

ABSTRACT
Japanese macaque (JM) rhadinovirus (JMRV) is a novel, gamma-2 herpesvirus that was recently isolated from JM with inflammatory demyelinating encephalomyelitis (JME). JME is a spontaneous and chronic disease with clinical characteristics and immunohistopathology comparable to those of multiple sclerosis in humans. Little is known about the molecular biology of JMRV. Here, we sought to identify and characterize the small RNAs expressed during lytic JMRV infection using deep sequencing. Fifteen novel viral microRNAs (miRNAs) were identified in JMRV-infected fibroblasts, all of which were readily detectable by 24 h postinfection and accumulated to high levels by 72 h. Sequence comparisons to human Kaposi's sarcoma-associated herpesvirus (KSHV) miRNAs revealed several viral miRNA homologs. To functionally characterize JMRV miRNAs, we screened for their effects on nuclear factor kappa B (NF-κB) signaling in the presence of two proinflammatory cytokines, tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β). Multiple JMRV miRNAs suppressed cytokine-induced NF-κB activation. One of these miRNAs, miR-J8, has seed sequence homology to members of the cellular miR-17/20/106 and miR-373 families, which are key players in cell cycle regulation as well as inflammation. Using reporters, we show that miR-J8 can target 3′ untranslated regions (UTRs) with miR-17-5p or miR-20a cognate sites. Our studies implicate JMRV miRNAs in the suppression of innate antiviral immune responses, which is an emerging feature of many viral miRNAs.
IMPORTANCE Gammaherpesviruses are associated with multiple diseases linked to immunosuppression and inflammation, including AIDS-related cancers and autoimmune diseases. JMRV is a recently identified herpesvirus that has been linked to JME, an inflammatory demyelinating disease in Japanese macaques that mimics multiple sclerosis. There are few large-animal models for gammaherpesvirus-associated pathogenesis. Here, we provide the first experimental evidence of JMRV miRNAs in vitro and demonstrate that one of these viral miRNAs can mimic the activity of the cellular miR-17/20/106 family. Our work provides unique insight into the roles of viral miRNAs during rhadinovirus infection and provides an important step toward understanding viral miRNA function in a nonhuman primate model system.
INTRODUCTION
Japanese macaque rhadinovirus (JMRV) is a novel simian herpesvirus that was recently isolated from a central nervous system (CNS) lesion of a Japanese macaque (JM) or snow monkey (Macaca fuscata) with Japanese macaque encephalomyelitis (JME) (1, 2). JME is an inflammatory, demyelinating disease that affects ∼1 to 3% of the JM colony each year at the Oregon National Primate Research Center (ONPRC) (1). At the clinical and histopathological levels, JME is comparable to multiple sclerosis (MS), an immune-mediated disease, and is the only known spontaneously occurring, natural MS-like disease in nonhuman primates (NHP). Animals with JME present with ataxia and progressive paralysis and at the pathological level exhibit demyelinated lesions in the CNS accompanied by macrophage infiltration and axonal loss (1). More recently, JME has been shown to also possess immunological similarities to MS, including oligoclonal bands in the cerebrospinal fluid and T cell infiltrates in the brain exhibiting Th17 phenotypes (3). Notably, JMRV has been detected and isolated from multiple JME lesions in different animals, suggesting that viral gene products and/or virus-influenced host factors may contribute to JME pathogenesis (1, 2).
DNA sequence analysis of JMRV revealed that the virus is member of the gammaherpesvirus family (1, 2), which includes the gamma-1 lymphocryptoviruses (such as Epstein-Barr virus [EBV]) and the gamma-2 rhadinoviruses. The rhadinoviruses are divided further into two lineages. Viruses of the rhadinovirus 1 (RV1) lineage include the only known human rhadinovirus (Kaposi's sarcoma-associated herpesvirus [KSHV]) and retroperitoneal fibromatosis-associated herpesvirus (RFHV), which infects Old World nonhuman primates. The rhadinovirus 2 (RV2) lineage includes JMRV; rhesus macaque (RM) rhadinovirus (RRV), which infects RM (Macaca mulatta); and Macaca nemestrina rhadinovirus 2 (MneRV2), which infects pig-tailed macaques (Macaca nemestrina) (2, 4, 5). JMRV is most closely related to RRV at the genetic level; the genomes are colinear, and the majority of viral open reading frames (ORFs) are homologous to a few unique ORFs that are specific to JMRV (2).
Multiple viral microRNAs (miRNAs) have been demonstrated for members of the gammaherpesvirus family and can be detected during both latent and lytic infections (6–12). miRNAs are small, ∼22-nucleotide (nt), noncoding RNAs that play key roles in the posttranscriptional regulation of gene expression. Expressed by all multicellular organisms, miRNAs regulate many important biological processes, including development, homeostasis, hematopoiesis, and the development of immune responses. Aberrant miRNA expression patterns have been linked to disease states and clinical outcomes (12–15). Pathogen encounters, such as those during virus infection, can perturb the cellular miRNA environment (13, 16). A number of viruses, particularly the herpesviruses, encode viral miRNAs that play important roles throughout the viral life cycle and can contribute to pathogenesis through the regulation of expression of both host and viral genes (7, 12, 13, 15).
Like their cellular counterparts, herpesvirus miRNAs utilize the host miRNA biogenesis machinery. Both viral and cellular miRNAs arise primarily from long, RNA polymerase II-mediated primary transcripts that fold into stem-loop structures, which are cleaved in the nucleus by the RNase III enzyme Drosha together with its binding partner DGCR8 (17, 18). This cleavage event liberates an ∼60-nt hairpin precursor miRNA (pre-miRNA) that is exported to the cytoplasm and subsequently cleaved by a second RNase III enzyme, Dicer, resulting in an ∼22-nt miRNA:miRNA* duplex (19). One arm of the duplex is bound by an Argonaute (Ago) protein to form the RNA-induced silencing complex (RISC) and functions as the mature miRNA to guide the RISC to sites on target RNAs (20, 21). Interactions between the RISC and target 3′ untranslated regions (UTRs) occur initially via nt 2 to 5 within the 5′ seed region (nt 2 to 8) of the mature miRNA, which become exposed when the miRNA is bound by Ago and facilitate target identification through sequence complementarity (20–22). Seed pairing is stabilized by compensatory binding throughout the rest of the miRNA to the RNA target. RISC binding predominantly represses mRNA translation; target sites with high sequence complementarity to a miRNA can result in direct cleavage and degradation of the target RNA (20).
By small-RNA sequencing experiments, a total of 15 viral pre-miRNAs have been experimentally demonstrated for the gamma-2 herpesvirus RRV (10, 23). Akin to the 12 viral pre-miRNAs encoded by KSHV (6, 8, 9), all 15 RRV miRNAs are encoded within the latency-associated region, flanked by ORF69 and ORF71 (23). Despite their similar positions within each viral genome, RRV and KSHV miRNAs generally lack sequence homology, with a notable exception being RRV miR-rR1-15 and KSHV miR-K10a, which share a 5′ seed sequence (10, 23). Interestingly, this seed is also conserved in the predicted retroperitoneal fibromatosis-associated herpesvirus Macaca nemestrina (RFHVMn) miRNA miRc-RF-9-3p and exhibits homology to the seed region of cellular miR-142-3p, offset by one nucleotide (24, 25). Recent biochemical studies demonstrate that KSHV miR-K10a can partly mimic miR-142-3p-mediated repression due to this offset seed sequence homology (25). KSHV also encodes functional mimics of the cellular miRNAs miR-23a/b/c (KSHV miR-K3-5p) and miR-155 (KSHV miR-K11) that enable the virus to tap into conserved host miRNA-regulatory networks (26–29).
To date, no targets for RRV or any other rhadinovirus 2 miRNAs have been demonstrated. Studies on KSHV and EBV indicate that many viral miRNAs contribute to persistent infection and viral latency by attenuating antiviral immune responses and enhancing intrinsic cell signaling pathways that promote the survival and proliferation of infected cells (12, 13, 15, 16). KSHV miR-K10a, for example, targets TWEAKR (tumor necrosis factor [TNF]-like weak inducer of apoptosis receptor), which can protect infected cells from apoptosis and dampen proinflammatory cytokine responses (30). Two other KSHV miRNAs, miR-K9 and miR-K5, target IRAK1 (interleukin-1 [IL-1] receptor-associated kinase 1) and MYD88 (myeloid differentiation primary response protein 88), respectively, thereby inhibiting signaling through Toll-like receptors (TLRs) and the IL-1 receptor (31). Consequently, these KSHV miRNAs can inhibit TLR agonist- or IL-1-induced activation of nuclear factor kappa B (NF-κB) and indirectly suppress the downstream expression of NF-κB-dependent proinflammatory cytokines such as IL-6 and IL-8 (31). Additional KSHV miRNAs have been shown to target components of NF-κB pathways that can contribute to viral latency or lytic replication through the regulation of viral immediate early gene expression (32, 33).
In this study, we aimed to define and characterize the viral miRNAs expressed by JMRV during lytic infection in vitro. Based on the hypothesis that the JMRV miRNAs likely function similarly to other gammaherpesvirus miRNAs, we then utilized a function-first approach and screened for their impact on proinflammatory cytokine-directed NF-κB activation. Through these approaches, we identified specific JMRV miRNAs that inhibit NF-κB activation, including one miRNA that has homology to cellular miR-17 family members and can target 3′ UTRs with miR-17 cognate sites.
MATERIALS AND METHODS
Prediction of JMRV miRNAs.
Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/) was used to analyze homologous sequences between the JMRV and RRV latency-associated regions and then between JMRV and the previously described RRV precursor miRNAs (23). JMRV regions bearing homology to RRV pre-miRNAs were extracted and folded with RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) and Mfold (http://unafold.rna.albany.edu/?q=mfold) to determine potential pre-miRNA structures.
Cell culture, infections, and transfections.
Primary rhesus fibroblasts (RFs), telomerase-immortalized RFs (tRFs), and HEK293T (293T) cells were maintained at 37°C in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Primary RFs were plated 1 day prior to infections and infected with a wild-type JMRV isolate (2) for 2 h at 37°C in a minimal volume of complete medium, followed by a medium change. For the initial identification of JMRV miRNAs, RFs were infected at a multiplicity of infection (MOI) of 2 and harvested at 72 h. A MOI of 0.85 was chosen for the time course experiments based on cytopathic effects (CPE) observed after 72 h postinfection (p.i.). Telomerase-immortalized RF-NF-κB reporter cells (tRF), maintained under puromycin selection, were a gift from Vic DeFilippis and constitutively express an NF-κB-responsive firefly luciferase reporter as well as renilla luciferase (under the control of a separate promoter) for normalization. Cell lines were transfected by using Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol.
RNA isolation and generation of miRNA sequencing (miR-Seq) libraries.
Total RNA from infected RFs was harvested in TRIzol (Life Technologies) and isolated according to the manufacturer's protocol, with minor modifications, primarily substitution of the final 70% ethanol wash for ice-cold 95% ethanol. For experiments where the JMRV MOI was 2, denoted by an asterisk (72 h*), small RNAs were enriched from the total RNA by using the miRVana miRNA isolation kit (Life Technologies, Carlsbad, CA) as described previously (34) and used for deep sequencing. In experiments where the MOI was 0.85, sequencing libraries were prepared from total RNA. Deep-sequencing libraries were generated by using the Illumina Tru-Seq v2.0 small-RNA kit and the bar-coded indexes according to the manufacturer's protocol. A pilot PCR was performed for each cDNA library to ensure that amplification occurred in the linear range. Sequencing was performed on an Illumina HiSeq2000 instrument by the Oregon Health & Science University (OHSU) Massive Parallel Sequencing Services Core.
miR-Seq bioinformatics analysis.
miR-Seq reads were obtained in fastq format and preprocessed to remove the Illumina adapter sequences and any low-quality reads by using scripts from the fastx toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Processed reads (>15 nt) were aligned concurrently with the rhesus macaque genome (rheMac3) and the JMRV genome (Macaca fuscata rhadinovirus; GenBank accession number AY528864.1) by using Bowtie (parameters used were -m 25, -v 2, –best, -strata) (35). Viral miRNAs were annotated based on (i) perfect alignment to the JMRV genome, (ii) size (∼20 to 23 nt), and (iii) evidence of a hairpin precursor via RNAfold. As an additional measure, we utilized the miRDeep2 pipeline to annotate macaque cellular miRNAs (miRBase v21) and any other potential viral miRNAs and to obtain read counts for each viral and cellular miRNA (36). To determine differentially expressed (DE) miRNAs, cellular miRNAs with >10 reads/miRNA (determined by miRDeep2) were analyzed using the Bioconductor package edgeR (37). Library sizes were normalized by using the edgeR default-weighted trimmed mean of M values (TMM) method. Since multiple conditions were tested (0 h, 24 h, 48 h, and 72 h postinfection), expression level differences were fitted by using the generalized linear model (GLM), and the likelihood ratio test was used to calculate P values in edgeR. Significant, DE miRNAs with read counts of >50 are reported.
Reverse transcription-PCR (RT-PCR) to detect JMRV transcripts.
DNase-treated RNA was reverse transcribed by using random hexamers and SuperScript III (SsIII) (Life Technologies) according to the manufacturer's protocol. cDNAs were PCR amplified for 30 cycles, and products were visualized on 1% Tris-acetate-EDTA (TAE) agarose gels. The following primers were used to amplify JMRV cDNAs: vMIP fwd (5′-CAGAAGCTGTGCGCCAATCC-3′), vMIP rev (5′-CTTCGTCGGAAGCATCGTCA-3′), ORF73 fwd (5′-GGGTCTGGGTTACAAAAGATGACT-3′), and ORF73 rev (5′-GAATTGGCAGTCCTCTGTCCAT-3′).
miRNA TaqMan RT-quantitative PCR (qRT-PCR) assays.
One hundred nanograms of total RNA was reverse transcribed in a 15-μl reaction mixture consisting of the RNA sample, 1 μl of the miRNA stem-loop RT primer, 1 μl MultiScribe reverse transcriptase, 1.5 μl 10× RT buffer, 0.15 μl 100 mM deoxynucleoside triphosphates (dNTPs), 0.19 μl RNase inhibitor, and nuclease-free water. The reaction mixture was incubated for 30 min at 16°C, 30 min at 42°C, and 5 min at 85°C and maintained at 4°C. Real-time quantitative PCR was performed by using 10% of each RT reaction mixture, TaqMan 2× universal PCR master mix (Life Technologies), 0.3 μl of a 6-carboxyfluorescein (FAM)-labeled TaqMan probe specific for a given miRNA, 0.3 μl of miRNA-specific forward PCR primer, 0.3 μl of universal reverse primer, and nuclease-free water. All PCR mixtures were prepared in triplicates in a PCR Clean Work Station. Primer and probe sequences are available upon request. Real-time PCR was carried out in 96-well optical plates by using an Applied Biosystems 7700 sequence detector. RNU6B, miR-16, or U6 was used as indicated as an endogenous reference control to compare the expression levels of viral miRNAs between samples; the 2−ΔΔCT[/teq] method was used to calculate relative expression levels.
Generation of JMRV miRNA expression vectors.
Approximately 150 to 200 nt surrounding each JMRV pre-miRNA were PCR amplified from genomic DNA isolated from JMRV-infected RFs. Primer sequences are available upon request. Amplified regions were cloned into the XhoI and XbaI sites of pcDNA3.1+, and expression vectors were confirmed by sequencing. To confirm miRNA expression, vectors were transfected into cells, total RNA was harvested after 48 to 72 h, and JMRV miRNA levels were assayed by TaqMan qRT-PCR.
Luciferase reporter assays.
tRF-NF-κB cells were transfected with 250 ng viral miRNA expression vectors in 96-well black-well plates. At 44 h posttransfection, cells were stimulated with 25 ng/ml TNF-α or 25 ng/ml recombinant IL-1β (Life Technologies) for 4 h or as stated otherwise. For 3′-UTR reporter assays, 20 ng pLSG-based vectors containing either the human CDKN1A 3′ UTR or tandem miR-17-5p or miR-20a-5p binding sites in the firefly luciferase 3′ UTR (as previously described [38]) was cotransfected into 293T cells with 20 ng the pLSR renilla control vector and 250 ng the pcDNA3-based miRNA expression vector; cells were harvested at 48 h posttransfection. For all luciferase assays, cells were lysed in 1× passive lysis buffer, and lysates were analyzed for luciferase activity by using a dual-luciferase reporter assay kit (Promega).
Accession number(s).
Raw fastq files for miRNA sequencing experiments have been deposited in the NCBI Sequence Read Archive (SRA) database under BioProject accession number PRJNA329121.
RESULTS
Prediction of JMRV miRNAs.
RRV encodes a total of 15 viral pre-miRNAs that are located in a latency region homologous to the 12 pre-miRNAs encoded by the only known human rhadinovirus, KSHV (8–10, 23, 39). One previous report suggested that at least nine of the RRV pre-miRNAs are conserved in JMRV (40). To predict possible miRNAs encoded in the JMRV genome, we used Clustal Omega to align the JMRV latency region, located between JMRV ORF69 and ORF71, with the homologous RRV latency region. This alignment showed that the regions of these two viruses are >85% identical at the genomic DNA level. The 15 RRV pre-miRNA sequences were then aligned to the JMRV latency region; all RRV pre-miRNAs exhibited significant sequence homology to JMRV, and 15 candidate JMRV pre-miRNAs were identified. JMRV pre-miRNAs were numbered based on their homology to the RRV miRNAs (Fig. 1A). JMRV sequences were then extracted and folded with RNAfold to define the stem-loop structures for JMRV pre-miRNA candidates (Fig. 1B).
FIG 1.

Candidate JMRV miRNAs. (A) Positions of the predicted 15 viral precursor miRNAs within the JMRV latency-associated region. miRNAs are numbered based on their sequence homology to the RRV miRNAs. (B) Predicted hairpin structures for all JMRV precursor miRNAs (including partial flanking regions). For miR-J3, the flanking regions that include the moRs are also shown.
Experimental identification of JMRV miRNAs.
To gain experimental evidence of JMRV miRNAs, we analyzed the small-RNA population in JMRV-infected primary RM fibroblasts (RFs) by deep sequencing. Small RNAs (<200 nt) were isolated 72 h after infection of primary RFs (MOI = 2) by using the miRVana small-RNA isolation kit and used to generate Illumina TruSeq cDNA libraries. Based on CPE and virus plaques determined by light microscopy, >75% of cells were infected at the time of RNA isolation. Deep sequencing was performed on the Illumina HiSeq2000 instrument. We obtained 15,468,541 reads (≥15 nt) that were aligned concurrently to the JMRV and RM genomes, allowing for up to two mismatches. The majority of reads (10,198,585; 65.9%) mapped to the 619 known RM miRNA hairpins present in miRBase v21 (Fig. 2A). An additional 25.5% of reads mapped elsewhere to the rheMac3 genome; many of these reads likely represent cellular miRNAs not yet annotated for RM. A total of 7.05% of reads aligned to the JMRV genome. Of those reads aligning to the JMRV genome, the majority mapped to the latency region (Fig. 2B); 96.8% of JMRV reads mapped to the 15 predicted JMRV pre-miRNAs (representing 6.83% of the total aligned reads). Based on read counts, miR-J3-5p was the most abundant viral miRNA present in RFs at 72 h p.i. (Table 1).
FIG 2.

Deep-sequencing analysis of small RNAs in JMRV-infected rhesus fibroblasts. (A) Distribution of small-RNA sequencing reads from JMRV-infected primary RFs (72 h postinfection; MOI = 2) falling within the following categories: JMRV miRNAs, RM miRNAs, the rhesus macaque genome (rheMac3; non-miRNAs), or the JMRV genome (non-miRNA region). One percent of reads did not match the listed categories and are classified as “unaligned.” (B) Small-RNA sequencing reads aligning to the JMRV genome (with up to two mismatches). Reads aligning to the plus strand are shown as positive values, while reads aligning to the minus strand are shown as negative values on the y axis. The strong peak at nt ∼111000 to 117000 corresponds to the JMRV latency-associated region where the viral miRNAs are encoded. (C) TaqMan qRT-PCR analysis of cellular and viral miRNA expression in JMRV-infected RFs (72 h*) or uninfected RFs (mock). Delta-cycle threshold (CT) values for each miRNA are reported relative to values for RNU6B detected in mock-infected or infected RFs. (D) Total read counts for sequences mapping to cellular RM miR-16-5p or JMRV miRNA miR-J7-3p, miR-J2-3p, or miR-J5-5p. (E) Comparison of rhadinovirus miRNA and cellular miR-142-3p sequences. Seed homology among miRNAs of JMRV, RRV, and KSHV and the predicted MneRV2 miRNA miRc-N15-3p is highlighted in gray. (F) JMRV miR-J6-3p undergoes A→I editing (depicted as “G” in boldface type) within the seed region, highlighted in gray. Also shown are the sequences of unedited JMRV miR-J6-3p and the RRV homolog. (G) Alignments of JMRV and KSHV pre-miRNA positional homologs (miR-J11/miR-K9 and miR-J9/miR-K3) and two other miRNAs with seed homology (miR-J4/miR-K12). The mature miRNA sequences for each virus are depicted above or below the pre-miRNA sequence. Regions of viral miRNAs with seed homology are boxed in gray.
TABLE 1.
Major isoforms of JMRV miRNAs and moRs identified by deep sequencing
| miRNA | Major sequence | Total no. of reads | Genome start position (nt) |
|---|---|---|---|
| miR-J7-3p | CGCACGTTAATTGCTTTCTAGT | 312 | 111683 |
| miR-J7-5p | TGGAGAGCAGTTAACGTGCGTTC | 737 | 111721 |
| miR-J6a-3p | CGATGTGCGACCTTTTGCGAT | 12,751 (74.3% edited) | 111807 |
| miR-J6b-3p | CGATGTACGACCTTTTGCGAT | 4,412 (25.7% unedited) | 111807 |
| miR-J6-5p | CGCGGAAAGGTGTGCACATCGTA | 5,510 | 111840 |
| miR-J15-3p | TAGTGTTGCTCGTGATCGTAGA | 9,822 | 111947 |
| miR-J15-5p | TGCGGTCACCAACAACGCTATT | 45 | 111983 |
| miR-J5-3p | GGCGTGTTCTTTGGATTCCGTT | 921 | 112117 |
| miR-J5-5p | CCCGGAACCCAAAGACACGTGCC | 197,910 | 112155 |
| miR-J14-3p | GGTGTTTTTTTGGGTCCGCTTGT | 2,090 | 112297 |
| miR-J14-5p | AAGGGACCCAAAGACACATTCTC | 25,513 | 112337 |
| miR-J13-3p | TAGTAACCTGACAGCAGTTAGT | 35,956 | 112485 |
| miR-J13-5p | TAATTGCAGTTGGTGTGCTACT | 3,764 | 112522 |
| miR-J4-3p | CTCGTTAACCGTCCTCCCGAGA | 300,989 | 112885 |
| miR-J4-5p | TGGGGAGGGTGGTCAGCGCGCG | 545 | 112935 |
| moR-J3-3p | ATTCGCTGCTTACAGTCGCC | 556 | 113332 |
| miR-J3-3p | CAGGGTCTGCGACGGACACTGT | 20,013 | 113352 |
| miR-J3-5p | GTGTTTAGTCGTGCTCCCTGTT | 307,880 | 113387 |
| moR-J3-5p | CTCAATGAGCAGTTAGTCT | 3,663 | 113409 |
| miR-J12-3p | ATTTCGTTTTTAGATTCCCTAAT | 8,128 | 113456 |
| miR-J12-5p | TAGGGAACTAAAGACAATTTCC | 1,076 | 113494 |
| miR-J2-3p | TATACGGCGCTGCACGGTTGGC | 44,503 | 114561 |
| miR-J2-5p | TACCATGCCGCGTCCCGTATTC | 329 | 114602 |
| miR-J11-3p | GGCGGGTCTGAGGTATGGGGAA | 117 | 114746 |
| miR-J11-5p | GACCCAGCCTACAGTCCCGCTG | 3,887 | 114779 |
| miR-J10-3p | ATTACATCTTGTTAGGGGGAT | 13,494 | 115053 |
| miR-J9-3p | GCGGTCATCAGAACATATCACC | 3,075 | 116040 |
| miR-J9-5p | GAATATGTTCCGCTGACACCGCTG | 17,277 | 116075 |
| miR-J1-3p | CCACCGAGGATGCGGTCAATG | 47 | 116234 |
| miR-J1-5p | CGATCGCACCTTTGGCCGGCCGG | 14,952 | 116266 |
| miR-J8-3p | AAAGTGCTCACAAGACATCCC | 7,596 | 116363 |
| miR-J8-5p | GATGTCTTGCCAGCACTTTCCT | 5,540 | 116403 |
Table 1 lists the major sequences, read counts, and positions in the viral genome for all JMRV miRNAs. We identified 23 mature viral miRNAs and 7 passenger miRNAs arising from 15 precursors. Similarly to what was reported previously for other herpesviruses (23, 41), we also detected abundant reads for several viral miRNA-offset RNAs (moRs) (Table 1). Primarily, the JMRV moRs mapped to regions immediately adjacent to the miR-J3 mature and passenger miRNAs, which is similar to the functional moRs that were reported previously for RRV that arise from the miR-J3 homolog miR-rR-3 and exhibit almost identical sequences (23).
To independently confirm JMRV miRNA expression, we used TaqMan-based miRNA stem-loop qRT-PCR assays to detect two abundant miRNAs (miR-J2-3p and miR-J5-5p) and one lowly expressed miRNA (miR-J7-3p), which differs from RRV miR-r7-3p by 3 nucleotides. Cellular miR-16-5p was assayed as a control. Expression levels determined by qRT-PCR for these three miRNAs recapitulated their expression patterns observed by deep sequencing (Fig. 2C and D).
We next compared the JMRV miRNA sequences to the KSHV miRNA sequences reported previously for other deep-sequencing experiments (11, 42). Similarly to what is observed for RRV and predicted for other rhadinoviruses, including RFHVMn and MneRV2 (4, 23–25), a KSHV miR-K10a/miR-142-3p seed homolog, miR-J15-3p, was identified for JMRV (Fig. 2E). KSHV miR-K10a is expressed as two isoforms that differ by 1 nucleotide at the 5′ end to generate miR-K10a and miR-K10a+1 (25, 42). Additionally, miR-K10 undergoes adenosine-to-inosine (A→I) editing by adenosine deaminases acting on RNAs (ADARs) to generate miR-K10b (6, 8, 9, 25, 42). We found no evidence of miRNA isoforms or editing events for miR-J15-3p. However, another JMRV miRNA, miR-J6-3p, exhibited evidence of significant A→I editing in the seed region at nucleotide position 7 (74.3% of the miR-J6-3p reads were edited), which presumably alters and expands the repertoire of targets for this miRNA (Fig. 2F and Table 1); this is unlike the RRV miR-6 homolog, which lacks any evidence of A→I editing (23). No other JMRV miRNAs showed signs of A→I editing.
Intriguingly, besides miR-J15, several other JMRV miRNAs exhibited partial sequence homology to the KSHV miRNAs. JMRV and KSHV pre-miRNA sequences were aligned by using Clustal Omega (Fig. 2G). The positional homologs JMRV miR-J9-3p and KSHV miR-K3-3p are >70% identical (including a 7-nt stretch that encompasses the seeds of these two miRNAs), while positional homologs miR-J11-5p and miR-K9-5p share offset 5′ and 3′ seed homology (highlighted in Fig. 2G). We also note that miR-J4-5p and miR-K12-3p share a stretch of 10 identical nucleotides in the 5′ regions of each mature miRNA (Fig. 2G). These observations indicate that the RV1 and RV2 miRNAs are more conserved than previously reported (10, 23, 40). Likely, rhadinovirus miRNA sequence conservation was overlooked in previous studies since the seed sequences of the miRNAs are not identical but rather are offset by 1 or 2 nucleotides. While it has been observed for some time that viral and cellular miRNAs with identical seed sequences can act as functional mimics (26, 29), recent examples of viral miRNA seed sequence mimicry highlight that offset seed homology is also effective in binding 3′ UTRs and repressing targets with cognate seed match sites (25, 28). Thus, we predict that homology pairs such as JMRV miR-J11-5p and KSHV miR-K9-5p will have a common set of miRNA targets conserved in macaques and humans.
Kinetics of JMRV miRNA expression in vitro.
For nearly all gammaherpesvirus miRNAs to date, viral miRNA expression has been examined in a latent model of infection or following virus reactivation from latency (8, 9, 23, 39, 43). JMRV undergoes lytic replication following de novo infection of cultured fibroblasts (2). To gain insight into the temporal kinetics of JMRV gene expression during lytic infection, we infected primary RM fibroblasts with JMRV (MOI of 0.85, compared to an MOI of 2 in Fig. 2) and harvested total RNA from uninfected cells (mock) or at select time points p.i. (24, 48, and 72 h). Significant cytopathic effects were evident after 72 h p.i.; therefore, we did not continue the experiments to 96 h.
We first examined the expression of two JMRV gene products, the lytic JM24 transcript encoding viral macrophage inflammatory protein (vMIP) (RRV R3 homolog) and the JM153 transcript (ORF73) encoding latency-associated nuclear antigen (LANA) (2), using RT-PCR (Fig. 3A). Genomic DNA from JMRV-infected fibroblasts was used as a positive control. Transcripts from both vMIP and LANA were detectable by as early as 24 h p.i., and we observed a modest increase in vMIP expression levels from 24 to 72 h, consistent with active viral replication. We next performed miRNA-specific stem-loop qRT-PCR to measure the levels of eight individual JMRV miRNAs and cellular miR-16-5p as a control (Fig. 3C). The detection of miR-J1-5p and miR-J12-3p plateaued at between 48 and 72 h; however, for the majority of the viral miRNAs, we observed steady accumulation throughout the course of infection.
FIG 3.

miRNA expression kinetics during JMRV lytic infection. (A) RT-PCR analysis of vMIP, LANA, and cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression in uninfected primary RFs (mock) or RFs infected with JMRV for the times indicated (MOI of 0.85 for the 24-, 48-, and 72-h samples and MOI of 2 for the 72-h* sample). PCR products were analyzed on a 1% TAE agarose gel and stained with ethidium bromide for visualization. +RT and no RT indicate analyses with or without reverse transcriptase. W, nuclease-free water (negative control); JMRV, genomic DNA from JMRV-infected cells (positive control). (B) Small-RNA sequencing analysis of JMRV-infected RFs at 24, 48, and 72 h postinfection. Shown are reads aligning across the JMRV genome. As described in the legend of Fig. 2, reads aligning to the plus strand are shown as positive values, while reads aligning to the minus strand are shown as negative values on the y axis. (C) TaqMan qRT-PCR analysis of cellular miR-16-5p and select JMRV miRNAs in uninfected (mock) or JMRV-infected RFs (24, 48, or 72 h postinfection). Expression values are normalized to values for RNU6B and are reported relative to miRNA levels at 24 h postinfection (set to “1”). (D) Normalized read counts for each JMRV miRNA (5p and 3p strands) at 24 h, 48 h, or 72 h postinfection as determined by deep sequencing. miRNAs are ordered from left to right based on their position in the JMRV genome. No reads were detected for miR-J10-5p. miR-J6-3p reads are shown separately in panel F. (E) Distribution of mapped reads for each of the four small-RNA sequencing libraries (mock- and JMRV-infected samples). (F) Normalized read counts for the edited form or nonedited form of miR-J6-3p as well as the 5p arm detected in each JMRV-infected sample. (G) Cellular miRNAs significantly altered (P value of <0.05 and FDR of <0.05) during JMRV lytic infection. Normalized miRNA read counts for six downregulated and four upregulated cellular miRNAs are shown.
To comprehensively explore small-RNA expression patterns following lytic infection, we then used deep sequencing to analyze the small RNAs. Reads aligning to the JMRV genome are shown in Fig. 3B. Consistent with the results shown in Fig. 2, the majority of reads aligning to JMRV (99.2% of JMRV reads at 72 h) mapped to the cluster of viral miRNAs encoded within the latency-associated region (nt ∼111650 to 116450 on the antisense strand). All JMRV miRNAs were detectable by 24 h p.i., representing 2.7% of the total miRNA population, and continued to accumulate throughout the course of infection (Fig. 3D and E). By 72 h p.i., the JMRV miRNAs accounted for 17.9% of all annotated miRNAs (13.4% of all reads). This pattern is akin to those of miRNAs expressed by other herpesviruses, such as cytomegaloviruses, that can undergo lytic replication in fibroblasts (44). Figure 3D shows normalized miRNA read counts for individual JMRV miRNAs, which are ordered by their position within the JMRV genome. We did not detect reads mapping to miR-J10-5p, and low read counts were detected for miR-J11-3p and miR-J15-5p; thus, we conclude that these represent passenger strands, at least in the context of lytic replication (Fig. 3D and Table 1). We looked for evidence of miR-J6-3p editing and found that while there was an overall increase in miR-J6-5p and -3p expression levels, the ratio of unedited to edited miR-J6-3p did not dramatically change over time (Fig. 3F). This pattern is consistent with A→I editing of the primary miRNA transcript and indicates that, unlike many cellular miRNAs, editing does not appear to interfere with miR-J6-3p biogenesis and is more likely to functionally alter target specificity (45).
While the majority of viral reads mapped to the viral miRNA locus, we observed other small peaks with read counts above the background noise that aligned to JMRV and appeared to increase over time. A small cluster of reads mapped to nt 22607 to 23566 of the JMRV genome (Fig. 3B). Presumably, these reads arise from a JMRV homolog of the KSHV polyadenylated nuclear (PAN) RNA, which is highly abundant during KSHV lytic infection (46). At 72 h, the JMRV PAN reads accounted for <0.11% of the total JMRV mapped reads and lacked uniformity in the 5′ ends, which implies that they are degradation products and do not have function as small RNAs.
Cellular miRNAs during JMRV infection.
To determine whether JMRV influences the expression of any specific cellular miRNAs during infection, deep-sequencing libraries were mapped to known RM cellular miRNAs (miRBase v21). Using miRDeep2 to obtain the read counts for each mature miRNA, we identified a total of 443 mature cellular miRNAs in the four samples (a miRNA was counted only if there was an average of 10 or more reads mapping to the miRNA). We then employed the edgeR Bioconductor package and performed GLM (generalized linear model) fitting to identify miRNAs differentially expressed throughout the time course of infection. edgeR utilizes weighted TMM (trimmed mean of M values) normalization to compensate for differences in library size. Of the 443 cellular miRNAs, we identified 75 miRNAs for which levels changed >2-fold during JMRV infection compared to mock infection. Of these changes, 14 were statistically significant (P < 0.05; false discovery rate [FDR] of <0.05); however, only 10 of the 14 miRNAs had >50 read counts in at least one library (representing ∼0.001% of the miRNA population), which is still 10-fold below the recommended cutoff (>0.01%) for functionally relevant miRNAs (47). Six miRNAs were downregulated in response to JMRV infection (miR-345, miR-122a-5p, miR-183-5p, miR-222-5p, miR-671-3p, and miR-370-5p), and four were modestly upregulated (miR-876-5p, miR-193a-3p, miR-324-5p, and miR-29b-3p) (Fig. 3F). Excluding miR-345, log fold change (FC) values ranged from −1.6 to 1.3 in comparisons of mock and JMRV infections. Thus, with the exception of miR-345 (log FC = −5.1), which had substantially reduced expression, JMRV did not dramatically alter the levels of cellular miRNAs during lytic replication.
NF-κB functional screen identifies anti-inflammatory JMRV miRNAs.
Cellular miRNAs are common components of many intricate signal transduction pathways, including NF-κB signaling, and can provide robustness to cellular gene expression as well as give feedback signals within cell signaling networks (48, 49). miR-155, for example, is an NF-κB target and also participates in NF-κB networks through the targeting of IκB kinases (IKKs) and other NF-κB signaling components (48, 50). Multiple studies demonstrate that NF-κB signaling is manipulated by gammaherpesvirus miRNAs (31–34, 51, 52). In addition to mimicking miR-155 activity via miR-K11, KSHV encodes four miRNAs (miR-K1, -K3, -K5, and -K9) that can alter NF-κB-dependent transcription through the targeting of IκBα and nuclear factor I/B (NFIB) as well as MYD88 and IRAK1, which mediate signaling from TLRs (31–33, 42). KSHV miR-K10a also targets TWEAKR, which can consequently suppress the expression of proinflammatory cytokines that recruit effector cells to sites of infection (30). Thus, viral miRNA manipulation of NF-κB signaling components and proinflammatory cytokine responses is thought to facilitate, in part, innate immune evasion (16). Here, we tested the hypothesis that JMRV manipulates proinflammatory cytokine-mediated responses through viral miRNAs.
To first demonstrate that NF-κB-mediated transcription is activated during JMRV infection, we assayed transcript levels of two NF-κB-responsive cytokine genes, IL-1A and IL-8. The levels of both transcripts were significantly upregulated in JMRV-infected cells compared to control cells (Fig. 4A). Next, we performed a small-scale screen to identify JMRV miRNAs that might influence proinflammatory cytokine signaling and NF-κB activity. Expression vectors for nine individual JMRV pre-miRNAs were generated, and viral miRNA expression from each vector was confirmed by qRT-PCR following transient transfection of 293T cells (Fig. 4B). To identify viral miRNA regulators of NF-κB, tRF-NF-κB-Luc cells expressing an NF-κB-responsive firefly luciferase reporter gene and a constitutive renilla luciferase gene for normalization were transfected with the individual JMRV miRNA vectors. Dual-luciferase activity was measured after 48 h (Fig. 4C). To induce canonical NF-κB signal transduction, transfected cells were treated with inflammatory cytokines, either 25 ng/ml TNF-α or 25 ng/ml IL-1β, for 4 h. With the exception of miR-J4, ectopic expression of each JMRV miRNA did not alter basal luciferase levels (Fig. 4D, mock). An ∼5-fold increase in luciferase activity was observed in vector control cells stimulated with TNF-α or IL-1β (Fig. 4D), and similar ∼5-fold increases were observed in the presence of JMRV miR-J3, miR-J7, miR-J10, or miR-J14, demonstrating that these miRNAs do not alter the NF-κB pathway. Notably, the ectopic expression of either miR-J2 or miR-J4 inhibited TNF-α-mediated NF-κB activation but not IL-1β-mediated activation, while the expression of miR-J13 or the KSHV miR-K10a seed homolog miR-J15 moderately suppressed IL-1β-mediated NF-κB activation (Fig. 4D to F). Thus, miR-J2, miR-J4, miR-J13, and miR-J15 suppress NF-κB activity, potentially through specific targets within each of the TNF-α and/or IL-1 signal transduction pathways. Intriguingly, the presence of miR-J8 suppressed NF-κB activity ∼2- to 3-fold following treatment with either cytokine (Fig. 4D to F), suggesting that miR-J8 targets a factor or set of factors common to both the TNF-α and IL-1 signaling pathways.
FIG 4.

JMRV miRNA screen for regulators of NF-κB activity. (A) IL-1A and IL-8 transcripts are upregulated in JMRV-infected cells. Shown are data from qRT-PCR analysis of NF-κB-dependent cytokine transcripts in mock- or JMRV-infected RFs (72 h*; MOI = 2). Expression values are normalized to cellular GAPDH levels and reported relative to values for mock infection. (B) TaqMan qRT-PCR analysis of JMRV miRNAs expressed from pcDNA3.1+. Individual JMRV miRNA expression vectors (miRNA) or the pcDNA3.1+ empty vector (V) were transfected into 293T cells, and RNA was harvested for analysis at 48 to 72 h posttransfection. RNA from JMRV-infected RFs (72 h*; MOI = 2) was assayed in parallel as a positive control. U6 and RNU6B small RNAs were tested as controls. CT, cycle threshold value. (C) Schematic of the experimental design for screening of JMRV miRNA activities in tRF-NF-κB reporter cells. NF-κB RE indicates NF-κB-responsive elements in the promoter region of firefly luciferase (F-Luc). Cells also stably express renilla luciferase (R-Luc) driven by a separate promoter. (D) tRF-NF-κB luciferase reporter cells were transfected with 250 ng the pcDNA3.1+ empty vector (V) or individual JMRV miRNA expression vectors (denoted with “J”), grown for 44 h, and then left untreated (mock) or treated for 4 h with 25 ng/ml TNF-α or 25 ng/ml IL-1β to induce luciferase expression. Lysates were harvested and analyzed by using a dual-luciferase reporter assay kit, and values are reported relative to values for renilla luciferase (internal control). Shown are the averages of data from five independent experiments performed in duplicates or triplicates. Values are reported relative to values for mock-treated empty-vector control cells (V). RLU, relative light units. (E and F) NF-κB activation data reported in panel D were normalized to basal luciferase levels (mock) for each independent experiment prior to averaging. *, P < 0.01; **, P < 0.02 (as determined by Student's t test).
To investigate this further, we varied the amounts of the JMRV miRNAs tested as well as the concentrations of TNF-α. tRF-NF-κB-Luc cells were transfected with increasing amounts of miR-J2, miR-J4, or miR-J8 and then stimulated with 10, 25, or 50 ng/ml TNF-α for 4 h. As shown in Fig. 4, miR-J2 modestly inhibited reporter activity in the presence of TNF-α (Fig. 5A); however, the inhibitory activities of miR-J2 could be overcome with higher doses of TNF-α, suggesting that the TNF-related target(s) for this miRNA is not a critical, core component of the signaling pathway and might be an ancillary factor. Increasing amounts of miR-J4 induced basal luciferase expression in unstimulated cells (Fig. 5B). At the lowest dose of TNF-α (10 ng/ml), miR-J4 enhanced NF-κB activity (Fig. 5B); we also observed enhanced activity with 25 ng/ml TNF-α and medium levels (125 ng) of miR-J4 but then low NF-κB activity with high levels (250 ng) of miR-J4 compared to the empty vector. Notably, luciferase activity from the NF-κB reporter peaks at between 10 and 25 ng/ml TNF-α and then declines at higher cytokine concentrations (i.e., 50 ng/ml TNF-α) (Fig. 5A to C). Thus, based on the dose-response pattern of NF-κB activation, we conclude that miR-J4 is actually an inducer of NF-κB activity in tRFs and that over a certain threshold, the addition of miR-J4 in the presence of TNF-α becomes inhibitory to the signaling pathway. This response pattern suggests that miR-J4 might tune NF-κB activation through host factors that provide both positive and negative feedback to cytokine-mediated signal transduction. In contrast to miR-J2 and miR-J4, the expression of miR-J8 blocked NF-κB reporter activity at all TNF-α concentrations (Fig. 5C).
FIG 5.

JMRV miRNAs inhibit cytokine-mediated NF-κB activation. (A to C) JMRV miR-J2, miR-J4, and miR-J8 alter TNF-α-induced NF-κB activity in tRFs. (D and E) JMRV miR-J13 and miR-J8 inhibit IL-1β-induced NF-κB activity in tRFs. For panels A to E, tRF-NF-κB reporter cells were transfected with increasing amounts of JMRV miRNA expression vectors using the pcDNA3.1+ empty vector as a filler. At 44 h posttransfection, cells were stimulated with either TNF-α at the doses indicated (10 to 50 ng/ml) or 25 ng/ml IL-1β. Lysates were analyzed for dual-luciferase activity as described in the legend of Fig. 4. Reported are the averages of data for two independent experiments with standard deviations.
We next varied the amounts of JMRV miRNAs tested during IL-1β treatment. Consistent with the results described above (Fig. 4), the expression of miR-J2 or miR-J4 had no effect on the induction of NF-κB reporter activity by IL-1β. The presence of miR-J13 moderately reduced the level of reporter activation (Fig. 5D), while the expression of miR-J8 potently blocked IL-1β-mediated NF-κB activation (Fig. 5E). Thus, we conclude that miR-J8 acts as an anti-inflammatory miRNA that counteracts multiple proinflammatory cytokine responses and inhibits canonical NF-κB activation.
RV2 herpesviruses encode potential mimics of host miRNAs.
miR-J8 was the only JMRV miRNA to strongly inhibit both TNF-α- and IL-1β-induced NF-κB activity, which prompted us to further examine the miR-J8 sequence. Intriguingly, the seed sequence (nt 2 to 8) of miR-J8-3p is identical to that of the cellular miRNAs miR-302c, miR-373, and miR-519a (Fig. 6A). Of further interest, nt 1 to 8 of miR-J8 are identical to nt 2 to 9 of miR-17/20/106/93. miR-302a and miR-373 are members of the miR-302/367 cluster (miR-302a/b/c) and the miR-371/373 cluster, while the miR-17 family includes miRNAs from the miR-17/92 cluster (miR-17 and miR-20a), the miR-106a-363 cluster (miR-106a and miR-20b), and the miR-106b-25 cluster (miR-106b and miR-93). Figure 6A compares the sequence of miR-J8-3p and the sequences of several M. fuscata miR-17 and miR-373 family members, as identified by deep-sequencing analysis of JM brain tissue (R. Skalsky, unpublished data). Notably, the RV2 homolog RRV miR-rR1-8-3p and the predicted RV2 homolog MneRV2 miRc-N1-3p also share this common miR-17 family seed sequence (Fig. 6B).
FIG 6.
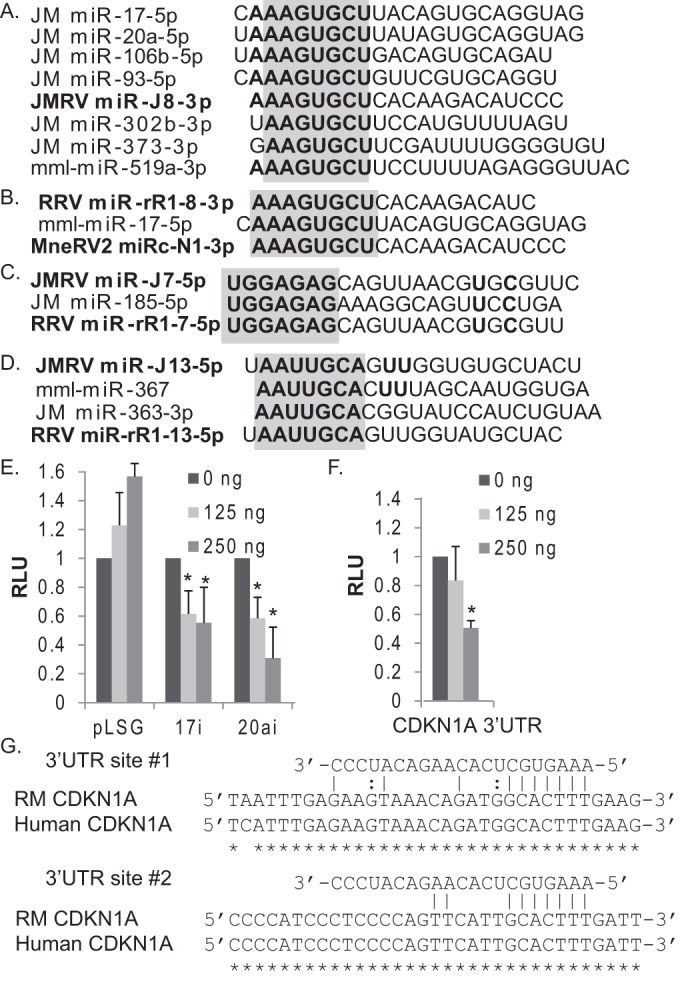
JMRV encodes a viral miRNA mimic of the miR-17 family. (A) Sequence alignments of JM cellular miR-17 family members (miR-17, miR-20a, miR-106b, miR-93, miR-302b, and miR-373) and JMRV miR-J8-3p. (B to D) Sequence alignments for other rhadinovirus and macaque cellular miRNAs indicate potential viral miRNA seed mimics. For panels A to D, miRNA seed sequences are in boldface type, and seed homology is highlighted in gray. (E) JMRV miR-J8 inhibits luciferase expression from miR-17 and miR-20a luciferase reporters (17i and 20ai, respectively). 293T cells were cotransfected with 20 ng pLSG-based firefly luciferase miRNA reporters, 20 ng pLSR (expressing renilla as an internal control), and increasing amounts of pcDNA3-miR-J8 using pcDNA3.1+ as a filler. Lysates were harvested at 48 to 72 h posttransfection and assayed for dual-luciferase activity. (F) JMRV miR-J8 targets the CDKN1A 3′ UTR, a known target of miR-17. 293T cells were cotransfected with 20 ng pLSG-CDKN1A, 20 ng pLSR (expressing renilla as an internal control), and increasing amounts of pcDNA3-miR-J8. Lysates were assayed as described above for panel E. For panels E and F, shown are the averages of data from three independent experiments performed in triplicates. *, P < 0.01, as determined by Student's t test. RLU, relative light units. (G) Alignments of RM and human CDKN1A 3′ UTRs illustrating the two predicted JMRV miR-J8-3p target binding sites that overlap cellular miR-17-5p target sites.
Given that several gamma-1 herpesvirus miRNAs exhibit perfect and offset seed homology to host miRNAs (26, 29, 41, 53), we explored the remaining JMRV miRNA sequences identified here as well as previously reported RRV miRNA sequences (23). Comparison of the RV2 miRNA sequences to all known cellular macaque miRNA sequences revealed that JMRV miR-J7-5p and RRV miR-rR1-7-5p have perfect 5′ seed sequence homology to miR-185-5p (Fig. 6C). Notably, JMRV miR-J13-5p (which inhibited IL-1β signaling) and the RRV homolog miR-rR1-13-5p exhibited offset seed homology to miR-367 and miR-363 (Fig. 6D). These cellular miRNAs arise from the same clusters that include the miR-17 family members highlighted above.
JMRV encodes a miRNA mimic of the miR-17/20/106 and miR-520/373 families.
To determine whether miR-J8 could indeed mimic the activity of miR-17 family members, we tested the effect of miR-J8 on two different luciferase reporters that contain two tandem, fully complementary binding sites for either miR-17-5p or miR-20a within the firefly luciferase 3′ UTR. Previous studies demonstrated that luciferase expression from either reporter is significantly reduced in the presence of miR-17-5p or miR-20a (38). As expected, both the miR-17-5p and miR-20a reporters were responsive to ectopic miR-J8 expression and could be inhibited in a dose-dependent manner (Fig. 6E).
One well-characterized target of miR-17-5p is the CDKN1A (cyclin-dependent kinase inhibitor 1A) transcript that encodes p21, which regulates cell cycle progression by preventing the G1/S transition. Interestingly, the CDKN1A 3′ UTR is also targeted by KSHV miR-K1 (a miR-J8 positional homolog); knockdown of p21 levels by KSHV miR-K1 enhances cell cycle progression (54). We interrogated the RM CDKN1A 3′ UTR for potential miR-J8/miR-17 binding sites and identified two seed match sites that were 100% conserved in the human CDKN1A 3′ UTR (Fig. 6G). This prompted us to then test a luciferase reporter containing the human CDKN1A 3′ UTR (pLSG-CDKN1A) (described in references 54 and 38) against JMRV miR-J8. Similarly to the miR-17-5p and miR-20a luciferase reporters, the expression of miR-J8 inhibited luciferase expression from the CDKN1A reporter in a dose-dependent manner (Fig. 6F). Together, these data demonstrate that miR-J8 can mimic the activity of miR-17 family members by binding cognate sites.
DISCUSSION
In this study, we provide the first experimental evidence of the viral miRNAs expressed by JMRV during lytic infection in vitro and demonstrate that several of these viral miRNAs can disrupt the cell signaling events induced by proinflammatory cytokines. Using evolutionary conservation in concert with deep-sequencing experiments, we identified over 25 mature JMRV miRNAs, arising from 15 precursors, which share significant homology to previously reported RRV miRNAs (10, 23). Intriguingly, at least four of the JMRV miRNAs (miR-J15-3p, miR-J11-5p, miR-J9-3p, and miR-J4-5p) and their RRV counterparts (RRV miR-rR1-15-3p, miR-rR1-11-5p, miR-rR1-9-3p, and miR-rR1-4-5p) exhibit some level of homology to RV1 KSHV miRNAs (miR-K10a-3p, miR-K9-5p, miR-K3-3p, and miR-K12-3p, respectively). This homology was more evident when the pre-miRNA terminal loop sequence and miRNA-flanking sequences were included in addition to the miRNA sequence itself. Three out of these four RV1 and RV2 miRNA pairs are also positional homologs, indicating their importance to the rhadinovirus life cycle.
While it may come as no surprise that the JMRV and RRV miRNAs have a high level of homology—in fact, several are fully conserved—our analysis identified a number of distinct, species-specific sequence differences. The most noteworthy differences are those affecting the major miRNA isoform seed sequences, which potentially influence target site recognition (20). These include JMRV miR-J3-3p and RRV miR-rR1-3-3p (differences in nt 7), miR-J7-3p and miR-rR1-7-3p (differences in nt 8), miR-J10-3p and miR-rR1-10-3p (5′ ends shifted by 2 nt), and miR-J9-5p and miR-rR1-9-5p (5′ ends shifted by 1 nt). Although the importance of these divergences in the viral miRNA seed regions cannot be fully realized until additional genomic or transcriptomic information is generated and available for JM, thereby allowing comparative genomics, we speculate that these are compensatory changes in RV2 miRNAs that arose as a result of critical target binding sites within host or viral RNAs. Given that gammaherpesviruses persist latently in cells with only limited viral gene expression and thus that the viral miRNAs, which are expressed during latency as well as in the lytic phase, primarily encounter cellular RNAs in this scenario, arguably, it is the divergent species-specific target interaction sites within the host cellular RNAs that have driven the RV2 miRNAs to acquire these changes. Future in-depth knowledge of the JM genome as well as knowledge of the miRNA targetomes in JMRV- or RRV-infected cells should provide important insight into the evolutionary relationships between key host/viral targets and viral miRNAs for rhadinoviruses.
Intriguingly, we observed evidence of significant A→I editing for one JMRV miRNA, miR-J6-3p, which results in an adenosine-to-inosine change at position 7 of the miRNA seed sequence. Posttranscriptional hydrolytic deamination events such as these are easily detected by deep-sequencing experiments since a portion of the miRNA reads contain a nontemplated “G” instead of an “A.” Up to 75% of the miR-J6-3p reads showed evidence of editing, and likely, this represents a species-specific event since no editing of the corresponding RRV homolog has been reported in vitro or in vivo (10, 23). We can rule out the possibility that JMRV infection induces global deamination events since we observed only very low levels of A-to-G transitions for a few cellular miRNAs (miR-22-3p [0.9% of reads], miR-222-3p [0.8% of reads], and miR-100-5p [0.5% of reads]); none of the A-to-G transitions were at such a high level as that seen for miR-J6-3p or considered significant to be truly edited. Inosine thermodynamically favors pairing with cytosine; thus, an A-to-I change in the seed region can redirect a miRNA to an entirely new set of target mRNAs. It is unknown at this point whether miR-J6-3p editing also occurs in vivo or during latent infection and how this contributes to the diversity of the JMRV miRNA targetome.
RRV miRNAs were first identified over 9 years ago (10) and are detectable in latently infected B cell lymphoma and retroperitoneal fibromatosis tissues in RM (23); however, no targets or functions for RRV miRNAs or any other RV2 miRNAs have been described to date. To start to elucidate the functions of RV2 miRNAs, we determined whether any JMRV miRNAs specifically altered responses to proinflammatory cytokines. Our screen identified multiple anti-inflammatory viral miRNAs, including miR-J8 and miR-J13, which dampened NF-κB activation in response to either IL-1β or TNF-α. With the exception of miR-J4, which upregulated NF-κB activity at low cytokine doses, none of the other viral miRNAs tested synergistically enhanced NF-κB activity during cytokine treatment. In vivo, cytokine induction of NF-κB-mediated transcription sets up a potent inflammatory response, which can recruit immune effector cells to sites of infection as well as facilitate viral clearance. Gammaherpesviruses must strategically evade such antiviral responses to establish and maintain long-term, persistent infection. Our work supports the hypothesis that JMRV miRNAs play an active role in innate immune evasion and may therefore provide protection to an infected cell by silencing cytokine signaling.
A closer look at the JMRV miRNA sequences provides us some insight into how these miRNAs might be manipulating cytokine pathways. Sequence comparisons to host miRNAs revealed that miR-J8 has significant seed sequence homology to members of the miR-17/20/106 and miR-373 families. Recent studies demonstrated that miR-17 contributes both positively and negatively to the life cycles of other viruses. For example, miR-17 regulates a number of interferon-responsive genes in gammaherpesvirus-infected B cells and can target the interferon regulatory factor 9 (IRF9) 3′ UTR directly, and when overexpressed in epithelial cells, it can promote herpes simplex virus 1 replication (38, 42, 55, 56). EBV miRNAs cotarget multiple miR-17 targets, which may positively contribute to B cell proliferation and/or differentiation during the establishment of latent infection (55). In contrast, human cytomegalovirus (hCMV) selectively inhibits miR-17 activity by inducing the degradation of miR-17 through a viral decoy RNA; deletion of the viral miR-17 decay element in the context of the virus limits hCMV lytic replication (57). Here, we show that miR-J8 can target 3′ UTRs harboring miR-17-5p or miR-20a binding sites, which strongly implies that miR-J8 can mimic the function of miR-17 family members and thereby also gain access to miR-17 regulatory networks. What is somewhat surprising is that the observed phenotype of miR-J8 in NF-κB signaling does not entirely fit with what has been described previously for miR-17, which is generally regarded as a proinflammatory miRNA. miR-17 has been observed to be upregulated in MS patients, and through targeting of IKZF4 (Ikaros family zinc finger 4) in T cells, it can enhance Th17 responses (58–60). In B cells, miR-17 targets multiple negative regulators of the NF-κB pathway, including TNFAIP3 (A20), and together with additional members of the miR-17/92 cluster drives NF-κB activation (38, 61). Consistent with the identified targets of miR-17 and the phenotypes observed from overexpression of miR-17/92 in B cells in vivo, we have also observed enhanced NF-κB activity following ectopic miR-17 expression in vitro (Skalsky, unpublished). Thus, while miR-J8 is certainly able to bind miR-17 cognate sites, the viral miRNA does not appear to functionally mimic miR-17 in the context of TNF-α or IL-1 cytokine signaling. Notably, miR-J8 also has sequence homology to members of the miR-373 family. In genome-wide miRNA screens to identify effectors of the NF-κB pathway in breast cancer cells, miR-373 family members (miR-373 and miR-520) strongly inhibited TNF-α signaling by directly targeting a core NF-κB signaling component, RELA (p65) (62). Additionally, overexpression of miR-20a in rheumatoid fibroblast-like synoviocytes suppressed IL-6 and CXCL10 release in response to lipopolysaccharide (LPS) treatment; in this context, miR-20a was shown to target apoptosis signal-regulating kinase 1 (ASK1), a critical mitogen-activated protein kinase kinase kinase (MAP3K) component of TLR and TNF signaling pathways (63). These observations raise the possibility that JMRV miR-J8 as well as RRV miR-rR1-8 may target macaque homologs of RELA and/or ASK1. miR-J8 silencing of RELA, in particular, would suppress NF-κB activation, which could explain the strong inhibition of NF-κB activity that we observed following cytokine treatment.
Like miR-J8, JMRV miR-J13 also inhibited IL-1β signaling, and closer examination of the miR-J13 sequence revealed seed homology to the cellular miRNAs miR-367 and miR-363. miR-363 is encoded within the miR-106a/363 cluster, while miR-367 is encoded within the miR-302/367 cluster, which plays an important role in cell cycle regulation, human embryonic stem cell pluripotency, self-renewal, and somatic cell reprogramming (64). Recent studies showed that miR-367 can target the 3′ UTR of IRAK4 in primary microglia, thereby decreasing NF-κB activity and inhibiting proinflammatory cytokine production (65). IRAK4 is a critical protein kinase component of TLR/IL-1 signal transduction and thus serves as an attractive candidate target for miR-J13, which may function similarly to miR-367 via cognate binding sites. Thus, JMRV encodes not one but potentially two miRNA mimics of cellular miRNAs, both of which display anti-inflammatory phenotypes in the context of cytokine signaling.
How miR-J8, miR-J13, and other JMRV miRNAs manipulate inflammatory responses and contribute to pathogenesis in vivo, particularly in the context of an immune-mediated disease, is an important question to address in the future. In short, our works provides initial insight into some of the regulatory functions of RV2 miRNAs and provides an essential step toward understanding viral miRNA functions in a NHP model system.
ACKNOWLEDGMENTS
We thank Victor DeFilippis at the Vaccine and Gene Therapy Institute, Beaverton, OR, for the tRF-NF-κB-Luc cells and members of Jay Nelson's laboratory for insightful discussions.
This work was supported by a pilot project award to R.L.S. from the Oregon National Primate Research Center, grant P51 OD011092, and Public Health Service grants 8P51OD011092-54 and CA075922 (S.W.W.).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Axthelm MK, Bourdette DN, Marracci GH, Su W, Mullaney ET, Manoharan M, Kohama SG, Pollaro J, Witkowski E, Wang P, Rooney WD, Sherman LS, Wong SW. 2011. Japanese macaque encephalomyelitis: a spontaneous multiple sclerosis-like disease in a nonhuman primate. Ann Neurol 70:362–373. doi: 10.1002/ana.22449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Estep RD, Hansen SG, Rogers KS, Axthelm MK, Wong SW. 2013. Genomic characterization of Japanese macaque rhadinovirus, a novel herpesvirus isolated from a nonhuman primate with a spontaneous inflammatory demyelinating disease. J Virol 87:512–523. doi: 10.1128/JVI.02194-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blair TC, Manoharan M, Rawlings-Rhea SD, Tagge I, Kohama SG, Hollister-Smith J, Ferguson B, Woltjer RL, Frederick MC, Pollaro J, Rooney WD, Sherman LS, Bourdette DN, Wong SW. 2016. Immunopathology of Japanese macaque encephalomyelitis is similar to multiple sclerosis. J Neuroimmunol 291:1–10. doi: 10.1016/j.jneuroim.2015.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruce AG, Thouless ME, Haines AS, Pallen MJ, Grundhoff A, Rose TM. 2015. Complete genome sequence of pig-tailed macaque rhadinovirus 2 and its evolutionary relationship with rhesus macaque rhadinovirus and human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J Virol 89:3888–3909. doi: 10.1128/JVI.03597-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Damania B, Desrosiers RC. 2001. Simian homologues of human herpesvirus 8. Philos Trans R Soc Lond B Biol Sci 356:535–543. doi: 10.1098/rstb.2000.0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci U S A 102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grundhoff A, Sullivan CS. 2011. Virus-encoded microRNAs. Virology 411:325–343. doi: 10.1016/j.virol.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat Methods 2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 9.Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J Virol 79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schafer A, Cai X, Bilello JP, Desrosiers RC, Cullen BR. 2007. Cloning and analysis of microRNAs encoded by the primate gamma-herpesvirus rhesus monkey rhadinovirus. Virology 364:21–27. doi: 10.1016/j.virol.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Umbach JL, Cullen BR. 2010. In-depth analysis of Kaposi's sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J Virol 84:695–703. doi: 10.1128/JVI.02013-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y, Haecker I, Yang Y, Gao SJ, Renne R. 2013. Gamma-herpesvirus-encoded miRNAs and their roles in viral biology and pathogenesis. Curr Opin Virol 3:266–275. doi: 10.1016/j.coviro.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grey F. 2015. Role of microRNAs in herpesvirus latency and persistence. J Gen Virol 96:739–751. doi: 10.1099/vir.0.070862-0. [DOI] [PubMed] [Google Scholar]
- 14.Nana-Sinkam SP, Croce CM. 2014. MicroRNA regulation of tumorigenesis, cancer progression and interpatient heterogeneity: towards clinical use. Genome Biol 15:445. doi: 10.1186/s13059-014-0445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skalsky RL, Cullen BR. 2015. EBV noncoding RNAs. Curr Top Microbiol Immunol 391:181–217. doi: 10.1007/978-3-319-22834-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullen BR. 2013. MicroRNAs as mediators of viral evasion of the immune system. Nat Immunol 14:205–210. doi: 10.1038/ni.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen TA, Jo MH, Choi YG, Park J, Kwon SC, Hohng S, Kim VN, Woo JS. 2015. Functional anatomy of the human microprocessor. Cell 161:1374–1387. doi: 10.1016/j.cell.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 20.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 22.Schirle NT, Sheu-Gruttadauria J, MacRae IJ. 2014. Structural basis for microRNA targeting. Science 346:608–613. doi: 10.1126/science.1258040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umbach JL, Strelow LI, Wong SW, Cullen BR. 2010. Analysis of rhesus rhadinovirus microRNAs expressed in virus-induced tumors from infected rhesus macaques. Virology 405:592–599. doi: 10.1016/j.virol.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruce AG, Ryan JT, Thomas MJ, Peng X, Grundhoff A, Tsai CC, Rose TM. 2013. Next-generation sequence analysis of the genome of RFHVMn, the macaque homolog of Kaposi's sarcoma (KS)-associated herpesvirus, from a KS-like tumor of a pig-tailed macaque. J Virol 87:13676–13693. doi: 10.1128/JVI.02331-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manzano M, Forte E, Raja AN, Schipma MJ, Gottwein E. 2015. Divergent target recognition by coexpressed 5′-isomiRs of miR-142-3p and selective viral mimicry. RNA 21:1606–1620. doi: 10.1261/rna.048876.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, Cullen BR. 2007. A viral microRNA functions as an orthologue of cellular miR-155. Nature 450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kincaid RP, Sullivan CS. 2012. Virus-encoded microRNAs: an overview and a look to the future. PLoS Pathog 8:e1003018. doi: 10.1371/journal.ppat.1003018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manzano M, Shamulailatpam P, Raja AN, Gottwein E. 2013. Kaposi's sarcoma-associated herpesvirus encodes a mimic of cellular miR-23. J Virol 87:11821–11830. doi: 10.1128/JVI.01692-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R. 2007. Kaposi's sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol 81:12836–12845. doi: 10.1128/JVI.01804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abend JR, Uldrick T, Ziegelbauer JM. 2010. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J Virol 84:12139–12151. doi: 10.1128/JVI.00884-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. 2012. Kaposi's sarcoma-associated microRNAs target IRAK1 and MYD88, two components of the Toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J Virol 86:11663–11674. doi: 10.1128/JVI.01147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. 2010. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol 12:193–199. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu CC, Li Z, Chu CY, Feng J, Sun R, Rana TM. 2010. MicroRNAs encoded by Kaposi's sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep 11:784–790. doi: 10.1038/embor.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skalsky RL, Kang D, Linnstaedt SD, Cullen BR. 2014. Evolutionary conservation of primate lymphocryptovirus microRNA targets. J Virol 88:1617–1635. doi: 10.1128/JVI.02071-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40:37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, Nusbaum JD, Feederle R, Delecluse HJ, Luftig MA, Tuschl T, Ohler U, Cullen BR. 2012. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog 8:e1002484. doi: 10.1371/journal.ppat.1002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai X, Cullen BR. 2006. Transcriptional origin of Kaposi's sarcoma-associated herpesvirus microRNAs. J Virol 80:2234–2242. doi: 10.1128/JVI.80.5.2234-2242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walz N, Christalla T, Tessmer U, Grundhoff A. 2010. A global analysis of evolutionary conservation among known and predicted gammaherpesvirus microRNAs. J Virol 84:716–728. doi: 10.1128/JVI.01302-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waidner LA, Morgan RW, Anderson AS, Bernberg EL, Kamboj S, Garcia M, Riblet SM, Ouyang M, Isaacs GK, Markis M, Meyers BC, Green PJ, Burnside J. 2009. MicroRNAs of Gallid and Meleagrid herpesviruses show generally conserved genomic locations and are virus-specific. Virology 388:128–136. doi: 10.1016/j.virol.2009.02.043. [DOI] [PubMed] [Google Scholar]
- 42.Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, Shamulailatpam P, Love CL, Dave SS, Tuschl T, Ohler U, Cullen BR. 2011. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10:515–526. doi: 10.1016/j.chom.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Hara AJ, Chugh P, Wang L, Netto EM, Luz E, Harrington WJ, Dezube BJ, Damania B, Dittmer DP. 2009. Pre-micro RNA signatures delineate stages of endothelial cell transformation in Kaposi sarcoma. PLoS Pathog 5:e1000389. doi: 10.1371/journal.ppat.1000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hook L, Hancock M, Landais I, Grabski R, Britt W, Nelson JA. 2014. Cytomegalovirus microRNAs. Curr Opin Virol 7:40–46. doi: 10.1016/j.coviro.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishikura K. 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17:83–96. doi: 10.1038/nrm.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun R, Lin SF, Gradoville L, Miller G. 1996. Polyadenylylated nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 93:11883–11888. doi: 10.1073/pnas.93.21.11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD. 2012. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods 9:840–846. doi: 10.1038/nmeth.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma X, Becker Buscaglia LE, Barker JR, Li Y. 2011. MicroRNAs in NF-kappaB signaling. J Mol Cell Biol 3:159–166. doi: 10.1093/jmcb/mjr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. 2005. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 50.Musilova K, Mraz M. 2015. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia 29:1004–1017. doi: 10.1038/leu.2014.351. [DOI] [PubMed] [Google Scholar]
- 51.Lo AK, To KF, Lo KW, Lung RW, Hui JW, Liao G, Hayward SD. 2007. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc Natl Acad Sci U S A 104:16164–16169. doi: 10.1073/pnas.0702896104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramalingam D, Kieffer-Kwon P, Ziegelbauer JM. 2012. Emerging themes from EBV and KSHV microRNA targets. Viruses 4:1687–1710. doi: 10.3390/v4091687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao Y, Xu H, Yao Y, Smith LP, Kgosana L, Green J, Petherbridge L, Baigent SJ, Nair V. 2011. Critical role of the virus-encoded microRNA-155 ortholog in the induction of Marek's disease lymphomas. PLoS Pathog 7:e1001305. doi: 10.1371/journal.ppat.1001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gottwein E, Cullen BR. 2010. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J Virol 84:5229–5237. doi: 10.1128/JVI.00202-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA. 2012. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J 31:2207–2221. doi: 10.1038/emboj.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, Cox JE, Hsiang TY, Krug RM, Sullivan CS. 2013. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 14:435–445. doi: 10.1016/j.chom.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee S, Song J, Kim S, Kim J, Hong Y, Kim Y, Kim D, Baek D, Ahn K. 2013. Selective degradation of host microRNAs by an intergenic HCMV noncoding RNA accelerates virus production. Cell Host Microbe 13:678–690. doi: 10.1016/j.chom.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 58.Kang SG, Liu WH, Lu P, Jin HY, Lim HW, Shepherd J, Fremgen D, Verdin E, Oldstone MB, Qi H, Teijaro JR, Xiao C. 2013. MicroRNAs of the miR-17∼92 family are critical regulators of T(FH) differentiation. Nat Immunol 14:849–857. doi: 10.1038/ni.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindberg RL, Hoffmann F, Mehling M, Kuhle J, Kappos L. 2010. Altered expression of miR-17-5p in CD4+ lymphocytes of relapsing-remitting multiple sclerosis patients. Eur J Immunol 40:888–898. doi: 10.1002/eji.200940032. [DOI] [PubMed] [Google Scholar]
- 60.Liu SQ, Jiang S, Li C, Zhang B, Li QJ. 2014. miR-17-92 cluster targets phosphatase and tensin homology and Ikaros family zinc finger 4 to promote TH17-mediated inflammation. J Biol Chem 289:12446–12456. doi: 10.1074/jbc.M114.550723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jin HY, Oda H, Lai M, Skalsky RL, Bethel K, Shepherd J, Kang SG, Liu WH, Sabouri-Ghomi M, Cullen BR, Rajewsky K, Xiao C. 2013. MicroRNA-17∼92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J 32:2377–2391. doi: 10.1038/emboj.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keklikoglou I, Koerner C, Schmidt C, Zhang JD, Heckmann D, Shavinskaya A, Allgayer H, Guckel B, Fehm T, Schneeweiss A, Sahin O, Wiemann S, Tschulena U. 2012. MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-kappaB and TGF-beta signaling pathways. Oncogene 31:4150–4163. doi: 10.1038/onc.2011.571. [DOI] [PubMed] [Google Scholar]
- 63.Philippe L, Alsaleh G, Pichot A, Ostermann E, Zuber G, Frisch B, Sibilia J, Pfeffer S, Bahram S, Wachsmann D, Georgel P. 2013. miR-20a regulates ASK1 expression and TLR4-dependent cytokine release in rheumatoid fibroblast-like synoviocytes. Ann Rheum Dis 72:1071–1079. doi: 10.1136/annrheumdis-2012-201654. [DOI] [PubMed] [Google Scholar]
- 64.Gao Z, Zhu X, Dou Y. 2015. The miR-302/367 cluster: a comprehensive update on its evolution and functions. Open Biol 5:150138. doi: 10.1098/rsob.150138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuan B, Shen H, Lin L, Su T, Zhong L, Yang Z. 2015. MicroRNA367 negatively regulates the inflammatory response of microglia by targeting IRAK4 in intracerebral hemorrhage. J Neuroinflammation 12:206. doi: 10.1186/s12974-015-0424-3. [DOI] [PMC free article] [PubMed] [Google Scholar]