

Abstract
Voltage‐gated calcium channels are essential players in many physiological processes in excitable cells. There are three main subdivisions of calcium channel, defined by the pore‐forming α1 subunit, the CaV1, CaV2 and CaV3 channels. For all the subtypes of voltage‐gated calcium channel, their gating properties are key for the precise control of neurotransmitter release, muscle contraction and cell excitability, among many other processes. For the CaV1 and CaV2 channels, their ability to reach their required destinations in the cell membrane, their activation and the fine tuning of their biophysical properties are all dramatically influenced by the auxiliary subunits that associate with them. Furthermore, there are many diseases, both genetic and acquired, involving voltage‐gated calcium channels. This review will provide a general introduction and then concentrate particularly on the role of auxiliary α2δ subunits in both physiological and pathological processes involving calcium channels, and as a therapeutic target.
Abbreviations
- AID
α‐interaction domain
- AP‐1
adaptor protein complex‐1
- BBS
bungarotoxin binding site
- BTX
α‐bungarotoxin
- DRG
dorsal root ganglion
- EM
electron microscopy
- ER
endoplasmic reticulum
- GK
guanylate kinase
- GPCR
G‐protein coupled receptor
- GPI
glycosyl‐phosphatidyl inositol
- HIV
human immunodeficiency virus
- MIDAS
metal ion‐dependent adhesion site
- PMCA
plasma membrane Ca2+‐ATPase
- RyR
ryanodine receptor
- SERCA
sarcoplasmic and endoplasmic reticulum Ca2+ ATPase
- SH3
src homology‐3
- SNP
single nucleotide polymorphism
- VWA
Von Willebrand Factor‐A domain
Introduction
Excitable cells contain functional voltage‐gated ion channels, including calcium channels. Neurons and muscle cells are conventionally excitable, but many other cell types show oscillatory changes in voltage, dependent on the interplay between voltage‐gated and calcium‐dependent channels (for example see Hu et al. 2012). Free intracellular Ca2+ is maintained at 10–100 nm in the cytoplasm, a low level relative to the extracellular milieu. Voltage‐gated calcium channels then react to membrane depolarization by opening, and thus allowing Ca2+ to move down its electrochemical gradient. Ca2+ entry, particularly but not exclusively through voltage‐gated calcium channels, provides an elevation of intracellular calcium ion concentration, to drive many processes. These include hormone secretion, neurotransmitter release, calcium‐dependent transcription of a variety of genes, and also spontaneous pacemaker activity in some neurons, muscles and secretory cells (Mangoni et al. 2003; Guzman et al. 2009; Putzier et al. 2009; Hu et al. 2012; Striessnig et al. 2015). The present review concentrates particularly on the roles of the accessory α2δ subunits. For more comprehensive coverage of calcium channel function, the reader is directed to other recent reviews (Striessnig et al. 2014; Zamponi et al. 2015; Zamponi, 2016).
Voltage‐gated calcium channel subunits
Functional voltage‐gated calcium channels are composed of pore‐forming α1 subunit proteins, encoded by the CACNA1x genes (for review see Catterall et al. 2003), of which there are 10 isoforms in the mammalian genome. In the case of the CaV1.1–CaV1.4 channels (known as L‐type channels), these are encoded by CACNA1S, ‐C, ‐D and ‐F, respectively, and also known as α1S, α1C, α1D and α1F. The CaV2.1–CaV2.3 channels (termed P/Q ‐, N‐and R‐type from physiological experiments: Nowycky et al. 1985; Mintz et al. 1992; Piedras‐Rentería & Tsien, 1998) are encoded by CACNA1A, ‐B and ‐E, respectively, and also known as α1A, α1B and α1E. The T‐type CaV3 channels (encoded by CACNA1G, ‐H and ‐I) are also termed α1G, α1H and α1I (Cribbs et al. 1998; Perez‐Reyes et al. 1998). They are much more similar to each other than to the CaV1 and CaV2 channels (Fig. 1).
Figure 1. Calcium channel α1 subunit homology .
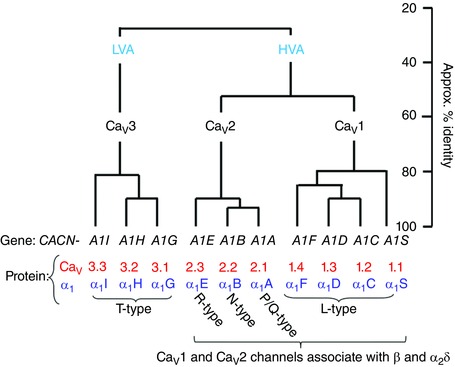
The relationship between the 10 mammalian voltage‐gated calcium channel α1 subunits, and their gene names (black) and protein nomenclature (red and blue). The calcium channels were historically first divided into high voltage‐activated (HVA) and low voltage‐activated (LVA).
Although the α1 subunits dictate the principal biophysical and pharmacological properties of these channels, their expression is enhanced and their properties are modified by the two main auxiliary (or accessory) subunits (Tanabe et al. 1987; Mikami et al. 1989; Mori et al. 1991; Varadi et al. 1991). The α2δ and β subunits also play important roles in channel folding and their subsequent transport to the cell surface, and into particular domains of polarized cells such as neurons. These processes are together known as trafficking, and involve multiple steps. Both the CaV1 and CaV2 classes of channels are able to form a heteromeric complex, co‐assembling with one of four β subunits (encoded by CACNB1—4; Fig. 2 A and B), and one of four α2δ subunits (encoded by CACNA2D1—4; Fig. 2 A and C). For the CaV3 channels, the α1 subunits can form functional channels alone, but may also associate with other proteins.
Figure 2. Domains in β and α2δ subunits and their interaction with the α1 subunit .
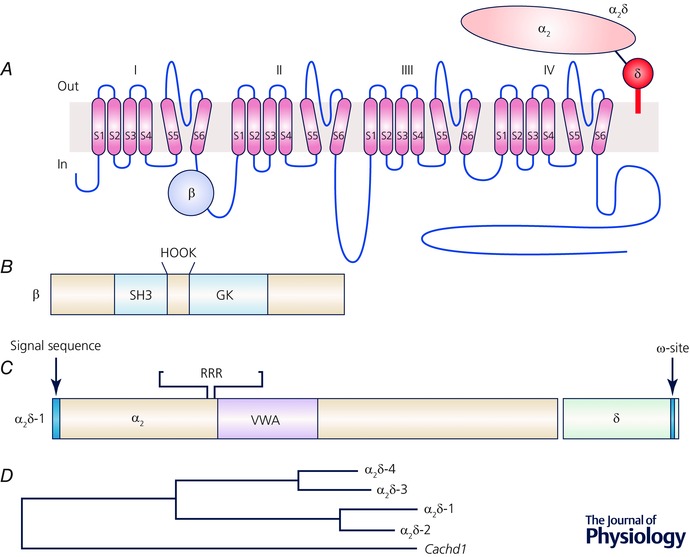
A, topology of the calcium channel complex. B, known domains in β subunits. C, known domains in α2δ subunits. D, approximate phylogenetic tree generated for mouse α2δ subunits using http://www.phylogeny.fr/. The VWA and Cache containing protein Cachd1 is included for comparison. The sequences AAI15872.1, AAH56389.1, EDL24740.1, AAI41092.1 and NP_932154.1 were used.
All of the α1, β and α2δ subunits form a large number of variants as a consequence of alternative splicing events. This opens the potential for a huge diversity of properties and function. A γ subunit also forms part of the skeletal muscle calcium channel complex, which comprises CaV1.1, β1a, γ1 and α2δ‐1(Jay et al. 1990). However, although multiple other γ subunits have been cloned (Letts et al. 1998; Moss et al. 2002; Tomita et al. 2003), no γ subunits have been shown to form an integral part of cardiac (Walsh et al. 2009) or neuronal (Moss et al. 2002; Müller et al. 2010) calcium channels. In contrast, they have well‐defined roles as transmembrane AMPA‐glutamate receptor modifying proteins (Tomita et al. 2003). Furthermore, for some CaV1 and CaV2 calcium channels, the tight binding of calmodulin to the so‐called ‘IQ’ domain in their C‐terminal tail allows calmodulin to be considered as a quasi‐subunit (Mori et al. 2008; Kim et al. 2010; Ben‐Johny et al. 2013).
Voltage‐gated calcium channel localization
CaV1.1 is the only isoform present in mammalian skeletal muscle t‐tubules, and shows very low expression elsewhere, including brain (Bannister & Beam, 2013). CaV1.2 is the main isoform in ventricular cardiac muscle, and is also present in smooth muscle cells, secretory tissue and the nervous system (Striessnig et al. 2014). CaV1.3 has a more limited localization than CaV1.2, playing a major role in sinoatrial node tissue, and in the auditory system (Platzer et al. 2000; Mangoni et al. 2003; Baig et al. 2011), although it is also present in brain. It is also present in some endocrine tissues, including aldosterone‐secreting cells of the adrenal gland, where somatic mutations give rise to resistant hypertension (Azizan et al. 2013; Scholl et al. 2013). CaV1.4 shows very restricted distribution, particularly in the visual system (Mansergh et al. 2005).
The CaV2.x channels show a primarily neuronal distribution and are involved in fast neurotransmitter release (Takahashi & Momiyama, 1993; Wu et al. 1999; Cao & Tsien, 2010). CaV2.1 channels are present throughout the brain, and are particularly prevalent in cerebellum (Ophoff et al. 1996), where they make up the predominant calcium current in Purkinje neurons (Mintz et al. 1992; Westenbroek et al. 1995). They are involved in neurotransmission in most mature mammalian central synapses (Westenbroek et al. 1995; Iwasaki et al. 2000, 2005; Nakamura et al. 2015). CaV2.2 is distributed throughout the central (Westenbroek et al. 1992) and peripheral nervous systems (Lipscombe et al. 1988; Boland et al. 1994; Wheeler et al. 1994). It is particularly important for neurotransmission early in mammalian development, although it co‐exists with CaV2.1 in most mature synapses (Iwasaki et al. 2000). CaV2.2 also plays a dominant role in the mature peripheral nervous system (Chaplan et al. 1994; Bowersox et al. 1996). CaV2.3, although originally described as being low voltage activated (Soong et al. 1993), is thought to correspond to residual R‐type calcium current (Zhang et al. 1993; Tottene et al. 2000; Wilson et al. 2000). It is present in many brain regions and is found both pre‐ and postsynaptically in neurons (Parajuli et al. 2012). CaV2.3 has been found to be involved in spontaneous release of glutamate (Ermolyuk et al. 2013), although the CaV2.3 blocker SNX‐482 also blocks some K+ channels, making dissection of its physiological functions more difficult (Kimm & Bean, 2014).
The CaV3 channels are extensively distributed in neurons and other excitable cells (Cribbs et al. 1998; Perez‐Reyes, 1998; Perez‐Reyes et al. 2009). For example, they are prevalent in the thalamus (Perez‐Reyes, 2003), and also have important roles in primary afferent pathways (Francois et al. 2015; Gadotti et al. 2015; for recent review see Zamponi et al. 2015). They have important roles in neuronal and cardiac excitability and in cardiac and neuronal pacemaker activity (Perez‐Reyes, 2003; Guzman et al. 2009; Putzier et al. 2009). In some synapses they also have a presynaptic function in transmitter release (Huang et al. 2011; Carbone et al. 2014).
Association of α1 subunits with auxiliary subunits
Biochemical isolation of calcium channels has indicated that native L‐, N ‐ and P/Q ‐type channels in muscle and brain are all associated with β and α2δ subunits (Tanabe et al. 1987; Witcher et al. 1993; Liu et al. 1996). However, it has been noted that the association of the α2δ subunit with the channel complex is more easily dissociated by the detergents used during purification than the interaction of the β subunit (Jay et al. 1991; Gee et al. 1996; Müller et al. 2010). It is also possible that not all native calcium channel complexes contain an α2δ subunit. By contrast the association between the α1 and β subunits is quite robust, and shows a high affinity for interaction with the intracellular loop between domains I and II of CaV1 and CaV2 channels (Pragnell et al. 1994; Canti et al. 2001; Van Petegem et al. 2004). Despite this difference, both the β and α2δ subunits increase the expression and function of these channels, as described below.
Structural information on voltage‐gated calcium channels
There is detailed structural information concerning the cytoplasmic β subunits. Initially a modelling study showed that β subunits contained a core SH3 and guanylate kinase‐like (GK) domain (Hanlon et al. 1999; Fig. 2 B). This was confirmed in X‐ray crystallographic studies of the SH3‐GK core domains of three calcium channel β subunits, in association with an interacting peptide derived from the I‐II linker (Chen et al. 2004; Opatowsky et al. 2004; Van Petegem et al. 2004). From these and other studies, the GK domain is seen to bind to the α‐interaction domain (AID) which is in the proximal part of the I‐II linker (Fig. 2 A). The β subunit is thought to promote the formation of an α‐helix, in the AID motif, extending back to the end of S6 in domain I (Opatowsky et al. 2004; for reviews see Richards et al. 2004; Buraei & Yang, 2010). This is likely to promote folding to form mature channels.
More recently, very valuable crystallographic information pertaining to the α1 subunit structure has come from studies of the bacterial single domain sodium channel NaVAb, whose structure was solved by X‐ray crystallography (Payandeh et al. 2011). Subsequently, key residues in the pore of this channel were mutated to render the channel Ca2+ permeable (Tang et al. 2014); this structure was able to provide detailed information about the Ca2+ permeation pathway. There are also structures of calmodulin interacting with the proximal C‐terminus of CaV1.2 (Kim et al. 2008, 2010), revealing the nature of this interaction and shedding light on the mechanism of Ca2+‐dependent inactivation.
The initial low resolution single particle electron micrographic (EM) structures of the L‐type calcium channel complex, also called the dihydropyridine receptor, from skeletal muscle (Serysheva et al. 2002; Wolf et al. 2003; Wang et al. 2004; Hu et al. 2015) and cardiac muscle (Walsh et al. 2009), showed an asymmetric structure, with a density identified as α2δ extending out from the complex. More recently a high resolution cryo‐EM structure of the CaV1.1 calcium channel complex purified from skeletal muscle has now provided us with much greater detail, at near atomic resolution, particularly regarding the transmembrane organization and pore of the α1 subunit, and the orientation of the α2δ subunit domains (Wu et al. 2015). It has shown a clockwise arrangement of the α1 subunit domains, and identified that there are multiple interactions of α2δ‐1 subunit with the extracellular loops of domains I‐III of the α1S subunit.
Modulation of calcium channel function by second messengers and G proteins
There is insufficient space in this review to cover the enormous amount of information on multiple second messenger effects on calcium channel function. Three key areas that can be highlighted are firstly: Ca2+‐dependent inactivation and facilitation of CaV2.1, CaV1.2 and CaV1.3 channels, by interaction with calmodulin associated with the C‐terminal tail of the α1 subunit (Dick et al. 2008; Minor & Findeisen, 2010; Ben‐Johny et al. 2013). Secondly, there is an important phosphorylation process that is responsible for β‐adrenergic stimulation of cardiac calcium currents (Reuter, 1987; Fuller et al. 2010). The mechanism involves enhancement of CaV1.2 currents by cyclic AMP‐dependent protein kinase, which results from phosphorylation‐induced relief of auto‐inhibition by a peptide cleaved from the channel C‐terminus (Fuller et al. 2010; Fu et al. 2013). Thirdly, there is a ubiquitous G‐protein coupled receptor (GPCR)‐mediated inhibition of the CaV2 class of channels mediated by Gβγ (Dolphin, 2003; Zamponi & Currie, 2013).
Regarding the interplay between second messenger modulation and auxiliary subunits, initial studies identified that Gβγ bound to a site on the I‐II linker of CaV2 channels that overlapped with the CaVβ subunit (Zamponi et al. 1997), opening the possibility that they compete for this binding site. We then identified that in the absence of a CaVβ subunit, Gβγ‐mediated inhibition is still present, but it is not voltage dependent, meaning that it cannot be removed by preceding depolarization. Therefore, the presence of the CaVβ subunit is required for Gβγ‐mediated G‐protein modulation to show voltage‐dependent properties (Meir et al. 2000; Zhang et al. 2008), and a simple competition for binding is not responsible for Gβγ‐mediated inhibition. Further to this, we identified key residues within the N‐terminus of CaV2 channels that are essential for G‐protein modulation (Page et al. 1998; Canti et al. 1999; Leroy et al. 2005), and this work was extended by others (Agler et al. 2005).
Interplay between the action of β and α2δ subunits in calcium channel function
For both CaV1 and CaV2 channels, the CaVβ subunits are extremely important for expression of functional channels in several heterologous expression systems (Varadi et al. 1991; Pragnell et al. 1994; Jones et al. 1998; Leroy et al. 2005). Interaction of the α1 subunit with a β subunit has a number of consequences. By binding via their guanylate kinase domain (Fig. 2 B) to the intracellular AID motif on the α1 subunits (Pragnell et al. 1994; Fig. 2 A), they increase folding of the channels and protect the channels from endoplasmic reticulum (ER)‐associated proteasomal degradation (Altier et al. 2011; Waithe et al. 2011); thus they allow more channels to reach the plasma membrane (Fig. 3 A). However, it is difficult to determine whether β subunits are absolutely essential for α1 subunits to reach the cell surface. This suffers from the problem that several expression systems, in particular Xenopus oocytes, express native β subunits (Canti et al. 2001). The α2δ subunits produce an additional increase in current density, described in more detail below (Fig. 3 B). However, because a number of expression systems, including Xenopus oocytes, HEK‐293 and the tsA‐201 cells derived from them, also contain some endogenous α2δ‐1 (Singer‐Lahat et al. 1992; Dolphin et al. 1999; Kadurin et al. 2012 a), this also complicates assessment of their role. Nevertheless, both α2δ and β subunits increase the expression at the plasma membrane of CaV1 and CaV2 channels, and where it has been investigated, some evidence suggests that α2δ subunits are poorly effective unless the CaVβ subunits are also expressed (Cassidy et al. 2014; Fig. 4).
Figure 3. Examples of effects of auxiliary subunits on CaV2.2 calcium channel currents .
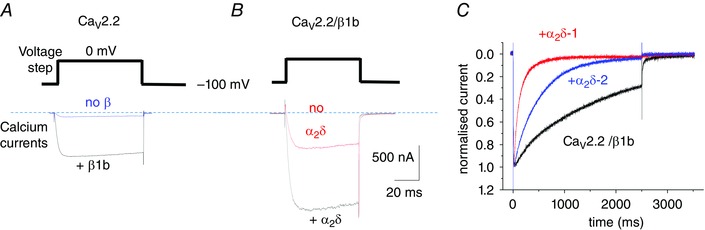
A, CaV2.2 calcium currents: effect of β subunits. Example of peak CaV2.2 current at 0 mV in the absence of β (blue) and presence of β1b (black). B, CaV2.2 calcium currents: effect of α2δ subunits. Example of peak CaV2.2/β1b current at 0 mV in the absence of α2δ (red) and presence of α2δ‐3 (black). Scale bars apply to both A and B. Charge carrier 1 mm Ba2+, expression in tsA‐201 cells, as in a previous study (Leroy et al. 2005). C, effect of different α2δ subunits on inactivation. Examples of normalized peak current for CaV2.2–β1b (black), CaV2.2–β1b–α2δ‐2 (blue) and CaV2.2–β1b–α2δ‐1 (red), over a 2.5 s timescale. Expression in Xenopus oocytes, as in a previous study (Canti et al. 2005).
Figure 4. CaV2.2 cell surface expression: effects of β1b and α2δ‐1 .
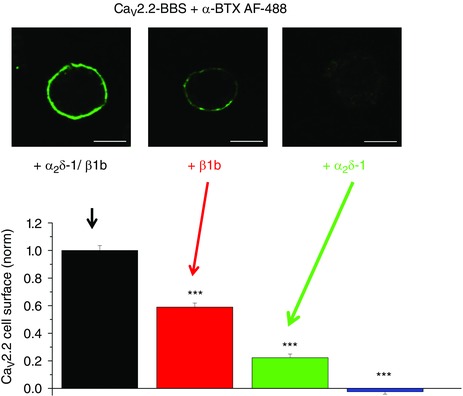
Cell surface expression of bungarotoxin binding site (BBS) tagged CaV2.2 labelled with α‐bungarotoxin (BTX) coupled to AF488 dye (green). Top panel: examples of N2a cells transfected with CaV2.2–β1b–α2δ‐1 (left), CaV2.2–β1b (middle) and CaV2.2–α2δ‐1 (right). Scale bar 20 μm. Bottom panel: mean (± SEM) data for cell surface expression of CaV2.2, for cells expressing CaV2.2–β1b–α2δ‐1 (black bar), CaV2.2–β1b (red bar) and CaV2.2–α2δ‐1 (green bar) or CaV2.2 alone (blue bar). Data are taken from a recent study (Cassidy et al. 2014).
Isoforms and topology of α2δ
All the α2δ proteins have a similar structure (for reviews see Felix, 1999; Davies et al. 2007; Dolphin, 2012; Fig. 2 C and D). The N‐terminus has a signal sequence directing the nascent polypeptide into the lumen of the ER, such that it becomes extracellular, once transported to the plasma membrane (Fig. 2 C). Several domains can be identified in the sequence of α2δ proteins, including a Von Willebrand Factor‐A (VWA) domain (Whittaker & Hynes, 2002; Fig. 2 C). These domains, as well as being present in von Willebrand factor itself, are generally involved in extracellular protein–protein interactions, dependent on divalent cations, particularly by integrins and extracellular matrix proteins. A key motif in VWA domains is the metal ion‐dependent adhesion site (MIDAS), which involves coordination of the divalent cation by a ring of up to five polar or charged residues (Whittaker & Hynes, 2002). If the MIDAS site is ‘perfect’, with the full complement of five residues, it is highly likely to be involved in such protein–protein interactions (Whittaker & Hynes, 2002), and this is the case in α2δ‐1 and α2δ‐2 (Canti et al. 2005), whereas α2δ‐3 and α2δ‐4 have one missing polar residue in the MIDAS motif. There is also a region in the α2δ subunits containing so‐called Cache domains, which have homology to bacterial chemosensory domains (Anantharaman & Aravind, 2000; Dolphin, 2012). The recent EM structure also identified a Cache domain, N‐terminal to the VWA domain (Wu et al. 2015). There are other identified genes with predicted similarity to α2δ subunits, such as CACHD1 (Whittaker & Hynes, 2002) (Fig. 2 D), whose functions remain to be determined.
The C‐termini of all α2δ subunits all have a hydrophobic region first identified to be a transmembrane domain (Ellis et al. 1988; Jay et al. 1991). This led to the α2δ proteins being described as single pass type I (C‐terminal cytoplasmic) transmembrane proteins. From prediction progams we found that at least two of the α2δ subunits (α2δ‐3 and α2δ‐4) are predicted with high likelihood to be glycosyl‐phosphatidyl inositol (GPI)‐anchored, partly because the C‐terminal hydrophobic domain is very short and present at the extreme C‐terminus, as well as the presence of a predicted GPI‐anchor ω‐site (Pierleoni et al. 2008; Davies et al. 2010). We have provided evidence for this post‐translational modification for α2δ‐1, α2δ‐2 and α2δ‐3 (Davies et al. 2010; Alvarez‐Laviada et al. 2014). The genes for all the α2δ subunits encode a single precursor protein, which is post‐translationally proteolytically processed into two polypeptides (Jay et al. 1991). The α2 and δ polypeptides remain disulfide‐bonded together. The cysteines residues involved in this disulfide bonding have been determined for α2δ‐1 (Calderon‐Rivera et al. 2012). We have recently been studying the relevance of proteolytic processing into α2 and δ to the physiological function of α2δ (Kadurin et al. 2012 b, and authors’ unpublished data).
Effect of α2δ subunits on calcium channels reaching the plasma membrane
In general, the α2δ subunits have been found to increase the expression (either functional expression or amount of protein on the plasma membrane) of several different CaV1 or CaV2 combinations with β subunits (Shistik et al. 1995; Gurnett et al. 1996; Felix et al. 1997; Wakamori et al. 1999; Gao et al. 2000; Yasuda et al. 2004; Canti et al. 2005; Davies et al. 2010). For example, for the CaV2.1–β4 combination, calcium currents were increased 3‐fold by α2δ‐2. This set of calcium channel subunits is found in cerebellar Purkinje cells, where α2δ‐2 is strongly represented (Barclay et al. 2001; Brodbeck et al. 2002). However, α2δ‐2 did not alter the single channel conductance, suggesting strongly that the large increase in whole cell current is solely due to an increase in the number of functional channels at the cell surface (Barclay et al. 2001; Brodbeck et al. 2002). However, the term ‘increased number of functional channels’ can indicate increased amount of channel protein in the plasma membrane and/or an increased proportion of the channels already in the plasma membrane able to respond to depolarization. There is strong evidence that the cell surface expression of CaV1 and CaV2 α1 subunits is increased by α2δ subunits (Fig. 4), although the mechanism(s) underlying this increase this are still being unravelled (Tran‐Van‐Minh & Dolphin, 2010; Cassidy et al. 2014).
The situation for CaV2.3 channels is less clear. It has been reported that CaV2.3 produces relatively large currents when expressed alone in Xenopus oocytes (Soong et al. 1993; Schneider et al. 1994; Qin et al. 1998), and α2δ‐1 subunits did not increase CaV2.3 currents in this expression system (Qin et al. 1998). Furthermore, in HEK‐293 cells α2δ‐1 produced a 2‐fold elevation of the maximum conductance for CaV2.3 alone, although it gave no additional increase beyond that of β subunits (Jones et al. 1998). Thus it is possible that CaV2.3 may be less affected by α2δ subunits, but this will require confirmation.
Increased trafficking of the calcium channels by these α2δ subunits is highly likely not to be their only mechanism of action. For example, liposomes containing skeletal muscle calcium channel protein exhibited greater calcium flux in the presence of α2δ subunits than in their absence (Gutierrez et al. 1991). Furthermore, the effect of α2δ‐1 on CaV2.2 channel density in the plasma membrane when expressed in N2a cells was at the most 2‐fold (Cassidy et al. 2014), whereas there was an approximately 10‐fold increase in CaV2.2 calcium currents in the presence of α2δ‐1 (Hoppa et al. 2012). It has been suggested that α2δ‐1 reduced the apparent turnover of CaV2.2, in studies using radiolabelled conotoxin (Bernstein & Jones, 2007), although Cassidy et al. (2014) did not find that α2δ‐1 reduced CaV2.2 endocytosis from the plasma membrane in N2a cells.
The perfect MIDAS motif present in the VWA domain of α2δ‐1 and α2δ‐2 subunits is required for increasing calcium currents (Canti et al. 2005; Hoppa et al. 2012), and also for cell surface expression of CaV2.2 (Cassidy et al. 2014). Mutation of the MIDAS motif also reduced the trafficking of the α2δ subunits themselves, when expressed alone (Canti et al. 2005; Cassidy et al. 2014). This mutation also abolished the capacity of both α2δ‐1 (Hoppa et al. 2012) and α2δ‐2 (Canti et al. 2005) subunits to increase calcium currents in several expression systems. However, α2δ‐3 and α2δ‐4 do not contain perfect MIDAS motifs (Whittaker & Hynes, 2002), and may therefore play a smaller trafficking role, despite increasing calcium currents (Davies et al. 2010), by what must be an additional mechanism.
In an early study, it was found that the α2 subunit of α2δ‐1 binds to of CaV1.1 domain III (Gurnett et al. 1997). However, the recent structural study shows interaction of α2δ‐1 with several extracellular loops in domains I–III of CaV1.1 (Wu et al. 2015). The α2δ‐1 MIDAS motif was found to be located immediately above the linker between the first two transmembrane segments in voltage‐sensing domain I (Wu et al. 2015). The limited structural evidence for other calcium channels also suggests extensive extracellular contact between CaV1.2 and α2δ‐1 (Walsh et al. 2009).
The CaV3 calcium channels produce large currents in the absence of co‐expressed accessory β or α2δ subunits, and therefore these proteins are not obligate auxiliary subunits for CaV3 channels. Nevertheless, both α2δ‐1 and α2δ‐2 were found to increase CaV3.1 currents and cell surface expression almost 2‐fold (Dolphin et al. 1999; Gao et al. 2000; Dubel et al. 2004); thus these channels may have the capacity to associate with α2δ subunits. In contrast, other studies found that α2δ‐1 and α2δ‐3 produced little change, whereas α2δ‐2 had a larger effect on CaV3.1 current density (Klugbauer et al. 1999; Lacinova et al. 1999; Hobom et al. 2000).
Trafficking of calcium channels to specific membrane domains
The auxiliary α2δ and β subunits play major roles in the trafficking of CaV1 and CaV2 channels not only to the cell surface, but also to specific domains of polarized cells, including muscle cells and neurons (Dolphin, 2012; D'Arco et al. 2015). We have postulated that the α2δ subunits are highly likely to interact with proteins involved in trafficking of membrane protein cargoes (Davies et al. 2006; Hendrich et al. 2008; Tran‐Van‐Minh & Dolphin, 2010). We have found that the α2δ subunits themselves purify with cholesterol‐rich lipid raft domains, and this may influence localization of the calcium channel complexes in plasma membrane microdomains (Davies et al. 2006, 2010; Kadurin et al. 2012 a). Interestingly, we have also found that a truncated α2δ subunit, from which we have removed the C‐terminal GPI‐anchor motif, is mainly secreted, but nevertheless exhibits some extrinsic plasma membrane association, via interactions that remain to be determined (Kadurin et al. 2012 a).
In recent work, we have found that the adaptor protein complex‐1 (AP‐1) is important for trafficking of CaV2.2 from the trans‐Golgi network to the plasma membrane, via an alternatively spliced exon 37 in the proximal C‐terminus. The splice variant of CaV2.2 containing exon 37a supports larger currents compared to that containing exon 37b (Castiglioni et al. 2006), and is selectively expressed in nociceptors (Bell et al. 2004). Our work revealed that AP‐1 binding motifs, YxxΦ and [DE]xxxL[LI], present only in exon 37a, increase the intracellular trafficking of exon 37a‐containing CaV2.2, both to the somatic plasma membrane and into the axons of dorsal root ganglion (DRG) neurons. The ability of exon37a to increase CaV2.2 currents and cell surface density are lost in the absence of α2δ subunits, suggesting that this auxiliary subunit promotes a particular step in the forward trafficking process (Macabuag & Dolphin, 2015).
Influence of α2δ subunits on biophysical properties of calcium channels
The α2δ subunits influence the voltage‐dependent and kinetic properties of the calcium currents; in particular they consistently increase the inactivation rate, although to different extents. The effects of α2δ subunits may also depend on the presence of a particular β subunit.
Activation
In the case of CaV1.2, it was found that α2δ‐1 subunits exerted little effect on the activation voltage dependence (Singer et al. 1991; Welling et al. 1993; Shistik et al. 1995; Bangalore et al. 1996; Shirokov et al. 1998). However, in other studies a hyperpolarization of activation was reported (Felix et al. 1997), and this was also observed from conductance‐voltage measurements (Platano et al. 2000). For CaV2.1, co‐expressed with β4 in mammalian cells, α2δ‐2 did not affect the voltage dependence of activation (Brodbeck et al. 2002). For CaV2.2 co‐expressed with β1b, α2δ‐1 increased the activation rate of currents, but had less effect on the voltage dependence of activation (Wakamori et al. 1999). Contrasting results were found for CaV2.3, which shows a greater capacity than CaV1.2 to produce currents in the absence of the auxiliary subunits (Stephens et al. 1997; Qin et al. 1998). For CaV2.3, α2δ‐1 was found to depolarize the activation, in the presence of either β1b or β2a, or in the absence of any β subunits (Qin et al. 1998). In contrast, in another study α2δ‐1 had no effect on the activation voltage dependence for CaV2.3 (Jones et al. 1998).
Inactivation
In some studies, it was found that the α2δ subunits hyperpolarized the steady‐state inactivation for several different calcium channel isoforms (Singer et al. 1991; Felix et al. 1997; Wakamori et al. 1999; Hobom et al. 2000; Canti et al. 2005; Hendrich et al. 2008; Davies et al. 2010), and in α2δ‐1 knockout mice there was a clear depolarization of the steady‐state inactivation curve for cardiac calcium channel currents (Fuller‐Bicer et al. 2009). However, for CaV2.3 it was found that, whereas β1b caused a hyperpolarization of the steady‐state inactivation, α2δ‐1 had no effect on this, either with or without a β subunit (Qin et al. 1998). The α2δ subunits also increased the rate of inactivation to varying extents, with the greatest effect being observed for α2δ‐1 (Fig. 3 C; for review see Canti et al. 2003).
Thus, although the α2δ subunits affect the kinetics and voltage‐dependent properties of the different calcium channel isoforms, there is no clear consensus for the different α1 and α2δ isoform combinations. One origin of this complexity may be that there are also usually more mature channels in the plasma membrane in the presence of α2δ subunits. Such a diversity of effects, although they may appear subtle when measured in isolation, can have important consequences in terms of calcium‐and voltage‐dependent events in cells, including action potential shape (Hoppa et al. 2012, 2014), and the firing properties of neurons (Margas et al. 2016).
Splice variants of α2δ subunits
The main α2δ‐1 subunit splice variant present in rat brain is different from that seen in skeletal muscle (Kim et al. 1992). Sequence alignments identified alternative splicing in three regions, called A, B and C (Angelotti & Hofmann, 1996). Our recent study (Lana et al. 2014) indicates that regions A and B are in separate exons, with region A in rat being encoded by exon 18a and region B representing an alternative 3′ splice acceptor site (start site) of exon 19. Region C is also a cassette exon. The main splice variant in rat skeletal muscle is +A +B ΔC, whereas α2δ‐1 (ΔA + B + C) is the principal brain splice variant (Angelotti & Hofmann, 1996; Lana et al. 2014). We have recently shown that it is also the main splice variant in DRG neurons (Lana et al. 2014). However, we also identified a novel minor splice variant (α2δ‐1 ΔA + B ΔC) in these neurons (Lana et al. 2014). Alternative splicing of other α2δ subunits has been described in other studies (Barclay & Rees, 2000; Qin et al. 2002).
Distribution of α2δ subunits in the peripheral and central nervous systems
The α2δ‐1, α2δ‐2 and α2δ‐3 subunits are widely expressed in both the peripheral and central nervous system, as documented in a comprehensive in situ hybridization study (Cole et al. 2005). α2δ‐1 is present in many neuronal cell types (Cole et al. 2005), including DRG neurons (Newton et al. 2001; Bauer et al. 2009). The α2δ‐1 protein is mainly situated in presynaptic terminals, as well as, to smaller extent, in neuronal somata, and also in dendrites (Taylor & Garrido, 2008; Bauer et al. 2009).
The α2δ‐1 transcript is expressed preferentially in excitatory compared to inhibitory neurons (Cole et al. 2005). In contrast, α2δ‐2 expression was found to be lower than α2δ‐1 in most brain regions, with restricted areas showing significant expression, such as the cerebellum (Cole et al. 2005). The distribution of α2δ‐2 partially correlates with expression in GABAergic neurons, including cerebellar Purkinje neurons (Barclay et al. 2001; Cole et al. 2005). The α2δ‐3 transcript is present throughout the brain, and is particularly prevalent in the caudate‐putamen (Cole et al. 2005). It is also present in the auditory system (Pirone et al. 2014) and in the retina (Perez de Sevilla et al. 2015). In contrast, α2δ‐4 protein is found in certain endocrine tissues, and is expressed at a low level in the brain (Qin et al. 2002). It also plays a key role in the retina (De Sevilla Muller et al. 2013).
Role of α2δ‐1 in neuropathic pain
Neuropathic pain is chronic pain resulting from nerve damage, which may have a number of different underlying causes. Neuropathic pain can be a result of trauma, either directly damaging or impinging on axons. Trigeminal neuralgia, which involves severe facial and jaw pain, is often caused by trapping or pressure on sensory nerves. Cancer‐induced neuropathic pain can be also result from direct damage to sensory nerves, or activation of nociceptors as a result of mediators secreted from tumours or in the inflammatory response (Schmidt et al. 2010). Neuropathic pain can also commonly be caused by direct damage to nerves by toxins and drugs. This would include diabetic neuropathy, due to axon damage as a direct result of chronic elevated plasma glucose concentration, and neuropathy caused by cancer chemotherapeutic drugs, for example platinum‐based drugs such as cisplatin, microtubule‐disrupting taxanes, such as paclitaxel, and vinca alkaloids including vincristine. Some older anti‐human immunodeficiency virus (HIV) drugs, such as 2′,3′‐dideoxycytidine, can also result in nerve damage and neuropathic pain (Joseph et al. 2004). Viral infection of DRGs can also cause neuralgia, including chronic post‐herpetic neuralgia (following shingles), or HIV‐induced neuropathic pain, which can be mimicked by injection of the viral coat protein HIV gp‐120 (Wallace et al. 2007; Schutz & Robinson‐Papp, 2013). Thus both HIV infection and some of the treatments used may initiate neuropathic damage.
Sensory nerve injury results in a change in transcription in those damaged neurons of many genes, which may be either up‐ or down‐regulated, often many‐fold (Newton et al. 2001; Wang et al. 2002; Xiao et al. 2002; Dawes et al. 2014). The mechanism of this effect has been investigated for the chemotherapeutic agent paclitaxel and may involve injury‐induced modulation of Ca2+ entry and neuronal calcium sensor‐1 degradation (Boehmerle et al. 2006, 2007).
Among the large number of genes whose expression is altered, there is a consistent elevation of α2δ‐1 mRNA, shown by in situ hybridization (Newton et al. 2001), quantitative PCR (Bauer et al. 2009), microarray analysis (Wang et al. 2002; Xiao et al. 2002) and RNAseq (Perkins et al. 2014). There is an equivalent increase in α2δ‐1 protein in DRGs and in the dorsal horn of the spinal cord, shown by immunoblotting (Luo et al. 2001) and immunohistochemistry (Bauer et al. 2009). The increase in α2δ‐1 appears to occur in every damaged DRG neuron (Bauer et al. 2009; Patel et al. 2013). In contrast, the levels of CaV2.2 mRNA and protein are not altered in these models (Wang et al. 2002; Li et al. 2006), although a change in splicing of exon 37 has been documented (Altier et al. 2007). This leads to the hypothesis that elevated α2δ‐1 results in increased CaV2.2 trafficking to terminals or localization to active zones, thus affecting presynaptic function. Nevertheless, α2δ‐1 may also have other roles, for example in neuronal sprouting.
Transgenic mice that overexpress α2δ‐1 exhibit a baseline phenotype of allodynia and hyperalgesia (Li et al. 2006), suggesting that the α2δ‐1 level in DRG neurons is important for determining the neuropathic response. In agreement with these results, we have shown that in α2δ‐1 knockout mice (Fuller‐Bicer et al. 2009), there is a marked reduction in baseline responses to mechanical and cold stimulation, and a very retarded hyperalgesic response to sciatic nerve injury, in comparison to wild‐type littermate mice (Patel et al. 2013). In agreement with this we found that DRGs from α2δ‐1 knockout mice showed strongly reduced ability to fire multiple action potentials (Margas et al. 2016).
We have also recently shown that heterologous over‐expression of α2δ‐1 in cultured DRG neurons (to mimic in vitro the neuropathic state) leads to increased calcium currents and prolonged cytoplasmic Ca2+ responses resulting from membrane depolarization (Fig. 5 A). These prolonged Ca2+ transients, once initiated, are not dependent on extracellular Ca2+ but are buffered by mitochondria. Thus, by controlling CaV2.2 channel density in the plasma membrane, possibly at sites where mitochondria and ER are also closely apposed, the α2δ‐1 subunit has a large effect on depolarization‐induced intracellular Ca2+ signalling in DRG neurons (D'Arco et al. 2015; Fig. 5 B).
Figure 5. Effect of α2δ‐1 on cytosolic Ca2+ levels .
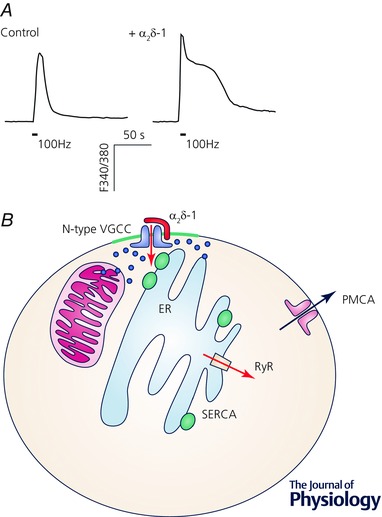
A, overexpression of α2δ‐1 in DRG neurons increased the width of depolarization‐induced intracellular calcium transients, measured using Fura‐2, induced by 100 Hz electrical stimulation (indicated by the bar). Data are taken from a recent study (D'Arco et al. 2015). B, cartoon of localization of CaV2.2 (N‐type) calcium channels in the plasma membrane near to ER and mitochondria. Ca2+ is taken up into ER via the sarcoplasmic–endoplasmic reticulum Ca2+‐ATPase (SERCA) pump, and can be released by ryanodine receptor (RyR) activation. Ca2+ is pumped out of cells by the plasma membrane Ca2+‐ATPase (PMCA). Cartoon adapted from from a recent study (D'Arco et al. 2015).
Regarding the involvement of other α2δ subunits in the pain pathway, the Drosophila melanogaster CACNA2D3 homologue, straitjacket, was identified as a gene involved in pain processing (Neely et al. 2010).
Role of α2δ subunits in epilepsies
Prior to the identification of α2δ‐1 as the receptor for gabapentin (see below), this drug was known to be of use in the treatment of some forms of epilepsy, as an adjunct drug to improve seizure control (Marson et al. 2000). Gabapentin binds to both α2δ‐1 and α2δ‐2, but not to the other α2δ subunits. Subsequently, we found, together with Michele Rees and Mark Gardiner, that the mutant mouse strains ducky and ducky2J involved disruption of the cacna2d2 gene (Barclay et al. 2001). These mice display paroxysmal dyskinesia and absence seizures. Although the mutations are different in the two mouse strains, being a complex rearrangement of the gene in ducky and a two base pair deletion in ducky2J, no full length α2δ‐2 protein is produced in either strain (Barclay et al. 2001; Brodbeck et al. 2002; Donato et al. 2006). Another mutant mouse, entla, with a similar epileptic phenotype, was then identified and found to have a duplication of exon 3 in cacna2d2 (Brill et al. 2004). Mice with a targeted gene deletion in cacna2d2 also show an epileptic and ataxic phenotype (Ivanov et al. 2004). The mutation in ducky and ducky2J mice is associated with abnormal morphology of the Purkinje cells (Brodbeck et al. 2002) and markedly attenuated spontaneous activity in these neurons (Donato et al. 2006).
Two human family pedigrees have recently been investigated, in which homozygous recessive mutations in CACNA2D2 resulted in infantile epileptic encephalopathy (Edvardson et al. 2013; Pippucci et al. 2013). The carriers of a single copy of the mutations had no phenotype, in agreement with the absence of phenotype in mice heterozygous for cacna2d2 expression (Barclay et al. 2001).
For α2δ‐1, no central phenotypes have been identified with any certainty in humans, possibly because most neurons contain more than one subtype of α2δ subunit, and these proteins may have a partially interchangeable function. However, CACNA2D1 has been identified as a candidate gene associated with some cases of West syndrome, an early‐onset epileptic encephalopathy (Hino‐Fukuyo et al. 2015). The CACNA2D1 locus has also been implicated in three patients investigated with intellectual disability and epilepsy, although these patients had deletions that also affected other genes (Vergult et al. 2015).
Night blindness
Mutations in the gene CACNA2D4, encoding α2δ‐4, produce photoreceptor dysfunction, resulting in a form of night blindness (Wycisk et al. 2006 b). A spontaneously occurring mouse mutation has also been identified in this gene, with a phenotype of autosomal recessive cone dystrophy, again causing night blindness (Wycisk et al. 2006 a,b). This emphasizes the importance of α2δ‐4 in photoreceptor function.
Neuropsychiatric disorders
As we have reviewed recently (Heyes et al. 2015), rare deleterious mutations in many of the calcium channel genes including CACNA2D1, CACNA2D2 and CACNA2D4 have been linked to both bipolar disorder and schizophrenia (Purcell et al. 2014). Furthermore, CACNA2D2 and CACNA2D4 have also been linked to these psychiatric disorders in Genome‐Wide Association Studies (Cross‐Disorder Group of the Psychiatric Genomics Consortium, 2013). However, most of the single nucleotide polymorphisms (SNPs) that are associated with these disorders are in introns or intergenic regions, and it remains unclear whether the SNPs have any effects to increase or decrease overall expression, or expression of particular splice variants, or otherwise alter the function of the gene with which they are associated (Heyes et al. 2015). Nevertheless, it has recently been found that expression of CACNA1S, CACNA2D4 and CACNA1F were increased in hippocampal‐like neurons derived from induced pluripotent stem cells in patients with bipolar disorder (Mertens et al. 2015). It is interesting that these particular calcium channel genes normally show very low expression in brain, so their physiological role in hippocampus is unclear.
A CACNA2D3 splice site mutation was identified as one of a large number of ‘likely gene‐disrupting mutations’ involved in autism specrum disorders (Iossifov et al. 2012). Other rare germline mutations, introducing premature stop codons or aberrant splicing, predicting truncated proteins, have also been found to be associated with autism (Girirajan et al. 2013; De Rubeis et al. 2014). Given the likelihood that autism involves synaptic dysfunction (Malhotra & Sebat, 2012; Ting et al. 2012), it is perhaps not surprising that mutations in α2δ‐3, which is present in presynaptic terminals, are found to be one of many potential genetic causes of autism.
Cardiac and endocrine dysfunction
The α2δ‐1 protein is strongly expressed together with the L‐type calcium channels in skeletal, cardiac and smooth muscle (Ellis et al. 1988; Jay et al. 1991; Klugbauer et al. 1999; Wolf et al. 2003; Walsh et al. 2009). CACNA2D1 mutations have been identified to cause human cardiac dysfunction, including short QT syndrome (Templin et al. 2011) and Brugada syndrome (Burashnikov et al. 2010). The mechanism of disruption resulting from mutations in α2δ‐1 has been probed (Bourdin et al. 2015). In agreement with this, disruption of the cacna2d1 gene in mice also caused a cardiac phenotype; the mice exhibited a reduction in basal ventricular myocardial contractility, associated with lower cardiac calcium current density (Fuller‐Bicer et al. 2009). Furthermore, mice lacking α2δ‐1 also showed reduced pancreatic β‐cell calcium currents, and an increased tendency to develop diabetes, particularly on one genetic background (Tuluc et al. 2014).
α2δ subunits as a therapeutic target
The α2δ subunits were discovered to be therapeutic targets completely fortuitously, by virtue of being the unexpected protein target for gabapentin binding. Otherwise they would not have been considered a priori as a relevant drug target, because of the absence of any known ligand or mechanism of action.
Identification of α2δ subunits as gabapentin receptors
Gabapentin and pregabalin were first synthesized as analogues of GABA, with the aim of developing novel antiepileptic drugs (Taylor et al. 2007; Silverman, 2008). After it was found that they did not act via GABA pathways, purification of the brain 3H‐gabapentin ‘receptor’ then resulted in the surprise identification of α2δ‐1 (Gee et al. 1996; Brown et al. 1998; Brown & Gee, 1998; Field et al. 2006; Li et al. 2011). 3H‐Gabapentin also binds to α2δ‐2 (Gong et al. 2001). Several residues in α2δ‐1 and α2δ‐2 are involved in the binding of the gabapentinoid drugs; one important motif involves three arginine residues, just proximal to the VWA domain (Brown & Gee, 1998; Davies et al. 2006; Field et al. 2006). The binding pocket for gabapentin in α2δ‐1 has been further elucidated in the cryo‐EM structure (Wu et al. 2015). One may speculate that the basis of the binding of these drugs to α2δ‐1 and α2δ‐2 subunits might stem from the presence of the Cache domains, and their ancestral role to sense nutrients in bacteria. Furthermore, it is likely that a low molecular weight endogenous ligand might also bind to α2δ‐1 and α2δ‐2, and be displaced competitively by gabapentin. The binding affinity for 3H‐gabapentin increases progressively as the α2δ protein is purified or dialysed, or when isolated in lipid raft fractions, suggesting that an endogenous bound substance that competes with gabapentin binding is being removed (Brown et al. 1998; Davies et al. 2006; Lana et al. 2016). It is also possible that gabapentin binding might disrupt the function(s) of the VWA domain or the Cache domains (Dolphin, 2012; Cassidy et al. 2014). It would be of great interest to determine the nature and function of this endogenous small molecule.
Use of gabapentinoid drugs for epilepsy
Gabapentin is licensed for use as an adjunct drug in several types of epilepsy (Marson et al. 2000) and as a monotherapy in some partial‐onset seizures (Glauser et al. 2006). Pregabalin is also effective in the therapy of some epilepsies (for review see Taylor et al. 2007). In order to determine whether α2δ‐1 or α2δ‐2 was responsible for the anti‐epileptic effects of these drugs, experiments were performed using knock‐in mice, engineered to contain a mutant α2δ‐1 or α2δ‐2 with reduced affinity for gabapentinoid drug binding (Field et al. 2006; Lotarski et al. 2011). Pregabalin was not found to be effective against electroshock‐induced seizures in mice in which α2δ‐1 subunits are mutated, whereas it was still effective in mice with an equivalent mutation in α2δ‐2 (Lotarski et al. 2014); thus it is likely that the anti‐seizure effect of these drugs is primarily via binding to α2δ‐1.
Neuropathic pain and the role of α2δ subunits
Gabapentin and pregabalin are licensed for use in the treatment of various forms of neuropathic pain (Taylor et al. 2007). In contrast, they have no effect on acute nociceptive pain (Dickenson et al. 2005; Moore et al. 2009). In neuropathic pain models in rodents, it has been shown that binding of the gabapentinoid drugs to α2δ‐1 subunits is required for their therapeutic effect (Field et al. 2006). This finding indicates that binding to α2δ‐2 is not important for the effect of gabapentin, and, indeed, α2δ‐2 was found to be reduced rather than up‐regulated in injured DRG neurons (Bauer et al. 2009). Pregabalin is also used in the treatment of fibromyalgia, defined as generalized widespread pain, which may also have a neuropsychiatric aspect (Smith & Moore, 2012).
In a recent study, we have documented changes in splicing, in addition to overall up‐regulation of α2δ‐1, in injured rat DRG neurons (Lana et al. 2014). There was elevated expression of a novel splice variant (α2δ‐1 ΔA + B ΔC), which has a lower affinity for gabapentin (Lana et al. 2014). It is interesting to speculate that variable up‐regulation of this, or other, splice variants in people who develop neuropathic pain might be relevant to the inconsistent efficacy of the α2δ ligand drugs within the patient population.
Calcium channel currents: effects of gabapentinoid drugs
Small acute inhibitory effects of gabapentin have been observed on calcium currents in several systems (Stefani et al. 1998; Martin et al. 2002; Sutton et al. 2002). However, in other studies no acute responses to gabapentin have been reported on native or heterologously expressed calcium channel currents (Schumacher et al. 1998; Davies et al. 2006; Heblich et al. 2008; Hendrich et al. 2008). In DRGs from α2δ‐1‐overexpressing mice, it was observed that the calcium currents were rapidly inhibited by gabapentin, whereas this was not the case in wild‐type mice (Li et al. 2006). These results imply either that gabapentin is not a direct channel blocker, which would indeed be predicted from the location of its binding site, or that α2δ‐1 is not associated with all the relevant calcium channels in DRGs from wild‐type mice.
Calcium channel trafficking: effects of gabapentinoid drugs
We have found that incubation of cultured cells for several hours or days, rather than acute application of gabapentin, produces a reduction of calcium currents, both in expression systems when α2δ‐1 or α2δ‐2 was co‐expressed, and also in DRG neurons (Heblich et al. 2008; Hendrich et al. 2008; Tran‐Van‐Minh & Dolphin, 2010). We observed a corresponding reduction in expression of the α2δ and associated α1 subunits on the cell surface (Hendrich et al. 2008; Tran‐Van‐Minh & Dolphin, 2010; Cassidy et al. 2014; Fig. 6). We also found that gabapentin reduced forward trafficking of α2δ‐2 by inhibiting a post‐Golgi trafficking step, in a process requiring Rab11, which is involved in trafficking of cargoes in the recycling endosome compartment (Tran‐Van‐Minh & Dolphin, 2010). When this pathway was isolated, a response to gabapentin could be observed on a time‐scale of 30 min. Furthermore, we observed that chronic administration to nerve‐injured rats of an anti‐hyperalgesic dosing regimen of pregabalin reduced the elevation in the dorsal horn of presynaptic α2δ‐1. We interpreted this effect as being due to reduced axonal trafficking in vivo (Bauer et al. 2009). It is possible that gabapentinoid drugs selectively target calcium channel populations that are rapidly turning over, thus sparing skeletal muscle and cardiac channels, but this will need further experimentation.
Figure 6. CaV2.2 cell surface expression: effect of gabapentin .
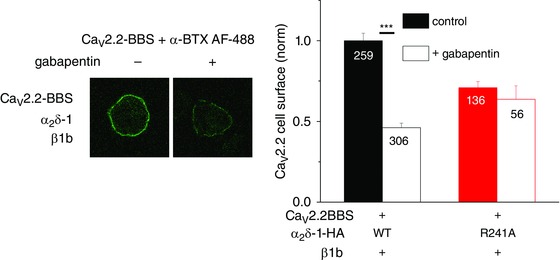
Cell surface expression of bungarotoxin binding site (BBS) tagged CaV2.2 labelled with α‐bungarotoxin (BTX) coupled to AF488 dye (green). Left panel: examples of N2a cells transfected with CaV2.2/β1b/α2δ‐1 in the absence (left), and presence (right) of gabapentin (100 μm for 24 h). Right panel: mean (± SEM) data for cell surface expression of CaV2.2, for cells expressing CaV2.2/β1b/α2δ‐1 (black‐filled and open bars), CaV2.2/β1b/α2δ‐1 R241A (a mutant α2δ‐1 that does not bind gabapentin; red‐filled and open bars) in the absence (filled bars), and presence (open bars) of gabapentin 100 μm for 24 h. Data are taken from a recent study (Cassidy et al. 2014). The number of cells measured is indicated on the bars, *** P<0.001, Student's t test.
Binding of α2δ subunits to other proteins: effects of gabapentinoid drugs
In various tissues it has been found that a proportion of α2δ subunits can be purified by biochemical means separately from α1 subunits (Gee et al. 1996; Müller et al. 2010), indicating that they may be only loosely associated with α1 subunits, or may exist separately. This suggests that they may have other functions in addition to being calcium channel subunits. For example, the α2δ‐3 proteins have a documented role in formation of synaptic boutons in Drosophila, which was found to be independent of their involvement with calcium channels, in that it was not mimicked by deletion of the relevant α1 subunit (Kurshan et al. 2009). However, since α2δ subunits play a role in trafficking calcium channels, as well as in calcium channel function, it may be that α2δ subunits directly influence the calcium transients which are involved in neurite outgrowth and synapse formation during development (Gu et al. 1994).
Furthermore, the α2δ‐1 protein has been found to co‐immunoprecipitate with thrombospondins, which are large multi‐domain extracellular matrix proteins (Eroglu et al. 2009); although it should be noted that thrombospondins also bind to many other proteins (Kazerounian et al. 2008). In the brain, specific thrombospondins are produced by astrocytes and promote the formation of silent excitatory synapses, lacking postsynaptic receptors (Christopherson et al. 2005). Thrombospondin‐induced synaptogenesis was found to require the postsynaptic presence of α2δ‐1 (Eroglu et al. 2009). Gabapentin was found to disrupt the in vitro interaction between α2δ‐1 and the synaptogenic domain of thrombospondin‐2, and also disrupted synaptogenesis, although it had no effect on pre‐formed synapses (Eroglu et al. 2009). This effect on synaptogenesis may not be relevant to the main mechanism of action of gabapentin either in neuropathic pain or as an antiepileptic drug, as much synaptic sprouting and remodelling would have taken place before the onset of therapy, although gabapentin could have a protective effect via this mechanism. Nevertheless, it should be emphasized that birth defects were found to be extremely uncommon in babies following chronic gabapentin exposure in the uterus of mothers who were taking the drug as an anti‐epileptic medication (Morrow et al. 2006; Molgaard‐Nielsen & Hviid, 2011), suggesting that it does not have any significant effect on synapse formation during development in utero.
As a corollary of a potential interaction between α2δ‐1 and thrombospondins, we have recently examined whether interaction of thrombospondins with α2δ‐1 might influence 3H‐gabapentin binding (Lana et al. 2016). We used thrombospondin‐4 as it is upregulated in neuropathic pain models (Pan et al. 2015). We found that in membranes from co‐transfected cells, thrombospondin‐4, significantly reduced the affinity for 3H‐gabapentin binding to α2δ‐1, in a divalent cation‐dependent manner. However, the effect on 3H‐gabapentin binding was not reproduced by the synaptogenic domain of thrombospondin‐4. Furthermore, we found only weak co‐immunoprecipitation of the two proteins, which could not be reproduced with the synaptogenic domain of thrombospondin‐4 (Lana et al. 2016). We also could not demonstrate any association between α2δ‐1 and thrombospondin‐4 on the cell surface of transfected cells, suggesting that the interaction between these two proteins to disrupt 3H‐gabapentin binding is occurring in an intracellular compartment of the transfected cells (Lana et al. 2016). It is nevertheless possible that such an interaction might reduce the efficacy of gabapentin in patients.
Conclusions and future directions
The α2δ subunits are important auxiliary subunits of the CaV1 and CaV1 voltage‐gated calcium channels. They play major roles in trafficking of these channels, both to the plasma membrane and to specific domains, as well as influencing the activation and biophysical properties of these channels. The mechanism of these effects, at a cell biological level, still remains to be determined in detail. They also play a role in the pathology of a number of genetic and other diseases, and represent an important therapeutic target site for drugs. Future therapeutic directions are likely to include identifying selective antagonists distinguishing CaV1.3 from CaV1.2 and other L‐type channels, finding selective antagonists for the different T‐type channels, and understanding better the mechanism of action of the α2δ ligands.
Additional information
Competing interests
None declared.
Acknowledgements
I would like to acknowledge the many students and postdoctoral associates who have contributed to the work from my laboratory, particularly those who have performed the studies described here. Much of our work has been funded by sequential grants from Wellcome Trust and Medical Research Council (MRC grants G0801756 and G0901758). My laboratory is currently funded by a Wellcome Trust Senior Investigator award (098360/Z/12/Z).
Biography
Annette Dolphin received her BA in Natural Sciences (Biochemistry) from the University of Oxford and her PhD from University of London, Institute of Psychiatry. She then held postdoctoral fellowships at the College de France in Paris with Joel Bockart, and at Yale University with Paul Greengard, before returning to UK to a staff scientist post at the National Institute for Medical Research, London, working with Tim Bliss. She then moved to an academic position as a lecturer in the Pharmacology Department of St George's Hospital Medical School, London University. She was appointed Chair of the Department of Pharmacology at Royal Free Hospital School of Medicine, London University in 1990, and moved to University College London in 1997. She is Professor of Pharmacology in the Department of Neuroscience, Physiology and Pharmacology at UCL. She was elected to the Academy of Medical Sciences in 1999, and the Royal Society in 2015. She is currently a Wellcome Trust Senior Investigator.

This is an Editor's Choice article from the 1 October 2016 issue.
References
- Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM & Yue DT (2005). G protein‐gated inhibitory module of N‐type (Cav2.2) Ca2+ channels. Neuron 46, 891–904. [DOI] [PubMed] [Google Scholar]
- Altier C, Dale CS, Kisilevsky AE, Chapman K, Castiglioni AJ, Matthews EA, Evans RM, Dickenson AH, Lipscombe D, Vergnolle N & Zamponi GW (2007). Differential role of N‐type calcium channel splice isoforms in pain. J Neurosci 27, 6363–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Garcia‐Caballero A, Simms B, You H, Chen L, Walcher J, Tedford HW, Hermosilla T & Zamponi GW (2011). The Cavβ subunit prevents RFP2‐mediated ubiquitination and proteasomal degradation of L‐type channels. Nat Neurosci 14, 173–180. [DOI] [PubMed] [Google Scholar]
- Alvarez‐Laviada A, Kadurin I, Senatore A, Chiesa R & Dolphin AC (2014). The inhibition of functional expression of calcium channels by prion protein demonstrates competition with α2δ for GPI‐anchoring pathways. Biochem J 458, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V & Aravind L (2000). Cache‐a signalling domain common to animal Ca channel subunits and a class of prokaryotic chemotaxis receptors. Trends Biochem Sci 25, 535–537. [DOI] [PubMed] [Google Scholar]
- Angelotti T & Hofmann F (1996). Tissue‐specific expression of splice variants of the mouse voltage‐gated calcium channel α2/δ subunit. FEBS Lett 397, 331–337. [DOI] [PubMed] [Google Scholar]
- Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W, Shaikh LH, Brighton CA, Teo AE, Davenport AP, Dekkers T, Tops B, Kusters B, Ceral J, Yeo GS, Neogi SG, McFarlane I, Rosenfeld N, Marass F, Hadfield J, Margas W, Chaggar K, Solar M, Deinum J, Dolphin AC, Farooqi IS, Striessnig J, Nissen P & Brown MJ (2013). Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 45, 1055–1060. [DOI] [PubMed] [Google Scholar]
- Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, Ali A, Ahmad I, Sinnegger‐Brauns MJ, Brandt N, Engel J, Mangoni ME, Farooq M, Khan HU, Nurnberg P, Striessnig J & Bolz HJ (2011). Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci 14, 77–84. [DOI] [PubMed] [Google Scholar]
- Bangalore R, Mehrke G, Gingrich K, Hofmann F & Kass RS (1996). Influence of L‐type Ca channel alpha 2/delta‐subunit on ionic and gating current in transiently transfected HEK 293 cells. Am J Physiol Heart Circ Physiol 270, H1521–H1528. [DOI] [PubMed] [Google Scholar]
- Bannister RA & Beam KG (2013). Cav1.1: The atypical prototypical voltage‐gated Ca2+ channel. Biochim Biophys Acta 1828, 1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, PerezReyes E, Lander ES, Frankel WN, Gardiner RM, Dolphin AC & Rees M (2001). Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci 21, 6095–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay J & Rees M (2000). Genomic organization of the mouse and human α2δ2 voltage‐dependent calcium channel subunit genes. Mamm Genome 11, 1142–1144. [DOI] [PubMed] [Google Scholar]
- Bauer CS, Nieto‐Rostro M, Rahman W, Tran‐Van‐Minh A, Ferron L, Douglas L, Kadurin I, Sri Ranjan Y, Fernandez‐Alacid L, Millar NS, Dickenson AH, Lujan R & Dolphin AC (2009). The increased trafficking of the calcium channel subunit α2δ‐1 to presynaptic terminals in neuropathic pain is inhibited by the α2δ ligand pregabalin. J Neurosci 29, 4076–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TJ, Thaler C, Castiglioni AJ, Helton TD & Lipscombe D (2004). Cell‐specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 41, 127–138. [DOI] [PubMed] [Google Scholar]
- Ben‐Johny M, Yang PS, Bazzazi H & Yue DT (2013). Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat Commun 4, 1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein GM & Jones OT (2007). Kinetics of internalization and degradation of N‐type voltage‐gated calcium channels: Role of the α2/δ subunit. Cell Calcium 41, 27–40. [DOI] [PubMed] [Google Scholar]
- Boehmerle W, Splittgerber U, Lazarus MB, McKenzie KM, Johnston DG, Austin DJ & Ehrlich BE (2006). Paclitaxel induces calcium oscillations via an inositol 1,4,5‐trisphosphate receptor and neuronal calcium sensor 1‐dependent mechanism. Proc Natl Acad Sci USA 103, 18356–18361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmerle W, Zhang K, Sivula M, Heidrich FM, Lee Y, Jordt SE & Ehrlich BE (2007). Chronic exposure to paclitaxel diminishes phosphoinositide signaling by calpain‐mediated neuronal calcium sensor‐1 degradation. Proc Natl Acad Sci USA 104, 11103–11108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland LM, Morrill JA & Bean BP (1994). ω‐Conotoxin block of N‐type calcium channels in frog and rat sympathetic neurons. J Neurosci 14, 5011–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdin B, Shakeri B, Tetreault MP, Sauve R, Lesage S & Parent L (2015). Functional characterization of CaVα2δ mutations associated with sudden cardiac death. J Biol Chem 290, 2854–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowersox SS, Gadbois T, Singh T, Pettus M, Wang YX & Luther RR (1996). Selective N‐type neuronal voltage‐sensitive calcium channel blocker, SNX‐111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J Pharmacol Exp Ther 279, 1243–1249. [PubMed] [Google Scholar]
- Brill J, Klocke R, Paul D, Boison D, Gouder N, Klugbauer N, Hofmann F, Becker CM & Becker K (2004). entla, a novel epileptic and ataxic Cacna2d2 mutant of the mouse. J Biol Chem 279, 7322–7330. [DOI] [PubMed] [Google Scholar]
- Brodbeck J, Davies A, Courtney J‐M, Meir A, Balaguero N, Canti C, Moss FJ, Page KM, Pratt WS, Hunt SP, Barclay J, Rees M & Dolphin AC (2002). The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated a2d‐2 protein with abnormal function. J Biol Chem 277, 7684–7693. [DOI] [PubMed] [Google Scholar]
- Brown JP, Dissanayake VU, Briggs AR, Milic MR & Gee NS (1998). Isolation of the [3H]gabapentin‐binding protein/α2δ Ca2+ channel subunit from porcine brain: development of a radioligand binding assay for α2δ subunits using [3H]leucine. Anal Biochem 255, 236–243. [DOI] [PubMed] [Google Scholar]
- Brown JP & Gee NS (1998). Cloning and deletion mutagenesis of the α2δ calcium channel subunit from porcine cerebral cortex. J Biol Chem 273, 25458–25465. [DOI] [PubMed] [Google Scholar]
- Buraei Z & Yang J (2010). The β subunit of voltage‐gated Ca2+ channels. Physiol Rev 90, 1461–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burashnikov E, Pfeiffer R, Barajas‐Martinez H, Delpon E, Hu D, Desai M, Borggrefe M, Haissaguerre M, Kanter R, Pollevick GD, Guerchicoff A, Laino R, Marieb M, Nademanee K, Nam GB, Robles R, Schimpf R, Stapleton DD, Viskin S, Winters S, Wolpert C, Zimmern S, Veltmann C & Antzelevitch C (2010). Mutations in the cardiac L‐type calcium channel associated with inherited J‐wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon‐Rivera A, Andrade A, Hernandez‐Hernandez O, Gonzalez‐Ramirez R, Sandoval A, Rivera M, Gomora JC & Felix R (2012). Identification of a disulfide bridge essential for structure and function of the voltage‐gated Ca2+ channel α2δ‐1 auxiliary subunit. Cell Calcium 51, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti C, Davies A, Berrow NS, Butcher AJ, Page KM & Dolphin AC (2001). Evidence for two concentration‐dependent processes for β‐subunit effects on α1B calcium channels. Biophys J 81, 1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti C, Davies A & Dolphin AC (2003). Calcium channel α2δ subunits: structure, function and target site for drugs. Curr Neuropharmacol 1, 209–217. [Google Scholar]
- Canti C, Nieto‐Rostro M, Foucault I, Heblich F, Wratten J, Richards MW, Hendrich J, Douglas L, Page KM, Davies A & Dolphin AC (2005). The metal‐ion‐dependent adhesion site in the Von Willebrand factor‐A domain of α2δ subunits is key to trafficking voltage‐gated Ca2+ channels. Proc Natl Acad Sci USA 102, 11230–11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti C, Page KM, Stephens GJ & Dolphin AC (1999). Identification of residues in the N‐terminus of α1B critical for inhibition of the voltage‐dependent calcium channel by Gβγ. J Neurosci 19, 6855–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YQ & Tsien RW (2010). Different relationship of N‐ and P/Q‐type Ca2+ channels to channel‐interacting slots in controlling neurotransmission at cultured hippocampal synapses. J Neurosci 30, 4536–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E, Calorio C & Vandael DH (2014). T‐type channel‐mediated neurotransmitter release. Pflugers Arch 466, 677–687. [DOI] [PubMed] [Google Scholar]
- Cassidy JS, Ferron L, Kadurin I, Pratt WS & Dolphin AC (2014). Functional exofacially tagged N‐type calcium channels elucidate the interaction with auxiliary α2δ‐1 subunits. Proc Natl Acad Sci USA 111, 8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiglioni AJ, Raingo J & Lipscombe D (2006). Alternative splicing in the C‐terminus of CaV2.2 controls expression and gating of N‐type calcium channels. J Physiol 576, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Striessnig J, Snutch TP & Perez‐Reyes E (2003). International Union of Pharmacology. XL. Compendium of voltage‐gated ion channels: calcium channels. Pharmacol Rev 55, 579–581. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Pogrel JW & Yaksh TL (1994). Role of voltage‐dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther 269, 1117–1123. [PubMed] [Google Scholar]
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L & Yang J (2004). Structural basis of the α1–β subunit interaction of voltage‐gated Ca2+ channels. Nature 429, 675–680. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, LAWLER J, Mosher DF, Bornstein P & Barres BA (2005). Thrombospondins are astrocyte‐secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. [DOI] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA & Gu G (2005). Differential distribution of voltage‐gated calcium channel alpha‐2 delta (α2δ) subunit mRNA‐containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491, 246–269. [DOI] [PubMed] [Google Scholar]
- Cross‐Disorder Group of the Psychiatric Genomics Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome‐wide analysis. Lancet 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs LL, Lee J‐H, Yang J, Satin J, Zhang Y, Daud A, Barclay J, Williamson MP, Fox M, Rees M & Perez‐Reyes E (1998). Cloning and characterization of α1H from human heart, a member of the T type Ca2+ channel gene family. Circ Res 83, 103–109. [DOI] [PubMed] [Google Scholar]
- D'Arco M, Margas W, Cassidy JS & Dolphin AC (2015). The upregulation of α2δ‐1 subunit modulates activity‐dependent Ca2+ signals in sensory neurons. J Neurosci 35, 5891–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A, Douglas L, Hendrich J, Wratten J, Tran‐Van‐Minh A, Foucault I, Koch D, Pratt WS, Saibil H & Dolphin AC (2006). The calcium channel α2δ‐2 subunit partitions with CaV2.1 in lipid rafts in cerebellum: implications for localization and function. J Neurosci 26, 8748–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A, Hendrich J, Van Minh AT, Wratten J, Douglas L & Dolphin AC (2007). Functional biology of the α2δ subunits of voltage‐gated calcium channels. Trends Pharmacol Sci 28, 220–228. [DOI] [PubMed] [Google Scholar]
- Davies A, Kadurin I, Alvarez‐Laviada A, Douglas L, Nieto‐Rostro M, Bauer CS, Pratt WS & Dolphin AC (2010). The α2δ subunits of voltage‐gated calcium channels form GPI‐anchored proteins, a post‐translational modification essential for function. Proc Natl Acad USA 107, 1654–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes JM, Antunes‐Martins A, Perkins JR, Paterson KJ, Sisignano M, Schmid R, Rust W, Hildebrandt T, Geisslinger G, Orengo C, Bennett DL & McMahon SB (2014). Genome‐wide transcriptional profiling of skin and dorsal root ganglia after ultraviolet‐B‐induced inflammation. PLoS One 9, e93338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih‐Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita‐Laza J, Jimenz GP, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei I, Lei J, Lehtimaki T, Lin CF, Ma'ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnstrom K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte‐Ruther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Li‐San W, Weiss LA, Willsey AJ, Yu TW, Yuen RK, Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, MW S, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ & Buxbaum JD (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sevilla Muller LP, Liu J, Solomon A, Rodriguez A & Brecha NC (2013). Expression of voltage‐gated calcium channel α2δ4 subunits in the mouse and rat retina. J Comp Neurol 521, 2486–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick IE, Tadross MR, Liang H, Tay LH, Yang W & Yue DT (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickenson AH, Bee LA & Suzuki R (2005). Pains, gains, and midbrains. Proc Natl Acad Sci USA 102, 17885–17886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2003). G protein modulation of voltage‐gated calcium channels. Pharmacol Rev 55, 607–627. [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2012). Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci 13, 542–555. [DOI] [PubMed] [Google Scholar]
- Dolphin AC, Wyatt CN, Richards J, Beattie RE, Craig P, Lee J‐H, Cribbs LL, Volsen SG & Perez‐Reyes E (1999). The effect of α2‐δ and other accessory subunits on expression and properties of the calcium channel α1G. J Physiol 519, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, Page KM, Koch D, Nieto‐Rostro M, Foucault I, Davies A, Wilkinson T, Rees M, Edwards FA & Dolphin AC (2006). The ducky2J mutation in Cacna2d2 results in reduced spontaneous Purkinje cell activity and altered gene expression. J Neurosci 26, 12576–12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubel SJ, Altier C, Chaumont S, Lory P, Bourinet E & Nargeot J (2004). Plasma membrane expression of T‐type calcium channel α1 subunits is modulated by high voltage‐activated auxiliary subunits. J Biol Chem 279, 29263–29269. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Oz S, Abulhijaa FA, Taher FB, Shaag A, Zenvirt S, Dascal N & Elpeleg O (2013). Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet 50, 118–123. [DOI] [PubMed] [Google Scholar]
- Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A, Schwartz A & Harpold MM (1988). Sequence and expression of mRNAs encoding the α1 and α2 subunits of a DHP‐sensitive calcium channel. Science 241, 1661–1664. [DOI] [PubMed] [Google Scholar]
- Ermolyuk YS, Alder FG, Surges R, Pavlov IY, Timofeeva Y, Kullmann DM & Volynski KE (2013). Differential triggering of spontaneous glutamate release by P/Q‐, N‐ and R‐type Ca2+ channels. Nat Neurosci 16, 1754–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF & Barres BA (2009). Gabapentin receptor α2δ‐1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix R (1999). Voltage‐dependent Ca2+ channel α2δ auxiliary subunit: Structure, function and regulation. Receptors Channels 6, 351–362. [PubMed] [Google Scholar]
- Felix R, Gurnett CA, De Waard M & Campbell KP (1997). Dissection of functional domains of the voltage‐dependent Ca2+ channel α2δ subunit. J Neurosci 17, 6884–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, Bramwell S, Corradini L, England S, Winks J, Kinloch RA, Hendrich J, Dolphin AC, Webb T & Williams D (2006). Identification of the α2δ‐1 subunit of voltage‐dependent calcium channels as a novel molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci USA 103, 17537–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois A, Schuetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, Mantilleri A, Nargeot J, Noel J, Wood JN, Moqrich A, Pongs O & Bourinet E (2015). The low‐threshold calcium channel Cav3.2 determines low‐threshold mechanoreceptor function. Cell Rep 10, 370–382. [DOI] [PubMed] [Google Scholar]
- Fu Y, Westenbroek RE, Scheuer T & Catterall WA (2013). Phosphorylation sites required for regulation of cardiac calcium channels in the fight‐or‐flight response. Proc Natl Acad Sci USA 110, 19621–19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MD, Emrick MA, Sadilek M, Scheuer T & Catterall WA (2010). Molecular mechanism of calcium channel regulation in the fight‐or‐flight response. Sci Signal 3, ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller‐Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, Muth JN, Mikala G, Petrashevskaya NN, Jordan MA, Zhang SP, Qin N, Flores CM, Isaacsohn I, Varadi M, Mori Y, Jones WK & Schwartz A (2009). Targeted disruption of the voltage‐dependent Ca2+ channel α2/δ‐1 subunit. Am J Physiol Heart Circ Physiol 297, H117–H124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadotti VM, Caballero AG, Berger ND, Gladding CM, Chen L, Pfeifer TA & Zamponi GW (2015). Small organic molecule disruptors of Cav3.2 – USP5 interactions reverse inflammatory and neuropathic pain. Mol Pain 11, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Sekido Y, Maximov A, Saad M, Forgacs E, Latif F, Wei MH, Lerman M, Lee JH, Perez‐Reyes E, Bezprozvanny I & Minna JD (2000). Functional properties of a new voltage‐dependent calcium channel α2δ auxiliary subunit gene (CACNA2D2). J Biol Chem 275, 12237–12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VUK, Offord J, Thurlow R & Woodruff GN (1996). The novel anticonvulsant drug, gabapentin (Neurontin), binds to the α2δ subunit of a calcium channel. J Biol Chem 271, 5768–5776. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, Mark K, Vu TH, Alkan C, Cheng Z, Biesecker LG, Bernier R & Eichler EE (2013). Refinement and discovery of new hotspots of copy‐number variation associated with autism spectrum disorder. Am J Hum Genet 92, 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauser T, Ben Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, Kalviainen R, Mattson R, Perucca E & Tomson T (2006). ILAE treatment guidelines: evidence‐based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 47, 1094–1120. [DOI] [PubMed] [Google Scholar]
- Gong HC, Hang J, Kohler W, Li L & Su TZ (2001). Tissue‐specific expression and gabapentin‐binding properties of calcium channel α2δ subunit subtypes. J Membr Bio 184, 35–43. [DOI] [PubMed] [Google Scholar]
- Gu X, Olson EC & Spitzer NC (1994). Spontaneous neuronal calcium spikes and waves during early differentiation. J Neurosci 14, 6325–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnett CA, De Waard M & Campbell KP (1996). Dual function of the voltage‐dependent Ca2+ channel α2δ subunit in current stimulation and subunit interaction. Neuron 16, 431–440. [DOI] [PubMed] [Google Scholar]
- Gurnett CA, Felix R & Campbell KP (1997). Extracellular interaction of the voltage‐dependent Ca2+ channel α2δ and α1 subunits. J Biol Chem 272, 18508–18512. [DOI] [PubMed] [Google Scholar]
- Gutierrez LM, Brawley RM & Hosey MM (1991). Dihydropyridine‐sensitive calcium channels from skeletal muscle. I. Roles of subunits in channel activity. J Biol Chem 266, 16387–16394. [PubMed] [Google Scholar]
- Guzman JN, Sanchez‐Padilla J, Chan CS & Surmeier DJ (2009). Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29, 11011–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon MR, Berrow NS, Dolphin AC & Wallace BA (1999). Modelling of a voltage‐dependent Ca2+ channel β subunit as a basis for understanding its functional properties. FEBS Lett 445, 366–370. [DOI] [PubMed] [Google Scholar]
- Heblich F, Tran‐Van‐Minh A, Hendrich J, Watschinger K & Dolphin AC (2008). Time course and specificity of the pharmacological disruption of the trafficking of voltage‐gated calcium channels by gabapentin. Channels 2, 4–9. [DOI] [PubMed] [Google Scholar]
- Hendrich J, Tran‐Van‐Minh A, Heblich F, Nieto‐Rostro M, Watschinger K, Striessnig J, Wratten J, Davies A & Dolphin AC (2008). Pharmacological disruption of calcium channel trafficking by the α2δ ligand gabapentin. Proc Natl Acad Sci USA 105, 3628–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ & Dolphin AC (2015). Genetic disruption of voltage‐gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol 134, 36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino‐Fukuyo N, Kikuchi A, Arai‐Ichinoi N, Niihori T, Sato R, Suzuki T, Kudo H, Sato Y, Nakayama T, Kakisaka Y, Kubota Y, Kobayashi T, Funayama R, Nakayama K, Uematsu M, Aoki Y, Haginoya K & Kure S (2015). Genomic analysis identifies candidate pathogenic variants in 9 of 18 patients with unexplained West syndrome. Hum Genet 134, 649–658. [DOI] [PubMed] [Google Scholar]
- Hobom M, Dai S, Marais E, Lacinova L, Hofmann F & Klugbauer N (2000). Neuronal distribution and functional characterization of the calcium channel α2δ‐2 subunit. Eur J Neurosci 12, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Hoppa MB, Gouzer G, Armbruster M & Ryan TA (2014). Control and plasticity of the presynaptic action potential waveform at small CNS nerve terminals. Neuron 84, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC & Ryan TA (2012). α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Rusin CG, Tan Z, Guagliardo NA & Barrett PQ (2012). Zona glomerulosa cells of the mouse adrenal cortex are intrinsic electrical oscillators. J Clin Invest 122, 2046–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Wang Z, Wei R, Fan G, Wang Q, Zhang K & Yin CC (2015). The molecular architecture of dihydropyrindine receptor/L‐type Ca2+ channel complex. Sci Rep 5, 8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, Dolphin AC & Shah MM (2011). Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci 14, 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR & Wigler M (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Kaneko M, Shin HS & Takahashi T (2005). Presynaptic N‐type and P/Q‐type Ca2+ channels mediating synaptic transmission at the calyx of Held of mice. J Physiol 568, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov SV, Ward JM, Tessarollo L, McAreavey D, Sachdev V, Fananapazir L, Banks MK, Morris N, Djurickovic D, Devor‐Henneman DE, Wei MH, Alvord GW, Gao B, Richardson JA, Minna JD, Rogawski MA & Lerman MI (2004). Cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 gene. Am J Pathol 165, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD & Takahashi T (2000). Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci 20, 59‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay SD, Ellis SB, McCue AF, Williams ME, Vedvick TS, Harpold MM & Campbell KP (1990). Primary structure of the gamma subunit of the DHP‐sensitive calcium channel from skeletal muscle. Science 248, 490–492. [DOI] [PubMed] [Google Scholar]
- Jay SD, Sharp AH, Kahl SD, Vedvick TS, Harpold MM & Campbell KP (1991). Structural characterization of the dihydropyridine‐sensitive calcium channel α2‐subunit and the associated δ peptides. J Biol Chem 266, 3287–3293. [PubMed] [Google Scholar]
- Jones LP, Wei SK & Yue DT (1998). Mechanism of auxiliary subunit modulation of neuronal α1E calcium channels. J Gen Physiol 112, 125–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Chen X, Khasar SG & Levine JD (2004). Novel mechanism of enhanced nociception in a model of AIDS therapy‐induced painful peripheral neuropathy in the rat. Pain 107, 147–158. [DOI] [PubMed] [Google Scholar]
- Kadurin I, Alvarez‐Laviada A, Ng SF, Walker‐Gray R, D'Arco M, Fadel MG, Pratt WS & Dolphin AC (2012. a). Calcium currents are enhanced by α2δ‐1 lacking its membrane anchor. J Biol Chem 1287, 33554–33566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadurin I, Bauer C, Lana B, Alvarez‐Laviada A, Nieto‐Rostro M, Douglas L, Troeberg L, Nagase H & Dolphin AC (2012. b). Voltage‐gated calcium channel α2δ subunits in lipid rafts: the importance of proteolytic cleavage into α2 and δ. Biophys J 102, 125a–125a. [Google Scholar]
- Kazerounian S, Yee KO & Lawler J (2008). Thrombospondins in cancer. Cell Mol Life Sci 65, 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Rumpf CH, Fujiwara Y, Cooley ES, Van Petegem F & Minor DL Jr (2008). Structures of Cav2 Ca2+/CaM‐IQ domain complexes reveal binding modes that underlie calcium‐dependent inactivation and facilitation. Structure 16, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Rumpf CH, Van Petegem F, Arant RJ, Findeisen F, Cooley ES, Isacoff EY & Minor DL Jr (2010). Multiple C‐terminal tail Ca2+/CaMs regulate Cav1.2 function but do not mediate channel dimerization. EMBO J 29, 3924–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H‐L, Kim H, Lee P, King RG & Chin H (1992). Rat brain expresses an alternatively spliced form of the dihydropyridine‐sensitive L‐type calcium channel α2 subunit. Proc Natl Acad Sci USA 89, 3251–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimm T & Bean BP (2014). Inhibition of A‐type potassium current by the peptide toxin SNX‐482. J Neurosci 34, 9182–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Marais E, Hobom M & Hofmann F (1999). Molecular diversity of the calcium channel α2‐δ subunit. J Neurosci 19, 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurshan PT, Oztan A & Schwarz TL (2009). Presynaptic α2δ‐3 is required for synaptic morphogenesis independent of its Ca2+‐channel functions. Nat Neurosci 12, 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacinova L, Klugbauer N & Hofmann F (1999). Absence of modulation of the expressed calcium channel α1G subunit by α2δ subunits. J Physiol 516, 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana B, Page KM, Kadurin I, Ho S, Nieto‐Rostro M & Dolphin AC (2016). Thrombospondin‐4 reduces binding affinity of [3H]‐gabapentin to calcium‐channel α2δ‐1‐subunit but does not interact with α2δ‐1 on the cell‐surface when co‐expressed. Sci Rep 6, 24531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana B, Schlick B, Martin S, Pratt WS, Page KM, Goncalves L, Rahman W, Dickenson AH, Bauer CS & Dolphin AC (2014). Differential up‐regulation in DRG neurons of an αδ‐1 splice variant with a lower affinity for gabapentin after peripheral sensory nerve injury. Pain 155, 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy J, Richards MS, Butcher AJ, Nieto‐Rostro M, Pratt WS, Davies A & Dolphin AC (2005). Interaction via a key tryptophan in the I‐II linker of N‐type calcium channels is required for β1 but not for palmitoylated β2, implicating an additional binding site in the regulation of channel voltage‐dependent properties. J Neurosci 25, 6984–6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, Mori Y, Campbell KP & Frankel WN (1998). The mouse stargazer gene encodes a neuronal Ca2+ channel gamma subunit. Nat Genet 19, 340–347. [DOI] [PubMed] [Google Scholar]
- Li CY, Zhang XL, Matthews EA, Li KW, Kurwa A, Boroujerdi A, Gross J, Gold MS, Dickenson AH, Feng G & Luo ZD (2006). Calcium channel α2δ1 subunit mediates spinal hyperexcitability in pain modulation. Pain 125, 20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Taylor CP, Weber M, Piechan J, Prior F, Bian F, Cui M, Hoffman D & Donevan S (2011). Pregabalin is a potent and selective ligand for α2δ‐1 and α2δ‐2 calcium channel subunits. Eur J Pharmacol 667, 80–90. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Madison DV, Poenie M, Reuter H, Tsien RY & Tsien RW (1988). Spatial distribution of calcium channels and cytosolic calcium transients in growth cones and cell bodies of sympathetic neurons. Proc Natl Acad USA 85, 2398–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, De Waard M, Scott VES, Gurnett CA, Lennon VA & Campbell KP (1996). Identification of three subunits of the high affinity w‐conotoxin MVIIC‐sensitive Ca2+ channel. J Biol Chem 271, 13804–13810. [PubMed] [Google Scholar]
- Lotarski S, Hain H, Peterson J, Galvin S, Strenkowski B, Donevan S & Offord J (2014). Anticonvulsant activity of pregabalin in the maximal electroshock‐induced seizure assay in αδ (R217A) and αδ (R279A) mouse mutants. Epilepsy Res 108, 833–842. [DOI] [PubMed] [Google Scholar]
- Lotarski SM, Donevan S, El Kattan A, Osgood S, Poe J, Taylor CP & Offord J (2011). Anxiolytic‐like activity of pregabalin in the Vogel conflict test in α2δ‐1 (R217A) and α2δ‐2 (R279A) mouse mutants. J Pharmacol Exp Ther 338, 615–621. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME & Yaksh TL (2001). Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve‐injured rats. J Neurosci 21, 1868–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macabuag N & Dolphin AC (2015). Alternative splicing in CaV2.2 regulates neuronal trafficking via adaptor protein complex‐1 adaptor protein binding motifs. J Neurosci 35, 14636–14652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D & Sebat J (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J & Nargeot J (2003). Functional role of L‐type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA 100, 5543–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansergh F, Orton NC, Vessey JP, Lalonde MR, Stell WK, Tremblay F, Barnes S, Rancourt DE & Bech‐Hansen NT (2005). Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet 14, 3035–3046. [DOI] [PubMed] [Google Scholar]
- Margas W, Ferron L, Nieto‐Rostro M, Schwartz A & Dolphin AC (2016). Effect of knockout of α2δ‐1 on action potentials in mouse sensory neurons. Phil Trans Roy Soc B 371, 20150430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson AG, Kadir ZA, Hutton JL & Chadwick DW (2000). Gabapentin add‐on for drug‐resistant partial epilepsy. Cochrane Database Syst Rev CD001415. [DOI] [PubMed] [Google Scholar]
- Martin DJ, McClelland D, Herd MB, Sutton KG, Hall MD, Lee K, Pinnock RD & Scott RH (2002). Gabapentin‐mediated inhibition of voltage‐activated Ca2+ channel currents in cultured sensory neurones is dependent on culture conditions and channel subunit expression. Neuropharmacology 42, 353–366. [DOI] [PubMed] [Google Scholar]
- Meir A, Bell DC, Stephens GJ, Page KM & Dolphin AC (2000). Calcium channel β subunit promotes voltage‐dependent modulation of α1B by Gβγ. Biophys J 79, 731–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, Zheng Y, Diffenderfer KE, Zhang J, Soltani S, Eames T, Schafer ST, Boyer L, Marchetto MC, Nurnberger JI, Calabrese JR, Odegaard KJ, McCarthy MJ, Zandi PP, Alba M, Nievergelt CM, Pharmacogenomics of Bipolar Disorder Study , Mi S, Brennand KJ, Kelsoe JR, Gage FH & Yao J (2015). Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S & Numa S (1989). Primary structure and functional expression of the cardiac dihydropyridine‐sensitive calcium channel. Nature 340, 230–233. [DOI] [PubMed] [Google Scholar]
- Minor DL Jr & Findeisen F (2010). Progress in the structural understanding of voltage‐gated calcium channel (CaV) function and modulation. Channels (Austin) 4, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Adams ME & Bean BP (1992). P‐type calcium channels in rat central and peripheral neurons. Neuron 9, 85–95. [DOI] [PubMed] [Google Scholar]
- Molgaard‐Nielsen D & Hviid A (2011). Newer‐generation antiepileptic drugs and the risk of major birth defects. JAMA 305, 1996–2002. [DOI] [PubMed] [Google Scholar]
- Moore RA, Straube S, Wiffen PJ, Derry S & McQuay HJ (2009). Pregabalin for acute and chronic pain in adults. Cochrane Database Syst Rev, CD007076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori MX, Vander Kooi CW, Leahy DJ & Yue DT (2008). Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high‐resolution mechanistic implications for channel regulation by Ca2+ . Structure 16, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Friedrich T, Kim M‐S, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, Mikoshiba K, Imoto K, Tanabe T & Numa S (1991). Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature 350, 398–402. [DOI] [PubMed] [Google Scholar]
- Morrow J, Russell A, Guthrie E, Parsons L, Robertson I, Waddell R, Irwin B, McGivern RC, Morrison PJ & Craig J (2006). Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. J Neurol Neurosurg Psychiatry 77, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss FJ, Viard P, Davies A, Bertaso F, Page KM, Graham A, Canti C, Plumpton M, Plumpton C, Clare JJ & Dolphin AC (2002). The novel product of a five‐exon stargazin‐related gene abolishes CaV2.2 calcium channel expression. EMBO J 21, 1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B & Schulte U (2010). Quantitative proteomics of the Cav2 channel nano‐environments in the mammalian brain. Proc Natl Acad Sci USA 107, 14950–14957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Harada H, Kamasawa N, Matsui K, Rothman JS, Shigemoto R, Silver RA, DiGregorio DA & Takahashi T (2015). Nanoscale distribution of presynaptic Ca2+ channels and its impact on vesicular release during development. Neuron 85, 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely GG, Hess A, Costigan M, Keene AC, Goulas S, Langeslag M, Griffin RS, Belfer I, Dai F, Smith SB, Diatchenko L, Gupta V, Xia CP, Amann S, Kreitz S, Heindl‐Erdmann C, Wolz S, Ly CV, Arora S, Sarangi R, Dan D, Novatchkova M, Rosenzweig M, Gibson DG, Truong D, Schramek D, Zoranovic T, Cronin SJ, Angjeli B, Brune K, Dietzl G, Maixner W, Meixner A, Thomas W, Pospisilik JA, Alenius M, Kress M, Subramaniam S, Garrity PA, Bellen HJ, Woolf CJ & Penninger JM (2010). A genome‐wide Drosophila screen for heat nociception identifies α2δ3 as an evolutionarily conserved pain gene. Cell 143, 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton RA, Bingham S, Case PC, Sanger GJ & Lawson SN (2001). Dorsal root ganglion neurons show increased expression of the calcium channel α2δ‐1 subunit following partial sciatic nerve injury. Brain Res Mol Brain Res 95, 1–8. [DOI] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP & Tsien RW (1985). Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316, 440–446. [DOI] [PubMed] [Google Scholar]
- Opatowsky Y, Chen CC, Campbell KP & Hirsch JA (2004). Structural analysis of the voltage‐dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron 42, 387–399. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD & Frants RR (1996). Familial hemiplegic migraine and episodic ataxia type‐2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552. [DOI] [PubMed] [Google Scholar]
- Page KM, Canti C, Stephens GJ, Berrow NS & Dolphin AC (1998). Identification of the amino terminus of neuronal Ca2+ channel α1 subunits α1B and α1E as an essential determinant of G protein modulation. J Neurosci 18, 4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Yu H, Park J, Yu YP, Luo ZD & Hogan QH (2015). Painful nerve injury upregulates thrombospondin‐4 expression in dorsal root ganglia. J Neurosci Res 93, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli LK, Nakajima C, Kulik A, Matsui K, Schneider T, Shigemoto R & Fukazawa Y (2012). Quantitative regional and ultrastructural localization of the Cav2.3 subunit of R‐type calcium channel in mouse brain. J Neurosci 32, 13555–13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Bauer CS, Nieto‐Rostro M, Margas W, Ferron L, Chaggar K, Crews K, Ramirez JD, Bennett DL, Schwartz A, Dickenson AH & Dolphin AC (2013). α2δ‐1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J Neurosci 33, 16412–16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N & Catterall WA (2011). The crystal structure of a voltage‐gated sodium channel. Nature 475, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E (1998). Molecular characterization of a novel family of low voltage‐ activated, T‐type, calcium channels. J Bioenerg Biomembr 30, 313–318. [DOI] [PubMed] [Google Scholar]
- Perez‐Reyes E (2003). Molecular physiology of low‐voltage‐activated T‐type calcium channels. Physiol Rev 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Perez‐Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M & Lee J‐H (1998). Molecular characterisation of a neuronal low‐voltage‐activated T type calcium channel. Nature 391, 896–900. [DOI] [PubMed] [Google Scholar]
- Perez‐Reyes E, Van Deusen AL & Vitko I (2009). Molecular pharmacology of human Cav3.2 T‐type Ca2+ channels: block by antihypertensives, antiarrhythmics, and their analogs. J Pharmacol Exp Ther 328, 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez de Sevilla ML, Sargoy A, Fernandez‐Sanchez L, Rodriguez A, Liu J, Cuenca N & Brecha N (2015). Expression and cellular localization of the voltage‐gated calcium channel α2δ3 in the rodent retina. J Comp Neurol 523, 1443–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins JR, Antunes‐Martins A, Calvo M, Grist J, Rust W, Schmid R, Hildebrandt T, Kohl M, Orengo C, McMahon SB & Bennett DL (2014). A comparison of RNA‐seq and exon arrays for whole genome transcription profiling of the L5 spinal nerve transection model of neuropathic pain in the rat. Mol Pain 10, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedras‐Rentería ES & Tsien RW (1998). Antisense oligonucleotides against α1E reduce R‐type calcium currents in cerebellar granule cells. Proc Natl Acad Sci USA 95, 7760–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierleoni A, Martelli PL & Casadio R (2008). PredGPI: a GPI‐anchor predictor. BMC Bioinformatics 9, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippucci T, Parmeggiani A, Palombo F, Maresca A, Angius A, Crisponi L, Cucca F, Liguori R, Valentino ML, Seri M & Carelli V (2013). A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS One 8, e82154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirone A, Kurt S, Zuccotti A, Ruttiger L, Pilz P, Brown DH, Franz C, Schweizer M, Rust MB, Rubsamen R, Friauf E, Knipper M & Engel J (2014). α2δ3 is essential for normal structure and function of auditory nerve synapses and is a novel candidate for auditory processing disorders. J Neurosci 34, 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platano D, Qin N, Noceti F, Birnbaumer L, Stefani E & Olcese R (2000). Expression of the α2δ subunit interferes with prepulse facilitation in cardiac L‐type calcium channels. Biophys J 78, 2959–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott‐Fischer A, Stephan K, Bova S, Chen H, Zheng H & Striessnig J (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L‐type Ca2+ channels. Cell 102, 89–97. [DOI] [PubMed] [Google Scholar]
- Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP & Campbell KP (1994). Calcium channel β‐subunit binds to a conserved motif in the I‐II cytoplasmic linker of the α1‐subunit. Nature 368, 67–70. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kahler A, Duncan L, Stahl E, Genovese G, Fernandez E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SG, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA & Sklar P (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzier I, Kullmann PH, Horn JP & Levitan ES (2009). Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J Neurosci 29, 15414–15419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Olcese R, Stefani E & Birnbaumer L (1998). Modulation of human neuronal α1E‐type calcium channel by α2δ‐subunit. Am J Physiol Cell Physiol 274, C1324–C1331. [DOI] [PubMed] [Google Scholar]
- Qin N, Yagel S, Momplaisir ML, Codd EE & D'Andrea MR (2002). Molecular cloning and characterization of the human voltage‐gated calcium channel α2δ‐4 subunit. Mol Pharmacol 62, 485–496. [DOI] [PubMed] [Google Scholar]
- Reuter H (1987). Calcium channel modulation by β‐adrenergic neurotransmitters in the heart. Experientia 43, 1173–1175. [DOI] [PubMed] [Google Scholar]
- Richards MW, Butcher AJ & Dolphin AC (2004). Calcium channel β subunits: structural insights AID our understanding. Trends Pharmacol Sci 25, 626–632. [DOI] [PubMed] [Google Scholar]
- Schmidt BL, Hamamoto DT, Simone DA & Wilcox GL (2010). Mechanism of cancer pain. Mol Interv 10, 164–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Wei X, Olcese R, Costantin JL, Neely A, Palade P, Perez‐Reyes E, Qin N, Zhou J, Crawford GD, Smith RG, Appel SH, Stefani E & Birnbaumer L (1994). Molecular analysis and functional expression of the human type E neuronal Ca2+ channel α1 subunit. Receptors Channels 2, 255–270. [PubMed] [Google Scholar]
- Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson‐Williams C, Libutti SK, Mane S, Hellman P, Westin G, Akerstrom G, Bjorklund P, Carling T, Fahlke C, Hidalgo P & Lifton RP (2013). Somatic and germline CACNA1D calcium channel mutations in aldosterone‐producing adenomas and primary aldosteronism. Nat Genet 45, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TB, Beck H, Steinhäuser C, Schramm J & Elger CE (1998). Effects of phenytoin, carbamazepine, and gabapentin on calcium channels in hippocampal granule cells from patients with temporal lobe epilepsy. Epilepsia 39, 355–363. [DOI] [PubMed] [Google Scholar]
- Schutz SG & Robinson‐Papp J (2013). HIV‐related neuropathy: current perspectives. HIV AIDS (Auckl ) 5, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serysheva II, Ludtke SJ, Baker MR, Chiu W & Hamilton SL (2002). Structure of the voltage‐gated L‐type Ca2+ channel by electron cryomicroscopy. Proc Natl Acad Sci USA 99, 10370–10375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov R, Ferreira G, Yi JX & Ríos E (1998). Inactivation of gating currents of L‐type calcium channels – Specific role of the α2δ subunit. J Gen Physiol 111, 807–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shistik E, Ivanina T, Puri T, Hosey M & Dascal N (1995). Ca2+ current enhancement by α2/δ and β subunits in Xenopus oocytes: contribution of changes in channel gating and α1 protein level. J Physiol 489, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman RB (2008). From basic science to blockbuster drug: the discovery of Lyrica. Angew Chem Int Ed Engl 47, 3500–3504. [DOI] [PubMed] [Google Scholar]
- Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F & Dascal N (1991). The roles of the subunits in the function of the calcium channel. Science 253, 1553–1557. [DOI] [PubMed] [Google Scholar]
- Singer‐Lahat D, Lotan I, Itagaki K, Schwartz A & Dascal N (1992). Evidence for the existence of RNA of Ca2+‐channel α2/δ subunit in Xenopus oocytes. Biochim Biophys Acta Mol Cell Res 1137, 39–44. [DOI] [PubMed] [Google Scholar]
- Smith MT & Moore BJ (2012). Pregabalin for the treatment of fibromyalgia. Expert Opin Pharmacother 13, 1527–1533. [DOI] [PubMed] [Google Scholar]
- Soong TW, Stea A, Hodson CD, Dubel SJ, Vincent SR & Snutch TP (1993). Structure and functional expression of a member of the low voltage‐activated calcium channel family. Science 260, 1133–1136. [DOI] [PubMed] [Google Scholar]
- Stefani A, Spadoni F & Bernardi G (1998). Gabapentin inhibits calcium currents in isolated rat brain neurons. Neuropharmacology 37, 83–91. [DOI] [PubMed] [Google Scholar]
- Stephens GJ, Page KM, Burley JR, Berrow NS & Dolphin AC (1997). Functional expression of rat brain cloned α1E calcium channels in COS‐7 cells. Pflügers Archiv 433, 523–532. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Ortner NJ & Pinggera A (2015). Pharmacology of L‐type calcium channels: novel drugs for old targets? Curr Mol Pharmacol 8, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Pinggera A, Kaur G, Bock G & Tuluc P (2014). L‐type Ca channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal 3, 15–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton KG, Martin DJ, Pinnock RD, Lee K & Scott RH (2002). Gabapentin inhibits high‐threshold calcium channel currents in cultured rat dorsal root ganglion neurones. Br J Pharmacol 135, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T & Momiyama A (1993). Different types of calcium channels mediate central synaptic transmission. Nature 366, 156–158. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T & Numa S (1987). Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature 328, 313–318. [DOI] [PubMed] [Google Scholar]
- Tang L, Gamal El‐Din TM, Payandeh J, Martinez GQ, Heard TM, Scheuer T, Zheng N & Catterall WA (2014). Structural basis for Ca2+ selectivity of a voltage‐gated calcium channel. Nature 505, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CP, Angelotti T & Fauman E (2007). Pharmacology and mechanism of action of pregabalin: the calcium channel alpha2‐delta (α2‐δ) subunit as a target for antiepileptic drug discovery. Epilepsy Res 73, 137–150. [DOI] [PubMed] [Google Scholar]
- Taylor CP & Garrido R (2008). Immunostaining of rat brain, spinal cord, sensory neurons and skeletal muscle for calcium channel alpha2‐delta (α2‐δ) type 1 protein. Neuroscience 155, 510–521. [DOI] [PubMed] [Google Scholar]
- Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, Sticht H, Rauch A, Puleo C, Hu D, Barajas‐Martinez H, Antzelevitch C, Luscher TF, Abriel H & Duru F (2011). Identification of a novel loss‐of‐function calcium channel gene mutation in short QT syndrome (SQTS6). Eur Heart J 32, 1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Peca J & Feng G (2012). Functional consequences of mutations in postsynaptic scaffolding proteins and relevance to psychiatric disorders. Annu Rev Neurosci 35, 49–71. [DOI] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA & Bredt DS (2003). Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol 161, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottene A, Volsen S & Pietrobon D (2000). α1E subunits form the pore of three cerebellar R‐type calcium channels with different pharmacological and permeation properties. J Neurosci 20, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran‐Van‐Minh A & Dolphin AC (2010). The α2δ ligand gabapentin inhibits the Rab11‐dependent recycling of the calcium channel subunit α2δ‐2. J Neurosci 30, 12856–12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuluc P, Mastrolia V, Drach M, Flucher SM, Renstrom E, Striessnig J & Flucher BE (2014). Calcium channel α2δ‐1 subunit knockout causes diabetes due to impaired insulin release. Biophys J 106, 331a–331a. [Google Scholar]
- Van Petegem F, Clark KA, Chatelain FC & Minor DL Jr (2004). Structure of a complex between a voltage‐gated calcium channel β‐subunit and an α‐subunit domain. Nature 429, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi G, Lory P, Schultz D, Varadi M & Schwartz A (1991). Acceleration of activation and inactivation by the β subunit of the skeletal muscle calcium channel. Nature 352, 159–162. [DOI] [PubMed] [Google Scholar]
- Vergult S, Dheedene A, Meurs A, Faes F, Isidor B, Janssens S, Gautier A, Le CC & Menten B (2015). Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur J Hum Genet 23, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waithe D, Ferron L, Page KM, Chaggar K & Dolphin AC (2011). β‐Subunits promote the expression of Cav2.2 channels by reducing their proteasomal degradation. J Biol Chem 286, 9598–9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamori M, Mikala G & Mori Y (1999). Auxiliary subunits operate as a molecular switch in determining gating behaviour of the unitary N‐type Ca2+ channel current in Xenopus oocytes. J Physiol 517, 659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace VC, Blackbeard J, Segerdahl AR, Hasnie F, Pheby T, McMahon SB & Rice AS (2007). Characterization of rodent models of HIV‐gp120 and anti‐retroviral‐associated neuropathic pain. Brain 130, 2688–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CP, Davies A, Butcher AJ, Dolphin AC & Kitmitto A (2009). 3D structure of CaV3.1 – comparison with the cardiac L‐type voltage‐gated calcium channel monomer architecture. J Biol Chem 284, 22310–22321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun H, Della PK, Benz RJ, Xu J, Gerhold DL, Holder DJ & Koblan KS (2002). Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience 114, 529–546. [DOI] [PubMed] [Google Scholar]
- Wang MC, Collins RF, Ford RC, Berrow NS, Dolphin AC & Kitmitto A (2004). The three‐dimensional structure of the cardiac L‐type voltage‐gated calcium channel: comparison with the skeletal muscle form reveals a common architectural motif. J Biol Chem 279, 7159–7168. [DOI] [PubMed] [Google Scholar]
- Welling A, Bosse E, Cavalie A, Bottlender R, Ludwig A, Nastainczyk W, Flockerzi V & Hofmann F (1993). Stable co‐expression of calcium channel α, β and α2/δ subunits in a somatic cell line. J Physiol 471, 749–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP & Catterall WA (1992). Biochemical properties and subcellular distribution of an N‐type calcium channel α1 subunit. Neuron 9, 1099–1115. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TVB, Snutch TP & Catterall WA (1995). Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. J Neurosci 15, 6403–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DB, Randall A & Tsien RW (1994). Roles of N‐type and Q‐type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264, 107–111. [DOI] [PubMed] [Google Scholar]
- Whittaker CA & Hynes RO (2002). Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell 13, 3369–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SM, Toth PT, Oh SB, Gillard SE, Volsen S, Ren D, Philipson LH, Fletcher CF, Tessarollo L, Copeland NG, Jenkins NA & Miller RJ (2000). The status of voltage‐dependent calcium channels in α1E knockout mice. J Neurosci 20, 8566–8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher DR, De Waard M, Sakamoto J, Franzini‐Armstrong C, Pragnell M, Kahl SD & Campbell KP (1993). Subunit identification and reconstitution of the N‐type Ca2+ channel complex purified from brain. Science 261, 486–489. [DOI] [PubMed] [Google Scholar]
- Wolf M, Eberhart A, Glossmann H, Striessnig J & Grigorieff N (2003). Visualization of the domain structure of an L‐type Ca2+ channel using electron cryo‐microscopy. J Mol Biol 332, 171–182. [DOI] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M & Yan N (2015). Structure of the voltage‐gated calcium channel Cav1.1 complex. Science 350, aad2395. [DOI] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JGG, Catterall WA & Sakmann B (1999). Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx‐ type synapses. J Neurosci 19, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wycisk KA, Budde B, Feil S, Skosyrski S, Buzzi F, Neidhardt J, Glaus E, Nurnberg P, Ruether K & Berger W (2006. a). Structural and functional abnormalities of retinal ribbon synapses due to Cacna2d4 mutation. Invest Ophthalmol Vis Sci 47, 3523–3530. [DOI] [PubMed] [Google Scholar]
- Wycisk KA, Zeitz C, Feil S, Wittmer M, Forster U, Neidhardt J, Wissinger B, Zrenner E, Wilke R, Kohl S & Berger W (2006. b). Mutation in the auxiliary calcium‐channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet 79, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao HS, Huang QH, Zhang FX, Bao L, Lu YJ, Guo C, Yang L, Huang WJ, Fu G, Xu SH, Cheng XP, Yan Q, Zhu ZD, Zhang X, Chen Z, Han ZG & Zhang X (2002). Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci USA 99, 8360–8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Chen L, Barr W, Mcrory JE, Lewis RJ, Adams DJ & Zamponi GW (2004). Auxiliary subunit regulation of high‐voltage activated calcium channels expressed in mammalian cells. Eur J Neurosci 20, 1–13. [DOI] [PubMed] [Google Scholar]
- Zamponi GW (2016). Targeting voltage‐gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov 15, 19–34. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J & Snutch TP (1997). Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature 385, 442–446. [DOI] [PubMed] [Google Scholar]
- Zamponi GW & Currie KP (2013). Regulation of CaV2 calcium channels by G protein coupled receptors. Biochim Biophys Acta 1828, 1629–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A & Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J‐F, Randall AD, Ellinor PT, Horne WA, Sather WA, Tanabe T, Schwarz TL & Tsien RW (1993). Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology 32, 1075–1088. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen YH, Bangaru SD, He L, Abele K, Tanabe S, Kozasa T & Yang J (2008). Origin of the voltage dependence of G‐protein regulation of P/Q‐type Ca2+ channels. J Neurosci 28, 14176–14188. [DOI] [PMC free article] [PubMed] [Google Scholar]