

Abstract
Key points
In the present study, we document the role of compact myelin in regulating the structural and functional properties of ion channels at the nerve terminals, using electrophysiology, dynamic Na+ imaging and immunohistochemistry.
The subcellular segregation of Na+ channel expression and intracellular Na+ dynamics at the heminode and terminal was lost in the dysmyelinated axon from Long–Evans shaker rats, which lack compact myelin.
In Long–Evans shaker rats, loss of the Navβ4 subunit specifically at the heminode reduced resurgent and persistent Na+ currents, whereas K+ channel expression and currents were increased.
The results of the present study suggest that there is a specific role for compact myelin in dictating protein expression and function at the axon heminode and in regulating excitability of the nerve terminal.
Abstract
Axon myelination increases the conduction velocity and precision of action potential propagation. Although the negative effects of demyelination are generally attributed to conduction failure, accumulating evidence suggests that myelination also regulates the structural properties and molecular composition of the axonal membrane. In the present study, we investigated how myelination affects ion channel expression and function, particularly at the last axon heminode before the nerve terminal, which regulates the presynaptic excitability of the nerve terminal. We compared the structure and physiology of normal axons and those of the Long–Evans shaker (LES) rat, which lacks compact myelin. The normal segregation of Na+ channel expression and dynamics at the heminode and terminal was lost in the LES rat. Specifically, NaV‐α subunits were dispersed and NaVβ4 subunit was absent, whereas the density of K+ channels was increased at the heminode. Correspondingly, resurgent and persistent Na+ currents were reduced and K+ current was increased. Taken together, these data suggest a specific role for compact myelin in the orchestration of ion channel expression and function at the axon heminode and in regulating excitability of the nerve terminal.
Keywords: Calyx of held, Kv channels, Myelin, Nav channels, Presynaptic terminal
Key points
In the present study, we document the role of compact myelin in regulating the structural and functional properties of ion channels at the nerve terminals, using electrophysiology, dynamic Na+ imaging and immunohistochemistry.
The subcellular segregation of Na+ channel expression and intracellular Na+ dynamics at the heminode and terminal was lost in the dysmyelinated axon from Long–Evans shaker rats, which lack compact myelin.
In Long–Evans shaker rats, loss of the Navβ4 subunit specifically at the heminode reduced resurgent and persistent Na+ currents, whereas K+ channel expression and currents were increased.
The results of the present study suggest that there is a specific role for compact myelin in dictating protein expression and function at the axon heminode and in regulating excitability of the nerve terminal.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- AIS
the axon initial segment
- AP
action potential
- HVA
high‐voltage activated
- LE
Long–Evans rats
- LES
Long–Evans shaker
- MBP
myelin basic protein
- MNTB
medial nucleus of the trapezoid body
- Nav
voltage‐activated Na+ channel
- PBS
phosphate‐buffered saline
- ROI
region of interest
- TEA
tetraethylammonium chloride
- UTHSCSA
University of Texas Health Science Center, San Antonio
- WT
wild‐type
Introduction
A high density of voltage‐activated Na+ channels (Nav) clustered at discrete subcellular regions along the axon is necessary for the reliability and temporal fidelity of action potential (AP) firing. In addition to its role as an electrical insulator, the myelin sheath contributes to ion channel clustering. A recent study described alterations in the structure of the node of Ranvier and the relocation of Nav clusters at the axon initial segment (AIS) during demyelination (Hamada and Kole, 2015). These structural changes in axons following demyelination affect neuronal excitability, AP propagation, and neurotransmission.
The final destination of an AP is the nerve terminal where neurotransmitter release occurs. One of the critical factors for presynaptic excitability is the type, density and location of Nav channels (Engel and Jonas, 2005, Leão et al. 2005, Kawaguchi and Sakaba, 2015). Specifically, transient, persistent and resurgent Na+ currents, originating from the axon heminode, are required for the reliability of APs firing at high frequency and proper high fidelity signalling (Leão et al. 2005, Huang et al. 2008, Kim et al. 2010). In previous studies, we found that dysmyelination of auditory afferent axons reduces the reliability of presynaptic firing and temporal fidelity of synaptic transmission during high‐frequency firing in the calyx of Held synapse (Kim et al. 2013 a; Kim et al. 2013 b). These observations raise the possibility that loss of compact myelin may alter the pattern of ion channel expression at the last heminode and in the nerve terminal. The role of myelin on the structural and functional properties of the last axon heminode is an essential part of a complete understanding of the consequence of myelin loss on presynaptic excitability and neurotransmission. Currently, the extent to which channel proteins underlying accurate signalling and high‐fidelity performance are disrupted in dysmyelinated nerve terminal is largely unknown. Here, we tested the idea that myelin is an active participant in setting the structural and functional properties at the last heminode, where propagated APs are finely tuned before invading the terminal.
To examine the impact of lack of compact myelin on the axon heminode and terminal, we took advantage of the calyx of Held in the auditory brainstem that allows direct access to the heminode and terminal for electrophysiology and live cell imaging. Nav channels form clusters at nodes of Ranvier and the last heminode adjacent to the calyx terminal, but not within the presynaptic terminal (Leão et al. 2005). This distribution of Nav channels may contribute to low membrane excitability of the terminal and to the short duration of presynaptic APs (Alle and Geiger, 2006, Kawaguchi and Sakaba, 2015). Previously, we found electrophysiological abnormalities in presynaptic firing and synaptic transmission at the calyx of Held in the Long Evans shaker (LES) rat (Kim et al. 2013 a,b). The LES rat has a genetic deletion of myelin basic protein (MBP) and fails to condense the myelin sheath (Kwiecien et al. 1998, O´Conner et al. 1999). Axons from young LES rats before postnatal 4 weeks are loosely covered by 2–3 thin layers of myelin, but then undergo progressive demyelination until, by three months of age, most axons are demyelinated (Smith et al. 2013). Using the LES rat, we investigated how myelin loss impacts the structural and physiological properties of ion channels at the heminode and the calyx of Held terminal, using presynaptic Na+ imaging, Na+ and K+ current recording, and immunostaining. We found that lack of compact myelin specifically impacts expression of Nav and Kv channels at the axon heminodes and consequently alters subcellular Na+ dynamics at heminodes and terminals. The loss of Navβ4 subunit at the last heminode underlies the subcellular specific disruption of Nav. These results suggest that myelination is essential for proper location of Navβ4 subunit and the clustering of Na+ and K+ channels at axon heminodes and terminals, to maintain presynaptic excitability at nerve terminals in the auditory nervous system.
Methods
Ethical approval
Long–Evans rats (LE; wild‐type) and Long‐Evans Shaker rats (LES; from Dr Kwiecien, McMaster University, Ontario, Canada) of both sexes between the ages of postnatal day 13 (P13) and P17 were used for electrophysiological recordings and Na+ imaging, and at P20–P25 or P50 for immunohistochemistry. All procedures followed the approved animal care protocols by the University of Texas Health Science Center, San Antonio (UTHSCSA) Institutional Animal Care and Use Committee. All authors understood the ethical principles that The Journal of Physiology operates under and the work complied with the animal ethics checklist reported by Grundy (2015).
Animals were maintained as a heterozygous stock. A fraction of offspring from the heterozygous animals exhibited the LES phenotype, a distinguishable tremor, starting ∼2 weeks following birth (Delaney et al. 1995; O'Connor et al. 1999; Kim et al. 2013). The LES phenotype is autosomal recessive, present only in animals that are homozygous for the MBP mutation, whereas heterozygous and wild‐type (WT) animals show no signs of locomotion disturbance (Kwiecien et al. 1998; O'Connor et al. 1999; Kwiecien 2010; Smith et al. 2013, Kim et al. 2013 a,b). In the present study, we used WT LE (mbp+/+) and LES (homozygous mutant, mbp–/–) rats for all experiments.
Slice preparation
Transverse brainstem slices were prepared from rat pups of P13–P17. All procedures followed approved UTHSCSA animal care protocols. After rapid decapitation following isoflurane anaesthesia, the brainstem was quickly removed from the skull and immersed in ice‐cold low‐Ca2+ artificial cerebrospinal fluid (aCSF) containing (in mm): 125 NaCl, 2.5 KCl, 3 MgCl2, 0.1 CaCl2, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 0.4 ascorbic acid, 3 myo‐inositol and 2 Na‐pyruvate, pH 7.3–7.4 when bubbled with carbogen (95% O2, 5% CO2; osmolarity of 310–320 mosmol l–1). The brainstem was dissected and secured in a chamber with glue, and 200 μm (for electrophysiology and Na+ imaging) or 100 μm (for immunostaining) sections were cut with a vibratome (VT1200S; Leica, Bannockburn, IL, USA). Slices were then transferred to an incubation chamber containing normal aCSF bubbled with carbogen and maintained at 35°C for 30 min and thereafter at room temperature. Normal aCSF was the same as the slicing aCSF, except that it contained 1 mm MgCl2 and 2 mm CaCl2.
Electrophysiology
Slices were perfused at 2 ml min−1 and visualized using an infrared differential interference contrast microscope (AxoExaminer; Zeiss, Oberkochen Germany) with a 60× water‐immersion objective and a CMOS camera (ORCA‐Flash2.8; Hamamatsu, Hamamatsu City, Japan). Whole‐cell patch clamp recordings were performed in normal aCSF at room temperature (22–24°C) using an EPC‐10 amplifier controlled by Patch master software (HEKA, Elektronik, Lambrecht/Pfalz, Germany). For presynaptic current clamp recordings, the pipette solution contained (in mm): 125 K‐gluconate, 20 KCl, 5 Na2‐phosphocreatine, 10 Hepes, 4 Mg‐ATP, 0.2 EGTA and 0.3 GTP, pH adjusted to 7.3 with KOH. Presynaptic Na+ current recordings used a pipette solution containing (in mm): 130 CsCl, 10 tetraethylammonium chloride (TEA)‐Cl, 5 Na2‐phosphocreatine, 10 Hepes, 4 Mg‐ATP, 10 EGTA and 0.3 GTP, pH adjusted to 7.3 with KOH, and the external solution contained 2 mm 4‐aminopyridine, 10 mm TEA‐Cl and 0.2 mm CdCl2 to block presynaptic K+ and Ca2+ channels. Recordings were not corrected for the predicted liquid junction potential of 4 mV (CsCl‐based internal solution) or 11 mV (K‐gluconate‐based internal solution). Patch electrodes had resistances of 3–4 MΩ. For voltage clamp experiments, series resistance was <15 MΩ and was compensated 80%. Current clamp recordings were continued only if the initial uncompensated series resistance was <20 MΩ (Kim et al. 2013). The presynaptic axons of the calyx of Held terminal were stimulated with a bipolar platinum‐iridium electrode (Frederick Haer, Bowdoinham, ME, USA) placed near the midline spanning the afferent fibre tract of the medial nucleus of the trapezoid body (MNTB). An Iso‐Flex stimulator driven by a Master 10 pulse generator (AMPI, Jerusalem, Israel) delivered 100 μs pulses at 1.2 times the threshold (<15 V constant voltage). Signals were filtered at 2.9 kHz and acquired at a sampling rate of 10–50 μs.
Presynaptic Na+ imaging
The calyx of Held terminals were whole‐cell patch clamped using a pipette containing K‐gluconate based solution (see above) supplemented with 100 μm CoroNa Green (a Na+ indicator) and 50 μm Alexa 568 (Invitrogen, Carlsbad, CA, USA). We used an LED illumination system (Calibri; Zeiss) to excite indicators at 488 and 555 nm. Na+ imaging was performed with simultaneous whole‐cell current clamp recordings at the calyx terminals. Presynaptic APs were generated at 180 Hz for 1.5 or 3 s. The use of Alexa 568 permitted us to image cellular structures along the axon without bleaching the Na+‐sensitive dye. Na+ transients were recorded from the axon heminodes and presynaptic terminals. Fluorescence signals were acquired at 10–15 Hz and analysed using Zen Pro software (Zeiss). For analysis of CoroNa signals, regions of interest (ROI) within the axon heminode or the nerve terminal were drawn manually. Background fluorescence was measured from an area outside the cell, and all data are presented after background subtraction. All values are expressed as ΔF/F, where ΔF is the change in fluorescence during or after stimulation and F is the basal fluorescence of the ROI measured before stimulation. Measurements of intracellular Na+ concentration were calibrated by perfusing the slices with aCSF containing different concentrations of Na+ (ranging from 5 to 125 mm) in the presence of the Na+ ionophore monesin (50 μm) and ouabain (100 μm) to block the Na+ pump (Meier et al. 2006; Huang and Trussel, 2014). When NaCl was reduced, it was replaced by equimolar NMDG‐Cl to maintain osmolality.
Immunohistochemistry
Rat brainstem slices (100 μm) were fixed with 4% (w/v) paraformaldehyde in phosphate‐buffered saline (PBS) for 30 min. Free‐floating sections were blocked in 4% goat serum and 0.3% Triton X‐100 in PBS for 1 h. For immunohistochemistry, slices were incubated with the primary antibody overnight at 4°C. The primary antibodies used were: mouse anti‐sodium channel (Pan; dilution 1:200; Sigma, St Louis, MO, USA), mouse anti‐Kv3.1 (dilution 1:50; Neuromab, Davis, CA, USA), mouse anti‐Kv1.2 (dilution 1:200; Neuromab), rabbit anti‐Nav1.6 (dilution 1:100; Alomone Labs, Jerusalem, Israel), mouse anti‐Nav β4 (dilution 1:50; Neuromab), rabbit anti‐calretinin (dilution 1:200; Invitrogen), guinea pig anti‐Caspr (dilution 1:1000; from Dr Manzoor Bhat, UTHSCSA) and guinea pig polyclonal anti‐vesicular glutamate transporter 1 (vGluT1; dilution 1:1000; Millipore, Billerica, MA, USA). Antibody labelling was reported by incubation with appropriate Alexa dye‐conjugated secondary antibodies (dilution 1:500; Invitrogen) for 2 h at room temperature. After five rinses with PBS, coverslips were mounted onto slides using mounting medium (Fluoroshield; Sigma). Stained slices were imaged with laser lines at 488, 568 and 647 nm using an 60× oil‐immersion objective on a confocal laser scanning microscope (IX81 Fluoview 1000; Olympus, Tokyo, Japan).
Statistical analysis
Data are reported as the mean ± SEM. Each result established from immunostaining data was based on an analysis of at least 20 cells in 10 slices from three animals. Experimental data were analysed and presented using Igor Pro (Wavemetrics, Lake Oswego, OR, USA). Statistical significance was determined using Student's t test (Excel; Microsoft Corp., Redmond, WA, USA). P < 0.05 was considered statistically significant.
Results
AP‐evoked Na+ transients at the normal myelinated heminode and calyx nerve terminal
To investigate AP‐evoked intracellular Na+ transients in the axon heminode and nerve terminal, we recorded intracellular Na+ dynamics from the calyx of Held terminal and axon using the Na+‐sensitive dye CoroNa green during whole‐cell patch clamp recordings. Brief injections of current generated AP trains (180 Hz, 1.5 or 3 s in duration) and increased CoroNa green fluorescence at the axon heminode and at the presynaptic terminal (Fig. 1 A and B).
Figure 1. Na+ transients during action potentials in the calyx of Held nerve terminal .
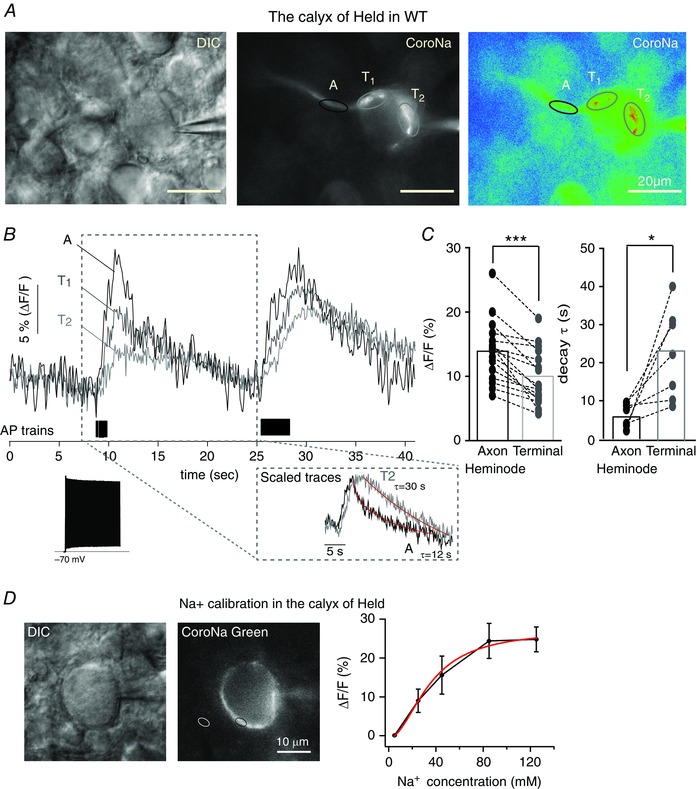
A, differential interference contrast and pseudocolour fluorescence images of the calyx of Held axon and terminal in the MNTB of the WT rat (P15) loaded with the Na+ indicator, CoroNa‐green (100 μm), during whole‐cell recording. Ovals indicate subcellular regions of interest [axon heminode (A, black) and terminal (T, grey)] from which fluorescence measurements were obtained. B, intracellular Na+ transients, measured as relative changes in CoroNa‐green fluorescence (ΔF/F), elicited by AP trains (180 Hz, 1.5 s and 3 s) recorded at the axon heminode (A, black trace) and presynaptic terminal (T1 and T2, grey traces). Note the rapid rise in Na+ at the axon heminode (A) and the slower and smaller rise at the presynaptic terminal (T1 and T2). Inset, representative trace of AP train at 180 Hz (1.5 s) and a scaled trace of the presynaptic terminal (grey) to the peak of signal from the axon heminode (black) to compare the decay time course of Na+ transient at the heminode (black, τ = 12 s) and terminal (grey, τ = 30 s). C, summary of the intensity (ΔF/F) and decay time constant (τ) of Na+ transients elicited by AP trains for (3 s, 180 Hz) at heminodes and terminals in the individual myelinated axon. Broken lines join paired data points obtained from the same axon. Data were analysed using a paired t test: *** P < 0.0001 and * P < 0.05, respectively. D, differential interference contrast and fluorescence images of the calyx of Held terminal, loaded with a Na+ indicator, CoroNa‐green (100 μm), using whole‐cell recordings. The black circle indicates the measuring area at presynaptic terminal and the white circle indicates the background area. Na+ calibration curve relating the change in fluorescence intensity as function of the applied Na+ concentrations ([Na+]): Fitting curve (red) was obtained using the Hill equation. [Colour figure can be viewed at wileyonlinelibrary.com]
Despite the smaller surface to volume ratio of the axon compared to the nerve terminal, the increase in CoroNa green fluorescence following an AP train (180 Hz, 3 s) was significantly higher at the axon heminode (ΔF⁄F = 15 ± 1.2%) than at the presynaptic terminal in the same axons (ΔF⁄F = 10 ± 1.1%) (n = 15, paired t test, P < 0.0001) (Fig. 1 B and C). Based on a calibration curve determined in the presence of monensin (50 μm; a Na+ ionophore) and ouabain (100 μm; to block the Na+ pump), the changes in CoroNa fluorescence correspond to intracellular Na+ concentration increases of ∼50 mm and 30 mm at the heminode and terminal, respectively (Fig. 1 D). No increases in fluorescence were observed when Na+ channels were blocked with TTX (1 μm; data not shown). In addition, the rise time of the fluorescence increase was faster at the heminode (t 10%–90% = 2.6 ± 0.17 s) than at the terminal (t 10%–90% = 3.7 ± 0.24 s), as was the exponential decay time constant (τ = 6.4 ± 1.2 s in the heminode vs. 22.3 ± 4.5 s in the terminal, n = 5, P = 0.02, paired t test) (Fig. 1 B and C). The earlier, steeper and faster Na+ transient recorded at the heminode, together with the fact that the rise time of the transient recorded in the terminal is longer than the AP train, suggests that, in the normal myelinated axon terminal, most of the Na+ currents enters at the heminode and then diffuse to the presynaptic terminal.
Lack of condensed myelination alters AP‐evoked Na+ transients at axon heminodes and presynaptic terminals
To examine the effect of the loss of condensed myelination on Na+ dynamics, we repeated the same experiments in the LES rat, a myelin‐deficient model (Fig. 2 A). By contrast to the normal axon, Na+ transients following APs in LES axons were significantly larger in the presynaptic terminal than at the heminode (Fig. 2 B and C). AP trains (180 Hz, 3 s) elevated intracellular Na+ (ΔF⁄F) by 10 ± 1.5% at the heminode and 13 ± 1.7% at the presynaptic terminal of the same axon (n = 11, paired t test, P = 0.04). The decay kinetics of Na+ transients along dysmyelinated axons were more variable than for the WT. Unlike that observed in the WT, there were no significant differences in the decay time constant of Na+ transients at axon heminodes and at presynaptic terminals, which were 38.6 ± 5.8 s and 44.4 ± 6.4 s, respectively (n = 9, P = 0.41, paired t test) (Fig. 2 C). Thus, in the LES, Na+ appears to enter simultaneously at both the heminode and the terminal, and the evidence for intercellular diffusion of Na+ described above for the normal axon terminal was not apparent in the dysmyelinated axon.
Figure 2. Lack of myelin alters the kinetics of Na+ transients at axon heminodes and terminals from LES rats .
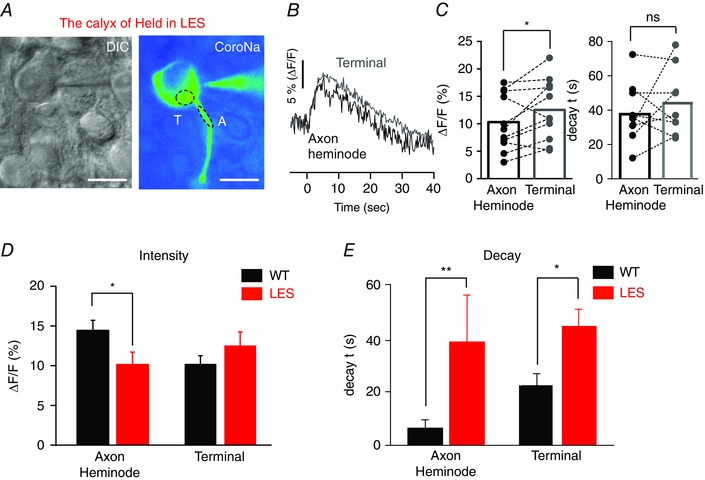
A, differential interference contrast and fluorescence images of the LES calyx axon and terminal (P16), which was loaded with CoroNa‐green (100 μm) during whole‐cell recordings. Ovals indicate heminode and terminal regions of interest in the dysmyelinated calyx axon from which fluorescence measurements were obtained. B, representative trace of Na+ transient (ΔF/F) in response to AP trains (180 Hz, 3 s) at the axon heminode (black) and presynaptic terminal (grey) in the LES rat. C, summary of changes in the intensity (ΔF/F) and decay time constant (τ) of Na+ transients at heminode and terminal in the same dysmyelinated axon from LES rats. Broken lines link data from the axon heminode and terminal of the same identified cell. Paired t test: * P < 0.05 and non‐significant (NS), respectively. D, comparison of Na+ transient (ΔF/F) in response to AP trains (180 Hz, 3 s) at axon heminodes and terminals from WT and LES rats. E, comparison of decay time constant (τ) of Na+ transients elicited by AP trains at axon heminodes and terminals from WT and LES rats. *** P < 0.0001, ** P < 0.001 and * P < 0.05, respectively. [Colour figure can be viewed at wileyonlinelibrary.com]
The differences in Na+ dynamics between WT and LES axons are summarized in Fig. 2(D) and (E). At axon heminodes, the increase in [Na+]i following APs was significantly higher in the WT than the LES (ΔF⁄F = 15 ± 1.2% and 10 ± 1.5%, n = 15 and 11, P = 0.03, unpaired t test). At presynaptic terminals, however, there was no significant difference in AP‐triggered increase in [Na+]i between myelinated and dysmyelinated axon terminals (ΔF⁄F = 10 ± 1.1% and 13 ± 1.7%, n = 15 and 11, P = 0.23, unpaired t test) (Fig. 2 D). Compared to myelinated axons, the decay of the Na+ transient was much slower at both axon heminodes and terminals in LES rats (n = 8) (Fig. 2 E). At heminodes, the decay time constant of the Na+ transients was 6.4 ± 1.2 s (n = 11) vs. 38.6 ± 5.8 s (n = 9) for WT and LES, respectively (P < 0.001), whereas, at the terminal, time constants were 22.3 ± 4.5 s (n = 7) and 44.4 ± 6.4 s (n = 9), respectively (P = 0.018). The level of myelination thus controls the intensity and kinetics of Na+ dynamics at axon heminodes and presynaptic terminals.
Loss of compact myelination disrupts Na+ channel clusters specifically at the last heminode next to the calyx terminal
Because Na+ influx via Na+ channels is one of the main sources for Na+ transients at the heminode and terminal, we examined the expression of Na+ channels by immunofluorescence microscopy of the AIS, nodes of Ranvier and the last heminode before the terminal (Fig. 3). The size of the Nav cluster in the AIS was similar in WT and LES axons for both the cochlear nucleus globular bushy cells (the projections of which form the calyx of held nerve terminal) and MNTB neurons (data not shown). Surprisingly, a lack of compact myelin did not change the immunohistochemical staining pattern of Nav cluster at nodes of Ranvier, nor was there any difference in the distance between nodes in the LES rat. The integrity of NaV channel pattering at nodes of Ranvier was confirmed in LES rats at age P50 (data not shown). The major alteration observed at nodes of Ranvier concerned the pattern of expression of Caspr, a prominent component of the paranodal axon glial junctions. Normal calyx axons, which were labelled with calretinin (a Ca2+‐binding protein expressed in nerve fibres), presented strong and well‐defined labelling for Caspr at both edges of the Nav channel cluster, whereas Caspr labelling was weak and disperse in nodes of Ranvier from the LES (Fig. 3 A and B). Despite the disruption of paranodal structure in LES axons, the length of the Nav cluster at nodes of Ranvier and the distance between nodes was not significantly different in WT and LES axons (Fig. 3 B). The lengths of nodes were 2.2 ± 0.08 μm (n = 54) in WT and 2.0 ± 0.09 μm (n = 55) in LES rats (unpaired t test, P = 0.27) and the internodal distances were 56 ± 3.8 μm in WT and 49± 2.7 μm in LES (n = 14 and 8, P = 0.248, unpaired t test) (Fig. 3 D and G).
Figure 3. Myelin loss specifically disrupts Na+ channels at the axon terminal heminode .
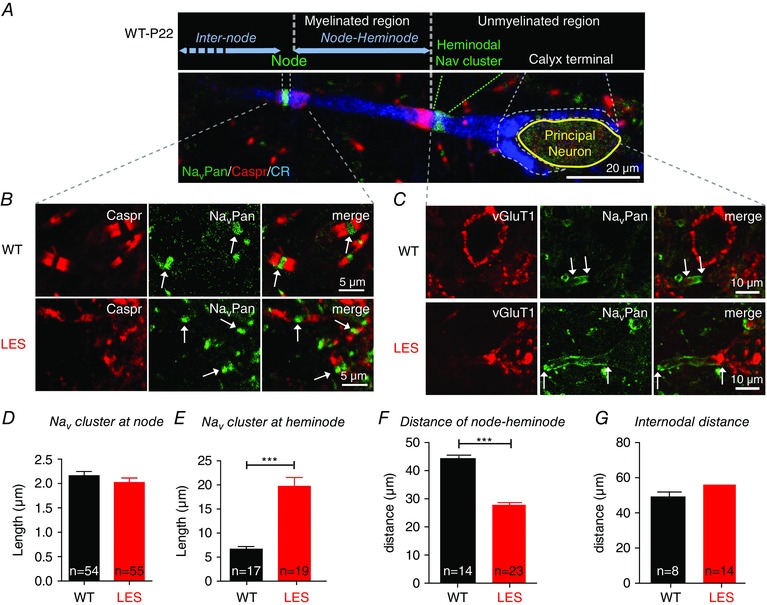
A, expression of NavPan (green), Caspr (red) and calretinin (CR, blue) immunoreactivity along the calyx of Held axon and terminal in the MNTB in the WT rat. The dashed lines delimit nodal and heminodal Nav cluster, as well as the calyx of Held. Blue horizontal arrows indicate the distance between the heminode and the next node, and the internodal distance, respectively. B, expression of Caspr (red) and NavPan (green) at axon nodes in the MNTB from WT and LES rats. Arrows indicate NavPan at nodes. C, expression of NavPan (green) and vGluT1 (red) at axon heminodes and calyx terminals. Arrows indicate the end of NavPan signals for measuring the length of Na+ channel cluster. D and E, summary of the length of Na+ channel cluster at nodes of Ranvier and axon heminodes. F and G, summary of the distance between the axon heminode and the next node, and between two nodes (internodal distance). *** P < 0.0001.
The last heminode separates the myelinated axon from the non‐mylinated nerve terminal. By contrast to the relatively normal clustering of Nav channels at the AIS and nodes of Ranvier, the expression of Nav channels was markedly altered in the last heminode and the calyx nerve terminal of the LES axon. In WT axons, Nav channels clustered in the heminode, which was located 14.1 ± 7.1 μm away from the neck of the calyx terminal (n = 28) (Fig. 3 A and C). In LES rats, the heminodal Nav cluster was much longer than in WT (19.7 ± 7.1 μm, n = 19, in the LES vs. 6.7 ± 2.2 μm, n = 17, in WT; P < 0.0001 unpaired t test) (Fig. 3 E). As a result of the dispersed Nav cluster at the heminode, the distance between the heminode and the next node was reduced in LES compared to WT (28 ± 4.5 μm vs. 44± 2.8 μm, n = 23 and 14, P < 0.001, unpaired t test) (Fig. 3 F). In addition, unlike for WT axons, in the LES axon, Nav channels invaded the calyx nerve terminal. This result indicates that compact myelination is essential for the expression pattern of Nav channels at the heminode adjacent to the presynaptic terminal.
These experiments were repeated using specific antibodies for Nav1.2 and Nav1.6 channel isoforms to test the possibility of differential or compensatory expression of two different Nav isoforms (Boiko et al. 2001, Vega et al. 2008). Expression of Nav1.6 followed the pattern described above both in WT and LES axons (Fig. 4 A), whereas Nav1.2 was not detected (data not shown), suggesting that the main subtype of Nav channels at axon heminodes and nodes is Nav1.6.
Figure 4. Dispersed Nav1.6 channel expression extends into the presynaptic terminal .
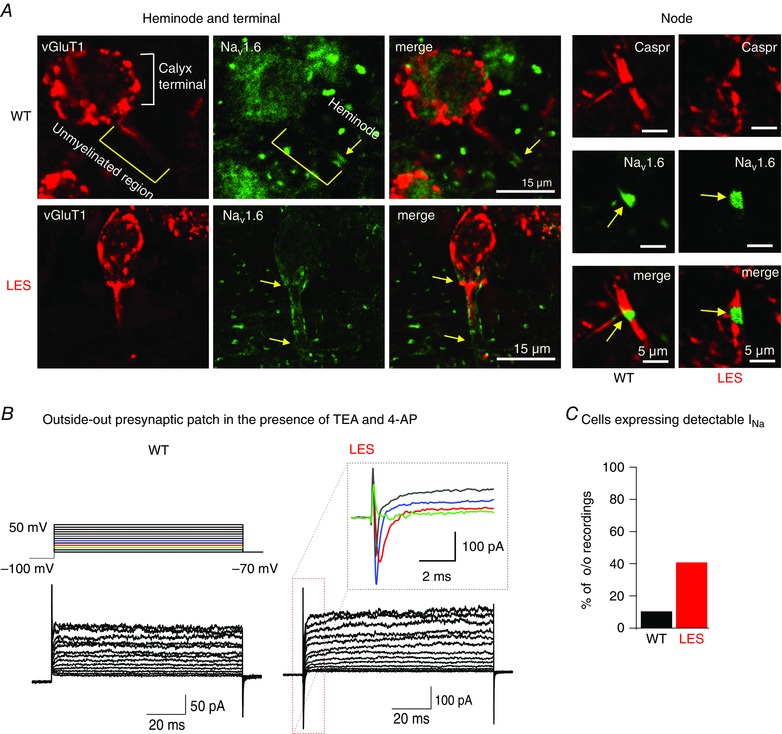
A, heminodal staining at calyx of Held presynaptic terminal with Nav1.6 (green) and vGluT (red) in WT and LES rats (left). Nodal and paranodal staining with Nav1.6 (green, yellow arrows) and Caspr (red) in WT and LES rats (right). B, representative traces of the outside‐out patch recordings from the calyx terminals in WT and LES rats. Voltage‐activated Na+ and K+ currents were evoked by step depolarization (from −100 mV to 50 mV; in the presence of TEA 10 mm and 4‐AP 2 mm) in the outside‐out patches. Inset, expanded scale of Na+ currents in the boxed area from the LES recording. C, summary of the proportion of the presynaptic terminals displaying detectable I Na in WT and LES rats in the outside‐out patch recordings.
The immunostaining data described above demonstrate that the Nav channel expression pattern extends into the presynaptic terminals in the LES axon, whereas, in WT axons, Nav channels are restricted to a cluster located 10–20 μm away from the terminal. We thus tested whether functional Na+ currents can be detected at presynaptic terminals using outside–out patch recording from the calyx terminal (Fig. 4 B). In WT rats, we rarely detected Na+ currents from outside‐out patch (one in 10 patches). By contrast, four of 10 patches from LES calyx terminals expressed measurable Na+ currents (−147 ± 16.8 pA at 0 mV, n = 4). Together with the immunostaining, our Na+ current recordings suggest that the lack of compact myelination disrupted the Nav cluster at the axon heminode and allowed Nav channels to invade the presynaptic terminal.
Dysmyelination reduces resurgent and persistent Na+ currents at the calyx of Held terminals
The detection of Na+ currents using outside‐out patches from LES terminals indicates the changes in the distribution of Nav channels, rather than an increase in the total whole‐cell Na+ currents at the LES heminode and tsierminal. Consistent with this idea, the Na+ imaging experiments and dispersed expression of Nav channels described above suggest smaller Na+ currents in the LES axon heminode. We tested whether alteration in Nav channel distribution affects Na+ currents under voltage clamp recordings in the presence of K+ and Ca2+ channel blockers (10 mm TEA, 2 mm 4‐AP and 0.2 mm CdCl2). A step‐depolarization from the holding potential of −80 mV to +30 mV induced large and rapidly inactivating transient Na+ currents with amplitudes of −4.2 ± 0.48 nA (n = 4) for WT and −4.1 ± 0.61 nA for the LES (n = 5, P > 0.05). Upon repolarization to potentials ranging from +20 to −70 mV, a voltage‐ and time‐dependent resurgent Na+ current (I NaR) was present (Fig. 5 A) (Raman and Bean, 1997, Kim et al. 2010, Lewis and Raman, 2014). The peak of resurgent Na+ current, measured at −40 mV, was significantly decreased in LES calyx terminals (WT: –310 ± 78.1 pA vs. LES: –61 ± 16.5 pA, n = 4 and 5, P < 0.001) (Fig. 5 B and C). We next examined persistent Na+ current (I NaP) using voltage ramps of 100 mV s−1 (from −90 mV to +50 mV) in the calyx of Held in WT and LES rats. During the voltage ramps, the fast transient current was fully inactivated to isolate I NaP, which was completely blocked by 1 μm TTX (data not shown). By subtracting a trace with 1 μm TTX from a control trace, the current–voltage relationship for I NaP was determined, revealing peak values of −376 ± 19.1 pA in WT calyces and −170 ± 26.8 pA in LES calyces (n = 4 and 6, P < 0.05) (Fig. 5 D). Thus, I NaR and I NaP are significantly reduced in the axon terminals from LES rats.
Figure 5. Reduced resurgent and persistent Na+ currents at axon terminal in the LES rat .
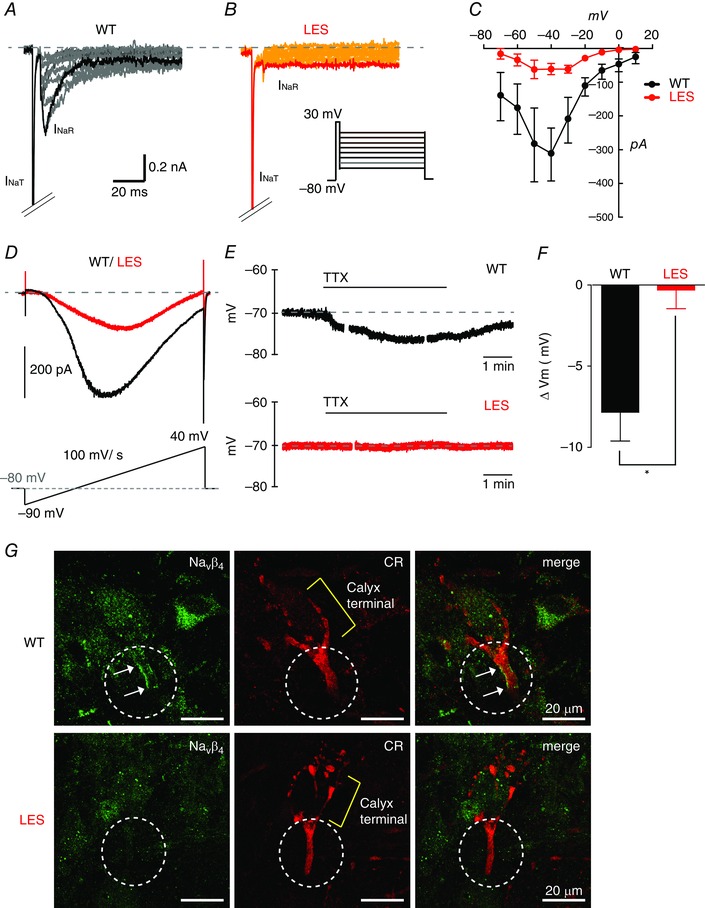
A and B, representative traces of resurgent Na+ currents evoked by step repolarization (from +20 mV to −70 mV) after a brief depolarization (+30 mV, 5 ms) to generate transient Na+ currents (INaT) at the calyx of Held terminals in WT (A) and LES rats (B). C, current–voltage (I–V) relationship of resurgent Na+ currents form WT (black) and LES rats (red). D, persistent Na+ currents were recorded by ramp depolarization from −90 mV to +40 mV (100 mV/s) at the calyx of Held terminals in WT (black) and LES rats (red). Representative traces of persistent Na+ currents after TTX subtraction. E, in current clamp recordings, TTX (1 μm) hyperpolarized the calyx terminal in the MNTB from WT but not LES rats. F, summary of changes in calyx membrane resting potential (ΔV m) by the application of TTX (1 μm) in the MNTB from WT and LES rats. G, Navβ4 (green) expression at the axon heminode with calretinin (CR, red) in WT and LES rats. Circles indicate the heminodal region and arrows indicate the Navβ4 cluster. Unpaired t test, * P < 0.05. [Colour figure can be viewed at wileyonlinelibrary.com]
To determine the function consequences of I NaP on the resting potential, we first set the resting potential to −70 mV by applying small holding currents (−18.7 ± 3.4 pA vs. −9.7 ± 1.8 pA for WT and LES calyx terminals, respectively, n = 9 and 9, P = 0.03). Then, membrane potential was recorded during the application of 1 μm TTX to inhibit I NaP. TTX hyperpolarized WT calyx terminals (ΔV m = −7.8 ± 3.5 mV, n = 5, P = 0.008, paired t test), whereas, in LES, TTX application had no significant effect on the resting membrane potential (ΔV m = −0.2 ± 2.3 mV; n = 5, P = 0.814) (Fig. 5 E and F).
Previous studies demonstrated that the Navβ4 subunit underlies the generation of I NaR and I NaP (Grieco et al. 2005, Aman et al. 2009, Bant and Raman, 2010, Kim et al. 2010). To test whether a lack of compact myelin selectively impacts on Navβ4 expression, we examined Navβ4 expression at axon heminodes and terminals from WT and LES rats. We found that Navβ4 clusters were located at the last axon heminodes but not at terminals in the WT, whereas LES heminodes did not exhibit detectable Navβ4 (n = 17 cells in four slices) (Fig. 5 G). It thus appears that reduction of Navβ4 expression in LES heminodes explains the observed reduction in I NaR.
Alterations in the distribution of Kv channels in the dysmyelinated axon terminal
In addition to Nav channels, axons depend on Kv channels to shape the AP waveform. We therefore examined the expression of low‐voltage activated Kv1.2 channels and high‐voltage activated (HVA) Kv3.1 channels because these two channel types play an important role in reliable AP firing at the calyx of Held terminal (Ishikawa et al. 2003, Johnston et al. 2010). Along the normally myelinated axon, clusters of Kv3.1 and Kv1.2 were located, respectively, at nodes and heminodes, and at juxtaparanodes (Fig. 6 A). In LES, Kv1.2 clusters located at juxtaparanodes were completely disrupted, whereas clusters of Kv3.1 could still be identified at nodes (Fig. 6 B). At the heminode, however, Kv3.1 expression was disperse and extended into the calyx nerve terminals (Fig. 6 C). To quantify Kv3.1 expression at presynaptic terminals, we calculated the ratio of immunofluorescence of Kv3.1 to that of vGluT1 (a presynaptic marker). This measure of Kv3.1 expression at the calyx terminal was higher in LES (0.93 ± 0.12, n = 8) than WT (0.6 ± 0.09, I = 7, P = 0.048, unpaired t test) (Fig. 6 D), indicating that Kv3.1 expression increases at the calyx terminal from the LES.
Figure 6. Lack of compact myelin disrupts the cluster of Kv1.2 at juxtaparanodes and Kv3.1 at heminodes .
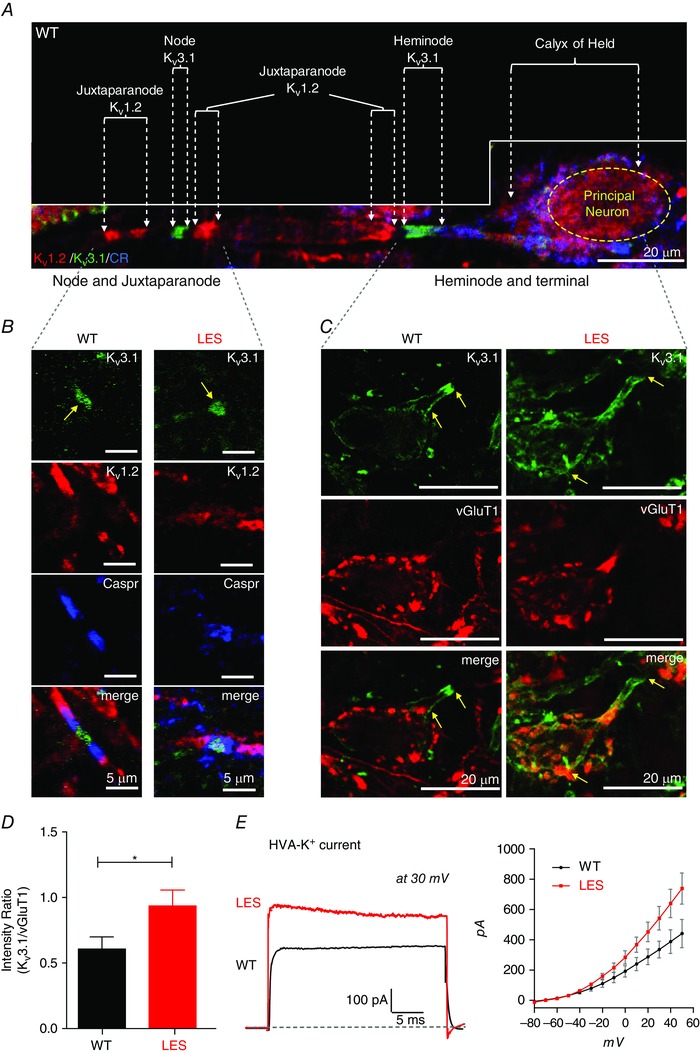
A, the calyx of Held axon and terminal were immunolabelled for Kv3.1 (green), Kv1.2 (red) and calretinin (CR, blue) in the WT rat brainstem. Dashed arrows indicate heminodes, nodes and juxtaparanodes, as well as the calyx terminal. Note, Kv3.1 was expressed at the node and the heminode and Kv1.2 was expressed at the juxtaparanode. B, expression of Kv3.1 (green), Caspr (blue) and Kv1.2 (red) at nodes, paranodes and juxtaparanodes along the axon in WT and LES rats. Arrows indicate Kv3.1 at nodes. C, at the axon heminode and terminal, expression of Kv3.1 (green) and vGluT1 (red) in WT and LES rats. Yellow arrows indicate the extent of Kv3.1 expression, which invades the presynaptic terminal in the LES. D, summary of the intensity ratio of Kv3.1 signal by vGluT1 at presynaptic terminals in WT and LES rats obtained in (C). E, representative trace of TEA‐sensitive HVA‐K+ (at +30 mV; sensitive to TEA 1 mm) at holding −70 mV recorded in the calyx terminal from WT and LES rats. Traces were obtained after TEA subtraction. The current–voltage (I–V) relationships of TEA‐sensitive HVA‐K+) currents in WT and LES rats obtained after the TEA subtraction. Unpaired t test, * P < 0.05. [Colour figure can be viewed at wileyonlinelibrary.com]
We examined how altered expression of Kv3.1 channels affects functional HVA K+ currents at the calyx terminals in LES rats. Kv3.1 channels are sensitive to a low concentration of TEA (1 mm) (Coetzee et al. 1999). TEA‐sensitive HVA K+ currents were significantly larger at presynaptic terminals in LES (542 ± 77 pA at 30 mV, n = 6) than in WT rats (335 ± 55 pA at 30 mV, n = 6, unpaired t test, P = 0.0012) (Fig. 6 E). This observed increase in HVA K+ current is consistent with and provides a mechanistic explanation for the larger after‐hyperpolarization after the action potential in calyx terminals from LES rats described previously (Kim et al. 2010, 2013 a).
We examined the expression and co‐localization patterns for Nav1.6 and Kv3.1 at nodes of Ranvier, the heminode and the nerve terminal for normal and LES axons in the MNTB. In the WT axon, Nav1.6 and Kv3.1 clusters are co‐localized to the axon node and heminode. At the last heminode, Nav1.6 clusters were located 10–20 μm from the terminal, with only sparse Nav1.6 channels at the presynaptic terminal, whereas Kv3.1 expression extended to the terminal. In the LES rat, expression of both Nav1.6 and Kv3.1 was dispersed and channels invaded the terminal (Fig. 7 A and C). By contrast, the overall structure of distal nodes of Ranvier in LES rats appeared to be less disrupted than the nerve terminal. Kv1.2 channels, which are normally clustered at the juxtaparanode, were dispersed throughout the node in the LES axon, although the co‐localization of Nav1.6 and Kv3.1 clusters at nodes from LES rats appeared to be similar to WT nodes (Fig. 7 B). This appears to be sufficient for low frequency firing of the presynaptic terminal because LES calyx terminals can follow action potential trains up to 100 Hz without conduction failure (Kim et al. 2013 a). However, the observation that the reliability and firing at high frequencies (>300 Hz) (as occurs physiologically) is severely compromised in the LES calyx nerve terminal (Kim et al. 2013 a) suggests that the precise distribution of ion channels is essential for its function.
Figure 7. Lack of condensed myelin affects the distribution of Nav and Kv clusters at the heminode and calyx of Held terminal in the MNTB .
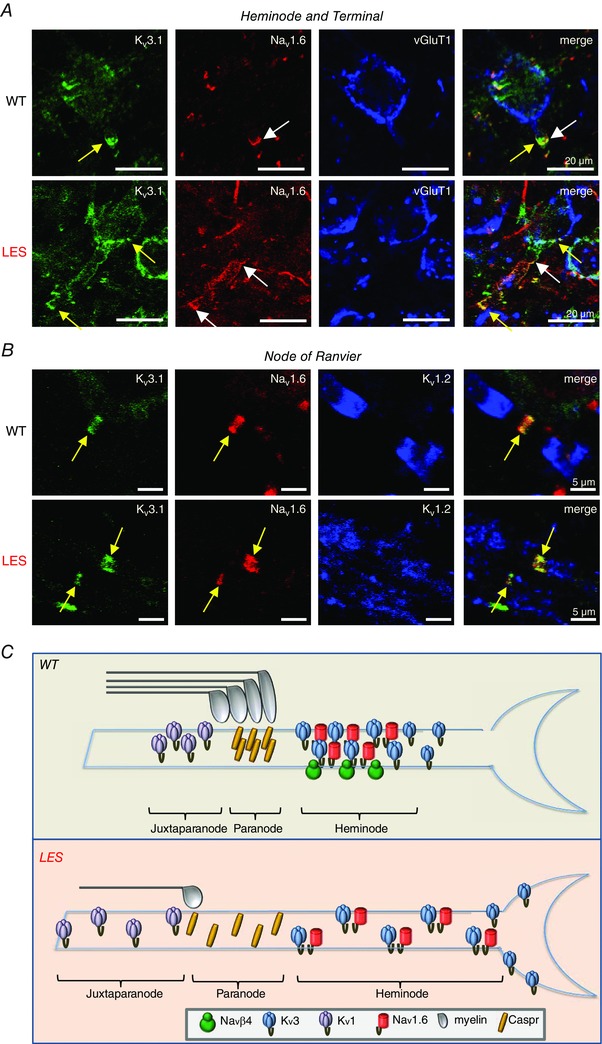
A, expression of Kv3.1 (green) and Nav1.6 (red) at the axon heminode and terminal marked with vGluT1 (blue) in WT and LES rats. Kv3.1 (yellow arrow) and Nav1.6 clusters (white arrow) were well co‐localized at the axon heminode in WT rats, whereas their clusters completely disrupted at heminode and dispersed to the terminal in LES rats (yellow arrows). B, nodal and juxtaparanodal staining with Kv3.1 (green), Nav1.6 (red) and Kv1.2 (blue) in WT and LES rats. Kv3.1 and Nav1.6 clusters are well preserved at the node, although juxtaparanodal structures stained with Kv1.2 are disrupted in LES axons. C, summary diagram of effects of myelin loss on the distribution of voltage‐gate activated ion channels (Nav1.6, Navβ, Kv1 and Kv3) at the last heminode. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Alterations in the structure of the node of Ranvier during demyelination have been described, although little is known about the role of myelination in regulating the structural and functional properties of the nerve terminal. In the present study, we provide evidence indicating that proper myelination plays a critical role in fine‐tuning presynaptic excitability and Na+ dynamics in the CNS nerve terminal. Specifically, compact myelination is required for the structural and functional properties of Nav and Kv channels located in the last axon heminode. Loss of compact myelin disrupts Nav channel clustering, and also changes Na+ dynamics and presynaptic excitability at the nerve terminal. We propose that the loss of Navβ4 (leading to the loss of resurgent Na+ current) and specific disruption of Nav and Kv clusters at the axon heminode and terminal underlies the loss of action potential reliability in axons and nerve terminals in the LES model of dys‐/demyelinating diseases.
Axon myelination regulates AP propagation to the calyx terminal and presynaptic excitability
Changes in ion channel expression in the last axon heminode critically affect presynaptic excitability at the nerve terminal. Previous studies indicate that small changes to the conductance or spatial distribution of voltage‐dependent Na+ and K+ channels can determine the success or failure of AP invasion from a myelinated axon into a non‐myelinated segment, such as the heminode (Waxman and Wood, 1984). The auditory nervous system depends on a high reliability and temporal fidelity of impulse propagation to process auditory information. The calyx of Held terminal, located in the auditory brainstem, can fire APs at rates approaching 1 kHz, and its postsynaptic partner, the MNTB neuron, can keep up with transmission at frequencies up to 600 Hz in vitro without failure (Wu and Kelly 1993; Taschenberger and von Gersdorff 2000; Kopp‐Scheinpflug et al. 2008; Kim et al. 2013 a). For this fast and accurate spiking performance, the projections from cochlear nucleus cells require highly myelinated axons, as well as Nav kinetics with fast recovery from inactivation and the presence of resurgent Na+ currents at the heminode next to the terminal.
LES rats present slower conduction to the calyx of Held with a higher failure rate at high frequency than WT (Kim et al. 2013 a). Conduction velocity is determined by several parameters, including axon diameter and internode length (internodal distance), as well as myelin thickness (Moore et al. 1978, Ford et al. 2015). In LES calyces, axon diameter increases from P18 (2.3 ± 0.19 μm, n = 10) to P38 (3.0 ± 0.26 μm, n = 14), whereas WT calyces showed no change in axon diameter (1.8 ± 0.1 μm at P18, n = 10 vs. 1.8 ± 0.1 μm at P38; data not shown). Therefore, changes to axon diameter probably do not account for slower conduction and increased AP failure in the LES rat. There was no significant difference with respect to internodal length in LES and WT axons, and the distance between the last node and the heminode was significantly reduced in LES calyces. Therefore, changes to internode distances also cannot account for the LES phenotype. In principle, a larger axon diameter and smaller internodal length would rather increase conduction velocity (Ford et al. 2015). Therefore, a reduced conduction velocity in the LES may be a direct consequence of dys‐/demyelination because modelling studies predict that the degree of myelination is one of the most important determinants of conduction velocity (Moore et al. 1978).
With respect to the frequency‐dependent AP failures observed in the LES rat, our recordings made in the nerve terminal cannot determine where these failures occur along the axon. In a previous study, we showed that the mature calyx of Held synapse relies on resurgent Na+ currents for reliable high‐frequency firing, and that AP failures during train in the immature calyx terminal at P5–7 could be rescued by dialysing the terminal with NaVβ4 peptide (Kim et al. 2010). Together with the lack of expression of NaVβ4 in the LES heminode, these data suggest that resurgent Na+ current in the heminode is essential for proper excitability of the nerve terminal. However, in the aforementioned study, we were unable to track the diffusion of the peptide back along the axon. Possibly, resurgent Na+ current is important for proper excitability along the entire axon and the site of rescue by the β4 peptide may have been distal to the heminode.
Another key element for determining presynaptic excitability is the expression pattern and kinetics of Kv channels, which were also affected by myelin loss. A stronger and more dispersed expression of Kv3.1 at the heminode and presynaptic terminal was accompanied by larger HVA‐K+ currents and a larger after‐hyperpolarization, whereas persistent and resurgent Na+ current was reduced. Together, these alterations are expected to reduce membrane excitability (Waxman and Wood, 1984, Kawaguchi and Sakaba, 2015).
In WT rats, calyces at the age studied can follow AP trains of up to several hundred Hz without failure. The LES rat displays AP failures at stimulations frequencies above 300 Hz but is still able to fire APs at up to 100 Hz (Kim et al. 2013 a). Although we cannot measure Na+ and K+ currents along the axon, our immunohistochemistry indicates apparently normal clustering and co‐localization of Nav1.6 and Kv3.1 at nodes (Fig. 7), which must be sufficient to maintain its limited excitability despite disruption of Kv1.2 at juxtaparanodes (Fig. 6). A recent study suggests that clustering of channel proteins in nodes increases conduction velocity, independent of the presence of myelin (Freeman et al. 2015). Another factor that probably helps maintain AP propagation in the LES is that the distance between the last node of Ranvier and the heminode was significant decreased in LES calyces (Fig. 3 F). The boundary between myelinated and non‐myelinated segments of an axon terminal is a potential site for AP failure and depends critically on the length of the non‐myelinated segment (Waxman and Brill, 1978). In the LES, the reduced intermodal length closest to the nerve terminal may facilitate conduction at the nerve terminal, allowing it to function without failure in response to 100 Hz stimulation (Kim et al. 2013).
Axon myelination regulates Na+ dynamics at the nerve terminal
Where is the main entry of presynaptic Na+ during AP firing? In WT, Na+ transients measured at the heminode are larger and faster and occur earlier than at the presynaptic terminal (Fig. 1). Immunostaining of Nav channels showed strong labelling in the heminode with little or no expression in the calyx terminal (Fig. 3). Together, these data indicate that, in the normal myelinated axon, most Na+ enters at the last heminode via Nav channels and then diffuses passively to the calyx terminal. This hypothesis is similar to the retrograde diffusion of Na+ from the AIS to the soma in cortical pyramidal neurons (Fleidervish et al. 2010). What determines Na+ dynamics at the nerve terminal? The Na+/K+ ATPase is the main mechanisms regulating presynaptic Na+ gradients and the resting potential because blocking of Na+/K+ ATPase with ouabain (100 μm) gradually increased the basal Na+ level at presynaptic terminals (data not shown). However, in myelinated axons, the rapid decay of the Na+ transient after a train of APs mainly depends on fast bilateral diffusion along the axon (Fleidervish et al. 2010). Axon dysmyelination impacts the intensity and decay time of the Na+ transient, as well as Nav expression at the heminode and the presynaptic terminal (Figs 1, 2, 3). In the LES, a reduced and dispersed expression pattern of Nav channels probably explains the smaller and slower Na+ transient measured in the heminode. Conversely, the increased expression of Nav channels in the nerve terminal probably underlies the increased size of the Na+ transient measured in the terminal of LES calyces. Moreover, a more spatially uniform Na+ entry, as a result of a more spatially uniform Nav channel distribution, reduces the ability of Na+ to clear by diffusion and is expected to increase the time for Na+ transients to decay. However, we cannot rule out the possibility that demyelization also affects Na+ extrusion mechanisms mediated by Na+/K+ ATPase and NCX (Kim et al. 2005, 2007; Lee and Kim, 2015) in the LES rat.
Compact myelination is essential for clustering of Nav and Kv at the axon heminode and terminal, rather than at distal nodes
In the present study, we revealed the role of myelin in the segregation of Nav channel expression at the nerve terminal to maintain a high density of Nav channels present at the heminode and a sparse density of Nav channels in the presynaptic terminal. This conserved structure around the nerve terminal was severely disrupted by a loss of compact myelination in the LES rat, whereas alterations at distal axon were mostly confined to an altered expression pattern for caspr and Kv1.2 at paranodes and juxtaparanodes (Figs 3 and 6).
Previous studies using cuprizone to induced demyelination (Dupree et al. 2004, Crawford et al. 2009, Hamada and Kole, 2015) or mice deficient for ceramide galactosyltransferase (Dupree et al. 2004) demonstrated disruption of the nodal Nav cluster. Unlike these previous studies, we observed that Nav1.6 and Kv3.1 at distal nodes remained well clustered in the LES rat and appeared quite similar to the WT, although paranodal and juxtaparanodal structures were disrupted. Nodal Na+ clusters require axon–oligodendrocyte interactions and axonal cytoskeletal mechanisms at nodes (Rasband et al. 1999, Susuki et al. 2013). The afferent fibres to the MNTB display Nav clusters at nodes of Ranvier in the first postnatal week, before MBP‐mediated formation of compact myelination, whereas the Nav channel is detectable at the last heminode only after the onset of myelination (after P7–8; data not shown). This suggests that MBP‐mediated compact myelination is required for Nav cluster formation specifically at the last heminode but not at nodes of Ranvier (Freeman et al. 2015). Instead, the interaction between axon and oligodendrocytes or the formation of axonglial junctions, prior to MBP expression, might play an important role in forming a nodal Nav cluster (Rasband et al. 1999, Susuki et al. 2013, Freeman et al. 2015).
In some demyelination models (e.g. experimental autoimmune encephalomyelitis and the cuprizone models), nodal clusters of Nav channels deteriorated during demyelination. In the LES model, we observed normal Na+ clustering at nodes. The discrepancy may depend on the underlying mechanisms of clustering and the cause of demyelination. If axon–oligodendrocyte interactions are the initial determinant for the location of Na+ channels at nodes, oligodendrocyte or axon degeneration could alter nodal structures in demyelinated models. In the LES model, we observed no strong effect on axon–oligodendrocyte interaction, and no axon or oligodendrocyte degeneration within the first month of age, although MBP deletion strongly inhibited the formation of compact myelination around axons. Our data suggest that the presence of compact myelination maintains the heminodal Nav cluster near the terminal. In the present study, nodal Nav channels maintained a normal cluster pattern along dysmyelinated LES axons in young adult animals (up to P50; data not shown). It remains to be determined whether nodal Nav clusters are maintained in ageing in the LES rat.
In summary, the results of the present study demonstrate that compact myelin is critical to maintain a proper location and function of Nav and Kv at the axon heminode and terminal.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
JHK designed the research. EB, SEK, SYL, and JHK collected data. EB, SEK, SYL, CK and JHK analysed and interpreted data. EB, CK and JHK wrote the manuscript. JHK provided financial support. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the NIDCD (R01DC013157) to JHK and by grants from CNPq (478663/2012‐9 and 306293/2012‐9) to CK.
Acknowledgements
We thank Mr Butch Moomaw for technical assistance with Na+ imaging, Dr Jacek Kwiecien (McMaster University) for providing the Long–Evans shaker rat and Dr Manzoor Bhat (UTHSCSA) for providing antibodies, respectively.
This is an Editor's Choice article from the 1 October 2016 issue.
References
- Alle H & Geiger JR (2006). Combined analog and action potential coding in hippocampal mossy fibers. Science 311, 1290–1293. [DOI] [PubMed] [Google Scholar]
- Aman TK, Grieco‐Calub TM, Chen C, Rusconi R, Slat EA, Isom LL & Raman IM (2009). Regulation of persistent Na current by interactions between beta subunits of voltage‐gated Na channels. J Neurosci 29, 2027–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo EJ, Xu T, Grinspan J, Lambert S, Levinson SR, Brophy PJ, Peles E & Scherer SS (2002). Genetic dysmyelination alters the molecular architecture of the nodal region. J Neurosci 22, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bant JS & Raman IM (2010). Control of transient, resurgent, and persistent current by open‐channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci USA 107, 12357–12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS & Matthews G (2001). Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30, 91–104. [DOI] [PubMed] [Google Scholar]
- Buffington SA & Rasband MN (2013). Na+ channel‐dependent recruitment of Navβ4 to axon initial segments and nodes of Ranvier. J Neurosci 33, 6191–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA et al (1999). Molecular diversity of K+ channels. Ann NY Acad Sci 868, 233–285. [DOI] [PubMed] [Google Scholar]
- Crawford DK, Mangiardi M, Xia X, López‐Valdés HE & Tiwari‐Woodruff SK (2009). Functional recovery of callosal axons following demyelination: a critical window. Neuroscience 164, 1407–1421. [DOI] [PubMed] [Google Scholar]
- Delaney KH, Kwiecien JM, Wegiel J, Wisniewski HM, Percy DH & Fletch AL (1995). Familial dysmyelination in a Long Evans rat mutant. Lab Anim Sci 45, 547–553. [PubMed] [Google Scholar]
- Dodson PD, Barker MC & Forsythe ID (2002). Two heteromeric Kv1 potassium channels differentially regulate action potential firing. J Neurosci 22, 6953–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Mason JL, Marcus JR, Stull M, Levinson R, Matsushima GK & Popko B (2004). Oligodendrocytes assist in the maintenance of sodium channel clusters independent of the myelin sheath. Neuron Glia Biol 1, 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel D & Jonas P (2005). Presynaptic action potential amplification by voltage‐gated Na+ channels in hippocampal mossy fiber boutons. Neuron 45, 405–417. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Lasser‐Ross N, Gutnick MJ & Ross WN (2010). Na+ imaging reveals little difference in action potential‐evoked Na+ influx between axon and soma. Nat Neurosci 13, 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman SA, Desmazières A, Simonnet J, Gatta M, Pfeiffer F, Aigrot MS, Rappeneau Q, Guerreiro S, Michel PP, Yanagawa Y, Barbin G, Brophy PJ, Fricker D, Lubetzki C & Sol‐Foulon N (2015). Acceleration of conduction velocity linked to clustering of nodal components precedes myelination. Proc Natl Acad Sci USA 112, E321–E328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried SI, Lasker AC, Desai NJ, Eddington DK & Rizzo JF 3rd (2009). Axonal sodium‐channel bands shape the response to electric stimulation in retinal ganglion cells. J Neurophysiol 101, 1972–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MC, Alexandrova O, Cossell L, Stange‐Marten A, Sinclair J, Kopp‐Scheinpflug C, Pecka M, Attwell D & Grothe B (2015). Tuning of Ranvier node and internode properties in myelinated axons to adjust action potential timing. Nat Commun 6, 8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR & Jonas P (2000). Dynamic control of presynaptic Ca2+ inflow by fast‐inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron 28, 927–939. [DOI] [PubMed] [Google Scholar]
- Grubb MS & Burrone J (2010). Activity‐dependent relocation of the axon initial segment fine‐tunes neuronal excitability. Nature 465, 1070–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL & Raman IM (2005). Open‐channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 45, 233–244. [DOI] [PubMed] [Google Scholar]
- Hamada MS & Kole MH (2015). Myelin loss and axonal ion channel adaptations associated with gray matter neuronal hyperexcitability. J Neurosci 35, 7272–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H & Jonas P (2014). A supercritical density of Na+ channels ensures fast signaling in GABAergic interneuron axons. Nat Neurosci 17, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H1 & Trussell LO (2008). Control of presynaptic function by a persistent Na(+) current. Neuron 60, 975–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H & Trussell LO (2014). Presynaptic HCN channels regulate vesicular glutamate transport. Neuron 84, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Nakamura Y, Saitoh N, Li WB, Iwasaki S & Takahashi T (2003). Distinct roles of Kv1 and Kv3 potassium channels at the calyx of Held presynaptic terminal. J Neurosci 23, 10445–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J, Forsythe ID & Kopp‐Scheinpflug C (2010). Going native: voltage‐gated potassium channels controlling neuronal excitability. J Physiol 588, 3187–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi SY & Sakaba T (2015). Control of inhibitory synaptic outputs by low excitability of axon terminals revealed by direct recording. Neuron 85, 1273–1288. [DOI] [PubMed] [Google Scholar]
- Kim JH, Sizov I, Dobretsov M & von Gersdorff H (2007). Presynaptic Ca2+ buffers control the strength of a fast post‐tetanic hyperpolarization mediated by the alpha3 Na(+)/K(+)‐ATPase. Nat Neurosci 10, 196–205. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kushmerick C & von Gersdorff H (2010). Presynaptic resurgent Na+ currents sculpt the action potential waveform and increase firing reliability at a CNS nerve terminal. J Neurosci 30, 15479–15490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Renden R & von Gersdorff H (2013. a). Dysmyelination of auditory afferent axons increases the jitter of action potential timing during high‐frequency firing. J Neurosci 33, 9402–9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SE, Turkington K, Kushmerick C & Kim JH (2013. b). Central dysmyelination reduces the temporal fidelity of synaptic transmission and the reliability of postsynaptic firing during high‐frequency stimulation. J Neurophysol 110, 1621–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Korogod N, Schneggenburger R, Ho WK & Lee SH (2005). Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J Neurosci 25, 6057–6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp‐Scheinpflug C, Tolnai S, Malmierca MS & Rübsamen R (2008). The medial nucleus of the trapezoid body: comparative physiology. Neuroscience 154, 160–170. [DOI] [PubMed] [Google Scholar]
- Kwiecien JM, O'Connor LT, Goetz BD, Delaney KH, Fletch AL & Duncan ID (1998). Morphological and morphometric studies of the dysmyelinating mutant, the Long Evans shaker rat. J Neurocytol 27, 581–591. [DOI] [PubMed] [Google Scholar]
- Kwiecien JM & Delaney KH (2010). Endpoints in myelin‐deficient (MD) rats. Comp Med 60, 343–347. [PMC free article] [PubMed] [Google Scholar]
- Leão RM, Kushmerick C, Pinaud R, Renden R, Li GL, Taschenberger H, Spirou G, Levinson SR & von Gersdorff H (2005). Presynaptic Na+ channels: locus, development, and recovery from inactivation at a high‐fidelity synapse. J Neurosci 25, 3724–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY & Kim JH (2015). Mechanisms underlying presynaptic Ca2+ transient and vesicular glutamate release at a CNS nerve terminal during in vitro ischaemia. J Physiolx1 593, 2793–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH & Raman IM (2014). Resurgent current of voltage‐gated Na+ channels. J Physiol 592, 4825–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier SD, Kovalchuk Y & Rose CR (2006). Properties of the new fluorescent Na+ indicator CoroNa Green: comparison with SBFI and confocal Na+ imaging. J Neurosci Methods 155, 251–259. [DOI] [PubMed] [Google Scholar]
- Moore JW, Joyner RW, Brill MH & Waxman SD (1978). Najar‐Joa M. Simulations of conduction in uniform myelinated fibers. Relative sensitivity to changes in nodal and internodal parameters. Biophys J 21, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor LT, Goetz BD, Kwiecien JM, Delaney KH, Fletch AL & Duncan ID (1999). Insertion of a retrotransposon in Mbp disrupts mRNA splicing and myelination in a new mutant rat. J Neurosci 19, 3404–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE & Shrager P (1999). Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci 19, 7516–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM & Bean BP (1997). Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 15, 4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Cooksey E & Duncan ID (2013). Myelin loss does not lead to axonal degeneration in a long‐lived model of chronic demyelination. J Neurosci 33, 2718–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki K, Chang KJ, Zollinger DR, Liu Y, Ogawa Y, Eshed‐Eisenbach Y, Dours‐Zimmermann MT, Oses‐Prieto JA, Burlingame AL, Seidenbecher CI, Zimmermann DR, Oohashi T, Peles E & Rasband MN (2013). Three mechanisms assemble central nervous system nodes of Ranvier. Neuron 78, 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H & von Gersdorff H (2000). Fine‐tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci 20, 9162–9173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega AV, Henry DL & Matthews G (2008). Reduced expression of Na(v)1.6 sodium channels and compensation by Na(v)1.2 channels in mice heterozygous for a null mutation in Scn8a. Neurosci Lett 442, 69–73. [DOI] [PubMed] [Google Scholar]
- Waxman SG & Brill MH (1978). Conduction through demyelinated plaques in multiple sclerosis: computer simulations of facilitation by short internodes. J Neurol Neurosurg Psychiatry 41, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG & Wood SL (1984). Impulse conduction in inhomogeneous axons: effects of variation in voltage‐sensitive ionic conductances on invasion of demyelinated axon segments and preterminal fibers. Brain Res 294, 111–122. [DOI] [PubMed] [Google Scholar]
- Wu SH & Kelly JB (1993). Response of neurons in the lateral superior olive and medial nucleus of the trapezoid body to repetitive stimulation: intracellular and extracellular recordings from mouse brain slice. Hear Res 68, 189–201. [DOI] [PubMed] [Google Scholar]