

Abstract
Alternative isoform regulation (AIR) vastly increases transcriptome diversity and plays an important role in numerous biological processes and pathologies. However, the detection and analysis of isoform-level differential regulation is difficult, particularly in the face of complex and incompletely-annotated transcriptomes. Here we have used Illumina short-read/high-throughput RNA-Seq to identify 55 genes that exhibit neurally-regulated AIR in the pineal gland, and then used two other complementary experimental platforms to further study and characterize the Ttc8 gene, which is involved in Bardet-Biedl syndrome and non-syndromic retinitis pigmentosa. Use of the JunctionSeq analysis tool led to the detection of several novel exons and splice junctions in this gene, including two novel alternative transcription start sites which were found to display disproportionately strong neurally-regulated differential expression in several independent experiments. These high-throughput sequencing results were validated and augmented via targeted qPCR and long-read Pacific Biosciences SMRT sequencing. We confirmed the existence of numerous novel splice junctions and the selective upregulation of the two novel start sites. In addition, we identified more than 20 novel isoforms of the Ttc8 gene that are co-expressed in this tissue. By using information from multiple independent platforms we not only greatly reduce the risk of errors, biases, and artifacts influencing our results, we also are able to characterize the regulation and splicing of the Ttc8 gene more deeply and more precisely than would be possible via any single platform. The hybrid method outlined here represents a powerful strategy in the study of the transcriptome.
Introduction
Transcriptome complexity is amplified by alternative isoform regulation (AIR), a broad category of regulatory phenomena which can involve alternative splice sites, alternative transcription start sites, intron retentions, exon skipping, methylation, nucleosome occupancy, internal promoters, nonsense mediated decay, and/or transcript switching. Through these changes, proteins encoded by different transcripts of a single gene can have very different biological functions.
This paper describes an initial effort aimed at understanding AIR in the rat pineal gland, the neuroendocrine structure responsible for the 24-hour rhythm in melatonin production [1, 2]. The pineal gland evolved from ancestral photoreceptors that also gave rise to the retina; these two organs share a common genetic pattern and develop from the same primordia [3–13].
The pineal transcriptome undergoes robust neurally-regulated circadian changes [5, 14], driven by the endogenous clock in the suprachiasmatic nuclei (SCN). Signals from the SCN pass through a multi-synaptic pathway that includes the superior cervical ganglia (SCG). At night, norepinephrine is released from the SCG projections to the pineal gland, initiating a chain of events that leads to changes in the transcription of thousands of genes [5, 14]. Accordingly, when the SCG is removed (SCGX) or decentralized (DCN) most of these night/day differences are not observed [14, 15]. The night/day changes can be reproduced in organ culture by treating the pineal gland with norepinephrine or an analog of cyclic AMP, a second messenger for norepinephrine in this tissue [5, 14].
In the literature there is evidence suggesting that neurally-regulated alternative isoform regulation may occur in the rat pineal gland. Firstly: a splicing regulator gene, Mbnl2, is strongly upregulated at night [5, 14, 16]. This gene contains an RNA-binding domain [17] and regulates alternative splicing in humans [18, 19], mice [20], zebrafish [21], and drosophila [22]. In addition, several genes have already been found to exhibit neurally-controlled, rhythmic, isoform-specific differentials in the rat pineal gland, including Crem [23–25], Pde4b [26], Slc15a1/Pept1 [1, 27], and Atp7b [28]. Accordingly, further study might reveal additional differentially regulated alternative isoforms of these and other genes, and provide further insight into the molecular biology of the regulatory mechanisms involved.
Although interest in AIR is high, progress in understanding its functional importance has been slowed by the limitations of next-generation high-throughput RNA sequencing technologies (RNA-Seq). A major factor influencing this progress is the low quality of the extant transcript annotations, which is particularly sparse for the rat transcriptome. The Ensembl transcript annotations (release 80) for rat, mouse, and human all have roughly the same number of protein coding genes (22093, 22114, and 22002, respectively). However, only 22% of known genes have more than one known protein coding transcript in the rat annotation, compared with 60% and 84% in mouse and human annotations, respectively. The sparse annotation of the rat transcriptome presents a substantial obstacle to the detection of alternative isoform regulation, as many of the most popular isoform-level tools (including Kallisto [29], eXpress [30], and RSEM [31]) implicitly assume that the supplied annotation is both correct and comprehensive, and generally cannot assess the expression of unknown isoforms.
While some tools such as CuffLinks [32] can rescue some of these unannotated isoforms, these methods are of limited utility due to the inherent difficulty in assembling long transcripts using short read data [33, 34]. This inherent difficulty has become increasingly obvious in recent years with the development of long-read RNA-Seq. These new technologies, including Pacific Biosciences (PacBio) single-molecule real-time (SMRT) sequencing, provide the unprecedented ability to sequence transcripts across their entire length at high coverage depth. Several recent studies using these technologies have shown that in many cases the existing transcript annotations (which are often at least partially based on high-throughput/short-read assemblies) are highly incomplete, missing dozens or even hundreds of expressed transcripts [33, 35–37].
In this paper we apply a multi-platform “hybrid” methodology to study the alternative isoform regulation of the Tetratricopeptide Repeat Domain 8 gene (Ttc8, also known as Bbs8) in the rat pineal gland. Short-read/high-throughput stranded RNA-Seq data from a recently-published study [14] was analyzed using JunctionSeq [38] to detect, assess, and characterize differentially regulated alternative splice variants across the entire genome. Based on these results the Ttc8 gene was selected for further investigation. The short-read RNA-Seq results were validated and augmented using quantitative PCR (qPCR) and SMRT long-read RNA sequencing.
JunctionSeq is a Bioconductor package that detects “differential usage” (DU) of exons and splice junctions, in which individual “sub-features” (exons and splice junctions) display patterns of expression that differ from the overall pattern of read-coverage across their respective genes. The detection of differential usage of these features is intended to serve as a proxy for the detection of differential alternative isoform regulation. JunctionSeq can query novel splice junctions without knowledge of the full-length isoforms to which they belong, and has been shown to provide powerful detection of differential isoform usage even when provided with incomplete annotations [38]. This trait makes it particularly suited for the analysis of the rat pineal transcriptome.
In this study we identified 55 genes that exhibit neurally-regulated differential usage of exons and splice junctions. We focused on the alternative isoform usage of the Ttc8 gene in part because mutations in Ttc8 have been implicated in various ciliopathies including non-syndromic retinitis pigmentosa (RP) [39, 40] and in Bardet-Biedl syndrome (BBS, which commonly includes RP among its symptoms) [41, 42]. At least one form of non-syndromic RP is caused by defective splicing of a retina-specific splice variant in humans [39, 40]. Given the similarities between the pineal gland and retinal cells, it is possible that an in-depth study of alternative splicing in the pineal gland might also shed light onto its biological role in vision.
Although the exact function of the Ttc8 gene is not well-understood, the gene appears to be involved in protein transport in and out of cilia as part of a large complex called the BBSome [43]. Ttc8 proteins are structurally characterized as containing 8 tetratricopeptide repeats (TPR) in the C-terminal half of the protein, forming an extensive protein-protein interaction domain [44, 45].
Our hybrid approach leverages the complementary strengths of our various experimental platforms, offering new insight into the complex isoform-level expression regulation taking place in the Ttc8 gene. This report provides a valuable template for future investigation of such regulatory phenomena.
Results
Detection of differential isoform usage via JunctionSeq
JunctionSeq detected more than 500 genes displaying statistically significant differential exon or splice-junction usage in data from each of two in vivo experimental groups (control and sham) comparing day and night (adjusted p-value < 0.05, see Table 1). In each of the two in vitro experiments (CN vs NE and CN vs DBcAMP), more than 200 genes showed similar statistical significance. We found strong concordance in the genes detected in each of these four analyses, particularly between the two in vivo experiments and the two in vitro experiments (see Table 1 and S1 Fig). Even at the (extremely conservative) adjusted p-value threshold of 0.0001, we found 18 genes that showed statistically significant differential usage in all four experiments (see Table 1 and Table 2). Three of these top genes were already known in the literature to exhibit neurally-controlled alternative isoform regulation.
Table 1. The number of genes detected in each JunctionSeq analysis at various p-value cutoffs.
Also shown is the overlap between the genes detected across multiple separate analyses. Day and night conditions were compared in the four in vivo groups: Control (Ctrl), Sham, decentralized (DCN), and ganglionectomized (SCGX). The two in vitro analyses compared untreated (Untr) pineal glands with pineal glands treated with norepinephrine (NE) or (Untr) vs DBcAMP (DB).
| p-adjust | In Vivo–Normal Day vs Night | In Vitro Untreated vs Treated | Shared All 4 | In Vivo–Neural Stimulus Removed | |||||
|---|---|---|---|---|---|---|---|---|---|
| Ctrl | Sham | Both | Untr/NE | Untr/DB | Both | DCN | SCGX | ||
| 0.05 | 620 | 557 | 224 | 219 | 260 | 120 | 55 | 18 | 50 |
| 0.01 | 394 | 303 | 156 | 140 | 188 | 87 | 40 | 8 | 36 |
| 0.001 | 263 | 189 | 108 | 86 | 124 | 60 | 28 | 4 | 21 |
| 0.0001 | 202 | 136 | 87 | 64 | 88 | 47 | 18 | 2 | 18 |
| 0.00001 | 168 | 109 | 75 | 49 | 73 | 38 | 15 | 2 | 12 |
| 0.000001 | 142 | 94 | 60 | 42 | 65 | 34 | 14 | 2 | 9 |
Table 2. Summary information on the 18 genes detected at p-adjust < 0.0001 in all four analyses.
Column 3 indicates the number of distinct exonic regions (exons), known splice junctions (SJ) and novel splice junctions belonging to this gene, and columns 4–7 indicate the number of each feature type with statistically significant differential usage (# sig, p-adjust < 0.0001). The rightmost column indicates the least significant gene-wise adjusted p-value across the four analyses. *Genes previously known in the literature to exhibit differential isoform regulation between night and day in the rat pineal gland: Crem [23–25], Pde4b [26], and Atp7b [28].
| Gene Symbol | Locus | # Features (exons/ known SJ/ novel SJ) | # Sig. (exons / known SJ / novel SJ) | Least sig. gene-wise p-adjust | |||
|---|---|---|---|---|---|---|---|
| Ctrl Day/Night | Sham Day/Night | Untr vs NE | Untr vs DB | ||||
| Arsg | chr10:97771263-97863311(+) | 12/11/3 | 5/4/1 | 3/3/1 | 4/1/0 | 3/1/0 | 0 |
| Commd1 | chr14:107664254-107760191(-) | 3/2/4 | 1/1/3 | 1/1/3 | 3/1/3 | 2/1/3 | 0 |
| Crem* | chr17:57031765-57090888(+) | 21/15/5 | 19/8/3 | 16/6/2 | 18/9/1 | 19/9/1 | 0 |
| Dclk1 | chr2:144646307-144939389(+) | 21/20/0 | 18/17/0 | 18/17/0 | 16/8/0 | 11/7/0 | 0 |
| Ggcx | chr4:100277390-100293250(+) | 20/16/1 | 7/6/0 | 4/5/1 | 6/2/0 | 6/4/0 | 0 |
| Alox15 | chr10:56953690-56962161(-) | 14/13/10 | 0/1/5 | 0/1/3 | 0/0/1 | 0/1/3 | 4.62E-13 |
| Atp7b* | chr16:74865515-74947084(+) | 23/21/2 | 7/3/0 | 6/3/0 | 8/1/0 | 7/0/0 | 1.13E-12 |
| Ugp2 | chr14:106208175-106249103(-) | 13/10/3 | 1/1/0 | 1/1/0 | 1/1/0 | 1/1/0 | 2.04E-10 |
| Pde4b* | chr5:121952976-122136814(+) | 19/14/1 | 7/1/1 | 6/4/1 | 5/4/1 | 5/3/1 | 9.08E-10 |
| Ttc8 | chr6:122920316-122977132(+) | 18/16/6 | 2/1/3 | 2/1/4 | 1/1/1 | 2/1/1 | 7.26E-08 |
| Tsc22d1 | chr15:58554373-58658153(+) | 5/3/2 | 2/0/1 | 0/0/1 | 0/0/1 | 3/1/1 | 2.24E-07 |
| Eprs | chr13:103300931-103371577(+) | 38/35/2 | 36/26/0 | 21/17/0 | 8/5/0 | 8/5/0 | 2.62E-07 |
| Map3k5 | chr1:15412602-15613746(+) | 32/32/0 | 26/10/0 | 13/6/0 | 4/2/0 | 3/1/0 | 4.43E-07 |
| Btrc | chr1:265157378-265269837(+) | 14/13/1 | 1/0/0 | 2/1/0 | 1/0/0 | 1/0/0 | 6.23E-07 |
| Slc38a1 | chr7:137975548-138039984(-) | 19/16/6 | 0/0/2 | 2/0/2 | 0/0/2 | 0/0/2 | 2.11E-06 |
| Arhgap24 | chr14:8383315-8600424(-) | 10/8/4 | 0/0/1 | 1/0/2 | 0/0/2 | 3/1/3 | 1.54E-05 |
| Cbx6 | chr7:121050301-121058029(-) | 5/4/2 | 5/2/1 | 4/2/0 | 5/3/0 | 5/3/0 | 2.86E-05 |
| Pla2g4a | chr13:66849776-67206693(-) | 20/18/1 | 0/0/1 | 0/0/1 | 0/0/1 | 0/0/1 | 7.00E-05 |
As expected, we found very few genes that displayed significant differential usage in the two “stimulus deprived” experiments (DCN and SCGX, see Table 1); the DCN experiment provided the fewest. The small number of genes detected in the DCN analysis may represent either false discoveries or real night/day differences caused by factors other than neural stimulation via the SCG.
Of the top 18 genes detected by JunctionSeq as exhibiting differential usage in all four neural stimulus experiments, Ttc8 was selected for further validation and investigation, because several of the statistically-significant splice junctions were novel, the effect was strong and consistent in all four experiments, and the gene had been previously identified in the literature as having an alternatively spliced isoform with a strong phenotypic effect in the retina [39, 40].
The complete JunctionSeq results for all six analyses and all genes that displayed significant differential usage can be found in the S4 Dataset and S5 Dataset.
Differential transcription start site usage of the Ttc8 Gene
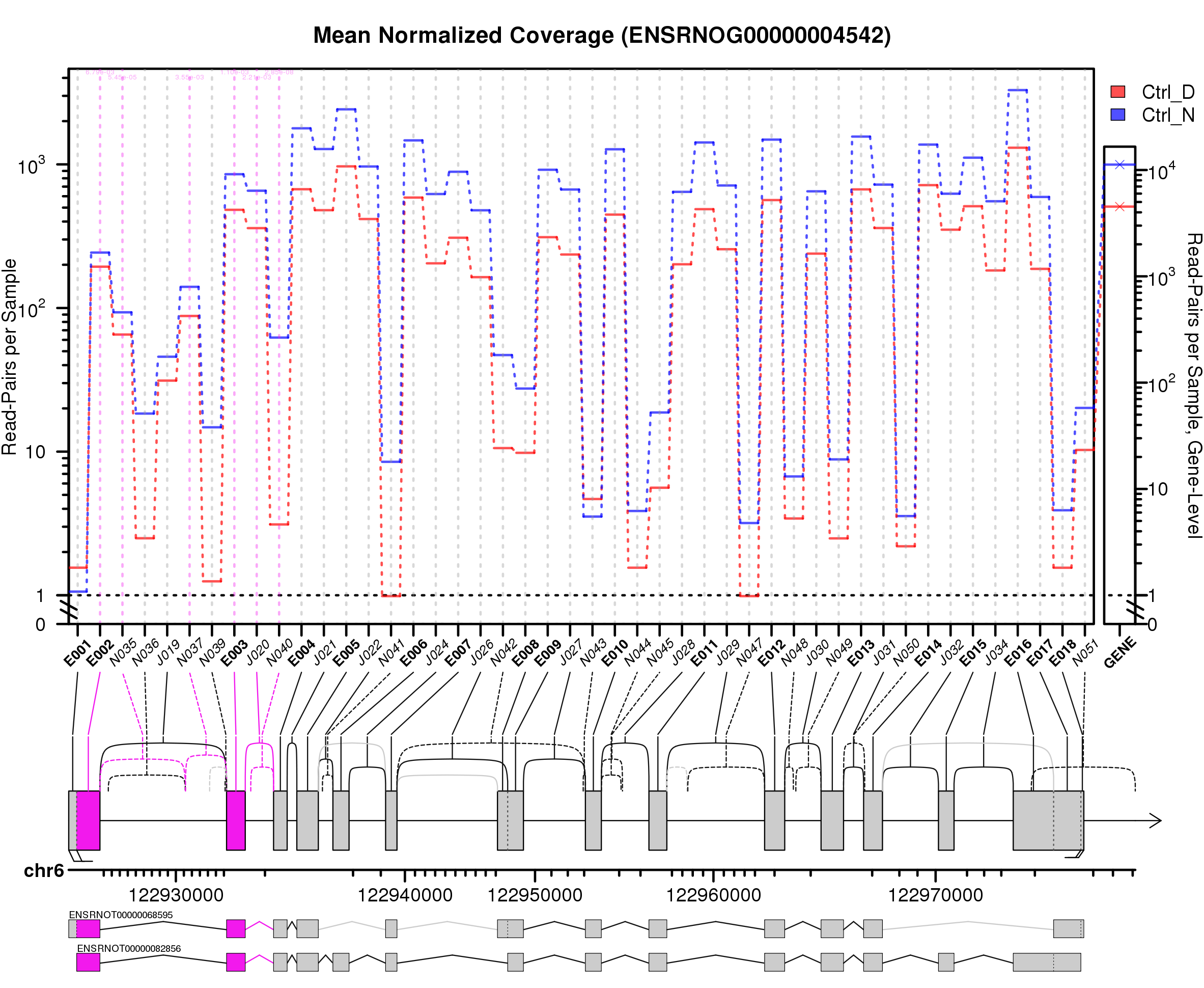
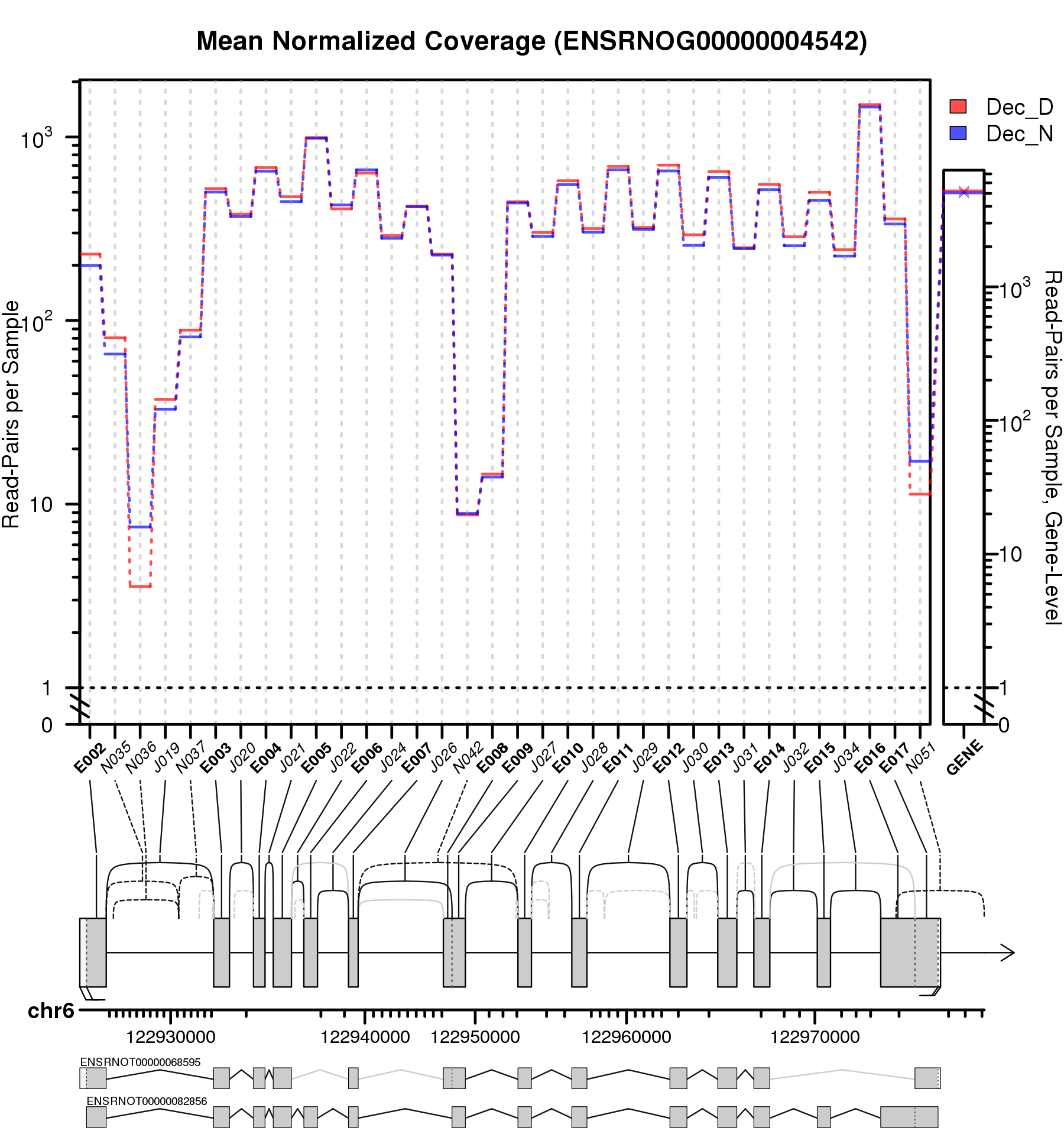
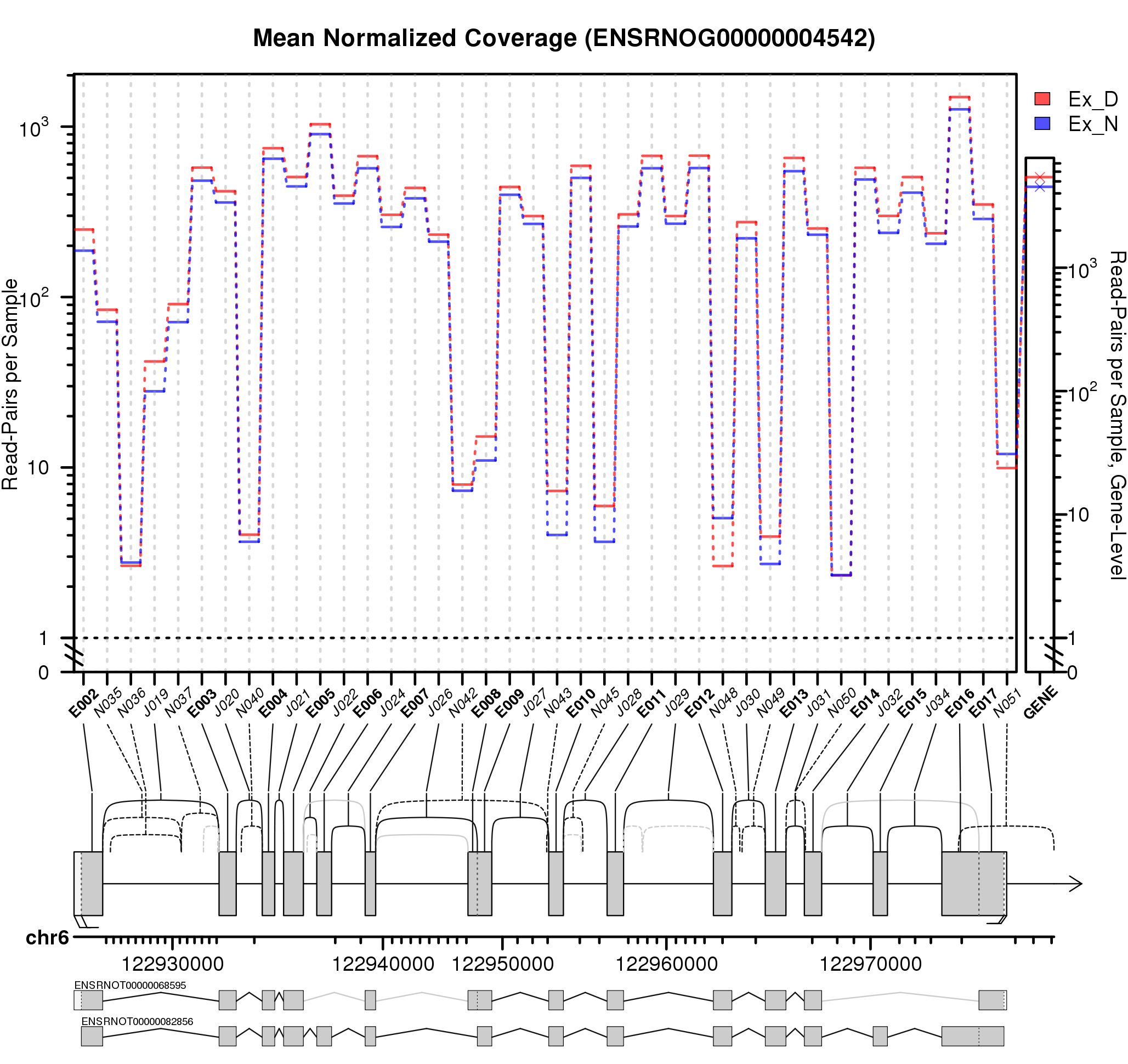
In the Ttc8 gene we detected 17 novel splice junctions that surpassed our initial coverage cutoff, all 17 of which used canonical GT-AG dinucleotide donor-acceptor sites. The following transcript annotation databases were checked to determine whether these exons and splice junctions were novel: Ensembl, RefSeq, the Mammalian gene Collection, and RGD. Only two of these novel splice junctions were present in any of these databases (see S13 Fig, S14 Fig and S15 Fig). Based on the gene profile plots (Fig 1 and Fig 2), we can separate the Ttc8 gene into three distinct regions based on their expression. The known 5’ start site displays roughly constant high-level coverage at both night and day. The novel splice junction N040 (which appears to splice from an alternative 5’ start site) displays an extreme differential with very low coverage during the day (~3 read-pairs per sample) and moderate coverage at night (~60 read-pairs per sample). Finally, the remainder of the gene appears to display a composite of these two, with higher overall coverage, but weaker relative night/day differences than the novel junction N040. Note that none of the novel exons or splice junctions appear in any of the transcript annotations. Note that the Rat Genome Database (RGD) is not included in this plot because it has not yet been lifted over to the rn6 rat genome build. See (S15 Fig) for the RGD annotation.
Fig 1. JunctionSeq gene profile plot for Ttc8 gene, sham night/day experiment.

This plot displays the estimates for the mean normalized read-pair coverage count for each exon and splice junction. The small plotting panel on the far right displays the total mean normalized read-pair count for the gene as a whole. The gene diagram below the main plotting frame displays the exonic regions (boxes, labelled E001-E018), known splice junctions (solid lines, labelled J019-J034) and novel splice junctions (dashed lines, labelled N035-N051). Significant features are drawn in pink, and features that did not pass the automatically-selected minimum count threshold were colored in light grey (and were not plotted). Beneath the gene diagram, the genomic positions are marked with ticks at each kilobase. Note that the scale is not uniform due to the nonlinear expansion of small features to improve readability. Beneath the scale the two Ensembl known isoforms are drawn. The upper isoform is the RefSeq transcript.
Fig 2. Summary browser tracks generated by QoRTs/JunctionSeq for the region surrounding exons 2 to 5 of Ttc8.

The top two tracks display the Ensembl and RefSeq annotations, respectively. The third track displays the names of the exons as they are referred to in the text and in previous literature, with our own names assigned to previously-unidentified exons. The fourth track, generated by QoRTs, displays the mean normalized read-pair coverage depth at each genomic position for night (blue) and day (red) in the sham group. The fifth and sixth tracks, generated by JunctionSeq, display the mean normalized read counts across each known exonic region and across each splice junction (respectively). The bottom track, also generated by JunctionSeq, displays the exons or splice junctions found to display statistically significant differential usage (at p-adjust < 0.01).
This lends itself to the hypothesis that splice junction N040 is associated with an internal promoter that is expressed almost exclusively at night, but still only at moderate levels. The novel exon associated with this internal promoter will be referred to as “exon 3a”. The annotated 5’ start site, on the other hand, appears to display consistently high expression at both day and night. Examination of the “wiggle” plots for Ttc8 further revealed that there also appears to be an unannotated 5’ transcription start site leading directly into exon 3 (Fig 2). This alternative exon form will be referred to as “exon 3b”. Like exon 3a, this region has almost no read coverage during the day but moderate read coverage at night.
Evidence of multiple novel splice junctions, transcription start sites, and terminal exons was found in the Illumina RNA-Seq data (see Fig 3). Combinatorially generating all possible combinations of these variable regions (or “sub-variants”) yielded 648 theoretical isoforms (see S2 Dataset). Most of these sub-variants were only detected at very low levels and did not display significant differential usage. Some additional features that appeared to lead to additional alternative start and end sites were not considered in this analysis.
Fig 3. All possible sub-variants detected in the Illumina RNA-Seq data.

Considering the three major 5’ ends and two major 3’ ends, there are 21 “sub-variants” across 8 “regions”, which can be combined to form 648 different possible combinations. Based on coverage in the Illumina and PacBio data, we narrowed this down to 9 “major” sub-variants that appear to be expressed in the pineal gland in substantial quantities. There are 24 possible combinations of these 9 major sub-variants.
For simplicity, a subset of the sub-variants which seemed to have good support in the data was selected, and will be referred to as the “major sub-variants” (see Fig 3, S3 Dataset). There are 24 different “major sub-variant combinations” that can be constructed using only these components (see Fig 4). Twelve use the known start site (exon 1: potential isoforms 1–12), six use one novel start site (exon 3a; potential isoforms 13–18) and six use the other novel start site (exon 3b; potential isoforms 19–24). Note that these potential isoforms represent all possible combinations of this set of start sites and splice junctions, not all of these combinations necessarily exist and we cannot directly determine which do and do not exist based purely on the short-read Illumina datasets.
Fig 4. Diagram of the 24 potential isoforms produced by the “major” sub-variants, with PacBio match counts.

Part (a) and (b) display the “standard” known isoform and a diagram of the whole gene, respectively. The wide portion of (a) indicates the coding DNA sequence (CDS) of the “standard” transcript, and the purple boxes in (a) and (b) indicate tetratricopeptide repeat (TPR) domains. Part (c) displays the 24 possible combinations formed by the “major” sub-variants from Fig 3. Novel features are displayed in pink, and the widened region in each transcript denotes the largest open reading frame (ORF). The data table on the right side of (c) displays the number of full-length ROI from the SMRT sequencing that were found to match each isoform. The first column indicates the number of perfect, base-for-base, full-length matches, and the second column lists “alignment matches”, in which the RNA-STAR aligner maps the ROI to the isoform. Note that the novel start sites were each covered by two primers that were less than 6 base-pairs apart, producing slightly different amplified molecules. Isoforms 21 and 24 were covered by two different primer pairs used in the PCR amplification. See S3 Dataset for more information on these theoretical isoforms and their PacBio coverage.
qPCR validation
Although qPCR cannot be used to accurately assess the expression of different loci relative to one another, it is possible to determine the relative expression of a specific exon or splice junction across multiple conditions. Accordingly, qPCR was used to confirm the differential usage of the three predicted start sites in Ttc8: exon 1 (using exon 2 as a proxy), exon 3a, and exon 3b (see Fig 4).
All three initial exons displayed a differential increase in expression at night in the neurally stimulated samples (see Fig 5). As expected, both exons 3a and 3b display much stronger differentials than exon 1. In the control group, exon 1 displays a 1.4-fold increase at night, whereas exons 3a and 3b display 2.3-fold and 16-fold increases, respectively. Similar increases in initial exon expression are apparent in the sham group, but not in the SCGX and DCN groups, indicating that the differential usage of these alternative transcription start sites is driven by neural activity.
Fig 5. Results of the start-site qPCR experiment.

Three primer pairs were used, corresponding to the three transcription start sites detected in the Illumina RNA-Seq data. The novel start sites that begin with exons 3a or 3b both display very strong upregulation at night in the control and sham groups.
This confirms the differential expression detected in the four RNA-Seq analyses. Furthermore: the fact that both exon 3a and 3b display substantially stronger differentials than exon 1 confirms the presence of isoform-specific differential usage.
Qualitative validation in an independent RNA-Seq dataset
Qualitative examination of a publicly-available RNA-Seq dataset generated from rat pineal glands collected at various times throughout the day [46] revealed that the novel exons 3a and 3b show visible coverage at night but not during the day (see S7 Fig). Hundreds of reads cover these novel regions at ZT19 and ZT23, whereas only a handful of reads cover the same regions at ZT3, ZT7, and ZT15. At the same time the other exons show moderate to high coverage at all time points, indicating that this effect is specific to these alternative initial exons.
Robust statistical analysis of this pattern is not possible because this dataset lacks replicates; however, the simple visual observation that these two novel alternative initial exons are present at night but not during the day is clearly consistent with the previous RNA-Seq results, and thus lend further credence to our conclusions. This dataset also confirms the existence of many of the other alternative splice sites and exonic regions seen in the discovery dataset. For example, we observe exon 2a, the novel skipping of exon 7, and the use of novel alternative terminal exons 9a and 9b.
Similar examination of rat retinal tissue RNA-Seq data over the same time points [46] reveals that there is little-to-no expression of the novel alternative initial exons 3a and 3b in the rat retina at either day or night. The rat retina does include a mixture of inclusion and skipping of exon 2a, and a very small number of reads (less than 10 per sample) appear to splice to exon 9a (see S12 Fig). Almost no reads splice across any of the other splice junctions, which is expected in view of the low coverage depth of these samples.
Isoform assembly via PacBio SMRT sequencing
Validating differential abundance across these splice junctions does not provide any information about the full-length isoforms to which these junctions belong, and distant variable-splicing regions cannot easily be “phased” using short-read RNA-Seq data. The CuffLinks transcript assembly tool [47] was used on this dataset, but failed to detect any of the novel splice junctions in Ttc8 despite hundreds of reads of coverage across numerous samples (see S1 Dataset).
To validate and identify the Ttc8 isoforms we ran a pooled set of transcript-length PCR-amplified products on a Pacific Biosciences RS II sequencer. This yielded a total of 410,981 “reads of insert” (ROI), which are the consensus sequences collapsed from repeated sub-reads across the same individual molecule (ROI are alternatively called “circular consensus sequence” (CCS) in previous versions of the protocol). Of these, 63% were “full length”, i.e. begin and end with one matched set of the seven PCR primer pairs. Many of the ROI (36%) did not match any primer pair on one or both ends (see Table 3). Using RNA-STAR, 74% of the ROI (including 94% of the full-length ROI) were successfully aligned uniquely to the rat genome. Of these aligned ROI, 99.5% aligned to the Ttc8 gene.
Table 3. Distribution of “reads of insert” (ROI) from the SMRT sequencing.
| # ROI | % ROI | ||
|---|---|---|---|
| Matches known, valid primer pair (“Full-Length”) | 257,018 | 62.5% | |
| Non Full length | 153,963 | 37.5% | |
| No primer match | 79,413 | 19.3% | |
| Matches known primer on one end | 69,817 | 17.0% | |
| Aligns uniquely to genome | 305,675 | 74.4% | |
| Aligns to location other than Ttc8 | 1,550 | 0.4% | |
| Aligns to Ttc8 and is full-length | 242,045 | 58.9% | |
| Full-length and matches predicted isoform | 180,479 | 43.9% | |
| Perfect full-length match to major isoform | 56,977 | 13.9% | |
| Alignment match to major isoform | 97,677 | 23.8% | |
| Matches major isoform with 8-9b intron retention | 24,064 | 5.9% | |
| Matches isoform with minor splicing variant | 1,761 | 0.4% | |
| Total Reads of Insert | 410,981 | 100.0% | |
More than 50,000 ROI were found to be perfect, base-for-base, full-length matches to one of the 24 primary predicted combination isoforms (see Table 3, S4 Table). We used QoRTs [48] to identify near-perfect matches to predicted isoforms and found that an additional 97,677 ROI matched one of the 24 major sub-variant combinations (see Table 3, S4 Table, S3 Dataset). In addition, a large number of reads (24,064) were found to match one of the 24 primary combinations except with an additional intron retention between exons 8 and 9a (see Fig 6, S5 Table). An additional 1302 reads matched one of the broader set of 648 predicted combinations (see Fig 6, S6 Table, S2 Dataset).
Fig 6. Additional novel isoforms discovered or validated via the PacBio SMRT sequence data.

Part (a) displays the gene annotation, including the TPR domains (purple) and all alternative splice sites initially discovered by the Illumina RNA-Seq data. Part (b) displays all possible isoforms based on combinations of the major sub-variants plus an extra intron retention between exons 8 and 9b. Part (c) displays the subset of the 624 isoforms that involve one or more “minor” sub-variants (see Fig 3) that had one or more “perfect” matches in the PacBio SMRT data. Novel features are displayed in pink, and the widened region in each transcript denotes the largest open reading frame (ORF). The data tables on the right side of parts (b) and (c) display the number of full-length ROI from the SMRT sequencing that were found to match each isoform. The first column indicates perfect base-for-base, full-length matches, and the second column lists the total number of matches, including alignment matches in which the RNA-STAR aligner maps the ROI to the isoform. For a complete listing of all possible combinations of all known splice variations see the online supporting information. See S2 Dataset for more information on these and all other theoretical isoforms.
Depending on the cutoff thresholds used, between 13 and 42 distinct isoforms were detected in the rat pineal gland. Thirteen distinct isoforms had more than 1000 perfect, base-for-base, primer-to-primer matches in the SMRT sequencing, 17 isoforms had 500 perfect matches, 31 had 50 perfect matches, and 42 isoforms had 1 or more perfect match. Note that the SMRT sequencing was conducted on a pool of pineal glands collected at a single time point (ZT19.5); additional isoforms may be present in this tissue at other time points. Thus, even with extremely conservative cutoff thresholds (500 perfect matches) we still detect more distinct isoforms (17 isoforms) than found in the Ensembl transcript annotations for rat (2 isoforms), mouse (6 isoforms), or human (15 isoforms).
It should be noted that since these isoforms were all based on splice junctions already detected in short-read high-throughput sequencing data, the chances that these isoforms are produced by sequencer artifacts is low. Short-read high-throughput RNA-Seq relies on fundamentally different chemistry compared with SMRT sequencing, and one would not expect these two methods to replicate one anothers’ biases or artifacts.
Characterization of Ttc8 isoforms
The Ttc8 gene is part of a larger complex that serves to transport proteins in and out of cilia in certain cells [43], such as the outer segments of rod and cone photoreceptor cells [49]. The primary known functional domain of the Ttc8 gene consists of eight tetratricopeptide repeat (TPR) domains located in the C-terminal half of the protein (see Fig 4 and Fig 6). These repeats form a series of alpha-helices that together function as a protein-binding domain related to pilF [42].
Near the 5’ end of the “standard” transcript, exon 2a is a 30-base-pair-long cassette exon that preserves the open reading frame (ORF). The function of these additional 10 amino acids is unknown, but they only seem to be expressed in the pineal gland and retina, which (at least evolutionarily) feature prominent cilia [50, 51]. At least one isoform containing this cassette exon (isoform-03) has been shown in the existing literature to display substantial protein translation in mouse photoreceptor cells [39].
Further downstream, the two novel start sites in exon 3a and 3b both lead to the same alternative start codon in exon 3. This codon is in a Kozak consensus sequence and is in-frame with the standard Ttc8 transcript. Thus, major sub-variant combinations that combine the known 3’ end with the alternative exons 3a or 3b (isoforms 15 and 21, see Fig 4) have open reading frames containing all eight TPR domains, though they are missing the first 124 amino acids from the N-terminal end. There are two novel alternative 3’ terminal exons (9a and 9b) that have much shorter open reading frames than the known transcripts, and are missing at least 7 of the 8 TPR domains. The novel splice junction that causes exon 7 to be omitted results in a frame shift that introduces a stop codon near the 5’ end of the following exon (exon 8), excluding or truncating all 8 TPR domains. This means that several distinct transcripts that exclude exon 7 all contain identical ORFs, as the stop codon occurs upstream of the point of divergence.
There are also several other novel exons and splice junctions that were detected only at very low levels in the Illumina and PacBio datasets (see Fig 6). An elongation of exon 4 (“exon 4a”) introduces a novel stop codon and thus has a truncated ORF. The novel cassette exons 10a and 11a both introduce similar stop codons, producing ORFs that cover only 2 out of 8 and 3 out of 8 TPR domains, respectively.
Finally, the intron retention between exons 8 and 9b includes a stop codon located 5 amino acids into the intron. It is unclear whether the retained intron reads are actually representative of mature transcripts or if they were derived from immature mRNAs that were still in the process of being spliced. The PCR amplification performed prior to library preparation included a series of PCR reactions performed on size-selected fractions. Thus, even a small quantity of these (much larger) mRNAs could produce a disproportionate number of reads.
Discussion
The results of this study provide interesting insights into the complex isoform population of the Ttc8 gene in the pineal gland and retina, and demonstrate the advantages of our hybrid, multi-platform sequencing strategy. These two points will be discussed sequentially in detail below.
Differential start site usage
The evidence of differential start site usage of the Ttc8 gene in the rat pineal gland is comprehensive, consistent, and robust. We have detected and validated start-site-specific night-time upregulation of two novel transcription start sites belonging to this gene. This upregulation appears to be neurally-controlled by the SCG via NE and cAMP, as evidenced by the fact that in vitro treatment with NE and DBcAMP replicates this effect, and the fact that the effect is abrogated by removal or decentralization of the SCG. Furthermore, the existence of the novel start sites was validated using the PacBio SMRT sequencing platform (along with the isoforms to which they belong).
The existence and differential regulation of these novel start sites have been consistently confirmed in several different independent analyses using multiple sequencing/assay methods and experimental conditions. This comprehensive, multi-platform approach ensures that our results are replicable and robust, and greatly reduces the possibility that these observed phenomena could be the product of artifacts or biases.
The Ttc8 gene
Although the exact function of this gene is not well understood, mutations in the gene have been implicated in non-syndromic retinitis pigmentosa (RP) [39, 40] and in Bardet-Biedl syndrome (BBS, which commonly includes RP among its symptoms) [41, 42]. At least one of the retina disease variants have been found in previous studies to be specifically related to the splicing of the gene’s transcripts. The cassette exon 2a is apparently expressed almost exclusively in the outer nuclear layer of the retina, and was absent from the ganglion cell layer, inner nuclear layer, and retinal pigment epithelium layers of the retina [40]. Similarly, this alternative exon was not found to be expressed in the brain, heart, kidney, liver, or testes. Despite the rarity of this splicing variant, previous studies have found that a specific point mutation just upstream of the 5’ acceptor site of this exon causes mis-splicing of the exon and results in nonsyndromic retinitis pigmentosa (RP) in humans [39, 40]. This particular mutation results in a frame-shift and premature stop codon in the 2a-inclusion isoforms, producing a non-functional protein. Other non-retinal tissues seem to be unaffected because they do not express exon 2a, resulting in RP rather than full BBS.
We found strong evidence that exon 2a is expressed in the pineal gland. In the discovery RNA-Seq dataset we find that around 2–3 times as many reads cover the splice junctions that include exon 2a (junctions N035 and N037, see Fig 1) compared with the junction that skips exon 2a (junction J019). Since reads are known to be non-uniform in their distribution these quantities may not be proportional to the actual expression levels, but the presence of these splice junctions in substantial quantities does at least indicate that the exon is expressed. The presence of this retina-specific exon is not totally unexpected given the phylogenetic and developmental relationship between the pineal gland and the photoreceptor cells in the retina. Our data suggests pedigrees affected by the nonsyndromic RP alleles may potentially have additional (undiagnosed) pleiotropic effects in the pineal gland, such as circadian timing issues related to reduced or impaired melatonin production. The two novel start site variants (exons 3a and 3b) contain a start codon that is in-frame with the known protein and lies upstream of the protein’s TPR domains. Thus, the hypothetical proteins produced by these isoforms would still include the full set of TPR domains and might thus still be functional.
We have also found numerous isoforms that use other downstream alternative splice junctions. Many of these isoforms have open reading frames that are missing most or all of these TPR domains, and thus could not possibly code for proteins that perform the same function as the standard Ttc8 isoform. It is possible that these isoforms code for proteins that use other (hitherto unknown) functional domains. Alternatively, these isoforms may actually represent long non-coding RNA’s, which have been found to have a substantial regulatory role in the rat endocrine system (and specifically in the rat pineal gland) [52, 53].
Combinatorial isoforms
A set of 31 distinct novel isoforms (and 1 known isoform) was found that had 50 or more perfect, base-for-base matches in the PacBio data and that included only the splice junctions and exonic regions already detected in the Illumina dataset.
This is more than twice the number of isoforms found in human annotation (15 isoforms), and more than ten times the number found in the rat annotation (two isoforms). Furthermore, this was only a limited search: we only studied a single tissue, and only amplified three initial exons and two terminal exons. In contrast, the human annotation lists three known initial exons and five known terminal exons (Ensembl annotation, release 75).
In fact, we observe perfect full-length ROI that match most of the possible combinations of major exons and splice-junctions found in the short-read data (see Fig 4). However, there are some potential transcripts that have zero perfect matches, such as isoform 6 and isoform 12 (see Fig 4). These two isoforms would combine exon 1 with exon 14 and the novel skip of exon 7. All of these components are observed in other isoforms, many of which have high read coverage: exon 1 appears in isoforms 1 through 12, the novel skip of exon 7 appears in isoforms 4–6, 10–12, 16–18, and 22–24, and exon 14 appears in isoforms 3, 6, 9, 12, 15, 18, 21, and 24. However, the particular combination of exon 1, skipping exon 7, and then the set of exons 9–14, appears to never occur in detectable quantities. This is unlikely to be due to differential PCR amplification, since isoform 6 uses the same primers as isoform 3 (which had many ROI matches), and the two 2kb isoforms only differ by the presence or absence of the 86bp exon 7 in the middle. Furthermore, we observe numerous reads both with and without exon 7 that begin with exon 3a or 3b and end with exon 14 (see isoforms 15, 18, 21, and 24), and we observe numerous reads both with and without exon 7 that start with exon 1 but end with one of the novel end sites 9a or 9b.
The fact that there are some specific combinations of exons and splice junctions that are completely absent suggests that there are splicing regulatory mechanisms that act to correlate or “link” specific distant variable regions. For example: there could be some mechanism preventing the “full” isoforms (i.e. the isoforms that use exons 1 through 14) from splicing out exon 7, or there could be regulatory mechanisms that specifically splice out exon 7 some percentage of the time for the transcripts that use the alternative start sites 3a or 3b and/or alternative end sites 9a or 9b.
Implications and relevance to future research
Although we have not established the function of the novel isoforms we have discovered (if any such function exists), this study has broad implications in the study of RNA expression and splicing. Even if these isoforms are not functional, knowledge of these isoforms is critical to the accuracy and validity of isoform-level abundance estimation methods. Such methods implicitly depend on the correctness and completeness of the isoform set (derived from an a priori transcript annotation and/or assembled from the data); the presence of a large number of unknown and/or undetectable isoforms will severely impact the accuracy of the estimates of the known isoforms.
Like several other studies that applied targeted, long-read sequencing of individual genes [36, 54], we found a very large number of novel isoforms. This strongly suggests that previous transcript discovery methods (predominantly based on high-throughput short-read RNA-Seq) were overly conservative and missed a large proportion of the true transcript population. This is consistent with the failure of the CuffLinks transcript assembly tool to detect any of the novel isoforms in our short-read RNA-Seq dataset (see S8 Fig, S1 Dataset). Long-read sequencing technologies offer a vast improvement over such methods, and as these technologies develop and become more widespread we may see a rapid increase in the number of known isoforms.
Furthermore, our results strongly suggest that numerous other genes also display differential transcript usage in the rat pineal gland between night and day, controlled by neural stimulation via the SCG. We found dozens of genes that displayed significant differential usage in all four “neurally-stimulated” experiments, whereas very few genes were detected in the SCGX and DCN experiments. Several of the neurally-regulated differentially-used genes were already well-known and extensively validated in the literature, including Crem [23–25], Pde4b [26], and Atp7b [28].
This differential transcript usage may be driven by the strong neurally-controlled night-time upregulation of the splicing regulator Mbnl2 [5, 14, 16]. This gene has been shown to regulate widespread but not universal alternative splicing in a wide range of species [18–22]. In fact, depletion of specific splicing factors, including Mbnl2, is thought to be a major cause of myotonic dystrophy because it results in dysregulation of splicing in specific transcripts [55]. It is possible that the daily 6-fold cycle of Mbnl2 in the pineal gland could regulate alternative splicing of many specific transcripts, including Ttc8.
The value of the multi-platform approach
In this report data from three major technologies were used: strand-specific Illumina RNA-Seq, qPCR, and PacBio SMRT sequencing. None of these methods could, taken individually, can fully explain the regulatory processes that are taking place. The Illumina next-generation RNA-Seq dataset provided a transcriptome-wide survey of differential expression and differential splicing; however, the short reads limited our ability to identify full-length isoforms, and the various potential artifacts and biases made further validation necessary. qPCR was used to confirm the differential usage of specific targeted exons and splice junctions, and PacBio SMRT sequencing was used to identify the complete full-length isoforms. Only by combining the results from all three platforms were we able to develop a complete and comprehensive understanding of the regulatory phenomena that influence the expression of Ttc8 in the rat pineal gland.
Methods
A more detailed descriptions of the precise methods used is available in the supplemental methods (see S1 Appendix). Animal use and care protocols were approved by the NIH Institutional Animal Care and Use Committee and followed the guidelines of the National Research Council's Guide for Care and Use of Laboratory Animals (Vol. 8) [56] and the Animal Research: Reporting In vivo Experiments (ARRIVE) guidelines [57].
RNA-Seq Analyses
Discovery dataset
The surgical techniques, gland culture, sample collection, and sequencing is described in detail in a previous publication [14]. Briefly: the in vivo dataset consisted of four surgical groups: no surgical intervention (Control); neonatal sham surgery (Sham); neonatal bilateral SCG decentralization (DCN); neonatal bilateral superior cervical ganglionectomy (SCGX) [58]. Each surgical group was further subdivided into two sets and euthanized at either mid-day or midnight, generating a total of 8 biological conditions. The in vitro dataset consisted of 3 biological conditions: untreated control (untreated), norepinephrine-treated (NE), and dibutyryl-cyclic-AMP-treated (DBcAMP). For each of the 11 biological conditions, pineal glands were grouped into 3 biological replicates producing 33 biological replicates in total.
All rat RNA-Seq data were aligned to the rn6 rat genome build using the RNA-STAR aligner (v2.4.0j) [59], with the Ensembl transcript annotation (release 80) [60]. Quality control was carried out with the QoRTs quality control tool and no quality issues were detected [48]. Read counts for genes, exons, and splice junctions were also generated via QoRTs. A complete description of the sequencing and data processing can be found online (see section 1.1 of S1 Appendix). This dataset is publicly available in the NCBI gene expression omnibus (GEO), accession number GSE63309.
Analysis with JunctionSeq
The detection of differential usage of exons and splice junctions was performed using the JunctionSeq Bioconductor package [38]. The statistical methods used by this tool are fully described in the JunctionSeq methodology paper. Briefly, this package applies statistical methods similar to those used in the DEXSeq Bioconductor package, with modifications to allow for the detection of differential usage of known and novel splice junction loci [61, 62]. JunctionSeq, like DEXSeq, applies a multivariate generalized linear model using a negative binomial distribution to detect exons or splice junctions that are disproportionately used relative to the expression of their respective genes [38, 61]. Because it can query both known and novel features, this method provides superior performance over similar methods when using flawed or incomplete transcript annotations [38]. Most alternatives such as Kallisto [29], eXpress [30], and RSEM [31] cannot assess differential splicing unless the full-length isoforms are known.
The four in vivo analyses compared day and night conditions within each of the four surgical groups. The two in vitro analyses compared treated samples versus the untreated controls for the NE treatment and the DBcAMP treatment, respectively. Thus, each JunctionSeq analysis had two biological conditions (either night/day or treated/untreated) with three biological replicates per condition.
Time-series dataset
The time series RNA-Seq dataset is publicly available online from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO), accession number GSE46069. The sample collection, extraction, and sequencing methods for this previously-published [46] dataset are described in full on the GEO project page. Briefly: rat pineal and retinal tissue samples were taken at six time-points: ZT1, 7, 13, 15, 19, and 23. Only one sample was sequenced for each time-point. Alignment and data processing was performed using the same pipeline used for the discovery dataset.
qPCR validation of differential transcription start site usage
qPCR was used to validate the day vs night difference in initial exon usage and whether this was affected by SCGX or DCN. Exon 2 was used as a proxy for usage of exon 1, since RNA-Seq showed that exon 2 is always included when exon 1 is used as the initial exon. A common reverse primer was used for all three alternative 5’ sub-variants: exon 2, exon 3a or exon 3b, each paired with a primer that matched exon 3 (see S1 Table). A more detailed description of the qPCR methods is available online (see section 1.3 of S1 Appendix).
Identification of full-length novel isoforms
Sample collection and preparation
To obtain representative samples of all the transcripts present in the pineal gland, a cDNA library constructed from rat pineal glands collected at ZT19.5 were amplified using seven different pairs of primers covering combinations of the three alternative initial exons and the two alternative terminal exons (S2 Table). The products of the seven PCR runs were pooled, prepared, and run on a Pacific Biosciences (PacBio) RS II sequencer in seven SMRT cells. “Sub-reads” were compiled into consensus “reads-of-insert” (ROI) using the SMRT Pipe software suite provided by Pac Bio. The PacBio SMRT sequencing data has been deposited online in the NCBI short read archive (SRA), accession ID SRX1736990. Further details on the PacBio SMRT sequencing methods are available online (see section 1.4.1–1.4.2 in S1 Appendix).
Analysis
We did not use the “Quiver” methodology proposed by PacBio to perform our transcript assembly, as this method was designed for use with different experimental designs and performed poorly under our conditions. Specifically: Quiver appears to have been designed for a different type of PCR amplification in which the primers were not expected to match the genomic template (and thus contigs are not required to begin and end with primer sequence), and where the “full-length” ROI were not required to begin and end with the start/end sites. This meant that in many cases it produced assemblies and read assignments that were obviously incorrect. For example: the major theoretical combination 15 (see Fig 4) was not detected, and the (numerous) reads that perfectly matched this isoform were instead matched to a partial segment of a larger isoform that began with exon 1. Our experimental design actually made the assembly much simpler. Our long-read sequence data had relatively low diversity (see S10 Fig), and we already had an a priori set of theoretical isoforms derived from the short read RNA-Seq data. Thus, in order to confirm the existence of these isoforms we did not need the advanced isoform assembly implemented by Quiver.
The ROI were matched to primer pairs using Phmmer [63], using the exact same method implemented by Quiver. However, unlike with Quiver, the primer sequences were not clipped off. The ROI were then aligned to the rat genome (build rn6) using the RNA-STARlong aligner (v2.4.2a), which was specifically designed for use on long reads of this type. These aligned ROI were then compared to the theoretical isoform set using the new “longReadClassifier” function of the QoRTs software package [48]. This tool counts the number of ROI that match by alignment to each isoform. An ROI was considered a “perfect match” if it was an exact, base-for-base, primer-to-primer match to the theoretical isoform. An ROI was considered an “alignment match” if and only if they covered all the isoform’s exons, spliced across all the isoform’s splice junctions, covered no other exons or splice junctions, and if both of the alignment ends were within 3 base pairs of the primer endpoints. Further details on the PacBio SMRT sequencing analyses are available online (see section 1.4.3–1.4.4 in S1 Appendix).
Supporting Information
(DOCX)
These transcripts were generated via a CuffLinks run on each sample in the discovery set, followed by the use of CuffMerge to merge these individual-sample assemblies. Note that CuffLinks failed to discover any of the novel transcripts, and there are no transcripts covering any of the 17 novel splice junctions observed in the Illumina and PacBio datasets.
(GZ)
It includes columns listing the ROI counts from the PacBio SMRT sequencing.
(TXT)
It includes columns listing the ROI counts from the PacBio SMRT sequencing. See also Fig 4C.
(TXT)
An excel file containing the JunctionSeq results for each analysis, for all genes that showed significant differential usage.
(XLSX)
This includes all exonic regions and known/novel splice junctions and their unique identifiers.
(GZ)
This includes all theoretical isoforms that had one or more perfect, full-length matches in the PacBio dataset. Also included is the GenBank accession numbers for each isoform.
(XLSX)
This Venn diagram displays the overlap between the genes detected as containing a differentially used feature in each of the four JunctionSeq “stimulus” analyses at the adjusted-p-value < 0.0001 level.
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
Rats were housed in a 14:10 light:dark cycle. ZT, Zeitgeber time.
(PNG)
{kind=link}
As you can see, there are 30–60 read pairs per sample covering junction N040 in the innervated sample groups (Ctrl_N, Sham_N, DBcAMP and NE). However, CuffLinks does not detect any transcripts containing this junction. It also fails to detect novel start site 3b despite substantial read coverage over this region. Subsequent validation proved that these novel splice sites are real.
(PNG)
{kind=link}
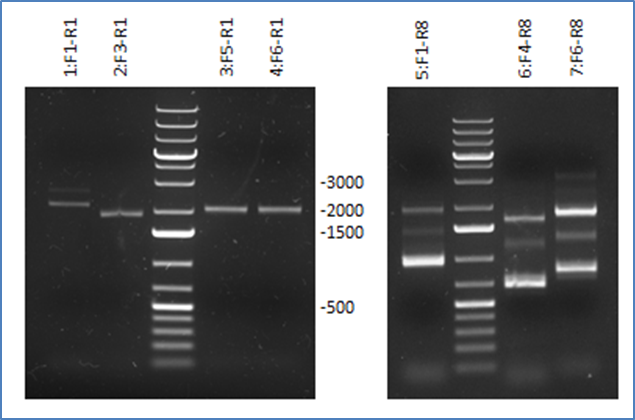
The label above each lane indicates the reaction number and the primer pair that was used for amplification. See S2 Table for primer sequences.
(PNG)
{kind=link}
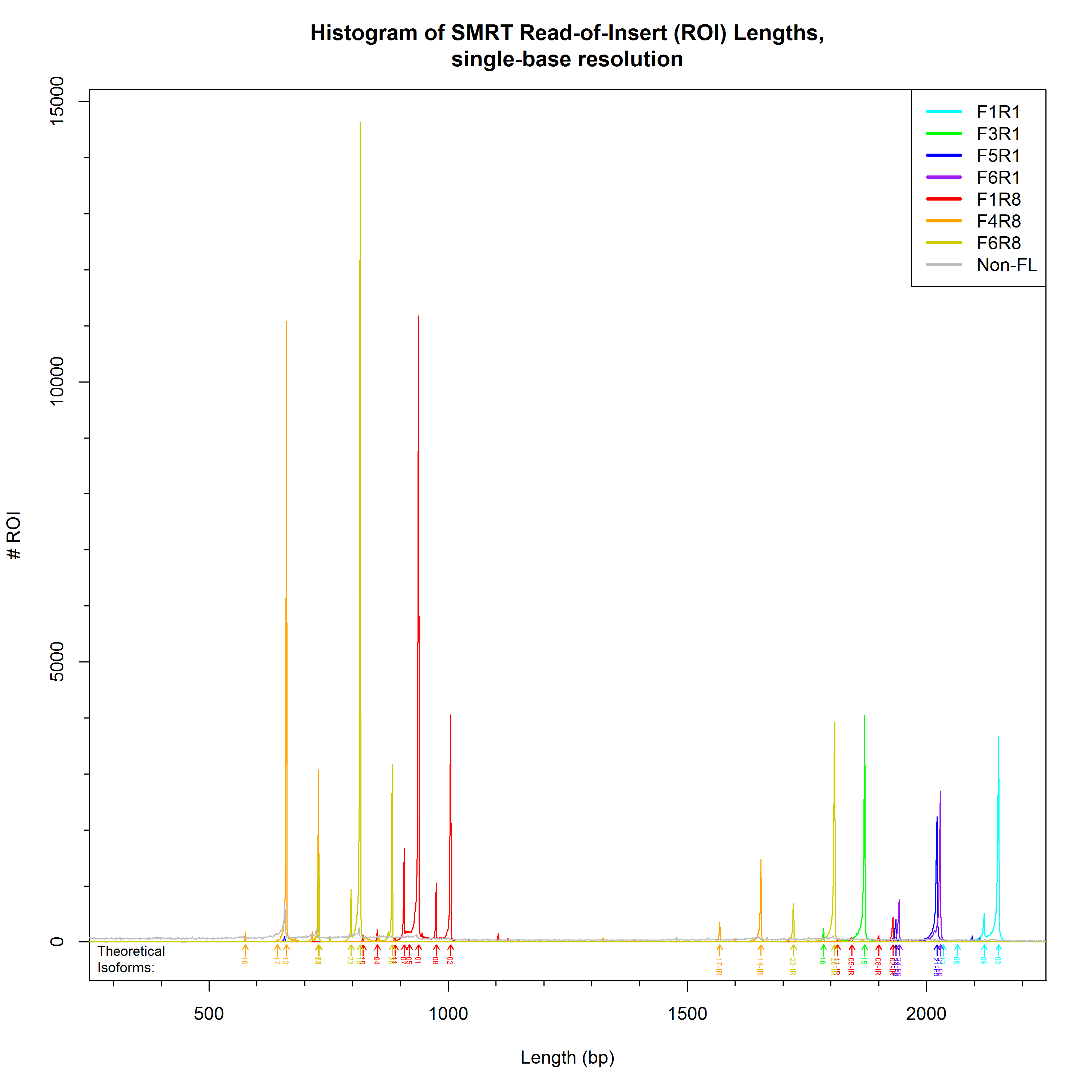
This plot shows a base-pair-resolution histogram of the read-of-insert (ROI) length produced by the PacBio SMRT sequencing of the Ttc8 gene. The reads are separated by primer pair and drawn separately in each color. The lengths of major potential predicted isoforms are marked at the bottom, along with the potential isoform ID. Reads of insert that did not match a legal primer pair at both ends were assumed to be non-full-length (non-FL) and are plotted in gray. Note that due to the accuracy of the SMRT sequencing platform, the presence of many of the predicted isoforms can be easily recognized based only on the high number of ROI at that exact length. Also note that this effect is less pronounced for the longer isoforms, due to the fact that the ROI accuracy is inversely associated with template length.
(PNG)
{kind=link}
(PNG)
{kind=link}
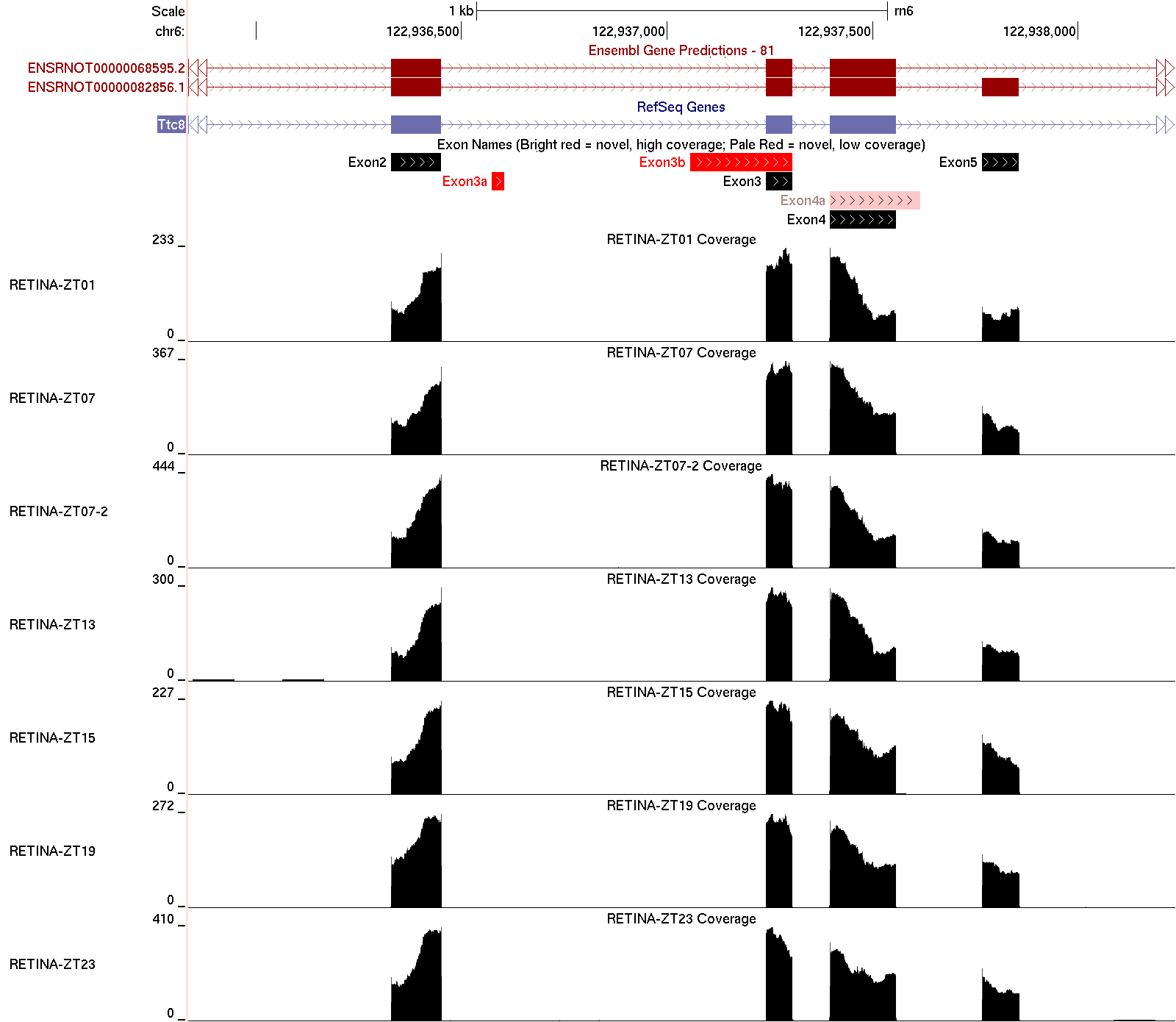
Rats were housed in a 14:10 light:dark cycle. Note that there are 2 samples at ZT7. Note that there are no reads covering the novel exon 3a, nor covering the region specific to novel exon 3b. ZT, Zeitgeber time.
(PNG)
{kind=link}
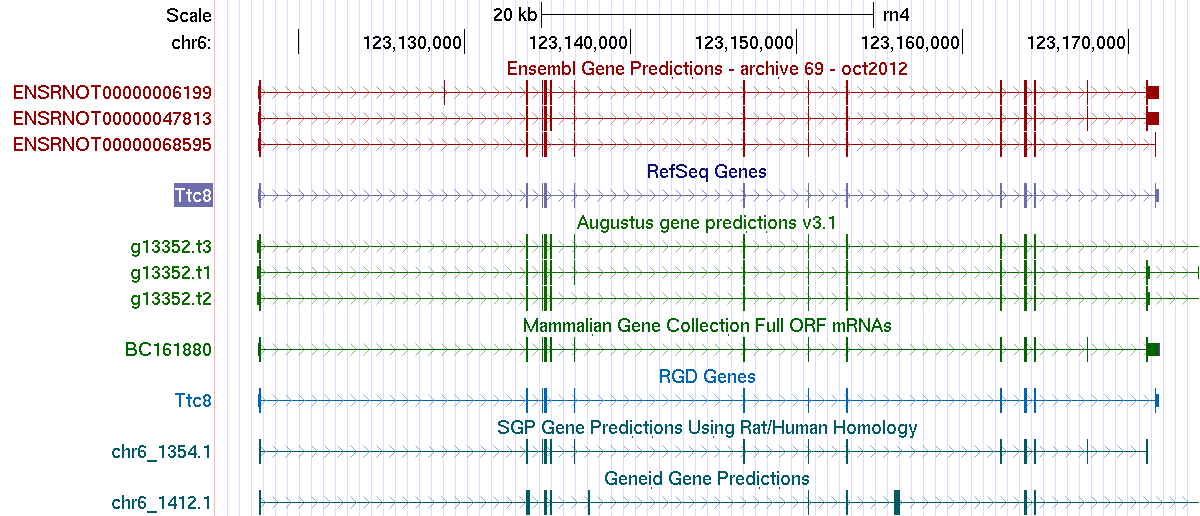
The above figure displays the gene annotations and gene predictions from Ensembl, RefSeq, Augustus, the mammalian gene collection (MGC), SGP, and geneid. Note that none of the novel exons or splice junctions appear in any of the transcript annotations. Note that the Rat Genome Database (RGD) is not included in this plot because it has not yet been lifted over to the rn6 rat genome build. See (S15 Fig) for the RGD annotation.
(PNG)
{kind=link}
This figure is identical to the previous, except zoomed in on the region containing the major novel exons 3a and 3b. Note that none of the novel exons or splice junctions appear in any of the transcript annotations.
(PNG)
{kind=link}
Note that this assembly includes the RGD curated gene database. Additionally, note the presence of transcript ENSRNOT00000006199 in the ensembl annotation (release 69), which was absent in subsequent ensembl releases. This transcript includes one of the “novel” exons detected in our analyses, exon 2a. The transcript appears to have been lost in the liftover to rn5 (possibly due to a nearby gap in the rn5 genome build), and was never re-added in subsequent releases. Note that none of the other novel exons or splice junctions appear in any of the transcript annotations.
(PNG)
{kind=link}
These primers were used to confirm and quantitate the differential expression of the three start sites of the Ttc8 gene. The first primer pair addresses the annotated Ttc8 start site, and primer pairs 2 and 3 address the two major novel start sites discovered in the Illumina RNA-Seq data.
(DOCX)
These primers were used to amplify the transcripts of the Ttc8 gene prior to sequencing with the PacBio SMRT platform. Note that primers F3 and F4 differ by only 4 bases, and primers F5 and F6 differ by only 6.
(DOCX)
In some cases additional size selection and amplification was necessary, as per the PacBio SMRT protocol. This table lists the size windows, primers, cycle counts, and total yield.
(DOCX)
These are the counts for ROI that match one of the 24 potential major isoforms (see Fig 3 and Fig 4).
(DOCX)
See also Fig 6B.
(DOCX)
See also Fig 3. Note that these counts are the number of ROI that perfectly match any of the theoretical potential isoforms that includes the given sub-variant. For example the first row (late donor, exon 4) compiles the counts from 8 different isoforms, all of which contain the late donor alternative splice junction on exon 4, as seen in Fig 6C.
(DOCX)
Acknowledgments
This work was supported by the Intramural Research Program, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892; and, the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892. We greatly appreciate the animal management contributions of Mr. Daniel Abebe, Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892. Library preparation, sequencing, and data processing was performed by the NISC Comparative Sequencing Program: Betty Barnabas, PhD; Robert Blakesley, PhD; Gerry Bouffard, PhD; Shelise Brooks, BS; Holly Coleman, MSc; Mila Dekhtyar, MSc; Michael Gregory, MSc; Xiaobin Guan, PhD; Jyoti Gupta, MSc; Joel Han, BS; Shi-ling Ho, BS; Richelle Legaspi, MSc; Quino Maduro, BS; Cathy Masiello, MSc; Baishali Maskeri, PhD; Jenny McDowell, PhD; Casandra Montemayor, MSc; James Mullikin, PhD; Morgan Park, PhD; Nancy Riebow, BS; Karen Schandler, MSc; Brian Schmidt, BS; Christina Sison, BS; Mal Stantripop, BS; James Thomas, PhD; Pam Thomas, PhD; Meg Vemulapalli, MSc; Alice Young, BA. A complete list of the NISC Comparative Sequencing Program can be found in the supporting information.
Data Availability
The sequences reported in this paper have been deposited in the National Center for Biotechnology Information Short Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra), accession ID SRX1736990. The 42 novel isoforms detected in the PacBio dataset were deposited in GenBank, accession numbers KX218320 through KX218361.
Funding Statement
This work was supported by the Intramural Research Program, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892; and, the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892.
References
- 1.Maronde E, Stehle JH. The mammalian pineal gland: known facts, unknown facets. Trends in endocrinology and metabolism: TEM. 2007;18(4):142–9. Epub 2007/03/22. 10.1016/j.tem.2007.03.001 . [DOI] [PubMed] [Google Scholar]
- 2.Klein DC, Bailey MJ, Carter DA, Kim JS, Shi Q, Ho AK, et al. Pineal function: impact of microarray analysis. Molecular and cellular endocrinology. 2010;314(2):170–83. Epub 2009/07/23. 10.1016/j.mce.2009.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein DC. The 2004 Aschoff/Pittendrigh lecture: Theory of the origin of the pineal gland—a tale of conflict and resolution. J Biol Rhythms. 2004;19(4):264–79. 10.1177/0748730404267340 . [DOI] [PubMed] [Google Scholar]
- 4.Klein DC. Evolution of the vertebrate pineal gland: the AANAT hypothesis. Chronobiol Int. 2006;23(1–2):5–20. 10.1080/07420520500545839 . [DOI] [PubMed] [Google Scholar]
- 5.Bailey MJ, Coon SL, Carter DA, Humphries A, Kim JS, Shi Q, et al. Night/day changes in pineal expression of >600 genes: central role of adrenergic/cAMP signaling. J Biol Chem. 2009;284(12):7606–22. Epub 2008/12/24. 10.1074/jbc.M808394200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babila T, Schaad NC, Simonds WF, Shinohara T, Klein DC. Development of MEKA (phosducin), G beta, G gamma and S-antigen in the rat pineal gland and retina. Brain Res. 1992;585(1–2):141–8. 10.1016/0006-8993(92)91199-o . [DOI] [PubMed] [Google Scholar]
- 7.Bernard M, Donohue SJ, Klein DC. Human hydroxyindole-O-methyltransferase in pineal gland, retina and Y79 retinoblastoma cells. Brain Res. 1995;696(1–2):37–48. 10.1016/0006-8993(95)00651-6 . [DOI] [PubMed] [Google Scholar]
- 8.Ho AK, Somers RL, Klein DC. Development and regulation of rhodopsin kinase in rat pineal and retina. J Neurochem. 1986;46(4):1176–9. 10.1111/j.1471-4159.1986.tb00634.x . [DOI] [PubMed] [Google Scholar]
- 9.Korf HW, Moller M, Gery I, Zigler JS, Klein DC. Immunocytochemical demonstration of retinal S-antigen in the pineal organ of four mammalian species. Cell Tissue Res. 1985;239(1):81–5. 10.1007/bf00214906 . [DOI] [PubMed] [Google Scholar]
- 10.Korf HW, White BH, Schaad NC, Klein DC. Recoverin in pineal organs and retinae of various vertebrate species including man. Brain Res. 1992;595(1):57–66. 10.1016/0006-8993(92)91452-k . [DOI] [PubMed] [Google Scholar]
- 11.Li X, Chen S, Wang Q, Zack DJ, Snyder SH, Borjigin J. A pineal regulatory element (PIRE) mediates transactivation by the pineal/retina-specific transcription factor CRX. Proc Natl Acad Sci U S A. 1998;95(4):1876–81. 10.1073/pnas.95.4.1876 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donoso LA, Merryman CF, Edelberg KE, Naids R, Kalsow C. S-antigen in the developing retina and pineal gland: a monoclonal antibody study. Invest Ophthalmol Vis Sci. 1985;26(4):561–7. Epub 1985/04/01. . [PubMed] [Google Scholar]
- 13.Lolley RN, Craft CM, Lee RH. Photoreceptors of the retina and pinealocytes of the pineal gland share common components of signal transduction. Neurochemical research. 1992;17(1):81–9. Epub 1992/01/01. 10.1007/bf00966868 . [DOI] [PubMed] [Google Scholar]
- 14.Hartley SW, Coon SL, Savastano LE, Mullikin JC, Program NCS, Fu C, et al. Neurotranscriptomics: The Effects of Neonatal Stimulus Deprivation on the Rat Pineal Transcriptome. PloS one. 2015;10(9):e0137548 10.1371/journal.pone.0137548 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savastano LE, Castro AE, Fitt MR, Rath MF, Romeo HE, Muñoz EM. A standardized surgical technique for rat superior cervical ganglionectomy. Journal of Neuroscience Methods. 2010;192(1):22–33. 10.1016/j.jneumeth.2010.07.007 [DOI] [PubMed] [Google Scholar]
- 16.Kim JS, Coon SL, Weller JL, Blackshaw S, Rath MF, Møller M, et al. Muscleblind‐like 2: circadian expression in the mammalian pineal gland is controlled by an adrenergic‐cAMP mechanism. Journal of neurochemistry. 2009;110(2):756–64. 10.1111/j.1471-4159.2009.06184.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He F, Dang W, Abe C, Tsuda K, Inoue M, Watanabe S, et al. Solution structure of the RNA binding domain in the human muscleblind‐like protein 2. Protein Science. 2009;18(1):80–91. 10.1002/pro.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho TH, Charlet‐B N, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. The EMBO journal. 2004;23(15):3103–12. 10.1038/sj.emboj.7600300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150(4):710–24. 10.1016/j.cell.2012.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charizanis K, Lee K-Y, Batra R, Goodwin M, Zhang C, Yuan Y, et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75(3):437–50. 10.1016/j.neuron.2012.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Machuca-Tzili LE, Buxton S, Thorpe A, Timson CM, Wigmore P, Luther PK, et al. Zebrafish deficient for Muscleblind-like 2 exhibit features of myotonic dystrophy. Disease Models and Mechanisms. 2011;4(3):381–92. 10.1242/dmm.004150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vicente-Crespo M, Pascual M, Fernandez-Costa JM, Garcia-Lopez A, Monferrer L, Miranda ME, et al. Drosophila muscleblind is involved in troponin T alternative splicing and apoptosis. 2008. [DOI] [PMC free article] [PubMed]
- 23.Foulkes NS, Borjigin J, Snyder SH, Sassone-Corsi P. Transcriptional control of circadian hormone synthesis via the CREM feedback loop. Proceedings of the National Academy of Sciences. 1996;93(24):14140–5. 10.1073/pnas.93.24.14140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korf H-W, Schomerus C, Maronde E, Stehle J. Signal transduction molecules in the rat pineal organ: Ca 2+, pCREB, and ICER. Naturwissenschaften. 1996;83(12):535–43. 10.1007/bf01141978 [DOI] [PubMed] [Google Scholar]
- 25.Schwartz WJ, Aronin N, Sassone-Corsi P. Photoinducible and rhythmic ICER-CREM immunoreactivity in the rat suprachiasmatic nucleus. Neuroscience letters. 2005;385(1):87–91. 10.1016/j.neulet.2005.05.018 . [DOI] [PubMed] [Google Scholar]
- 26.Kim J-S, Bailey MJ, Ho AK, Møller M, Gaildrat P, Klein DC. Daily rhythm in pineal phosphodiesterase (PDE) activity reflects adrenergic/3′, 5′-cyclic adenosine 5′-monophosphate induction of the PDE4B2 variant. Endocrinology. 2007;148(4):1475–85. 10.1210/en.2006-1420 [DOI] [PubMed] [Google Scholar]
- 27.Gaildrat P, Møller M, Mukda S, Humphries A, Carter DA, Ganapathy V, et al. A Novel Pineal-specific Product of the Oligopeptide Transporter PepT1 Gene CIRCADIAN EXPRESSION MEDIATED BY cAMP ACTIVATION OF AN INTRONIC PROMOTER. Journal of Biological Chemistry. 2005;280(17):16851–60. 10.1074/jbc.M414587200 [DOI] [PubMed] [Google Scholar]
- 28.Borjigin J, Payne AS, Deng J, Li X, Wang MM, Ovodenko B, et al. A novel pineal night-specific ATPase encoded by the Wilson disease gene. The Journal of neuroscience. 1999;19(3):1018–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bray N, Pimentel H, Melsted P, Pachter L. Near-optimal RNA-Seq quantification. arXiv preprint arXiv:150502710. 2015.
- 30.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nature methods. 2013;10(1):71–3. 10.1038/nmeth.2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28(5):511–5. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Au KF, Sebastiano V, Afshar PT, Durruthy JD, Lee L, Williams BA, et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proceedings of the National Academy of Sciences. 2013;110(50):E4821–E30. 10.1073/pnas.1320101110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayer KE, Pizarro A, Lahens NF, Hogenesch JB, Grant GR. Benchmark analysis of algorithms for determining and quantifying full-length mRNA splice forms from RNA-seq data. Bioinformatics. 2015:btv488 10.1101/007088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Treutlein B, Gokce O, Quake SR, Südhof TC. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proceedings of the National Academy of Sciences. 2014;111(13):E1291–E9. 10.1073/pnas.1403244111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schreiner D, Nguyen T-M, Russo G, Heber S, Patrignani A, Ahrné E, et al. Targeted combinatorial alternative splicing generates brain region-specific repertoires of neurexins. Neuron. 2014;84(2):386–98. 10.1016/j.neuron.2014.09.011 [DOI] [PubMed] [Google Scholar]
- 37.Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome. Nature biotechnology. 2013;31(11):1009–14. 10.1038/nbt.2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartley SW, Mullikin JC. Detection and visualization of differential splicing in RNA-Seq data with JunctionSeq. Nucleic acids research. 2016. Epub 2016/06/04. 10.1093/nar/gkw501 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy D, Singh R, Kolandaivelu S, Ramamurthy V, Stoilov P. Alternative splicing shapes the phenotype of a mutation in BBS8 to cause nonsyndromic Retinitis Pigmentosa. Molecular and cellular biology. 2015;35(10):1860–70. 10.1128/MCB.00040-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riazuddin SA, Iqbal M, Wang Y, Masuda T, Chen Y, Bowne S, et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. The American Journal of Human Genetics. 2010;86(5):805–12. 10.1016/j.ajhg.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bin J, Madhavan J, Ferrini W, Mok CA, Billingsley G, Héon E. BBS7 and TTC8 (BBS8) mutations play a minor role in the mutational load of Bardet‐Biedl syndrome in a multiethnic population. Human mutation. 2009;30(7):E737–E46. 10.1002/humu.21040 [DOI] [PubMed] [Google Scholar]
- 42.Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet–Biedl syndrome. Nature. 2003;425(6958):628–33. 10.1038/nature02030 [DOI] [PubMed] [Google Scholar]
- 43.Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–13. Epub 2007/06/19. 10.1016/j.cell.2007.03.053 . [DOI] [PubMed] [Google Scholar]
- 44.Blatch GL, Lässle M. The tetratricopeptide repeat: a structural motif mediating protein‐protein interactions. Bioessays. 1999;21(11):932–9. 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N [DOI] [PubMed] [Google Scholar]
- 45.Das AK, Cohen PT, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR‐mediated protein–protein interactions. The EMBO journal. 1998;17(5):1192–9. 10.1093/emboj/17.5.1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Falcón J, Coon SL, Besseau L, Cazaméa-Catalan D, Fuentès M, Magnanou E, et al. Drastic neofunctionalization associated with evolution of the timezyme AANAT 500 Mya. Proceedings of the National Academy of Sciences. 2014;111(1):314–9. 10.1073/pnas.1312634110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27(17):2325–9. 10.1093/bioinformatics/btr355 . [DOI] [PubMed] [Google Scholar]
- 48.Hartley SW, Mullikin JC. QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC bioinformatics. 2015;16:224 10.1186/s12859-015-0670-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nachury MV, Seeley ES, Jin H. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annual review of cell and developmental biology. 2010;26:59 10.1146/annurev.cellbio.042308.113337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zimmerman BL, Tso M. Morphologic evidence of photoreceptor differentiation of pinealocytes in the neonatal rat. The Journal of cell biology. 1975;66(1):60–75. 10.1083/jcb.66.1.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pévet P, Kappers JA, Voûte A. Morphologic evidence for differentiation of pinealocytes from photoreceptor cells in the adult noctule bat (Nyctalus noctula, Schreber). Cell and tissue research. 1977;182(1):99–109. 10.1007/bf00222058 [DOI] [PubMed] [Google Scholar]
- 52.Wang F, Li L, Xu H, Liu Y, Yang C, Cowley AW Jr, et al. Characteristics of long non-coding RNAs in the Brown Norway rat and alterations in the Dahl salt-sensitive rat. Scientific reports. 2014;4:7146 10.1038/srep07146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coon SL, Munson PJ, Cherukuri PF, Sugden D, Rath MF, Moller M, et al. Circadian changes in long noncoding RNAs in the pineal gland. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(33):13319–24. 10.1073/pnas.1207748109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509(7500):371–5. 10.1038/nature13173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nature communications. 2016;7 10.1038/ncomms11067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Care IoLARCo, Animals UoL, Resources NIoHDoR. Guide for the care and use of laboratory animals: National Academies; 1985. [Google Scholar]
- 57.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. British journal of pharmacology. 2010;160(7):1577–9. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Savastano LE, Castro AE, Fitt MR, Rath MF, Romeo HE, Munoz EM. A standardized surgical technique for rat superior cervical ganglionectomy. J Neurosci Methods. 2010;192(1):22–33. Epub 2010/07/20. 10.1016/j.jneumeth.2010.07.007 . [DOI] [PubMed] [Google Scholar]
- 59.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, et al. Ensembl 2013. Nucleic acids research. 2013;41(Database issue):D48–55. 10.1093/nar/gks1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome research. 2012;22(10):2008–17. 10.1101/gr.133744.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic acids research. 2011:gkr367 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
These transcripts were generated via a CuffLinks run on each sample in the discovery set, followed by the use of CuffMerge to merge these individual-sample assemblies. Note that CuffLinks failed to discover any of the novel transcripts, and there are no transcripts covering any of the 17 novel splice junctions observed in the Illumina and PacBio datasets.
(GZ)
It includes columns listing the ROI counts from the PacBio SMRT sequencing.
(TXT)
It includes columns listing the ROI counts from the PacBio SMRT sequencing. See also Fig 4C.
(TXT)
An excel file containing the JunctionSeq results for each analysis, for all genes that showed significant differential usage.
(XLSX)
This includes all exonic regions and known/novel splice junctions and their unique identifiers.
(GZ)
This includes all theoretical isoforms that had one or more perfect, full-length matches in the PacBio dataset. Also included is the GenBank accession numbers for each isoform.
(XLSX)
This Venn diagram displays the overlap between the genes detected as containing a differentially used feature in each of the four JunctionSeq “stimulus” analyses at the adjusted-p-value < 0.0001 level.
(PNG)
(PNG)
(PNG)
(PNG)
(PNG)
(PNG)
Rats were housed in a 14:10 light:dark cycle. ZT, Zeitgeber time.
(PNG)
As you can see, there are 30–60 read pairs per sample covering junction N040 in the innervated sample groups (Ctrl_N, Sham_N, DBcAMP and NE). However, CuffLinks does not detect any transcripts containing this junction. It also fails to detect novel start site 3b despite substantial read coverage over this region. Subsequent validation proved that these novel splice sites are real.
(PNG)
The label above each lane indicates the reaction number and the primer pair that was used for amplification. See S2 Table for primer sequences.
(PNG)
This plot shows a base-pair-resolution histogram of the read-of-insert (ROI) length produced by the PacBio SMRT sequencing of the Ttc8 gene. The reads are separated by primer pair and drawn separately in each color. The lengths of major potential predicted isoforms are marked at the bottom, along with the potential isoform ID. Reads of insert that did not match a legal primer pair at both ends were assumed to be non-full-length (non-FL) and are plotted in gray. Note that due to the accuracy of the SMRT sequencing platform, the presence of many of the predicted isoforms can be easily recognized based only on the high number of ROI at that exact length. Also note that this effect is less pronounced for the longer isoforms, due to the fact that the ROI accuracy is inversely associated with template length.
(PNG)
(PNG)
Rats were housed in a 14:10 light:dark cycle. Note that there are 2 samples at ZT7. Note that there are no reads covering the novel exon 3a, nor covering the region specific to novel exon 3b. ZT, Zeitgeber time.
(PNG)
The above figure displays the gene annotations and gene predictions from Ensembl, RefSeq, Augustus, the mammalian gene collection (MGC), SGP, and geneid. Note that none of the novel exons or splice junctions appear in any of the transcript annotations. Note that the Rat Genome Database (RGD) is not included in this plot because it has not yet been lifted over to the rn6 rat genome build. See (S15 Fig) for the RGD annotation.
(PNG)
This figure is identical to the previous, except zoomed in on the region containing the major novel exons 3a and 3b. Note that none of the novel exons or splice junctions appear in any of the transcript annotations.
(PNG)
Note that this assembly includes the RGD curated gene database. Additionally, note the presence of transcript ENSRNOT00000006199 in the ensembl annotation (release 69), which was absent in subsequent ensembl releases. This transcript includes one of the “novel” exons detected in our analyses, exon 2a. The transcript appears to have been lost in the liftover to rn5 (possibly due to a nearby gap in the rn5 genome build), and was never re-added in subsequent releases. Note that none of the other novel exons or splice junctions appear in any of the transcript annotations.
(PNG)
These primers were used to confirm and quantitate the differential expression of the three start sites of the Ttc8 gene. The first primer pair addresses the annotated Ttc8 start site, and primer pairs 2 and 3 address the two major novel start sites discovered in the Illumina RNA-Seq data.
(DOCX)
These primers were used to amplify the transcripts of the Ttc8 gene prior to sequencing with the PacBio SMRT platform. Note that primers F3 and F4 differ by only 4 bases, and primers F5 and F6 differ by only 6.
(DOCX)
In some cases additional size selection and amplification was necessary, as per the PacBio SMRT protocol. This table lists the size windows, primers, cycle counts, and total yield.
(DOCX)
These are the counts for ROI that match one of the 24 potential major isoforms (see Fig 3 and Fig 4).
(DOCX)
See also Fig 6B.
(DOCX)
See also Fig 3. Note that these counts are the number of ROI that perfectly match any of the theoretical potential isoforms that includes the given sub-variant. For example the first row (late donor, exon 4) compiles the counts from 8 different isoforms, all of which contain the late donor alternative splice junction on exon 4, as seen in Fig 6C.
(DOCX)
Data Availability Statement
The sequences reported in this paper have been deposited in the National Center for Biotechnology Information Short Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra), accession ID SRX1736990. The 42 novel isoforms detected in the PacBio dataset were deposited in GenBank, accession numbers KX218320 through KX218361.