


Abstract
A whole-cell biosensor utilizing a transcription factor (TF) is an effective tool for sensitive and selective detection of specialty chemicals or anthropogenic molecules, but requires access to an expanded repertoire of TFs. Using homology modeling and ligand docking for binding pocket identification, assisted by conservative mutations in the pocket, we engineered a novel specificity in an Acinetobacter TF, PobR, to ‘sense’ a chemical p-nitrophenol (pNP) and measured the response via a fluorescent protein reporter expressed from a PobR promoter. Out of 107 variants of PobR, four were active when dosed with pNP, with two mutants showing a specificity switch from the native effector 4-hydroxybenzoate (4HB). One of the mutants, pNPmut1 was then used to create a smart microbial cell responding to pNP production from hydrolysis of an insecticide, paraoxon, in a coupled assay involving phosphotriesterase (PTE) enzyme expressed from a separate promoter. We show the fluorescence of the cells correlated with the catalytic efficiency of the PTE variant expressed in each cell. High selectivity between similar molecules (4HB versus pNP), high sensitivity for pNP detection (∼2 μM) and agreement of apo- and holo-structures of PobR scaffold with predetermined computational models are other significant results presented in this work.
INTRODUCTION
A reporter assay utilizing a transcription factor (TF) is a very elegant technique to detect metabolites and intermediates and can be used for optimizing pathways and enzymes (1,2,3). Examples of improved enzymatic function via connecting the product accumulation (input) to a suitable reporter such as fluorescent protein expression (output) have started to emerge (4). The approach is limited by the availability of a specific TF that can ‘sense’ the molecule of interest. A specialty metabolite or a product of engineered enzymatic reaction may not have an available specific TF. Attempts to redesign existing TFs for molecules of interest are underway (5) with several promising outcomes (6,7,8). We previously demonstrated broadened specificity in PobR TF (9) that in addition to the native 4-hydroxybenzoate (4HB) could ‘sense’ 3,4-dihydroxybenzoate (34DHB) as an effector molecule. Conservative mutations were introduced into the predicted inducer binding pocket based on a structure-based homology model. These mutations resulted in a PobR variant capable of responding to 34DHB in a dose-dependent manner while retaining the original function and showing enhancement in sensitivity (9). In this study, we used the pobR gene library designed in our previous work (9), to select for a PobR variant responding to p-nitrophenol (pNP), a product of hydrolysis of chemical agents such as organophosphates (OPs).
OP insecticides, paraoxon (PXN) and parathion are potent inhibitors of the cholinesterase enzyme. Unintentional or deliberate exposure can lead to acute neurotoxic poisoning in humans and animals (10). pNP is one of the products of hydrolysis of these OPs, hence, a whole-cell biosensor for pNP can serve as a sensor for OP degradation. Our microbial library consisted of cells, each carrying a unique variant of pobR gene and a fluorescent reporter under the control of PobR native promoter. After four rounds of fluorescence-activated cell sorting (FACS) of pNP induced cell population for positive response and one round of negative selection of cell population grown in absence of pNP to eliminate constitutive expression, the library fetched four variants that responded to pNP. Interestingly, even though we did not perform any negative design or selection against native activity (negative selection), we identified two variants that showed a complete specificity switch, displayed by their specific detection of pNP and their inability to respond to 4HB (the native effector molecule of PobR), when used in a whole-cell system.
Our whole-cell biosensor for pNP that expressed a PXN-hydrolyzing enzyme, phosphotriesterase (PTE) from Pseudomonas diminuta, hydrolyzed exogenously provided PXN and showed a fluorescence signal that could be correlated with the activity of enzyme variants. Our ‘smart’ cells can ‘catalyze’ and ‘sense’ and are promising for the evolution of organophosphatases as well as other hydrolases that yield pNP as a hydrolytic product from real or surrogate substrates.
MATERIALS AND METHODS
Computational modeling
Structure prediction (11) and ligand docking (12) using Rosetta and DNA library construction have been discussed in our earlier work with 34DHB ligand and PobR TF (9). In this work, we chose PobR double mutant (ΔL141/L220V) called DoubleMut (9) as a scaffold for protein engineering due to its higher sensitivity for native effector molecule 4HB than PobR. In order to predict the structure of the inducer binding domain of the scaffold (DoubleMut-IBD), we used a structural template (PDB code: 2IA2) for homology modeling. The template showed a moderate level of sequence identity (35%) with the scaffold sequence. A total of 20 000 homology models were created and the best scoring model was selected for further computational experiments. For each ligand of interest, 5000 independent docking trajectories were performed in the putative ligand binding pocket of DoubleMut-IBD model and the ‘holo’ model with the best ligand–protein interface score was selected. Partial charges on the molecules were determined using AM1-BCC application in QuacPac package, version 1.3.1 of OpenEye Scientific Software (Santa Fe, NM).
Cloning, culture growth and flow cytometry
PobR cloning, transformation, media, Escherichia coli culture growth and sorting using flow cytometry were performed as described in our previous work (9) with some modifications. Our laboratory attained pobR library had a diversity of ∼14 million, which was approximately 30% of the aimed theoretical library size of 4.5 × 107 and estimated based on the number of transformants obtained experimentally. A plasmid construct (sensor plasmid) consisted of a pobR gene variant and gfp gene (encoding for green fluorescent protein) expressed under the control of PobR promoter. The library was transformed into BL21-Gold(DE3) (Agilent Technologies) competent cells and first grown in 50 ml growth media consisting of Luria-Bertani (LB) and Carbenicillin at 100 μg/ml (Carb100) with at least 109 cells as inoculum. The culture was induced with 200 μM pNP when the culture reached an optical density at 600 nm (OD600) of ∼0.6. The induction was then carried out overnight (14–18 h) at 18°C. Approximately 50 μl culture diluted in 5 ml flow buffer (1 × phosphate buffered saline (PBS) + 1% Sucrose) was then analyzed and sorted for brightest cells using FACSAria III (BD Biosciences) with excitation wavelength at 488 nm and emission measured at 530/30 nm. We passed 60 million cells (∼4-fold of laboratory attained library size of 14 million) through the flow cytometer in the first round and collected top 5% of the bright cells in LB. The sorted cells were recovered by vigorous shaking at 37°C for 30 min and then supplemented with Carb100 and the culture was grown to saturation before made into glycerol stocks (20% glycerol as final concentration) and stored in −80°C. Subsequent rounds, used glycerol stocks from the previous round and diluted 100-fold in 3 ml fresh growth media. In the second round, cells were again induced with 200 μM pNP and 20 million cells were passed through the flow cytometer to collect top 1% based on fluorescence intensity. Selection of bottom 50% out of 20 million cells of uninduced culture in the third round, top 1% of 6 million induced cells (with 200 μM pNP) in fourth round and finally top 1% of 6 million induced cells (with 400 μM pNP) in the fifth round was performed before being plated on solid media (LB + agar + Carb100) and testing the population monoclonally.
Ninety six colonies were picked from the plate and grown in 1 ml growth media in a 96-deep well plate at 37°C under vigorous shaking. After 2 h of growth, 500 μl of culture from each well was moved to another plate with 400 μM pNP as final concentration. The plates were moved to 18°C under vigorous shaking for overnight growth and induction. Next day, 5 μl of culture from each plate (uninduced and induced) was diluted in 200 μl flow buffer and analyzed using LSR II (BD Biosciences) with excitation wavelength at 405 nm and emission measured at 525/50 nm. The top eleven clones were selected based on contrast ratio [(Induced fluorescence signal)/(Uninduced fluorescence signal)], regrown, plasmid extracted and sequenced using primers specific for pobR.
For functional evolution of a pNP inducible PobR variant (pNPmut1), a sensor plasmid library with a theoretical diversity of 5 × 103 and consisting of diversification in the operator region and base insertion or deletion between operator and promoter was achieved by overlapping oligonucleotide polymerase chain reaction (PCR) assembly (13) and transformed into BL21-Gold(DE3) (Agilent Technologies) chemical competent cells. Two rounds of FACS were performed on cells induced with 200 μm pNP and 300 μm pNP in the first and second round respectively. In each round 12 million cells were passed through BD FACSAria III flow cytometer (BD Biosciences) and top 0.5% fluorescent cells were collected. Post second round of sorting, cells were plated on solid media and 95 colonies were picked and grown in 500 μl growth media in a 96-deepwell plate. As a control, cells harboring pNPmut1 sensor plasmid were also grown in a single well in the same plate. Post 2 h of growth at 37°C and under vigorous shaking (1000 rpm in a Micro Bio Shaker), cultures were split into two equal volumes, the first set representing uninduced sample and the second set induced with 300 μM pNP and grown overnight at 18°C under vigorous shaking. After 14 h of growth, 2 μl of the culture was diluted in 200 μl of flow buffer and analyzed using BD LSR II flow cytometer (BD Biosciences) with excitation wavelength at 405 nm and emission read at 525 nm. The top ten clones were selected based on contrast ratio, regrown, plasmid extracted and sequenced using suitable primers for pobR and operator region.
Paraoxon aliquots
PXN was purchased from Sigma-Aldrich. A total of 8 mM working stocks were made by dissolving the whole quantity assigned by the vendor in appropriate volume of water, and stored in −80°C. Freshly thawed aliquots were used for each experiment.
Two plasmid system and paraoxon hydrolysis
Seed cultures were initiated from fresh colonies or glycerol stocks and grown in LB media with Carbanecillin (60–100 μg/ml) and Kanamycin (35–50 μg/ml) overnight at 32°C or for ∼6 h at 37°C, to avoid over saturation of the culture. Seed cultures were diluted 100-fold in fresh media (LB with antibiotics) and grown for 1.5–2 h at 37°C and 200–250 rpm until they reached an OD600 of ∼0.6 and were moved on ice for ∼10 min to slow down the growth in exponential phase while preparation for next step of the assay was carried out. Each culture from ice was then distributed into 96-deep well plates in a volume of 470 μl and remaining 30 μl was compensated using 0.2–0.5 mM Zinc Sulfate, 1 mM of IPTG and 400 μM of PXN as final concentrations. The cells were grown for 14–16 h at 18°C and 1000 rpm in a deep well shaker, before diluted 100-fold in the flow buffer (1 × PBS + 1% Sucrose) and analyzed using a flow cytometer.
Protein cloning, expression and purification for crystallization
The Acinetobacter sp ADP1 pobR gene sequences, the full-length native, the double mutant (DoubleMut), the PNP responsive mutant (pNPmut1) and their effector binding domains, pobR-IBD, DoubleMut-IBD and pNPmut1-IBD, all adapted for E. coli were amplified with KOD DNA polymerase using conditions and reagents provided by the vendor (Novagen, Madison, WI, USA). The genes were cloned into a pMCSG68 vector by using a modified ligation-independent cloning protocol (14,15). The pMCSG68 vector bearing a Tobacco Etch Virus (TEV) protease cleavage site creates a construct with cleavable His6-tag fused into N-terminus of the target protein and adds three artificial residues (Ser-Asn-Ala) on that end. The gene was overexpressed in E. coli BL21 (DE3) carrying plasmids pMAGIC that encode one rare E. coli tRNA [Arg (AGG/AGA)] and pRK1037 (Scientific Reagents, Inc.).
The cells were grown using selenomethionine (SeMet) containing enriched M9 medium and under conditions known to inhibit methionine biosynthesis. The cells were grown at 37°C to an OD600 of ∼0.6 and protein expression was induced with 0.5 mM IPTG at 18°C. The cells were grown overnight with shaking at 18°C. The harvested cells were resuspended in five volumes of lysis buffer (50 mM HEPES pH 8.0, 500 mM NaCl, 20 mM imidazole, 10 mM β-mercaptoethanol and 5% v/v glycerol) and stored at −20°C. Cells were thawed and lysed by sonication after the addition of protease inhibitors (Sigma, P8849) and 1 mg/ml lysozyme. The lysate was clarified by centrifugation at 30 000 × g (RC5C-Plus centrifuge, Sorval) for 60 min, followed by filtration through 0.45 and 0.22 μm in-line filters (Gelman). Immobilized metal affinity chromatography (IMAC-I) using a 5-ml HiTrap Chelating HP column charged with Ni2+ ions followed by buffer-exchange chromatography on a HiPrep 26/10 desalting column (both GE Healthcare Life Sciences) were performed using an ÄKTAxpress system (GE Healthcare Life Sciences). The His6-tag was cleaved using the recombinant TEV protease expressed from the vector pRK508. The protease was added to the target protein in a ratio 1:30 and the mixture was incubated at 4°C for 48 h. Each PobR protein was then purified further using a 5 ml HiTrap Chelating HP column charged with Ni2+ ions. The protein in the flow-through and wash fractions was then dialyzed in a buffer consisting of 20 mM HEPES pH 8.0, 250 mM NaCl and 2 mM dithiothreitol and concentrated to 40 mg/ml using an Amicon Ultra Centrifugal Filter (Millipore).
Protein crystallization and data collection
Initial crystallization screens were set up with Mosquito robot (TTP Labtech) using sitting drop vapor diffusion technique in a 96-well CrystalQuick plate (Greiner) by mixing 0.4 μl of the protein solution with 0.4 μl of crystallization reservoir solution (130 μl in the well) equilibrated at 16°C. Multiple commercially available crystallization matrices including MCSG suites (MCSG1-4, from Anatrace) were screened. The DoubleMut-IBD was crystallized in the condition of MCSG1 F7 (0.1 M sodium/potassium phosphate pH 5.6) and the DoubleMut-IBD–3HB complex was crystallized in the condition of MCSG4 H8 (1.0 M Lithium sulfate 2.0% (w/v) PEG 8000) both with incubation at 16°C. Crystals were flash-cooled in liquid nitrogen with the reservoir solution plus 25% glycerol and 25% ethylene glycol as cryoprotectant for DoubleMut-IBD and DoubleMut-IBD–3HB crystal structure respectively prior to data collection.
Diffraction data were collected at 100 K on an ADSC quantum Q315r charged coupled device detector in the 19-ID beamline of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory. Single wavelength anomalous dispersion (SAD) data near the selenium absorption peak was collected from a SeMet-substituted protein. The diffraction data were processed using HKL3000 suite of programs. Data collection statistics are summarized in Supplementary Table S1.
Structure determination, refinement and analysis
DoubleMut-IBD was solved by SAD method using selenium near absorption peak data (0.97934 Å). All procedures for SAD phasing, phase improvement by density modification and initial protein model building were done using the structure module of HKL3000 software package (16). The mean figure of merit and the phasing power of the phase set for DoubleMut-IBD was 0.272 and 1.61 respectively, for 50–3.33 Å data. After phasing and density modification, HKL Build built 528 out of 534 residues and traced 522 residues. For DoubleMut-IBD–3HB complex, initially SAD phasing was tried but failed due to low figure of merit. Next, molecular replacement was used with the DoubleMut-IBD structure without solvent molecules (water molecules, phosphates, glycerols and a chloride) as a search model. The molecular replacement solution consisted of four protein chains and well-defined extra density, which was fit with a 3-hydroxybenzoate (3HB) later, in the binding pocket for each protein chain.
All models were rebuilt with graphics program COOT (17) and between each cycle of rebuilding, the models were refined by PHENIX (18) against the corresponding highest resolution datasets until they converged. The final refined structure of DoubleMut-IBD contained three protein chains with residues 96–271 (original PobR numbering), four phosphate molecules, six glycerol molecules, a chloride and 118 water molecules with R/Rfree of 0.170/0.215 to 2.31 Å. Similarly, DoubleMut-IBD–3HB contained four protein chains (residues 96–271 for chain A, 99–271 for chain B, 97–271 for chain C and 96–271 for chain D), four 3HB molecules (one per protein chain), six sulfates and 13 well-ordered water molecules with R/Rfree of 0.197/0.258 to 2.96 Å. [The data for DoubleMut-IBD-3HB were collected up to 2.95 Å, however, after removing reflections with negative intensities (by PHENIX), the resolution limit was adjusted to 2.96 Å]. The geometrical properties of the model were assessed by PROCHECK (19) and MolProbity (20). The refinement statistics are summarized in Supplementary Table S1. The atomic coordinates and the experimental structure factors of the apo- DoubleMut-IBD and 3HB bound DoubleMut-IBD have been deposited in the PDB under the codes 5HPF and 5HPI.
RESULTS
Choice of scaffold
A pNP specific TF can be used to design a coupled reporter system to detect hydrolytic reactions with pNP as a leaving group (21). Since pNP and the native effector molecule of PobR, that is 4HB, are similar in size and charge distribution (Figure 1A and B) and since a double mutant of PobR (ΔL141 and L220V), called DoubleMut (9), showed enhanced sensitivity to the native effector molecule, we used DoubleMut as a scaffold for mutagenesis and selection to create a pNP responsive TF.
Figure 1.
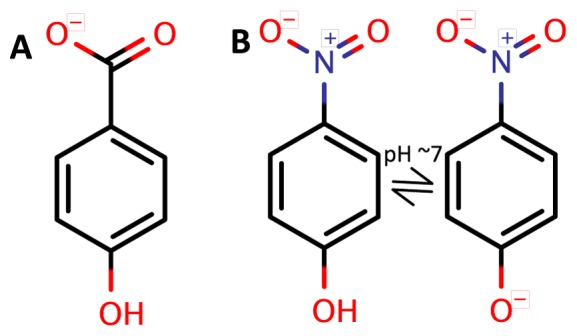
Native and targeted effector molecules for PobR transcription factor (TF). (A) 4HB (native); (B) pNP (targeted). At around neutral pH, pNP is expected to be in equilibrium with a deprotonated phenoxide state.
Homology modeling, ligand docking and structure determination
Homology modeling of DoubleMut as a symmetric homodimer was accomplished as described in our previous work (9) using a putative IclR TF (PDB code: 2IA2) (22) as a structural template. The putative IBD of the structural template showed 35% sequence identity with the DoubleMut sequence. In the IclR template structure, and a few other IclR TFs (23), the biological assembly is a dimer of dimers, forming a functional tetramer. The interactions in the tetramer (of chains A, B, C and D) are complex because the IBD of chain A interacts with that of chain C and similarly the IBDs of B and D interact, while the DNA binding domains (DBDs) of chains A and B form a dimer and the DBDs of C and D form the second dimer that ultimately bind to the tandem sites of the DNA operator (24). In the case of PobR also, the biological assembly is likely to be a dimer of dimers as also indicated by three repeated sequences in the DNAse protected region (25), two repeated sequences occurring as a palindrome and a direct repeated sequence separated by ∼12 bases from the palindrome that is proximal to the promoter region (24). A DBD might be critical to the protein assembly as can be seen from an obligate dimer interface between the DBDs of the template (PDB code: 2IA2) and some other IclR TFs (PDB codes: 2XRO, 2G7U, 3R4K). Our template for homology modeling was an IBD dimer from 2IA2 consisting of chains A and C in near C2 symmetry chosen from a tetrameric biological assembly. Based on the dimer conformation of the template IBDs, the model of DoubleMut was also produced as a dimer with each domain in near C2 symmetry with each other. In order to understand the protein–ligand interaction in each IBD, we used RosettaLigand (12) protocol to dock several aromatic molecules into an expected inducer binding pocket of a selected homology model of the DoubleMut-IBD dimer, where an inter-domain loop (L1) also makes an interaction with the inducer molecule in the model as observed in our previous work (9). Ligand docked DoubleMut-IBD models with 4HB (the native effector molecule of PobR with high sensitivity) (9), 34DHB (a non-native effector molecule with low sensitivity) (9) and pNP (undetectable response at sub-toxic levels for E. coli) were created.
We determined crystal structures of DoubleMut-IBD (residues 96–271) and DoubleMut-IBD in complex with 3-hydroxybenzoate (3HB), a molecule very similar to the native effector molecule, 4HB. Both structures show an asymmetric homodimer. In the IclR tetramer structures, the IBDs interact in a head-to-head fashion and are 2-fold related (24), which is also seen in our template for homology modeling (PDB code: 2IA2). In DoubleMut structure, the IBDs interact head-to-tail and are asymmetric. Nevertheless the α/β rich monomeric IBD crystal structure of DoubleMut, and the DoubleMut homology model closely align over each other with an RMSD of ∼1.8 Å over 175 residues of the IBD sequence and ∼1 Å over the domain excluding loop L1 (Figure 2A). The loop L1 consisting of residues 132–149 in the DoubleMut crystal structure is different from the homology model, where loop L1 also participates in head-to-head homodimerization (9). Structural comparison of apo- and holo-forms of DoubleMut showed distinct conformational changes in side chains (H216, M241) in the binding pocket and perturbed loop L1 (residues 132–152), which is also a part of the pocket (Figure 2B). This suggests that the structure must adjust to accommodate the ligand and consequently could present a changed surface conformation implicating its regulatory role as a TF upon induction.
Figure 2.

DoubleMut-IBD homology model and crystal structure. (A) Monomer of DoubleMut-IBD crystal structure (light gray) overlaid on DoubleMut homology model (black) and the template (PDB code: 2IA2, green). (B) Overlay of apo- (light gray) and holo- (magenta) crystal structures of DoubleMut, showing conformational changes in H216 and M241 side chains and rearrangement of loop L1 (residues 132–152). (C) DoubleMut-3HB crystal structure (magenta) overlaid on pre-determined ligand-docked homology models of DoubleMut with 4HB (light gray), 34DHB (black) and pNP (green). Carboxylate and nitro group of ligands showing polar interaction with backbone of S160, and side chains of T161 and N239 are highlighted with dotted lines. Figures were created using PyMOL (Version 0.99, Schrödinger, LLC).
Ligand-docked DoubleMut models showed placement of each ligand in a similar orientation and in close proximity to each other (Figure 2C). Crystal structure of DoubleMut-IBD with a 3HB molecule closely aligned the aromatic ring of the ligand with our predicted 4HB pose. (4HB is the native effector molecule of PobR). In the crystal structure, the ligand 3HB binds into a well-defined pocket that provides both hydrophobic and hydrophilic interactions. The aromatic ring of the ligand makes hydrophobic interaction with V220, L154 and M241. The carboxylate group of 3HB is positioned against N-terminus of helix α2, which provides partial positive charge to neutralize carboxylate negative charge. This carboxylate group interacts with amide of S160 and forms H-bonds with side chains of S160, T161 and N239 (Figure 2C). The hydroxyl moiety of 3HB interacts with H216 (Figure 2C). Interestingly, the hydroxyl group of the native effector 4HB is most likely hydrogen bonded to E217 located in the short loop between β3 and β4.
A genetically diversified pobR library, sensor plasmid and fluorescence-activated cell sorting
A library consisting of focused diversification of amino acids in the putative ligand binding pocket of PobR-ΔL141 using conservative mutations was accomplished in our previous work (9). In the current work, we found that DoubleMut (PobR-ΔL141/L220V) also presents a similar binding interface for variety of ligands (4HB, 34DHB, pNP). The first shell amino acids for all these ligands are quite consistent with the 16 positions that were selected in our previous work (9) and hence, the diversified library based on conservative mutations (Table 1) finds a direct relevance for optimization of binding for our target molecule pNP. The crystal structure of DoubleMut bound to 3HB that was obtained later further validates the putative pocket chosen and our selection of positions for diversification could be valid for optimization of pNP binding as well.
Table 1. PobR variants and sequence-function relationship.
| Native PobR position | Targeted mutationsb | 4HB specialist DoubleMutc (PobR-wt)d | Generalist 1 | Generalist 2 | pNP specialist pNPmut1 (pNPmut2)d |
|---|---|---|---|---|---|
| 118 | S, T | S | T | T | T |
| 120 | V, M, L | V | V | V | V |
| 127 | V, M, L | V | V | V | V(M) |
| 141 | deletion | Δ(L) | Δ | Δ | Δ |
| 143 | V, I, L, F | V | L | F | L |
| 148 | M, V, L | M | M | M | M |
| 154 | L, M, V | L | L | L | L(M) |
| 159 | T, N, S, I | T | T | T | I |
| 160 | S, T, A | S | T | A | A |
| 161 | T, N, S, I | T | S | T | T |
| 212 | S, T, A | S | S | T | T |
| 216 | H, Y | H | Y | Y | Y |
| 220 | V, M, L | V(L) | V | V | V |
| 222 | A, V, G | A | A | G | G |
| 224 | A, V, G | A | G | G | A |
| 239 | N, D, H | N | N | N | N |
| 241 | M, V, L | M | L | L | L |
| 245a | None | N | N | Y | N |
aPosition 245 was not a targeted position, and the mutation (N→Y) found in one of the variants was most likely an artifact of PCR amplification.
bTheoretical library size based on targeted mutations (excluding the artifact at position 245) is 2 × 3 × 3 × 4 × 3 × 3 × 4 × 3 × 4 × 3 × 2 × 3 × 3 × 3 × 3 × 3 ≈ 45 million. Diversified codons to achieve specific mutations at each position are described in our previous work (9).
cDoubleMut sequence identities are represented in bold characters.
dPobR-wt and pNPmut2 differ from DoubleMut and pNPmut1 respectively at two positions with amino acid identities shown in parentheses.
Our sensor plasmid was derived from pGLO plasmid (Bio-Rad), where araC gene and pBAD promoter were replaced by a diversified pobR gene sequence and the regulatory elements specific for PobR (9). PobR regulated the expression of gfp reporter gene encoding for green fluorescent protein. Theoretical diversity of our library was around 45 million and we had successfully obtained ∼30% of the diversity in our laboratory based on the number of E. coli transformants obtained with the sensor plasmid. Post five rounds of growth and FACS for selection of bacterial cells with maximum fluorescent intensity from an induced population (with 200 μM pNP in the first, second and fourth round and with 400 μM in the fifth round) with inclusion of a negative sorting in the third round to minimize the occurrence of high background phenotypes or constitutive expression, we performed monoclonal evaluation for pNP inducibility. Even though 400 μM pNP was found to be prohibitory due to toxicity of pNP for E. coli resulting in substantially reduced growth (pNP is toxic to the cells beyond 300 μM, with appreciable reduction in cell density over time, even when grown slowly at 18°C), the concentration could be used in later rounds and during monoclonal testing: Toxic concentrations in earlier rounds could have resulted in loss of library diversity and hence was avoided. Out of 96 clones tested monoclonally, top eleven clones were selected based on their contrast ratio [(induced fluorescence signal)/(uninduced fluorescence signal)]. Sequence analyses showed only four unique mutant protein sequences, indicating that another round of sorting was actually not required.
Transcription activity and inducer specificity
Four PobR variants showed a range of constitutive activity. Background signal of the variants was found to be 3- to 15-fold higher in intensity than PobR-wt or DoubleMut background signal. One sequence (Generalist 1 in Table 1) out of the eleven sequenced monoclonals had a maximum constitutive expression and showed response with both 4HB and pNP. The variant was seven mutations away from DoubleMut (4HB-specialist). Another variant (Generalist 2 in Table 1) with eight mutations at the intended positions of DoubleMut (four mutations overlapped with Generalist 1) and an N245Y mutation (an artifact introduced during PCR) was represented in five clones out of the sequenced eleven. This variant showed reduced constitutive activity compared to Generalist 1, low activity with the native effector molecule (4HB) and an appreciable gain in activity with pNP. A complete specificity switch was observed in case of the remaining two sequence variants (one of them represented four times in eleven sequences and another with a single representation), which showed activity with pNP only. One of the pNP-specialists showed eight mutations in the DoubleMut sequence. The other pNP-specialist had two mutations added to the first pNP-specialist and showed visible decrease in background signal but the benefit came at an expense of reduced signal amplitude and thus reduced contrast ratio. We named the first pNP-specialist pNPmut1 and used it for further experimental analyses.
Specificity switch and sensitivity of pNPmut1
The pNP-specialist pNPmut1 showed a specificity switch from 4HB to pNP in terms of transcription activation response to an effector molecule. pNPmut1 showed higher background signal than PobR-wt or DoubleMut (Figure 3A), but the lowest detection concentration was comparable for pNPmut1 (for pNP) and DoubleMut (for 4HB). The lowest detection limit (sensitivity) was ∼1.5 μM for both DoubleMut and pNPmut1 for their respective inducer molecules. At 200 μM concentration of an inducer molecule, the maximum contrast ratio for pNPmut1 was around 18-fold when treated with pNP, while for DoubleMut it was greater than 60-fold in the presence of 4HB, while these variants were non-responsive toward 4HB and pNP, respectively (Figure 3A–C). Ignoring the contrast ratio change for pNPmut1/4HB and DoubleMut/pNP (both of which show a change is <20% above background at the highest concentration), it showed ∼1000-fold switch in specificity from native inducer (4HB) to a non-native molecule (pNP), despite the high similarities between the two inducer molecules. Notably, it took only eight conservative mutations (S118T, V143L, T159I, S160A, S212T, H216Y, A222G, M241L) to completely transform a 4HB responding protein to become selectively sensitive to pNP.
Figure 3.

Engineered specificity switch in PobR TF. (A) Raw green fluorescent protein fluorescence of cells (405 nm excitation and 525 nm emission) expressing different variants of PobR and grown in the presence of pNP. (B) Switched response of PobR variants with 4HB. (C) Contrast ratio [(Induced fluorescence signal)/(Uninduced fluorescence signal)] of cells expressing PobR-wt, DoubleMut or pNPmut1. Error bars represent standard deviation from two sets of experiments performed on different days, sensor plasmid extracted from the cells after first set of experiments and retransformed in fresh competent cells to perform new experiments. (D) Comparison of the background signal (cells grown in the absence of pNP) of the sensor plasmid variants with a mutation in the possible protein dimer interface region (pNPmut1-Int), in the operator region (pNPmut1-O) or both combined (pNPmut1-1). (E) Contrast ratio of different variants of the sensor plasmid. Error bars represent standard deviation from experiments performed in triplicates.
Evolving contrast ratio of pNPmut1
In an attempt to increase the signal contrast ratio of pNPmut1, we created sequence diversification in the promoter proximal operator repeat and a deletion or insertion of one or two bases between the operator and promoter (Supplementary Table S2) based on the experience from our previous work (26). A combinatorial library of diversified plasmids (theoretical diversity 5 × 103) was transformed into E. coli and grown in the presence of pNP. Two rounds of positive selection (top 0.5% of cell population grown at 200 and 300 μM respectively) resulted in an enrichment of sensor plasmid variants with gain-of-function in terms of reduced constitutive activity and increased contrast ratio. Of 95 colonies selected post second round of sorting and plating, ∼75% showed reduced background fluorescence when compared to pNPmut1 and around 50% showed higher contrast ratio than cells harboring pNPmut1 and wild-type intergenic sequence between pNPmut1 and gfp genes. Subsequent sequencing of ten clones with the highest improvement in contrast ratio, showed same sequence for all of them confirming a convergence once again. The sequence had a single base change at the operator and a single unprecedented base change resulting in a mutation aspartate to asparagine at position 139 of PobR sequence. In order to elucidate the contribution of each mutation, we recreated pNPmut1, pNPmut1-O (for a single base change at the operator, A→C), pNPmut1-Int (D139N mutation) and pNPmut1-1 (where both mutations were combined). Very subtle effect was observed for mutation at the operator, which contributed to small but consistent drop in the background signal (Figure 3D). The effect of Asp→Asn mutation was very dramatic, resulting in ∼80% increase in contrast ratio at 100 μM pNP (Figure 3E). Combination of the two mutations gave an additive effect, with lower background signal and higher amplitude, which resulted in an advanced pNP sensor named pNPmut1-1 (Figure 3E).
When we align the DoubleMut-IBD homology model with the IBD of the template 2IA2, D139 is located on the interdomain interface (Supplementary Figure S1). The template is shorter than DoubleMut by four amino acids in loop L1, and a representative amino acid for D139 is absent in the template (Supplementary Figure S2). It is expected that change in charge due to D139N mutation will have strong impact on IBDs interactions, as suggested by a few positively charged residues in the proximity of D139 in the DoubleMut-IBD homology model (Supplementary Figure S1). When we look at the crystal structure of Pseudomonas putida IclR complex with DNA (PDB code: 2XRO) (27), an equivalent residue to D139 in 2XRO is E127, though this TF also shows shorter L1 loop. E127 in 2XRO forms an intradomain salt bridge with R131. In DoubleMut-IBD, D139 and R132 can form a similar intradomain salt bridge as found in the crystal structure, (however, not in the homology model) (Supplementary Figure S3).
Creation of ‘smart’ cells that can catalyze and sense hydrolysis of paraoxon
One of our objectives was to use our whole-cell biosensor to execute hydrolysis of the OP insecticide paraoxon (PXN) and to sense the product formation with a correlated fluorescence response, which can be detected using a flow cytometer. In order to create this ‘smart’ cell, we double transformed E. coli cells with the sensor plasmid and another plasmid consisting of a gene for PTE enzyme from Pseudomonas diminuta (28) and under the control of a T7 promoter and lacI regulation. With an experimental set up as shown (Figure 4A and Supplementary Figure S4), we observed development of a ‘yellow color’ (characteristic of pNP) in the liquid culture upon hydrolysis of PXN (Figure 4B). This was accompanied by a gain in cell fluorescence intensity due to the activation by pNPmut1-1 regulator of the gfp gene present on the sensor plasmid. Interestingly, the gain in cell fluorescence could also be correlated with IPTG concentration in the media, which controlled the expression level of PTE in the cell (Supplementary Figure S5). A variant of PTE with two mutations distal from the enzyme active site, K185R/I274N, shown to improve the whole cell activity (29) probably due to increase in expression level of the PTE enzyme or subtle structural perturbation beneficial to function, also showed higher cell fluorescence intensity compared to the wild-type PTE (Supplementary Figure S6). In order to estimate the activity range of PTE, which could be detected using the ‘smart’ cell, we created three variants of PTEK185R/I274N (from here onward called Native), by mutating F306 to histidine, lysine or glutamate. These mutants, named F306H, F306K and F306E were earlier shown to have a catalytic efficiency in a decreasing order (105, 104, 103 M−1s−1 respectively, while Native activity is 107 M−1s−1) when zinc is used as a divalent metal cofactor (30). When these mutants were expressed in a ‘smart’ cell, their respective background fluorescence intensity was indistinguishable (Figure 4C, top panel). When PXN substrate was added to the media, each mutant population showed very distinct cell fluorescence depending on the activity of enzyme inside the cell. In case of Native PTE, the contrast ratio [(Fluorescence intensity in the presence of PXN)/(Fluorescence intensity in the absence of PXN)] was ∼30, while the F306H variant showed a contrast ratio of ∼6 (Figure 4C, bottom panel). Weaker variants of PTE, namely F306K and F306E, showed <2-fold change in fluorescence intensity and were indistinguishable from each other, even though their published catalytic efficiencies differ by an order of magnitude (30). An important point to note here is that the enzymatic activity of ‘smart’ cells is a combined effect of the folded enzyme quantity and the catalytic efficiency, and a mutation can result in poorer expression or slower folding rate of an enzyme masking the difference in catalytic efficiencies measured in vitro.
Figure 4.

‘Smart’ microbial cells that hydrolyze paraoxon (PXN) and sense the product pNP. (A) Two plasmid system consisting of an Escherichia coli cell with a ‘sensor plasmid’ and an ‘enzyme plasmid’. Functioning of a smart cell is shown in steps I–VIII. IPTG induced expression of a phosphotriesterase (PTE) enzyme (steps I-III) hydrolyses exogenously supplied PXN substrate (steps IV–V), releasing pNP that activates the engineered TF, pNPmut1-1 (step VI) to express the reporter gene, gfp (step VII). Intracellular green fluorescent protein accumulation results in fluorescence of the cell (step VIII). The figure has been derived from our previous publication (26). (B) Substrate PXN and hydrolysis product pNP. (C) PTE variants (Native, F306E, F306H and F306K) expressed in ‘smart’ cells in the absence of substrate (top panel) and presence of 0.4 mM PXN (bottom panel, showing contrast ratio). Published catalytic efficiency of each PTE variant (30) is displayed in a box. Error bars represent standard deviation from two independent experiments performed on different days and divalent metal concentration between 0.2 and 0.5 mM. (D) Fluorescence histograms of ‘smart’ cells expressing native PTE and grown in the absence and presence of PXN (top panel). Fluorescence histograms of a mix of population with Native % as indicated and F306E, F306H and F306K in equal proportion, grown to an OD (600 nm) of 0.6 and then spiked with IPTG and PXN (bottom panel).
We further probed the response of a mixed population consisting of cells expressing different variants of the PTE enzyme. Cells expressing only Native PTE showed well-resolved fluorescence histograms, when grown in the absence and presence of PXN (Figure 4D, top panel). The contrast ratio of fluorescence intensities was calculated to be ∼30. Population mixes consisting of all PTE variants with Native at 25, 5 and 1% and remaining genotypes in equal proportion, at the time of induction with IPTG and addition of substrate PXN, showed a progressive decrease in mean fluorescence intensity of the population. The peak width at half height in each histogram was indistinguishable from the monoclonal population, confirming the population distribution was homogeneous. Sorting the top 1% population from each histogram using FACS and plating to isolate monoclonals followed by sequencing did not convincingly show the brightest cells consist of genotype of most efficient enzyme (that is Native), demanding for either optimization of the assay conditions or trapping the product pNP intracellularly or extracellularly in order to prevent contamination across cells with varying genotype in a mixed culture.
DISCUSSION
DoubleMut models and experimentally determined crystal structures
We successfully crystallized apo and holo forms of DoubleMut-IBD, and it showed an asymmetric dimer instead of a conventional near symmetric dimer observed in IclR TFs (24) (Supplementary Figure S7). We speculate that the absence of DBD could have resulted in a weaker driving force required to attain native biological assembly. But comparison of monomers of the crystal structure and the homology model shows a very encouraging accuracy at the atomic level. The crystal structure of DoubleMut-IBD overlaid on the DoubleMut model with a low RMSD (0.92 Å over 154 backbone Cα atoms), which is also better than the RMSD of the crystal structure with the template (PDB code: 2IA2) used for homology modeling (1.08 Å over 150 backbone Cα atoms) (Figure 2A). Our accuracy in modeling was further assessed by the crystal structure of 3HB bound to DoubleMut-IBD, which showed the key ligand–protein interactions were conserved in the 4HB docked DoubleMut model. Rosetta modeling for apo structure, determination of ligand binding pocket and the first shell residues chosen for mutagenesis, hence, were all native-like and expedited the protein evolution process circumventing need for any predetermined experimental structures of the scaffold.
Sensitivity of the designed transcription factor
Native PobR and DoubleMut are specialists of 4HB. Though minimum concentration of 4HB for PobR response is ∼25 μM, our engineered TF, DoubleMut shows a high sensitivity with a detection limit of <2 μM (Figure 3B). Our engineered specialist for pNP also shows a high sensitivity to pNP molecule, the lower ‘knee’ of the dose-response curve being <2 μM (Figure 3A). Comparing this sensitivity of pNPmut1 with gain-of-function in other engineered TFs, where an araC mutants showed novel specificity at ∼10 mM of D-arabinose (31), >10 mM of mevalonate (6), >20 mM of ectoine (32), ∼1 mM of TAL (4), qacR variant showed sensitivity to vanillin at ∼100 μM (7), PobR variant at >100 μM of dihydroxybenzoate (9) and LacI variants in the range of 100 μM to >1 mM depending on ligand (8), our engineered TF is 50- to 10 000-fold more sensitive to a non-native molecule.
Permeability of these ligands and actual intracellular concentration of the molecule may result in poor ‘apparent’ sensitivity. For example, in one case, co-expression of a permease resulted in one to two orders of improvement in sensitivity for a new effector molecule (31).
Route from one specialist to another via a generalist
A route to gaining a new function with a loss of native, usually includes a generalist activity and direct conversion of one specialist to another is rare unless a positive selection for new activity is pursued in conjunction with negative selection for elimination of old native activity (33). Our selection scheme for gain of target activity (and elimination of constitutivity) directly picked up both ‘generalists’ and ‘specialists’ without inclusion of any negative selection for native activity. Three variants of PobR shown in Table 1, DoubleMut, Generalist 1 and pNPmut1 show a stepwise change from 4HB response to pNP response. DoubleMut is a 4HB specialist, Generalist 1 has high ‘leaky’ response with broadened specificity for both 4HB and pNP, and finally pNPmut1 showed a lack of sensitivity to 4HB and gain in response to pNP. Though an elaborate mutational analysis is required to rationalize the specific role of each mutation, the stepwise transformation from one specialist to another via two generalists indicates that S118T, V143L, H216Y and M241L could be responsible for ‘seeding’ of broadened specificity, since three out of four mutations were also observed prominently in PobR variants for 34DHB (9), S160A, S212T and A222G could be boosting the pNP response and ultimately T159I mutation would have resulted in a complete specificity switch.
Specificity switch using conservative mutations
It took eight conservative mutations for ∼1000-fold specificity switch from 4HB to pNP. The two effector molecules consist of a single aromatic ring and a single negative charge at physiological pH. While the carboxylate group on 4HB is deprotonated to give a negative charge to the molecule, at physiological pH pNP is in equilibrium with a deprotonated hydroxyl group to give p-nitrophenolate anion. Hence, even though the native and targeted effector molecules (Figure 1) appear similar in size and global charge, they are quite different in terms of local negative charge distribution. This difference could explain the specificity switch we observed in the current work. In an earlier work involving evolution of TetR TF, a specificity switch of >4000-fold was shown between a native effector molecule (tetracycline) and a derivative with three functional groups missing (34). With structural knowledge available for close to a decade (35), it was quite intuitive to evolve TetR for the new effector molecule, by creating space-filling mutations, at the positions that interacted with the tetracycline functional groups. The approach took four rounds of evolution to create a quadruple mutant of TetR and three out of four mutations were actually conservative mutations.
Earlier work with XylR TF showed that while new ligand specificity was introduced to the TF, it was likely that the variant retained its original function (36), a feature we also observed earlier with PobR variant for 34DHB (9). Specificity switch observed in our present work for two similar molecules in that way does form a rare gain-of-function in TF engineering efforts.
‘Smart’ cells that catalyze and sense
We anticipated differential fluorescence intensity in a mixed population with varying genotypes coding for enzymes with variable activity. In our previous work, where the substrate for an enzyme was made intracellularly, we were able to show bimodality in population with contrasting enzymatic activity in a mixed population (26). This helped us enrich the population with more efficient catalyst using flow cytometry. Failure to see non-resolved fluorescence histogram peaks in a mixed population makes us speculate that the residence time of pNP inside a cell is very low and efforts to contain the mass transport using a ‘trap’ or a water-oil emulsion (37) or a gel-shell bead (38) will be necessary to exploit the sensitive pNP sensor for high throughput enzyme activity detection and evolution. Considering pNP is a leaving group in a diverse hydrolytic reactions, the caged ‘smart’ cells will be useful in evolution of several other industrial enzymes (21).
ACCESSION NUMBERS
PDB codes: 5HPF and 5HPI.
Supplementary Material
Acknowledgments
The authors wish to thank members of the Structural Biology Center at Argonne National Laboratory for their help with data collection at the 19-ID beamline. We thank Dr Frank Raushel from TAMU for gift of PTE gene from Pseudomonas diminuta, Dr Robert Williams and Dr Geoffrey Waldo, LANL for guidance throughout the project, Dr David Fox and Dr Andrew Bradbury, LANL for providing reagents and laboratory equipment and Dr Subhendu Chakraborti for initiating crystallography work for the project. We are grateful to OpenEye (http://www.eyesopen.com) for a free license to use their tools.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Defense Threat Reduction Agency [CBCALL12-LS-6-0622 to C.E.M.S.]; LANL Institutional Computing [W13_SynBio to C.E.M.S.]; UC Lab Fees research program [118766 to C. E.M.S.]; National Institutes of Health [GM094585, GM115586 to A.J.]; U.S. Department of Energy, Office of Biological and Environmental Research [DE-AC02-06CH11357 to A. J.]. Funding for open access charge: DTRA [CBCALL12-LS-6-0622].
Conflict of interest statement. Los Alamos National Laboratory is filing a patent application for pNP sensor and its use in evolution of hydrolytic enzymes.
REFERENCES
- 1.Raman S., Rogers J.K., Taylor N.D., Church G.M. Evolution-guided optimization of biosynthetic pathways. Proc. Natl. Acad. Sci. U.S.A. 2014;111:17803–17808. doi: 10.1073/pnas.1409523111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eggeling L., Bott M., Marienhagen J. Novel screening methods—biosensors. Curr. Opin. Biotechnol. 2015;35:30–36. doi: 10.1016/j.copbio.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 3.Rogers J.K., Church G.M. Genetically encoded sensors enable real-time observation of metabolite production. Proc. Natl. Acad. Sci. U.S.A. 2016;113:2388–2393. doi: 10.1073/pnas.1600375113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang S.-Y., Qian S., Akinterinwa O., Frei C.S., Gredell J.A., Cirino P.C. Screening for enhanced triacetic acid lactone production by recombinant Escherichia coli expressing a designed triacetic acid lactone reporter. J. Am. Chem. Soc. 2013;135:10099–10103. doi: 10.1021/ja402654z. [DOI] [PubMed] [Google Scholar]
- 5.Raman S., Taylor N., Genuth N., Fields S., Church G.M. Engineering allostery. Trends Genet. 2014;30:521–528. doi: 10.1016/j.tig.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang S.-Y., Cirino P.C. Design and application of a mevalonate-responsive regulatory protein. Angew. Chem. Int. Ed. 2011;50:1084–1086. doi: 10.1002/anie.201006083. [DOI] [PubMed] [Google Scholar]
- 7.de los Santos E.L.C., Meyerowitz J.T., Mayo S.L., Murray R.M. Engineering transcriptional regulator effector specificity using computational design and in vitro rapid prototyping: developing a vanillin sensor. ACS Synth. Biol. 2016;5:287–295. doi: 10.1021/acssynbio.5b00090. [DOI] [PubMed] [Google Scholar]
- 8.Taylor N.D., Garruss A.S., Moretti R., Chan S., Arbing M.A., Cascio D., Rogers J.K., Isaacs F.J., Kosuri S., Baker D., et al. Engineering an allosteric transcription factor to respond to new ligands. Nat. Methods. 2016;13:177–183. doi: 10.1038/nmeth.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jha R.K., Chakraborti S., Kern T.L., Fox D.T., Strauss C.E.M. Rosetta comparative modeling for library design: engineering alternative inducer specificity in a transcription factor. Proteins Struct. Funct. Bioinforma. 2015;83:1327–1340. doi: 10.1002/prot.24828. [DOI] [PubMed] [Google Scholar]
- 10.Costa L.G. Current issues in organophosphate toxicology. Clin. Chim. Acta. 2006;366:1–13. doi: 10.1016/j.cca.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Raman S., Vernon R., Thompson J., Tyka M., Sadreyev R., Pei J., Kim D., Kellogg E., DiMaio F., Lange O., et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins Struct. Funct. Bioinforma. 2009;77:89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis I.W., Baker D. Rosetta ligand docking with full ligand and receptor flexibility. J. Mol. Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Stemmer W.P.C., Crameri A., Ha K.D., Brennan T.M., Heyneker H.L. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 1995;164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 14.Stols L., Gu M., Dieckman L., Raffen R., Collart F.R., Donnelly M.I. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 15.Kim Y., Dementieva I., Zhou M., Wu R., Lezondra L., Quartey P., Joachimiak G., Korolev O., Li H., Joachimiak A. Automation of protein purification for structural genomics. J. Struct. Funct. Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. HKL -3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 17.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 18.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 20.Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi S.-L., Rha E., Lee S.J., Kim H., Kwon K., Jeong Y.-S., Rhee Y.H., Song J.J., Kim H.-S., Lee S.-G. Toward a generalized and high-throughput enzyme screening system based on artificial genetic circuits. ACS Synth. Biol. 2014;3:163–171. doi: 10.1021/sb400112u. [DOI] [PubMed] [Google Scholar]
- 22.Cymborowski M.T., Chruszcz M., Skarina T., Onopriyenko O., Savchenko A., Edwards A., Joachmiak A., Minor W., Midwest Center for Structural Genomics The crystal structure of a putative transcriptional regulator RHA06195 from Rhodococcus sp RHA1. 2006. doi:10.2210/pdb2ia2/pdb.
- 23.Zhang R., Kim Y., Skarina T., Beasley S., Laskowski R., Arrowsmith C., Edwards A., Joachimiak A., Savchenko A. Crystal structure of thermotoga maritima 0065, a member of the IclR transcriptional factor family. J. Biol. Chem. 2002;277:19183–19190. doi: 10.1074/jbc.M112171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molina-Henares A.J., Krell T., Eugenia Guazzaroni M., Segura A., Ramos J.L. Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 2006;30:157–186. doi: 10.1111/j.1574-6976.2005.00008.x. [DOI] [PubMed] [Google Scholar]
- 25.Dimarco A.A., Ornston L.N. Regulation of p-hydroxybenzoate hydroxylase synthesis by PobR bound to an operator in Acinetobacter calcoaceticus. J. Bacteriol. 1994;176:4277–4284. doi: 10.1128/jb.176.14.4277-4284.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jha R.K., Kern T.L., Fox D.T., Strauss C.E.M. Engineering an Acinetobacter regulon for biosensing and high-throughput enzyme screening in E. coli via flow cytometry. Nucleic Acids Res. 2014;42:8150–8160. doi: 10.1093/nar/gku444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu D., Fillet S., Meng C., Alguel Y., Kloppsteck P., Bergeron J., Krell T., Gallegos M.-T., Ramos J., Zhang X. Crystal structure of TtgV in complex with its DNA operator reveals a general model for cooperative DNA binding of tetrameric gene regulators. Genes Dev. 2010;24:2556–2565. doi: 10.1101/gad.603510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dumas D.P., Caldwell S.R., Wild J.R., Raushel F.M. Purification and properties of the phosphotriesterase from Pseudomonas diminuta. J. Biol. Chem. 1989;264:19659–19665. [PubMed] [Google Scholar]
- 29.Cho C.M.-H., Mulchandani A., Chen W. Functional analysis of organophosphorus hydrolase variants with high degradation activity towards organophosphate pesticides. Protein Eng. Des. Sel. 2006;19:99–105. doi: 10.1093/protein/gzj007. [DOI] [PubMed] [Google Scholar]
- 30.Watkins L.M., Mahoney H.J., McCulloch J.K., Raushel F.M. Augmented hydrolysis of diisopropyl fluorophosphate in engineered mutants of phosphotriesterase. J. Biol. Chem. 1997;272:25596–25601. doi: 10.1074/jbc.272.41.25596. [DOI] [PubMed] [Google Scholar]
- 31.Tang S.-Y., Fazelinia H., Cirino P.C. AraC regulatory protein mutants with altered effector specificity. J. Am. Chem. Soc. 2008;130:5267–5271. doi: 10.1021/ja7109053. [DOI] [PubMed] [Google Scholar]
- 32.Chen W., Zhang S., Jiang P., Yao J., He Y., Chen L., Gui X., Dong Z., Tang S.-Y. Design of an ectoine-responsive AraC mutant and its application in metabolic engineering of ectoine biosynthesis. Metab. Eng. 2015;30:149–155. doi: 10.1016/j.ymben.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Khersonsky O., Tawfik D.S. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 34.Scholz O., Köstner M., Reich M., Gastiger S., Hillen W. Teaching TetR to recognize a new inducer. J. Mol. Biol. 2003;329:217–227. doi: 10.1016/s0022-2836(03)00427-3. [DOI] [PubMed] [Google Scholar]
- 35.Hinrichs W., Kisker C., Duvel M., Muller A., Tovar K., Hillen W., Saenger W. Structure of the Tet repressor-tetracycline complex and regulation of antibiotic resistance. Science. 1994;264:418–420. doi: 10.1126/science.8153629. [DOI] [PubMed] [Google Scholar]
- 36.Galvão T.C., Mencía M., de Lorenzo V. Emergence of novel functions in transcriptional regulators by regression to stem protein types. Mol. Microbiol. 2007;65:907–919. doi: 10.1111/j.1365-2958.2007.05832.x. [DOI] [PubMed] [Google Scholar]
- 37.Agresti J.J., Antipov E., Abate A.R., Ahn K., Rowat A.C., Baret J.-C., Marquez M., Klibanov A.M., Griffiths A.D., Weitz D.A. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl. Acad. Sci. U.S.A. 2010;107:4004–4009. doi: 10.1073/pnas.0910781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischlechner M., Schaerli Y., Mohamed M.F., Patil S., Abell C., Hollfelder F. Evolution of enzyme catalysts caged in biomimetic gel-shell beads. Nat. Chem. 2014;6:791–796. doi: 10.1038/nchem.1996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.