data from SMRT sequencing to obtain the mean IPDR and the read coverage at each genomic position. Let ri and denote the mean IPDR associated with position i of the forward and reverse strands, respectively, and let ci and denote the read coverage at position i of the forward and reverse strands, respectively. To achieve a better prediction, we derive a modified IPDR vector from the raw read coverage and the IPDRs within 10 bases around pi. For this purpose, we consider that the property that any CpG site in one strand is reverse complementary to the CpG in the other strand, and the methylation status of Cs at a pair of CpG sites in both strands is consistent in most cases, making it meaningful to combine IPDR information for both strands to predict the methylation status. To represent positions in the minus strand, we note that since we set pi to the position of C of the focal CpG in the plus strand, the position of C of the CpG in the minus strand is , and the surrounding positions are for . We attach more importance to the IPDR values associated with a higher read coverage and we quantify this as in the plus strand ( in the minus strand). We then take the sum of all the products and normalize it by dividing it by the total coverage. Finally, we obtain the 21-dimensional modified IPDR vector for 21 genomic positions around CpG site pi:
We are now in a position to define a classifier that uses as explanatory variables and predicts the methylation status at pi. We attempted to use linear discriminant analysis (LDA) with the discriminant function
where we optimized the values of coefficient vector and variable γ using bisulfite sequencing data as the training dataset to improve the prediction. Supplementary Figure S1A and D shows the optimized vector that we used in this study. We do not claim this vector is the simplest one since excluding the low-contributing components from the parameter degraded the accuracy only by a little (Supplementary Fig. S3G). If the sign of the discriminant function, , is positive, the methylation status at pi is defined as ‘methylated’; otherwise, it is defined as ‘unmethylated’. Our goal is to achieve a higher accuracy using a lower read coverage in order to reduce the cost.
3.3 Predicting the methylation status of CpG blocks
In vertebrates, unmethylated CpG dinucleotides are rare (∼10%) and do not always exist in isolation, but they are likely to range over long hypomethylated regions. This motivates us to integrate low-coverage reads around CpGs in a region to yield high-coverage for estimating the methylation status in the entire region, rather than at a single-base resolution. Let A denote a region. The following formula expresses the average IPDR vector for 21 genomic positions around all the CpG sites in region A and its associated discriminant function:
Processing a longer region with sufficient CpG sites can improve the accuracy of the prediction, although it may overlook smaller regions. In our analysis, we imposed the constraint that each region contained at least b CpG sites. For example, we can set b to 50 because the average length of hypomethylated regions in five human cell types is approximately 2 kb (Su et al., 2012) and the average distance between neighboring CpG sites in the medaka genome is 53.5 bases, although this constraint should be adjusted according to each individual situation. The possibility of the hypermethylation (hypomethylation, respectively) of A increases with a larger positive (negative) value of , as well as for a larger total coverage,
A with a larger magnitude of is better for prediction.
3.4 Decomposing the genome into hypomethylated/hypermethylated CpG blocks
Now, we must consider how to decompose n CpG sites in the whole genome into hypermethylated regions and hypomethylated regions such that all regions are disjoint from each other, their union covers all CpG sites, and the two types of regions occur alternatingly along the genome. We calculate the optimal decomposition of regions that maximizes the following objective function:
To simplify this problem, we here mention one important characteristic of SMRT sequencing, that is, read coverage is not affected by the sequence composition (Bashir et al., 2012; English et al., 2012; Koren et al., 2012; Loomis et al., 2012; Zhang et al., 2012). Thus, the average coverage in A is constant at any position within 10 bp relative to CpGs. Technically, we can assume that the average of coverages at the jth position around all the CpG sites in region A is a constant that is dependent of A but is independent of j:
where denotes the number of CpG sites in A. This allows us to transform w(A) into a simpler form:
Subsequently, we simplify the objective function:
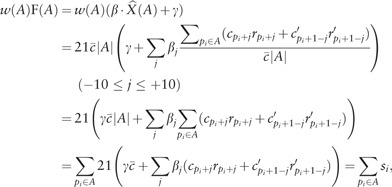 |
where si denotes
Finally, the objective function is a linear combination of si:
Although we set si to a score calculated from weighted IPDR information, we can set si to a log-likelihood function of the form for some likelihood function Qi. This simple form motivates us to design an O(n)-time dynamic programming algorithm for calculating the optimal value efficiently. We consider the sub-problem involving the first i CpG sites among all n sites, and let and be the maximum values of the objective function when the last ith CpG site is methylated and unmethylated, respectively. and meet the following recurrences:
The first max term implies extension of the running region by one CpG site, while the second term means a switch from the previous methylation status and the initiation of a new region with ≥ b CpG sites. For example, we can set b to 50, but one can change the requirement for the minimum number of CpG sites in a region by making an appropriate adjustment to the second term. Of and, the larger value gives the maximum value, and tracing back the optimal path from the maximum value provides all the boundaries between neighboring methylated and unmethylated regions. To calculate regions satisfying the constraint on the minimum number of CpG sites, we generalized the dynamic programming idea proposed by Csűrös (2004). One might wonder if the hidden Markov Model (HMM) can be used for computing hypomethylated and hypermethylated regions; however, it is not obvious that using HMM guarantees the requirement that each range has CpG sites.
4 Results
4.1 SMRT sequencing and bisulfite data benchmark
We collected 31.06-fold coverage SMRT subreads from the testes of medaka Hd-rR (assuming an estimated genome size of 800 Mb) using P6-C4 reagents (Supplementary Methods). We also collected 22.45-fold and 13.06-fold coverage SMRT reads from human peripheral blood of two Japanese individuals. Thus, we have three datasets in total, 1 for medaka and 2 for human. For sequencing two human samples, we employed the P6-C4 reagents and the P4-C2 or C2-C2 reagents, respectively (Supplementary Methods). In total 2848641, 7279594 and 19115712 subreads mapped to the medaka genome and the human genome, respectively. The mean mapped sub-read lengths were 8722 bases for medaka and 9254 and 2049 bases for 2 human samples (Supplementary Table S1).
As CpG methylation status reference data, we used the testes methylome of the medaka Hd-rR inbred strain by way of Illumina bisulfite sequencing (Qu et al., 2012). In this dataset, most of the CpG sites in the medaka genome are either unmethylated or methylated, and methylation at non-CpG sites is very rare (∼0.02%), allowing us to focus on CpG sites only. We evaluated the prediction accuracy of our integration method using the methylation scores calculated from bisulfite-treated Illumina reads as the answer set. Let S be the set of bisulfite-treated Illumina reads covering the ith CpG site, x be the number of methylated CpGs in S at i, and y be the coverage of S at i (the size of S). We then defined the methylation status as ‘unmethylated’ if the score x/y was less than 0.5; otherwise, it was defined as ‘methylated’. We need to carefully constrain the value of the coverage y. Allowing a lower value of y is likely to produce more erroneous methylation scores, while using y greater than a higher threshold would reduce the number of CpGs associated with their methylation scores. The average coverage was 9.40-fold in our bisulfite-treated reads collected from testes of the Hd-rR medaka inbred strain; however, the coverage at individual CpG sites varied to some extent. We defined the methylation score only when the CpG site was covered by 10 or more reads (i.e. ) in order to make sure the scores were robust enough.
4.2 Computational performance
Our linear-time algorithm allows us to handle vertebrate-scale genomes with millions of CpG sites in a reasonable amount of time. It took 2.265 s on average to process 1 Mbp (1191 s to handle 525.7 Mb of medaka genome v.1) using a laptop PC (Intel i7-3612QM processor with a clock rate of 2.10 GHz and 7.8 GB of main memory).
4.3 Predicting the methylation state from kinetic data
We implemented our method using linear discrimination of the vectors of (average) IPDR profiles around the CpG sites. We represented the vectors as points residing in the Euclidean space of the appropriate dimension and attempted to separate the points by a decision hyperplane (Fig. 1A). For better accuracy, we optimized two parameters of the decision hyperplane, the orientation and the intercept. Supplementary Figure S1A (for P6-C4 reagents) and D (for P4-C2 reagents) shows the optimized orientation. Our method divides the genome into regions containing CpG sites, such that each region is either hypomethylated or hypermethylated. While setting lower bound b to 50 is supported by the plausible heuristics with biological grounds, a looser bound (b < 50) allows us to detect shorter regions. We, therefore, examined when we could use a smaller value of without degrading the accuracy of prediction.
We predicted the methylation status of each CpG site by checking whether the CpG site was located in an hypomethylated or hypermethylated region output by our method. We measured the accuracy of the prediction by checking the consistency between the prediction and the methylation score associated with each CpG site. CpG sites without methylation score (due to the lack of bisulfite-treated reads) were ignored. We treat an unmethylated status as positive and a methylated status as negative because we are more interested in identifying rare hypomethylated regions accounting for a small portion (e.g. ∼10%) of CpG sites.
To evaluate the accuracy of our method, we used the chromosome 1 of length 34 959 811 bp in the medaka genome (version 2) that we assembled from SMRT sub-reads. For predicting CpG methylation accurately, we guaranteed that each CpG site was covered by at least three sub-reads, and set the coverage to 0 otherwise, which slightly reduced the original average read coverage, 31.06-fold, to 29.9-fold on the chromosome 1. We calculated various accuracy measures, such as sensitivity (recall), specificity (1–false-positive rate) and precision by comparing our prediction on each CpG site with the methylation level determined in a bisulfite sequencing study (Qu et al., 2012). As most CpG sites in the medaka genome are methylated consistently, there are only a small number of positive examples of unmethylated CpGs, and therefore, precision is more informative than specificity in evaluation. We made the tradeoff between sensitivity and precision through the selection of the intercept of the decision hyperplane () (Fig. 2A and Supplementary Figs. S2–S3). When we used 100% of 29.9-fold sub-reads, setting b to 35 outperformed the other values (Fig. 2A). Our prediction achieved 93.7% sensitivity and 93.9% precision, or 93.0% sensitivity and 94.9% precision, depending on the selection of the intercept. To examine the coverage effect, we used five subread sets of coverage 20, 40, 60, 80 and 100% of 29.9-fold. For coverages of 20 and 40% of 29.9-fold, setting b to 50 performed best (Supplementary Fig. S3). Both sensitivity and precision were for b = 45 even if the coverage is relatively small, 60% of 29.9-fold (Supplementary Fig. S3C). In selecting b, it was suggested to use a larger value (b = 50) when the read coverage is small (15–20-fold) so that the cumulative coverage (750–1000-fold) is large enough. One can use a smaller value (b = 35) with sufficient read coverage (∼30-fold), and b can be decreased gradually with deeper coverage. Setting b to 1 corresponds to the case where the methylation state of each CpG is predicted independently, but it could not achieve a good accuracy, which confirmed the merit of our aggregating approach (Supplementary Fig. S3F). The ROC curve, the tradeoff between false-positive rate and sensitivity, is also shown in Figure 2B. Overall, sensitivity and precision of our method are substantially high using a reasonable coverage of SMRT subreads.
Fig. 2.
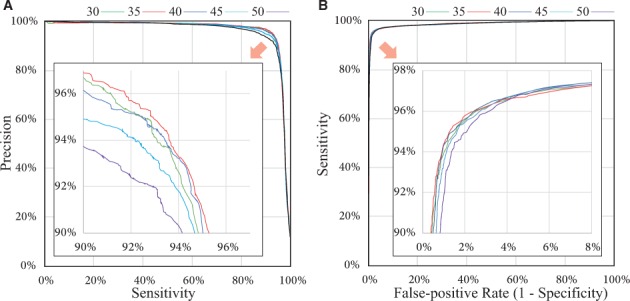
Accuracy of our method. (A) The sensitivity and precision (proportion of true-positives among the predicted positives) are evaluated on individual CpG sites when we change the intercept of the hyperplane (between -8.0 and 5.0) and set the minimum number of CpG sites in a region, b, to 30, 35, 40, 45 and 50. (B) The ROC curve of false-positive rate and sensitivity
4.4 Handling intermediate methylation states
We have introduced the two-class model of our prediction that assigns all of the CpG sites into either hypomethylated or hypermethylated regions; however, such a dichotomous model is rather unrealistic, and more refined predictions involving multi-level methylation states or even continuous methylation levels are desirable. For example, an intermediate level of CpG methylation could result from the distinct methylation states of two DNA molecules of diploid cells, although each cytosine must be either methylated or unmethylated in a single DNA molecule. More generally, a cell population can be epigenetically heterogeneous, which would possibly show a spectrum of methylation levels according to its composition. Finally, prediction allowing intermediate states can represent the ambiguity of the prediction, and exclusion of such ambiguous predictions should improve the overall prediction accuracy.
Thus we extended our method in order to achieve more informative multi-class prediction and quantify the methylation level of each CpG, which we call discrete methylation level (DML, Supplementary Methods). Specifically, DML is calculated as the average prediction over the set of 10 parameters with different sensitivity-specificity combinations, thus it measures the robustness of the prediction. We checked the accordance between our DML and intermediate or ambiguous methylation level captured by two other quantitative methods, bisulfite sequencing and Illumina BeadChip. On the medaka sample, we observed a strong correlation () between our DML and methylation level calculated from bisulfite sequencing (Supplementary Fig. S4C and E), and we confirmed that measurements on 92.0% of CpG sites were in concordance within an acceptable difference 0.25. We also compared our DML on the human sample to the beta value (an indicator of methylation level expressed as a value ranging over [0,1]) obtained from Illumina BeadChip after normalizing the beta values (Supplementary Methods). We observed a weaker correlation (, Supplementary Fig. S4D) and a smaller fraction (75.4%) of CpG sites in concordance within 0.25 presumably because the beta value is less quantitative than the methylation level calculated from bisulfite sequencing (Wang et al., 2015). With the sequencing depth in our case, CpG sites with intermediate methylation were more difficult to predict than completely methylated/unmethylated cases (Supplementary Fig. S4E). Therefore, excluding the prediction with intermediate levels improved the accuracy of the binary prediction (Supplementary Table S2). We concluded that DML serves to reflect the quantitative nature of methylation status in the samples to some extent, and is informative in achieving more accurate prediction as well.
4.5 Genome-wide methylation pattern of repetitive elements in the human genome
We investigated how individual occurrences of repetitive elements (REs) were methylated in the human genome (Fig. 3A). Since some occurrences of REs contain no or very few CpG sites, we only consider those occurrences with at least 10 CpGs to exclude less informative cases. First, we checked whether SMRT reads could address the repetitive regions in a useful manner for methylation analysis. Specifically, we considered a repeat occurrence to be covered by uniquely mapped SMRT reads if the IPD ratio was available on 50% of CpGs. We found that >96% were covered for every repeat type. To draw robust conclusions, we further applied a stringent quality control to each repeat occurrence so that the average read coverage be >5. Although this step reduced the number of repeat occurrences to be analyzed by 3–18%, this could be mitigated simply by producing more data. Finally, we treated an occurrence as hypomethylated if 50% of CpGs were predicted as unmethylated. Similarly, we considered an occurence as methylated intermediately if 50% of CpGs were predicted as 0.3–0.7 in DML measurement. Fractions of hypomethylated repeat occurrences vary considerably among different classes of REs, from ∼1% for L1 and Alu to ∼50% for MIR and >70% for simple repeats and low-complexity regions. The fraction of intermediately methylated repeats was 1.4% among all the repeat classes.
Fig. 3.

Epigenetic landscape of repetitive elements in the human genome. (A) The table shows a summary of methylation status on repetitive elements (REs) that we select using the Repeat Library 20140131 (Smit, A., Hubley, R. and Green, P. Repeatmasker open-4.0 at http://www.repeatmasker.org). (B, C) Distribution of CpG density (B) and sequence divergence from the representative in each repeat class (C) for methylated (lower box) and hypomethylated (upper box) repeat occurrences. The asterisks indicate statistical significance (P < 1%) determined by the U-test. (D) Genome-wide distribution of hypomethylated REs. The ratio of hypomethylated repeat occurrences to all occurrences in each 5-Mb bin is indicated by color shadings. We used the Ideographica web server to generate the image (Color version of this figure is available at Bioinformatics online.)
To validate our prediction regarding the repeat occurrences, we selected 21 regions for bisulfite Sanger sequencing, designed primers for nested PCR (Supplementary Table S3), and could amplify six regions (Supplementary Methods), indicating the difficulty in observing DNA methylation of REs using traditional bisulfite Sanger sequencing. In five (1 L1, 3 LTRs, 1 MIR) among the six amplified regions, we confirmed the consistency between our prediction and the methylation state observed by bisulfite Sanger sequencing (Supplementary Fig. S5). The other one L1 element was predicted hypomethylated. In this region, however, five unmethylated CpG sites were followed by five methylated CpG sites, which showed our method was not reliable in determining the precise boundary and the individual calls should be interpreted carefully.
We then examined the features for characterizing the differences between hypermethylated and hypomethylated REs. First, CpG density was significantly higher in the hypomethylated occurrences in almost all classes of REs (P < 1%, Fig. 3B). This observation was consistent with the known association between CpG-rich regions and unmethylation because methylation leads to depletion of CpG sites through deamination (Cooper and Krawczak, 1989). Second, sequence divergence from the representative in each repeat class showed a correlation with methylation status (Fig. 3C). For most classes, with the apparent exception of simple repeats, low-complexity regions and MIR elements, hypomethylated occurrences were significantly more divergent than were hypermethylated occurrences (P < 1%, Fig. 3C), presumably because younger copies of a repeat element are less divergent and are likely to be targets of DNA methylation. Kernel principal component analysis (PCA) using spectrum kernel suggested, for some repeat types, that the methylation statuses were correlated partly with sequence features (Supplementary Fig. S6).
Next, we examined whether the hypomethylated repeat occurrences were distributed uniformly or non-uniformly throughout the entire genome. We selected three major classes (LINE, Alu and LTR) of REs for this analysis. We calculated the ratios of hypomethylated copies to all REs in individual non-overlapping bins 5 Mb in size (Fig. 3D). The non-random distribution patterns were more evident for LINE and LTR than for Alu. For example, we found hypomethylated LINEs to be enriched in the p-arm of chromosome 1 and in chromosomes 17 and 19. There were hypomethylation ‘hot spots’ of LTR elements, e.g. in chromosomes 6 and 9 (Supplementary Fig. S7). It is intriguing that some of these hypomethylation hot spots, such as those in the p-arms of chromosomes 6 and Y, seem to be shared among different classes of REs.
We further investigated the methylation states of LINE/L1 elements, the only known active autonomous retrotransposons in mammals (Furano, 2000). Although most of LINE/L1 insertions contain many mutations, Penzkofer et al. (2005) categorize full-length L1 elements into three classes according to the conservation of two open reading frames (ORFs); namely (i) L1s with intact in the two ORFs that are likely to exhibit retro-transposition activity, (ii) L1s with an intact ORF2 but a disrupted ORF1 and (iii) non-intact L1s with two ORFs disrupted. We obtained the positions of these human LINE/L1 elements from L1Base (Penzkofer et al., 2005) and analyzed their methylation stateses (Supplementary Table S4). Although 0.61% of non-intact L1s were hypomethylated, all of L1s with intact in two ORFs and L1s with an intact ORF2 were hypermethylated. We also checked the presence of LINE insertions that were novel to the hg19 reference genome. We assembled the SMRT reads using the FALCON assembler and searched the assembly for novel LINE insertions that matched a hot L1 element (GenBank: M80343.1) of size 6050 bp with identity . The hot L1 element was used as the representative according to the procedure of L1Base (Penzkofer et al., 2005). We identified two novel instances covered by sufficient depth of SMRT reads that allowed us to call their methylation statuses confidently. Both of the two LINE insertions (their locations are in Supplementary Fig. S8) were estimated to be methylated. These results confirmed putatively active LINE/L1 elements with intact ORFs were preferentially methylated.
4.6 Tol2 transposable element in medaka
Medaka has an innate autonomous transposon known as Tol2, which is one of the first examples of autonomous transposons in vertebrate genomes and a useful tool for genetic engineering of vertebrates, such as zebrafish and mice (Kawakami, 2007). The excision activities of Tol2 are promoted when DNA methylation is reduced by 5-azacytidine treatment, which suggests that DNA methylation is one of the mechanisms regulating the Tol2 transposition (Iida et al., 2006). Nevertheless, observing the methylation status of each Tol2 copy using short reads is difficult, because Tol2 is 4682 b in length, and ∼20 highly similar copies of Tol2 exist in the genome (Koga et al., 2000).
To elucidate the methylation status of each Tol2 copy, we applied our method to a new assembly of the Hd-rR genome obtained exclusively from SMRT reads. BLAST search identified 17 copies of Tol2 contained entirely within this assembly, all of which were essentially identical (>99.8% sequence identity). We then called the methylation status of these Tol2. For comparison, we mapped the publicly available bisulfite-treated reads from the testes of the Hd-rR strain to these contigs and determined the methylation level on every 100-bp window using Bismark software.
The methylation status of these Tol2, observed by SMRT reads and bisulfite-sequencing, are shown in Figure 4. While virtually no Tol2 copies were mapped by bisulfite reads, as expected from their extremely high fidelity, 16 of 17 copies were anchored by SMRT reads, and all were predicted to be hypermethylated by our method. For the regions examined by both SMRT reads and bisulfite-treated short reads, our prediction was consistent with the methylation level calculated from the bisulfite-treated reads. For example, one Tol2 copy was surrounded by hypomethylated regions (number 14). From the bisulfite data, it appeared that the body of Tol2, from which data were missing, was hypomethylated. Nevertheless, our prediction estimated this region to be hypermethylated. These results demonstrate the ability of our method using SMRT reads to clarify DNA methylation states of highly identical REs such as active transposons.
Fig. 4.

Methylation analysis of Tol2, a 4682-bp long autonomous transposon, in medaka. The new genome assembly of SMRT reads had 17 regions (contigs) that contained complete Tol2 copies. The circles show our prediction of the methylation state of CpG sites, while the rectangles show the methylation states within each 100 bp window obtained from bisulfite sequencing. For both tracks, open indicates unmethylation and filled indicates methylation. The arrow above indicates the region of Tol2 insertions. As the eleventh 11th region was located at the extreme of the contig, Tol2 was not observed successfully by either SMRT sequencing or bisulfite sequencing. For the other 16 regions, methylation of Tol2 was observed consistently by SMRT sequencing, while virtually no information was available on the Tol2 region from bisulfite sequencing (Color version of this figure is available at Bioinformatics online.)
5 Discussion
In this study, we addressed the problem of uncovering the landscape of DNA methylation of repetitive elements (REs). To this end, we developed a unique application of SMRT sequencing to epigenetics. This direction had been already explored in the research community for bacterial and viral species. However, this application in large vertebrate genomes has been largely unexplored because of the subtle cytosine methylation signals in the kinetic information. Therefore, we proposed a new method to utilize relatively small amounts of kinetic information by incorporating a model reflecting our prior knowledge on the regional patterns of CpG methylation of vertebrate genomes. We confirmed the validity of our strategy by comparing the prediction to bisulfite sequencing data on medaka and to BeadChip analysis on human samples. These two datasets had very different characteristics, which seemed to be partly because of the methods used (i.e. BeadChip was designed to observe mainly CpG islands that are often hypomethylated, while bisulfite sequencing is used for genome-wide methylation analysis) and partly because of the nature of the samples used (i.e. the medaka samples were derived from an inbred strain, while the human samples were from diploid cells). Despite such differences in characteristics, our method using the same parameters performed almost equally well for both datasets. These observations suggested that the choice of parameters is robust for a wide variety of samples, which is a desirable feature for any method. We also presented an extension of our method to accommodate intermediate methylation states, the discrete methylation level (DML) and confirmed a high correlation (R = 0.884) between DML and bisulfite methylation level.
We explored the epigenetic landscape of REs within the human genome. Using the hg19 reference genome is an apparent limitation. By assembling individual personal genomes instead of the reference genome, new insertions of these REs are expected to be found, and such active occurrences should be of interest. Indeed, we detected two novel LINE insertions that were estimated to be methylated. Importantly, the more recent the insertion event, the less divergent it would be from the original copy, and therefore, there would be less likelihood of it being anchored by short reads. In such cases, long SMRT reads shed new light on the ecosystem of active REs in personal human genomes.
We demonstrated the use of long SMRT reads can increase the potential comprehensiveness of the epigenetics study. In addition, our method can substantially reduce laboratory work. For example, in the projects of resequencing or de novo assembly using SMRT sequencing, you can call the methylation statuses of the sample as well, completely in silico, without any additional experiment. This is another important strength compared to conventional bisulfite sequencing or affinity-based assays.
6 Data access
The sequence data (SMRT reads) from the medaka sample are deposited at the NCBI Archive (Accession No. SRP020483). Sequence data from a Japanese individual are available under controlled access through the National Bioscience Database Center (NBDC, accession number JGAS00000000003).
Supplementary Material
Funding
This work was supported in part by JSPS (Grant-in-Aid for JSPS Fellows 15J03645 [YS]), by MEXT (Grants-in-Aid for Scientific Research 22129008 and 23241058 [SM]) and by JST (CREST [SM]).
Conflict of Interest: none declared.
References
- Anway M.D. et al. (2005) Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science, 308, 1466–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir A. et al. (2012) A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol., 30, 701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck C.R. et al. (2010) LINE-1 retrotransposition activity in human genomes. Cell, 141, 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann N.D. et al. (2014) Detecting epigenetic motifs in low coverage and metagenomics settings. BMC Bioinformatics, 15, S16.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C. et al. (2008) Inter-individual variation of DNA methylation and its implications for large-scale epigenome mapping. Nucleic Acids Res., 36, e55.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T.A. et al. (2011) Direct detection and sequencing of damaged DNA bases. Genome Integrity, 2, 10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T.A. et al. (2012) Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res., 40, e29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T. et al. (2013) Enhanced 5-methylcytosine detection in single-molecule, real-time sequencing via tet1 oxidation. BMC Biology, 11, 4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus S.J. et al. (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature, 452, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper D., Krawczak M. (1989) Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum. Genet., 83, 181.. [DOI] [PubMed] [Google Scholar]
- Csűrös M. (2004) Maximum-scoring segment sets. IEEE/ACM Trans. Comput. Biol. Bioinf., 1, 139–150. [DOI] [PubMed] [Google Scholar]
- Eckhardt F. et al. (2006) DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet., 38, 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J. et al. (2009) Real-time DNA sequencing from single polymerase molecules. Science (New York, N.Y.), 323, 133–138. [DOI] [PubMed] [Google Scholar]
- English A.C. et al. (2012) Mind the gap: upgrading genomes with pacific biosciences RS long-read sequencing technology. PLoS ONE, 7, e47768.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G. et al. (2012) Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol, 30, 1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z. et al. (2013) Detecting DNA modifications from SMRT sequencing data by modeling sequence context dependence of polymerase kinetic. PLoS Comput. Biol., 9, e1002935.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg B.A. et al. (2010) Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods, 7, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furano A. (2000) The biological properties and evolutionary dynamics of mammalian line-1 retrotransposons. Prog. Nucleic Acid Res. Mol. Biol., 64, 255–294. [DOI] [PubMed] [Google Scholar]
- Gifford C.A. et al. (2013) Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell, 153, 1149–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier J.L. (2014) Retrotransposition in tumors and brains. Mobile DNA, 5, 11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer E.L. et al. (2015) DNA methylation on n 6-adenine in C. elegans. Cell, 161, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R.A. et al. (2010) Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol., 28, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida A. et al. (2006) Targeted reduction of the DNA methylation level with 5-azacytidine promotes excision of the medaka fish Tol2 transposable element. Genet. Res., 87, 187–193. [DOI] [PubMed] [Google Scholar]
- Kawakami K. (2007) Tol2: a versatile gene transfer vector in vertebrates. Genome Biol., 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga A. et al. (2000) Evidence for recent invasion of the medaka fish genome by the tol2 transposable element. Genetics, 155, 273.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S. et al. (2012) Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol., 30, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach J. et al. (2008) Selective aluminum passivation for targeted immobilization of single DNA polymerase molecules in zero-mode waveguide nanostructures. Proc. Natl Acad. Sci. U. S. A., 105, 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. et al. (2012) Landscape of somatic retrotransposition in human cancers. Science, 337, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R. et al. (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell, 133, 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R. et al. (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature, 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis E.W. et al. (2012) Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile x gene. Genome Res., 23, 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A. et al. (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature, 454, 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. (2010) Epigenetics. The seductive allure of behavioral epigenetics. Science, 329, 24–27. [DOI] [PubMed] [Google Scholar]
- Miura F. et al. (2012) Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res., 40, e136–e136.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaro A. et al. (2011) Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell, 146, 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri A.R. et al. (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature, 435, 903–910. [DOI] [PubMed] [Google Scholar]
- Muotri A.R. et al. (2010) L1 retrotransposition in neurons is modulated by mecp2. Nature, 468, 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nautiyal S. et al. (2010) High-throughput method for analyzing methylation of CpGs in targeted genomic regions. Proc. Natl Acad. Sci. U. S. A., 107, 12587–12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzkofer T. et al. (2005) L1base: from functional annotation to prediction of active line-1 elements. Nucleic Acids Res., 33, D498–D500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu W. et al. (2012) Genome-wide genetic variations are highly correlated with proximal DNA methylation patterns. Genome Res., 22, 1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J.P. et al. (2010) Hypomethylation of repeated DNA sequences in cancer. Epigenomics, 2, 245–269. [DOI] [PubMed] [Google Scholar]
- Schadt E.E. et al. (2012) Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res., 23, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R.J. et al. (2011) Transgenerational epigenetic instability is a source of novel methylation variants. Science (New York, N.Y.), 334, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R. et al. (2010) Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res., 20, 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z.D. et al. (2012) A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature, 484, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J. et al. (2012) CpG_mps: identification of CpG methylation patterns of genomic regions from high-throughput bisulfite sequencing data. Nucleic Acids Res, 41, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio J.M.C. et al. (2014) Extensive transduction of nonrepetitive DNA mediated by l1 retrotransposition in cancer genomes. Science, 345, 1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. et al. (2015) A systematic study of normalization methods for Infinium 450 K methylation data using whole-genome bisulfite sequencing data. Epigenetics, 10, 662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver I.C. et al. (2004) Epigenetic programming by maternal behavior. Nat. Neurosci., 7, 847–854. [DOI] [PubMed] [Google Scholar]
- Wilson A.S. et al. (2007) DNA hypomethylation and human diseases. Biochim. Biophys. Acta, 1775, 138–162. [DOI] [PubMed] [Google Scholar]
- Xie W. et al. (2013) Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell, 153, 1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. et al. (2015) Quantitative and multiplexed DNA methylation analysis using long-read single-molecule real-time bisulfite sequencing (SMRT-BS). BMC Genomics, 16, 350.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A. et al. (2010) Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science, 328, 916–919. [DOI] [PubMed] [Google Scholar]
- Zhang X. et al. (2012) Improving genome assemblies by sequencing PCR products with PacBio. BioTechniques, 53, 61–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.