

Abstract
Parkinson's disease (PD) is a progressive neurological condition caused by the degeneration of dopaminergic neurons in the basal ganglia. It is the most prevalent form of Parkinsonism, categorized by cardinal features such as bradykinesia, rigidity, tremors, and postural instability. Due to the multicentric pathology of PD involving inflammation, oxidative stress, excitotoxicity, apoptosis, and protein aggregation, it has become difficult to pin-point a single therapeutic target and evaluate its potential application. Currently available drugs for treating PD provide only symptomatic relief and do not decrease or avert disease progression resulting in poor patient satisfaction and compliance. Significant amount of understanding concerning the pathophysiology of PD has offered a range of potential targets for PD. Several emerging targets including AAV-hAADC gene therapy, phosphodiesterase-4, potassium channels, myeloperoxidase, acetylcholinesterase, MAO-B, dopamine, A2A, mGlu5, and 5-HT-1A/1B receptors are in different stages of clinical development. Additionally, alternative interventions such as deep brain stimulation, thalamotomy, transcranial magnetic stimulation, and gamma knife surgery, are also being developed for patients with advanced PD. As much as these therapeutic targets hold potential to delay the onset and reverse the disease, more targets and alternative interventions need to be examined in different stages of PD. In this review, we discuss various emerging preclinical pharmacological targets that may serve as a new promising neuroprotective strategy that could actually help alleviate PD and its symptoms.
Keywords: dopaminergic, Parkinson's disease, pharmacological targets, neuroprotection, preclinical
INTRODUCTION
Parkinson's disease (PD) is the most common neurodegenerative disorder after Alzheimer's disease (AD), affecting > 1.5% of the global population. PD is characterized by progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) region of the brain. The incidence of PD increases with age, and mean age of onset is about 65 years. Fresh evidences are insinuating towards epigenetic changes such as histone modifications as one of the leading factors behind brain aging and PD [1]. The lack of dopamine (DA) causes the classical motor symptoms of bradykinesia, rigidity, and resting tremors [2]. However, other neuronal fields and neurotransmitter systems including the locus coeruleus, the dorsal motor nucleus, the substantia innominata, the autonomic nervous system, and the cerebral cortex are also involved in PD [3]. As a consequence of this other symptoms including cognitive decline, sleep abnormalities, depression, and gastrointestinal and genitourinary disturbances are also developed. These “non-motor” symptoms progress and dominate in advanced stages of PD. It is projected that the number of patients with PD will increase to 8.7 million by 2030. Men are more susceptible to PD than women [2]. Years of genetic investigations in PD have led to the discovery of several heritable and sporadic forms of this disorder. The typical form of PD occurs in a sporadic idiopathic fashion [4] and hereditary factors are minimally associated with early-onset PD [5]. Comprehensive analyses of patients with mutations in PINK1, DJ1, LRRK2, UCHL1, MAPT, GBA, NAT2, INOS2A, GAK, HLA-DRA, APOE, and SNCA have significantly increased our understanding regarding the morphological, and pathological aspects seen in clinical and preclinical stages of PD [6, 7]. Other anticipated causes include environmental toxins, medications, and viruses that result in increased oxidative stress [8, 9]. Patients are classically diagnosed with PD based on thorough neurological and physical examinations. The diagnosis is made when two of the four classical symptoms of PD are present [10]. An accurate PD diagnosis is aided by DA transporter single photon emission computed tomography [11]. However, sensitivity and accuracy of this scan for diagnosing PD are equal to a clinical diagnosis; thus, raising doubts about the benefits of using a brain scan to confirm the diagnosis [12]. Despite these advances, a post-mortem neuropathological examination of brain tissue is the ultimate choice to precisely confirm the diagnosis of PD [13].
Levodopa (L-DOPA) was introduced almost 40 years ago and remains the best functional therapy for decreasing PD symptoms. However, the effects of L-DOPA tend to decrease as the disease advances. It gradually becomes difficult to manage symptoms and patients invariably develop motor impediments that comprise of motor fluctuations, dyskinesia, freezing, and fall [14]. Numerous other drugs from different classes are available to treat PD, and usually involve layering different treatments in a polypharmaceutical approach. More advanced DA agonists, such as pramipexole and ropinirole approved in 1997, were designed to selectively stimulate DA 2 receptors. Other key drug classes including the catechol-O-methyltransferase (COMT) inhibitors and monoamine oxidase (MAO) inhibitors have been approved to treat PD. As such, no single treatment is considered completely efficacious throughout the progression of PD; therefore the treatment is addressed according to the patient's disease severity and progression [15]. However, due to the side-effects associated with L-DOPA, nearly half of PD market sales are driven by advanced DA agonists along with COMT and MAO inhibitors. Nevertheless, the availability of generic versions of the DA agonists' pramipexole and ropinirole will limit growth across all major markets. Competition from generic compounds is most prominent in the United States, and it will remain the biggest market for PD until 2019. Germany is projected to produce the highest sales for PD drugs in 2019, whereas France will remain the smallest PD market until 2019 [15]. Symptomatic therapies will not take the place of existing drugs even though the market will rise due to new therapies in the pipeline. However, the existing landscape of therapeutic options will be revolutionized with development of neuroprotective candidates. Disease-modifying therapies for PD will change once the target genes and proteins that mediate the degeneration of dopaminergic neurons in the SN are identified. Nevertheless, it will take another decade until significant disease-modifying targets are identified [15]. Several emerging targets including AAV-hAADC gene therapy, phosphodiesterase-4, potassium channels, myeloperoxidase, acetylcholinesterase, MAO-B, DA, A2A, mGlu5, and 5-HT-1A/1B receptors are in different stages of clinical development. In this review, we discuss various emerging preclinical pharmacological targets that may serve as a new promising neuroprotective strategy that could actually help alleviate PD and its symptoms.
IMPORTANCE OF TARGET IDENTIFICATION AND VALIDATION FOR BETTER DRUG DISCOVERY
Discovering a new target from an original idea and launching the final product is a multifaceted process that involves around $1 billion and can take 12-15 years. Awareness of a target can come from various sources including academic, clinical, and commercial sectors. Once the target is selected, the therapeutic industry and academic centers develop a number of early procedures to identify lead molecules that possess appropriate characteristics for acceptable drugs [16]. Two main reasons contribute to drug failure. The first is a lack of safety and the second is ineffectiveness in humans. Therefore, a key step for developing a new drug is identifying and validating the target. A target is a comprehensive term applied to a variety of biological entities, including enzymes, substrates, metabolites, receptors, ion channels, transport proteins, DNA, RNA, ribosome, monoclonal antibodies, and various physiological mechanisms [17]. A target must be safe, potent, and efficacious to be functional; it should also satisfy clinical and commercial needs and should be druggable. A “druggable” target means it is available for developing a putative drug molecule. The molecule can be a small or large biological molecule but should elicit a biological response that can be measured in vitro and in vivo. Certain target classes are more amenable to discovering small molecule drugs, such as G-protein-coupled receptors, whereas antibodies are better at blocking protein/protein interactions. Proper identification and validation of the target allows us to learn the role of target modulation during mechanism-based side effects. Bioinformatics is one of the prime technique that helps to identify, choose, and prioritize probable disease targets [18]. The literature has a variety of sources, including publications, patent information, proteomics data, gene expression data, transgenic phenotyping and compound profiling data. Additional approaches for identification include investigating mRNA/proteins and their role under pathological conditions. Another excellent method is to look for genetic polymorphism and its consequence on disease progression. [19]. Furthermore, mutations in human phenotypes can nullify or exacerbate certain receptor, such as mutations in voltage-gated sodium channel NaV1.7; results in insensitivity or oversensitivity to pain [20, 21]. Phenotypic screening is another method that can be used to recognize pertinent disease targets [22].
Validation techniques for drug targets include using in vitro and in vivo tools and modulating a preferred target under disease conditions [23]. Reliability of these results increases significantly using a multi-validation approach [16]. Antisense technology is a powerful method that uses chemically modified RNA-like oligonucleotides designed to be complimentary to a region of a desired mRNA molecule [24]. The effects of antisense oligonucleotides are reversible compared with the gene knockout approach, and sustained presence of the antisense is required to inhibit the desired protein [25]. Nevertheless, the chemistry associated with synthesizing oligonucleotides has resulted in molecules with restricted bioavailability and strong toxicity, making in vivo use problematic [16]. As an alternative, transgenic animal are an attractive option for validation, as phenotypic changes resulting from gene manipulation can be observed. However, using transgenic animals is time-consuming and expensive. Hence, small interfering RNA (siRNA) has become popular for target validation. However, delivery to the target cell can be a problem with siRNA methodology [26]. Monoclonal antibodies are an another brilliant target validation tool, as they bind with a major region of the desired molecule surface, and thus help to selectively bind the target with high affinity. The only disadvantage of this technique is that antibodies cannot cross the cell membrane and cannot bind intracellular targets. Recently, chemical genomics has emerged as a systemic application of molecules to target identification and validation. Chemical genomics is nothing but the study of genomic responses to chemical compounds. This approach helps early identification of novel targets and drug [27].
EMERGING NEW PRECLINICAL EVIDENCE OF TENTATIVE PD TARGETS
Existing PD therapy include re-formulating conventional drugs already approved for PD, chemically modifying compounds that are accepted for other indications, developing novel small-molecules and gene therapy-based approaches. The pipeline for the therapeutic development of new targets appears to be dynamic on the surface. However, if conventional dopaminergic therapy is removed from the picture, the existing landscape is far less promising [28]. Most of the drugs approved for PD do not have adequate efficacy nor do they avert disease progression. Consequently, there is a critical need to discover novel therapies to overcome the disadvantages associated with current therapies. In the following section, we will discuss some emerging pharmacological PD targets with substantial preclinical evidence (as also depicted in Table 1) that can be further considered for clinical investigations to develop new PD therapeutic.
Table 1. Emerging preclinical pharmacological targets for Parkinson's disease (PD).
| Target | Pharmacological Model | Experimental Outcomes | Ref. |
|---|---|---|---|
| BAG1 | SH-SY5Y cells and C57BL/6J mice | BAG1 protects against mutant α-syn and rotenone-induced cell death. BAG1 also protects dopaminergic cells in the SN against MPTP-induced toxicity in vivo. | [279, 280] |
| Adenosine A2A Receptor | MPTP-intoxicated Marmosets | Istradefylline (Adenosine A2A Receptor antagonist) effectively alleviates motor impairments in combination with low dose dopaminergic drug without aggravating dyskinesia. | [281, 282] |
| 5Alpha-Reductase | MPTP-intoxicated Mice | Dutasteride (5Alpha-Reductase inhibitor) significantly prevents the demise of dopaminergic neurons in MPTP-intoxicated mice. | [283, 284] |
| CB2 Receptor | Intrastriatal injections of LPS into C57BL/6J, male wild-type or CB2 knockout C57BL/6J mice | Genetic deletion of CB2 receptors exacerbates LPS-induced inflammation. Stimulating CB2 receptors with HU-308 decreases the LPS-induced proinflammatory response. | [285, 286] |
| Cyclin D3/CDK6 | 6-OHDA-induced toxicity in SH-SY5Y cells | Sodium butyrate and suberoylanilide hydroxamic acid stabilizes the proliferative activity of PD lymphoblasts and decreases 6-OHDA-induced cell death in neuronal cells by preventing over-activation of the cyclin D3/CDK6/pRb cascade. | [287] |
| PDE7 | Ex vivo cultures obtained from male Wistar rats | Inhibiting PDE7 induces proliferation and growth of embryonic ventral mesencephalic-derived neurospheres and adult progenitor cells in vivo in the SNpc, as well as increases Nurr1 and TH MAP-2 expression in neural stem cells obtained from ventral mesencephalon. | [288, 289] |
| SIRT2 | LUHMES cells and MPTP model of PD | AK7 (SIRT2 inhibitor) protects dopaminergic neurons against α-syn-induced neurotoxicity in differentiated LUHMES cells and in MPTP model of PD. AK7 prevents dopamine loss, encourages long-term endurance of dopaminergic neurons, and conserves functional performance. |
[290, 291] |
| Trib3 | PC12 cells and rat dopaminergic ventral midbrain neurons exposed with 6-OHDA, MPP+, or α-syn fibrils | Toxin-induced upregulation of Trib3 protein increases neuronal cell death. Trib3 knockdown protects PC12 cells and ventral midbrain dopaminergic from all toxins. | [292, 293] |
| SK channel | Rotenone intoxication of human post mitotic dopaminergic neurons | Stimulating SK channels decreases mitochondrial membrane potential and maintains cell viability, the dendritic network, and ATP levels after rotenone insult. | [294, 295] |
| Sigma-1 Receptors | Intrastriatal injection of 6-OHDA in mice | PRE-084 (sigma-1 receptor agonist) facilitates steady and substantial development of impulsive forelimb use and augmented density of dopaminergic fibers in the utmost denervated striatal regions. | [296, 297] |
| Ribosomal protein s15 | Drosophila and human neuronal PD models | Phosphorylation of ribosomal protein s15, a substrate of LRRK2, is essential for the toxicity-related effects of the common G2019S LRRK2 mutation in human dopamine neurons and in G2019S the LRRK2 mutated Drosophila model of PD. | [298, 299] |
| Prolyl Hydroxylase Domain | Inducible genetic dopaminergic glutathione depletion model | Antagonizing the prolyl hydroxylase domain pharmacologically via 3,4-dihydroxybenzoate substantially lessens mitochondrial dysfunction and damage to dopaminergic neurons in the SNpc. | [300, 301] |
| VDR and NMDAR | Haloperidol-induced Parkinsonism in mice |
Vitamin D3 treatment enhances neural activity, motor-cognitive function, glia/neuron survival, and expression of neurofilaments. Inhibiting NMDAR and co-treatment enhances motor-cognitive functions but not as much as values detected post VDR stimulation. | [302, 303] |
| LHb | 6-OHDA-induced PD model | LHb lesions decrease apomorphine-induced rotational behavior. The lesions also increase dopamine levels in the striatum of PD model of rats. | [304, 305] |
| HIF-1α | MPP+-intoxicated SH-SY5Y cells | Orexin-A is an inducer of HIF-1α that diminishes MPP+-induced cell injury. | [306, 307] |
| ATF4 | Overexpressed or silenced ATF4 in cellular models of PD | Silencing ATF4 in neuronal PC12 cells boosts cell death in response to either 6-OHDA or MPP+. Overexpression of ATF4 decreases cell death caused by dopaminergic neuronal toxins. | [308, 309] |
| Hsp70 | H4 cells transfected with α-syn | CBX treatment activates heat shock factor 1 and thereby induces Hsp70. Hsp70 abates α-syn aggregation and prevents α-syn-induced cytotoxicity. | [310, 311] |
| LAMP2A | SH-SY5Y neuroblastoma cell line stably expressing LAMP2A; primary cortical cultures with high CMA activity and a rat synucleinopathy model | Overexpressing LAMP2A enhances CMA activity and protects against neurotoxicity caused by α-syn. Co-injection of LAMP2A with α-syn reverses α-syn neurotoxicity | [312, 313] |
| TFEB | Induction of autophagy by CCI-779 | CCI-779 inhibits mTOR, which increases nuclear translocation of TFEB, stimulates clearance of toxic oligomeric α-syn, and confers protection of nigral dopamine neurons against α-syn toxicity. Similarly, Rapamycin was observed to decrease Tau phosphorylation by inhibiting mTOR in senescence-accelerated OXYS rats. | [33, 314, 315] |
| PGC-1α | PGC-1α transgenic mice | Over-expressing PGC-1α in mice protects against MPTP-induced neuronal degeneration. | [316, 317] |
| T-type Ca2+ channels | 6-OHDA lesioned rats | Local administration of a T-type Ca2+ channel antagonist significantly decreases locomotor deficits. | [318, 319] |
| HDAC6 | PC-12 cells overexpressing human mutant (A53T) α-syn and SH-SY5Y cells intoxicated with MPP+ | Overexpressing α-syn upregulates HDAC6 expression in close association with α-syn to form aggresome-like bodies. HDAC6 deficiency obstructs formation of aggresome-like bodies and restricts autophagy in response to MPP+-induced stress. | [320, 321] |
| PI3K/Akt signaling pathway | MPP+-intoxicated PC12 cells | Treatment with tetrahydroxystilbene glucoside attenuate loss of cell viability, release of lactate dehydrogenase (LDH), and inhibits apoptosis in a dose-dependent manner probably by activating the PI3K/Akt signaling pathway | [322, 323] |
Abbreviations: BAG1: Bcl-2-associated athanogene-1, α-syn: α-synuclein, SN: substantia nigra, MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, CB2: cannabinoid type-2, LPS: lipopolysaccharide, 6-OHDA: 6-hydroxy dopamine, PD: Parkinson's disease, PDE7: phosphodiesterase-7, SNpc: substantia nigra pars compacta, SIRT2: Sirtuin-2, Trib3: Tribbles pseudokinase-3, MPP+: 1-methyl-4-phenylpyridinium, SK channel: small-conductance Ca2+-activated K+channel, VDR: vitamin D3 receptor, NMDAR: N-methyl-D-aspartate receptor, LHb: lateral habenula, HIF-1α: hypoxia inducible factor 1-alpha, ATF4: activating transcription factor 4, Hsp70: Heat shock protein-70, CBX: carbenoxolone, CMA: chaperone-mediated autophagic, TFEB: transcription factor EB, mTOR: mechanistic target of Rapamycin, PGC-1α: peroxisome proliferator-activated receptor-gamma coactivator-1α, HDAC6: histone deacetylase-6, PI3K/Akt: phosphatidylinositol 3-kinase/ protein kinase B
Micro-RNA as a PD target
Micro-RNAs (miRNAs) are small non-coding, single stranded RNA molecules consisting of 22 nucleotides. miRNAs control gene expression by base pairing to mRNA and trigger translation repression [29]. Abnormal miRNA expression has been associated with various neurological disorders, such as AD, PD, Huntington's disease, amyotrophic lateral sclerosis, schizophrenia, and autism. Un-regulated miRNAs in patients suffering from PD could be used as biomarkers for early identification and monitoring disease progression. Ascertaining the role of miRNAs in cell processes and learning how disorganized miRNA expression accounts for neurological effects is crucial for discovering new therapeutic strategies for PD. miRNAs have great therapeutic potential, particularly if it can be demonstrated that a single miRNA can activate or inhibit several desired genes, making it possible to modify a whole disease phenotype by modifying a single miRNA molecule. Hence, understanding the mechanisms by which miRNAs participates in the pathogenesis of PD may offer novel targets to researchers to develop pioneering therapies [30]. Certain miRNAs are highly elevated in brain tissues of patients with PD. miRNA-301b, miRNA-373, miRNA-26b, miRNA-224, miRNA-21, and miRNA-106b are few of the miRNAs found in PD brain tissues that actively participate in the autophagy pathway carried out by chaperone [31]. Malfunctioning of this pathway has been suggested to disorder degradation of the alpha-synuclein (α-syn) protein, which contributes to Lewy Body (LB) pathology [32]. The effect of miRNA-128 overexpression, a negative regulator of transcription factor EB (TFEB), was examined by Decressac and Bjorklund. In this study, the AAV vector was used to overexpress miRNA-128 in midbrain dopaminergic neurons. They demonstrated that miRNA-128-facilitated suppression of TFEB, aggravated the toxicity of α-syn by inhibiting autophagy, and consequently favored formation of toxic oligomers [33].
Since, quantifying specific pathogenic proteins in the neuronal population is essential for survival of neurons involved in PD pathogenesis, evaluating the role of miRNAs is important for treating PD [34]. Choi et al. demonstrated that miRNA-7, confers protection in 1-methyl-4-phenylpyridinium (MPP+)-induced cytotoxicity to differentiated human neural progenitor ReNcell VM cells, primary mouse neurons and also to dopaminergic SH-SY5Y cells. With the help of quantitative proteomic analysis, Choi and colleagues also determined that RelA, which is a constituent of nuclear factor-kappaB (NF-κB), was downregulated by miRNA-7. Latter on RelA mRNA was confirmed as a target for miRNA-7 and is essential for MPP+-induced cell death. These outcomes describe a novel mechanism by which suppression of NF-κB is responsible for the cell death mechanism following MPP+-induced toxicity, and, thus, propose miRNA-7 as a therapeutic target for PD [35]. In a very recent study, kabaria and colleagues demonstrated that miRNA-7, decreases expression of Keap1 by targeting the 3′-untranslated region (UTR) of its mRNA in SH-SY5Y cells which consequently amplifies nuclear factor erythroid 2-related factor 2 (Nrf2) activity. In addition, miRNA-7 was found to augment the level of reduced form of glutathione and decrease the intracellular hydroperoxides level, suggesting its anti-oxidative effect. These conclusions signify to a novel mechanism by which miRNA-7 exhibits its cytoprotective effects by activating the Nrf2 pathway [36]. In vitro and in vivo data have confirmed that dopaminergic neurons depend profoundly on a functional miRNA network. In a study that examined the role of miRNAs in mammalian midbrain dopaminergic neurons, miRNA-133b was found to be precisely expressed in midbrain dopaminergic neurons of healthy individuals but midbrain tissue from patients with PD entirely lacked this type of miRNA. It was proposed that the development and operation of midbrain dopaminergic neurons is regulated by miRNA-133b through a negative-feedback mechanism, which contains paired-like homeodomain transcription factor Pitx3 [37].
Sequence analysis of human α-syn showed that the entire UTR of the α-syn gene is highly conserved, insinuating a role for miRNA regulation [38]. Until now, miRNA-153 and miRNA-7 are the only two miRNAs that have been shown to directly aim α-syn. These two miRNAs downregulate α-syn mRNA and protein levels by binding to the 3′-UTR of α-syn [39]. Additional cellular studies have demonstrated that miRNA-7 decreases α-syn-induced neurotoxicity in a cellular model [40]. Additionally, miRNA-29a, miRNA-1, and miRNA-22 are less expressed in patients with PD compared to control subjects [41]. Thus, indicating that specific miRNAs could serve as effective biomarkers in patients with PD. A similar study performed using plasma from patients with untreated PD and control subjects found seven upregulated miRNAs, such as miRNA-454, miRNA-125a-3p, miRNA-137, miRNA-181c, miRNA-193a-3p, miRNA-196b, and miRNA-331-5p in PD patients. Based on these evidences, miRNAs have tremendous potential to be developed as biomarkers or therapeutics for PD [42].
Alpha7 nicotinic receptor as a PD target
Neuronal acetylcholine receptors (nAChR) are pentameric ligand-gated ion channels that consist of different combinations of α and β transmembrane subunits [43–45]. The α7 receptor is membrane-bound receptor and consists of five identical α subunits, with five agonist binding sites. α7 nAChRs are highly expressed in the hippocampus, medial habenula, thalamus, hypothalamus, geniculate nuclei, colliculi cortex, and amygdala, but are scarcely expressed in forebrain, striatum, medulla, and numerous brain nuclei [46]. The notion that nicotine might be useful to treat PD initially came from epidemiological data [47, 48]. The outcome of these experiments, combined with the discovery that nicotine enhances dopamine release [49, 50] suggested that nicotine might be responsible for the beneficial results of smoking in patients with PD. A protective role for α7 nAChRs against degeneration of dopaminergic neurons originated from experiments that used nicotine as a nAChR agonist [51–53]. Early reports by Janson et al. showed that nicotine dosed at or before the time of lesioning considerably increases nigral and striatal dopaminergic markers in rats [54, 55]. Later on, the neuroprotective ability of nicotine was established in several rodent models simulating damage to the dopaminergic nigrostriatal pathway. The evidence includes neuroprotection against 6-hydroxydopamine (6-OHDA)-induced nigrostriatal damage in rats [56] and protection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced degeneration of nigrostriatal neurons in mice. Nevertheless, these results were not consistent enough to establish the neuroprotective potential of nicotine [57–59]. Subsequent experiments in parkinsonian nonhuman primates revealed that nicotine increased striatal dopaminergic components, including the DA transporter, tyrosine hydroxylase (TH), the vesicular monoamine transporter and DA [60, 61]. Data obtained from nonhuman primates with simulated PD strongly indicated the neuroprotective potential of nicotine. Other nAChRs subtypes, besides α7 nAChRs, are also protective against nigrostriatal damage. The α4β2 nAChR agonist ABT-089 confers neuroprotection in 6-OHDA-induced damage nigrostriatal neurons in rats [62]. In contrast, nicotine assisted neuroprotection against nigrostriatal damage was not reproduced in α4β2 nAChR knockout mice [63]. This result pinpoints the importance of β2 nAChRs for the effect of nicotine. One possible mechanism by which nicotine exhibits its protective role might be through chaperoning β2 nAChRs to the cell surface. Chaperoning could change the structures and functions of the endoplasmic reticulum, the secretory vesicles, and the Golgi apparatus of cells thereby reducing endoplasmic stress and enhancing cell survival [64, 65]. Drugs with different levels of agonistic efficacy, such as the allosteric α7 nAChR modulator galantamine and the α7 agonists ABT-107 and DMXB, are neuroprotective against 6-OHDA-induced damage to nigrostriatal neurons in rats [62, 66, 67]. Moreover, the α7 agonist PNU-282987 is also neuroprotective for MPTP-induced damage to nigrostriatal neurons in mice [68]. In contrast, the α7 nAChR antagonist methylycaconitine blocks the neuroprotective effect of nicotine [69]. These data indicate an important role for these receptors in protection against nigrostriatal damage. Nicotine may also be useful to manage levodopa-induced dyskinesias (LIDs). Experimental data from nonhuman primates and rodents indicate that drugs interacting with nAChRs may reduce LIDs [70]. Preliminary experiments with the general nAChR agonist nicotine showed a 60% decrease in LIDs in parkinsonian rodents and monkeys, indicating efficacy across species [52, 71–74]. Similarly varenicline, another general nAChR agonist [75], reduces LIDs by 50% [76]. Notably, pharmacological effects of nicotine were still observed in chronically treated nonhuman primates for over 1 year [73]. Nicotine decreases LIDs, regardless of route of administration. At least 1 month is required to observe the peak anti-dyskinetic effect of nicotine [74]. However, it also took 1 month to completely abolish the anti-dyskinetic effect of nicotine [76]. These observations suggest that long-term molecular changes are most likely responsible for mediating the reduction in LIDs by nicotine. For example, the α7-agonist ABT-107 decreases LIDs by 60%, which continued for several months [76]. Interestingly, the ABT-107-induced anti-dyskinetic effect persisted for about 1 month after termination, suggesting long-term molecular changes. Another α7 agonist ABT-126 produced similar results [77]. Dosing the α7 nAChR agonist AQW051 to Macaca fascicularis also yielded a 60% reduction in LIDs, with no further deterioration of parkinsonism [78], indicating the effectiveness of α7 nAChR agonists in nonhuman primates. In conclusion, drugs that are agonistic to α7 and β2 nAChR diminish LIDs by up to 60% with no harmful effects on parkinsonism. Since α7 nAChR might signify ideal drug target to improve LIDs in PD. Numerous intracellular mechanisms have been presented that facilitate the beneficial effects of activating α7 nAChRs against noxious insults. Mitogen-activated protein kinases have been associated with α7 nAChR assisted neuroprotection against a range of toxicities to PC12 cells, spinal cultures, and keratinocytes [79–82]. Further downstream mechanisms related with α7 nAChR assisted neuroprotection include increases heme oxygenase [83], phospholipase C [80], nerve growth factor [84], and proinflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) [85]. In contrast, nitric oxide (NO) [86], caspases, and reactive oxygen species (ROS) [83] are associated with the toxic effects of α7 nAChR. Taken together, these results suggest α7 nAChRs are important targets for developing therapeutic approaches for PD.
Alpha synuclein as a PD target
Growing evidence from various experimental studies demonstrates that the α-syn protein, which is the main constituent of LBs, aggregate and accumulate intraneuronally in the brains of patients with PD. However, the association between this protein and the beginning of symptomatic behaviors linked with PD remains to be explored [87]. Reports suggest that expression of α-syn participates in dopamine biosynthesis by decreasing the action of TH or altering its phosphorylation [88, 89]. The functional ability of α-syn is very strictly related to its structure. Hence, understanding the structure and normal function of α-syn will help to learn about its involvement in PD. Human α-syn is a small acidic protein composed of 140 amino acids and is coded by the α-syn gene [90]. Recent data has revealed that α-syn at Ser129 is prominently phosphorylated in patients with PD [91]. Mutation in the α-syn gene, reduced rate of degradation or likely modifications of α-syn, such as missense mutations, truncations, or chemical modifications due to oxidative stress are some of the prime reasons behind misfolding and aggregation of α-syn. These mutations are focused at the α-syn N-terminus, indicating that they hinder regular cellular function [87]. Current in vivo outcome [92] have indicated that suppressing α-syn may protect dopaminergic neurons. Silencing the human α-syn gene with miRNA-30-hSNCA (a miRNA-30 transcript) at striatal dopaminergic terminals decreases the motor abnormalities seen in α-syn expressing rats and guards against damage to SN dopaminergic neurons. Conversely, efforts to silence the human α-syn gene with a small hairpin (sh)RNA in rat SN protected only against human α-syn-induced forelimb dysfunction but had no protective effect on DA neurons [93]. Moreover, high levels of shRNA α-syn are toxic to DA neurons, while lower levels of human α-syn gene silencing protect neighboring neurons. Taken together, these findings suggest that silencing the human α-syn gene may be a new therapeutic tool to control behavioral dysfunctions in PD. A number of clinical studies have shown the potential use of α-syn as a biomarker for PD in cerebrospinal fluid (CSF) [94].
Bearing in mind the several advantages of using biomarkers, researchers have begun to measure α-syn in peripheral organs of patients with PD. Wang, Gibbons et al. reported that patients with PD have increased levels of α-syn in cutaneous autonomic fibers [95]. This might clarify the incidence of autonomic dysfunction in patients with advanced PD [96]. Fresh data reveal that α-syn protein, its gene expression, and its signaling pathways is a challenging but effective way to alter motor behavioral insufficiencies and physiological deficits related with synucleinopathies. Additionally, many original compounds have been acknowledged as feasible to hinder or reverse the aggregation process [97]. Toth and coworkers used a combination of experimental and computational techniques [98] and discovered a small-molecule drug-like phenylsulfonamide compound (ELN484228) with the capability to bind monomeric α-syn and, therefore, reduce its transfer to mature synapses. These findings suggest that changing the properties of α-syn may have potential therapeutic benefits for battling PD. An alternative method to targeting this protein is to diminish Ser129 phosphorylation by obstructing the relevant kinases. Phosphorylation of α-syn appears to play an important role in the formation of the fibrillar aggregates causing PD pathology, since increased phosphorylation of α-syn is directly proportional to aggregation and toxic buildup in neurons [99]. Nevertheless, this tactic presents a potential restriction as numerous kinases are capable of phosphorylating α-syn. To tackle this issue, improvement of protein dephosphorylation has also been projected by Braithwaite and associates [100]. Hence, change in α-syn phosphorylation holds tremendous potential as a strategy to develop disease-modifying therapeutic interventions. Lately, numerous reports have described prion-like dispersal of misfolded α-syn [101–103]. Hence, there has been increased development of immunotherapies targeting clearance of α-syn aggregates and oligomers, [104, 105].
As toxic oligomeric forms of α-syn can enter and gather in the plasma membrane, get secreted and circulate extracellularly, it provides us a strong evidence for immunotherapy [104, 106] and also encourages development of oligomer specific antibodies [107]. Vaccination against α-syn can occur either by active or passive immunity. Active immunity involves stimulating the host to produce antibodies against α-syn aggregates, while passive immunization involves external administration of anti-α-syn antibodies to patients. The reason for the immunization to α-syn is to wash out these neurotoxic aggregates and impede neuron-to-neuron propagation [108]. In recent evidence, monoclonal antibody against α-syn was found to antagonize entry and cell-to-cell transfer of α-syn in primary neurons [109]. Further, Games and associates found that passively immunizing mThy1-α-syn transgenic mice with the truncated α-syn C-terminus mitigated axonal and synaptic pathology, salvaged loss of TH fibers in the striatum, and regulated motor and memory dysfunction [110]. Immunotherapy targeting α-syn remains puzzling, as fundamental mechanisms are not fully understood. Thus, further clinical work is needed. Several in vivo models of PD have revealed that neuroinflammation is not only an initial event but also fast-tracks the progression of nigral cell death. Yan and colleagues confirmed that dopaminergic neurodegeneration prompted by misfolded α-syn was intensified by activation of microglia through α-syn phagocytosis and release of cytokines and ROS [111]. α-syn released from injured dopaminergic neurons activates microglia and stimulates the release of proinflammatory mediators, causing the chronic and progressive dopaminergic neural degeneration linked with PD [112, 113]. Nevertheless, the relationship between PD-associated α-syn aggregation and microglial-induced neuroinflammation remains obscure. A recent article indicates that Toll-like receptor (TLR) 4 and TLR2 was observed to be essential for α-syn assisted activation of microglia [111, 114, 115]. Also, α-syn fibrils induced release of IL-1β from monocytes is facilitated by TLR2 [116]. Furthermore, the ability of microglia to detect misfolded α-syn increases the neurotoxic effect through the production of proinflammatory cytokines and ROS [117, 118]. Additionally, α-syn triggers constituents of innate and adaptive immune systems in patients with PD, which can alter pathological processes in animal models of PD [119, 120]. In fresh evidence by Thome and colleagues fractalkine signaling was observed to increase the inflammatory response in α-syn model of PD. Hence this report indicates that fractalkine is essential in the development of synucleinopathies, and could tentatively be a target for neuroprotective therapies for PD [121]. Therefore, studies are now focused on delineating the mechanisms and pathways linking neuroinflammation and α-syn.
Rho kinase as a PD target
Rho is a small GTP-binding protein that plays a crucial role in many cellular functions. RhoA is a member of the Rho family and participates in cellular mechanisms that act on its direct downstream effector Rho-associated kinase (ROCK) [122]. Irregular activation of the RhoA/ROCK pathway is seen in models of inflammatory demyelinating diseases, stroke, spinal cord injury, AD, and other diseases [29, 123]. A number of key mechanisms associated with activation of ROCK also play a major role in the degeneration of dopaminergic neurons [122]. Inhibiting ROCK confers protection to dopaminergic neurons in a MPTP/MPP+-induced in vitro and in vivo model of PD [124–127]. ROCK inhibitors protect dopaminergic neurons in a primary neuroglia mesencephalic culture intoxicated with MPP+ [125–127]. Labandeira-Garcia and associates reported that NADPH-oxidase through NF-κB, triggers ROCK, which, in turn, stimulates NADPH-oxidase [122]. This finding is congruent with earlier experiments in peripheral cells showing that NADPH-oxidase-derived superoxide actuates NF-κB [128], and NF-κB stimulates ROCK [129]. More than a few experimental studies have shown that activating microglia and generating ROS with NADPH-oxidase represent the early phases of dopaminergic cell death and that both aspects act synergistically with other elements to induce dopaminergic cell death as the primary stage of PD pathology [130]. Additionally, ROCK and the angiotensin (AT) system help deciding the fate of dopaminergic neurons. AT1 and AT2 receptors and NADPH oxidase have been found in dopaminergic neurons and nigral glial population [131]. In line with these observations a decrease in AT1 receptor levels, decreased MPTP-induced expression of RhoA and ROCK activity in the mouse SN. In cultured cells, the additive effect of MPP+ and AT-II-induced dopaminergic neuron death was also antagonized by the ROCK inhibitor Y-27632, indicating a vital interaction between the AT-II/AT1 and the RhoA/ROCK II pathway in MPTP-induced dopaminergic neuron death [126]. This evidence reveals that activating ROCK and NADPH may be strongly involved in the AT-II-induced inflammatory response, which is blocked by ROCK inhibitors.
Labandeira-Garcia and coworkers demonstrated that estrogen impedes the MPTP or 6-OHDA-induced neuroinflammatory response and dopaminergic cell death [122]. Also, inhibiting the nigral rennin-AT system works in favor with the anti-inflammatory and neuroprotective effects of estrogen [132–134]. Reports by Rodriguez and Dominguez reconfirmed these findings by demonstrating the protective effect of the ROCK inhibitor Y-27632 in dopaminergic cell death induced by estrogen depletion [135]. In addition, candesartan an AT1 receptor antagonist, blocks the estrogen-induced increase in ROCK activity [135]. Taken together, these outcomes recommend that stimulating ROCK might play a crucial role in increased susceptibility of dopaminergic neurons after the reduction in estrogen. Latter it was found that this outcome was facilitated by stimulating the AT-II/AT1 pathway. Several mechanisms seem to be associated in ROCK-induced dopaminergic susceptibility and the neuroprotective effects induced by the ROCK inhibitors [122]. Many prospective ROCK targets in apoptotic signaling have been proposed, including interactions with the pro-apoptotic factors glycogen synthase kinase 3 beta, Bcl-2 proteins, and protein kinase B [136]. The axon-stabilizing effects in injured neurons may denote another mechanism of neuroprotection for dopaminergic neurons after inhibiting ROCK [137]. Concurrent treatment with a ROCK inhibitor substantially augmented the number of surviving dopaminergic neurons [125, 127]. Nevertheless, glial cells play an important role in the neuroprotective effects of ROCK inhibition on dopaminergic [124, 125, 127] and other neurons [138]. Inhibiting ROCK decreases cell size and number of filopodia associated with microglial activation. In addition, ROCK inhibitor subdues activation of NADPH-oxidase and blocks the release of inflammatory cytokines, such as TNF-α and IL-β [76, 127]. In conclusion, ROCK is associated with a large number of cellular processes; thus, several mechanisms may be responsible for its protective effect on dopaminergic neurons.
Leucine-rich repeat kinase 2 as a PD target
Non-synonymous point mutations in the leucine-rich repeat kinase 2 (LRRK2/PARK8) gene is the leading genetic reason for autosomal dominant PD. Mechanisms specific to LRRK2 are being revealed with fresh discoveries of LRRK2 substrates [139]. The LRRK2 gene encodes a 286 kDa protein and is located on human chromosome 12. Cellular LRRK2 carries out numerous functions including modulation of protein translation [140, 141], changing microtubule dynamics [142], participating in endocytosis [143] and autophagy [144]. G2019S is the most common PD-associated LRKK2 mutation identified compared to other mutations. Majority of patients with LRRK2 PD lose SN neurons having fluctuating levels of LB inclusions, tau neurofibrillary tangles, or a combination of both [145]. Consequently, targeting LRRK2 is a striking option for developing PD therapeutics. Communication between the Roc-COR domains is significant for controlling LRRK2 GTPase activity because of the enzymatic capacity of LRRK2 mutants. Decreased GTPase activities in Y1699C and R1441C mutants suggest that the GTP-bound state is related with the disease. Hence, variations in LRRK2 GTPase activity either by obstructing the GTP-binding site of the ROC domain or by increasing GTPase activity embraces the therapeutic potential for LRRK2-associated PD. Nevertheless, no such study has attained this objective [146].
Mutations in the LRRK2 kinase domain change kinase activities, indicating that the pathogenicity of LRRK2 is facilitated through a non-kinase mechanism [146–148]. Therefore, the current attention on LRRK2 is based on inhibition of its kinase domain. Oxidative stress induced by LRRK2 was salvaged by DJ-1 in a neuroblastoma cell line [149], suggesting that antioxidants are potential inhibitors of LRRK2 kinase toxicity. The selection of small molecule inhibitors mostly depends on their capability to antagonize LRRK2 phosphorylation sites, such as, constitutive phospho sites, LRRK2 auto-phosphorylation sites, and those altered in the inhibitor-resistant mutant [150]. Numerous lead molecules have been identified for LRRK2 that are specific, potent, brain penetrating and have druggable attributes that will lead to a clinical trial [151]. Another promising possibility is to search for mutation-specific inhibitors that have no effect on wild-type LRRK2. However, the pathogenic role of mutant LRRK2 is not understood making the clinical endpoint difficult to predict [151]. Moreover, destabilizing microtubules has been proposed as a junctional point of idiopathic and genetic forms of Parkinsonism. As microtubule dysfunction occurs in patients with PD and mitigating this flaw restores control of the PD phenotype [142], controlling microtubule dynamics could serve as a potential therapeutic target. LRRK2 is also strongly associated with autophagy [152] and endocytosis [153]. Therefore, the endolysosomal and autophagosome-lysosome pathways are prospective target points to correct the effects of LRRK2. Use of LRRK2 inhibitors as a therapeutic option needs to be investigated in-depth, as LRRK2 significantly participates in immune function, metabolism, and kidney homeostasis [154].
Nuclear receptor related 1 protein as a PD target
Nuclear receptor related 1 protein (Nurr1) is a member of the ligand-stimulated transcription factors called nuclear receptors. Nurr1 does not have a hydrophobic site for ligand binding as in other nuclear receptors; thus, nuclear receptor function is ligand-independent [155, 156]. Nurr1 plays an important role in the development and specification of midbrain DA neurons throughout life [157]. Deficiency of Nurr1 in developed DA neurons results a decrease in DA neuron markers and motor impairments simulating early symptoms of PD. Removing Nurr1 from grown-up rodents results [158] in decreased expression of genes related with oxidative phosphorylation and mitochondrial function, suggesting a role for Nurr1 in the preservation of midbrain dopaminergic neurons [159–161]. Puigserver and colleagues performed a large meta-analysis of genome-wide gene expression studies and revealed that genes encrypting proteins participating in oxidative phosphorylation are highly dysfunctional in the remaining dopaminergic neurons of patients with PD [162]. The precise mechanism by which Nurr1 activates or inhibits these mitochondrial genes is not clearly known. However, other transcription factors essential for expressing nuclear respiratory genes, such as the nuclear respiratory factors NrF1 and NrF2, or the transcriptional co-activator peroxisome proliferator-activated receptor-γ co-activator 1α, which is the chief controller of mitochondrial biogenesis and cellular respiration, might functionally bind with Nurr1 [162]. Neuroprotective effect by CREB in neurons exposed to oxidative stress and the regulation of neuroprotective genes is facilitated by Nurr1 [163]. Also, increased expression of survival-promoting brain-derived neurotrophic factor is facilitated by increased Nurr1 expression induced by the NMDA receptor [164]. Nurr1 agonist increases transcriptional activation of dopaminergic specific genes and decreases the expression of proinflammatory genes in microglia [165]. Nurr1 is identified to be activated in mouse microglia in vivo after stereotaxic injection of lipopolysaccharide (LPS). Apart from having vital roles within dopaminergic neurons, Nurr1 has been projected to be part of an anti-inflammatory pathway in astrocytes and microglia that guard dopaminergic neurons from inflammation induced cytotoxicity [166]. In line with these reports, treatment with a Nurr1 agonist conferred neuroprotective and anti-inflammatory effects in 6-OHDA lesion model of PD [167]. Lately, pathologic α-syn aggregates were found to downregulate Nurr and its transcriptional targets in midbrain DA neurons [119, 168]. Numerous additional genes may be downregulated due to the toxicity of α-syn. α-Syn may impede glial cell-derived neurotrophic factor (GDNF)-signaling by inhibiting Nurr1 and its transcriptional target Ret [169]. Overexpressing α-syn blocks GDNF signaling and inhibits the ability of GDNF to protect dopaminergic neurons against α-syn-induced toxicity [170]. Taken together, these findings suggest that the GDNF-Ret-Nurr1 pathway is an exciting target for PD therapeutic interventions. Outcomes from various experiments have suggested that nuclear α-syn may be responsible, at least in part, for the pathological process, and that Nurr1 is one of its targets that is affected directly or indirectly [171, 172]. Therefore, decrease in expression of Nurr1 could spur dysfunction of dopaminergic neurons and collude the progression of PD; thereby making this protein a promising therapeutic target for PD [169].
Glucagon-like peptide 1 receptor as a PD target
Currently there has been a gush of interest in glucagon-like peptide 1 receptor (GLP-1R) as a prospective target for PD therapies. Under normal physiological conditions, the GLP-1 is released from intestinal epithelial cells in anticipation of high glucose and acts on pancreatic β-cells, liver, and muscle to reduce glucose levels. Several GLP-1R agonists have been used to treat type-II diabetes. Various experiments have indicated a connection between PD and type-II diabetes, as both diseases share molecular networks, such as inflammation [173]. GLP-1R agonist exenatide has growth factor-like properties and numerous positive effects in animal models of neurodegenerative disease and acute brain injury, such as preserving synapse plasticity, stimulating neurogenesis, decreasing protein aggregation and inflammation [174]. Although the fundamental mechanisms are not fully clarified, one effect of GLP-1R stimulation is to trigger the transcription factor cyclic adenosine monophosphate response element binding protein, which is associated with neuronal survival and synaptic plasticity. A recent clinical trial involving patients with PD administered daily injections of exenatide for 12 months. Exenatide was found to improve motor and cognitive functions [175] over 12 months even though the drug was eliminated from the system [176]. Although these data hints at a disease-modifying effect, the results should be considered cautiously, as control patients were not given placebo. Presently, a controlled phase II trial in patients with PD is ongoing, using a slow-release form of exenatide (Bydureon), which is taken in a once/week injection. Even if exenatide has shown encouraging results, several other pharmacological approaches targeting GLP-1R are marketed for type-II diabetes and possibly have a more promising profile. For example, the GLP-1R agonists liraglutide and lixisenatide have lengthy half-lives, are effective in in vivo AD models [177–179], and can normalize Ca2+ levels in human neuroblastoma cells [180]. In the immediate future, several trials will aim to achieve disease modification in PD through chronic dosing of different GLP-1R agonist.
Acid-sensing ion channel as a PD target
Conserving the physiological pH of interstitial fluid is critical for normal cellular functions. Tissue acidosis is a common pathological variation causing abnormal activation of acid-sensing ion channels (ASICs), which may considerably contribute to mitochondrial dysfunction, inflammation, and other pathological mechanisms, including stroke, pain, and psychiatric conditions. Hence, it has become clear that ASICs are important in the development of neurological diseases [181]. Intra and extracellular pH is sustained between 7.3-7.0 under normal physiological conditions. However, increased neuronal excitability changes cellular pH to trigger various ion channels and receptors in cell membranes, including voltage-gated and ligand-gated ion channels [182, 183]. ASICs, which were first cloned by Waldmann and associates [184], are stimulated in response to acidic extracellular pH [185]. Nevertheless, the practical roles of ASICs in central and peripheral components of the nervous system remain to be determined. At present, neurodegenerative diseases including PD and AD, are treatable but can't be cured by existing mode of treatment. Pre-clinical and clinical studies have suggested that overload of Ca2+, oxidative stress, mitochondrial dysfunction, energy metabolism, and acidosis are involved in neurodegenerative processes. These mechanisms frequently result in tissue acidification causing lactic acidosis, which further exacerbates neuronal damage. Similar observations have also been reported in the brains of patients with PD and in the typical MPTP-induced PD animal model [186, 187].
One study showed that mitochondrial ASIC1a might be a significant modulator of mitochondrial permeability transition pores and increase neuronal death due to oxidative stress [188]. Furthermore, Arias et al. discovered that MPTP-treated mice have brain acidosis and that treatment with the ASIC inhibitors amiloride and PcTx-1 confers protection against degeneration of SN neurons by preventing apoptosis and by decreasing DA and its transporter [189]. Moreover, the absence of endogenous parkin protein or mutations in the parkin gene cause irregular ASIC currents resulting in protein degradation and dopaminergic neuronal injury, indicating that ASIC currents may facilitate the essential pathology in PD [190]. Exposing rat microglial cells to LPS also increases ASIC1 and ASIC2a expression levels and stimulates production of inflammatory cytokines [170]. These reports indicate that controlling microglial ASIC function might control the disease pathology in PD.
Dopamine heteroreceptor complexes as PD target
The finding that DA-1 receptor (D1R) and D2R exists in the brain in the form of heteroreceptor complexes helped in understanding the therapeutic actions and side effects of L-DOPA and DA receptor agonists in the treatment of PD [191, 192]. Various heteroreceptors, including adenosine-A2A-D2R (A2AR-D2R), A2AR-D2R-metabotrophic glutamate receptor (A2AR-D2R-mGluR5), D2R-N-methyl-D-aspartate receptor (D2R-NMDAR), D1R-D3R, A1R-D1R, D1R-NMDAR, and A1R-D1R-D3R heteroreceptor complexes could be potential targets for alleviating the side effect effects associated with L-DOPA. The finding that coactivation of the D1R and D2R protomers leads to calcium signaling in the striatum [193] suggested that the mental side effects of DA agonist treatment, such as gambling, psychosis, and hallucinations, can include stimulation of the D1R and D2R protomers of this heteroreceptor complex.
D1R-D3R heteroreceptor complexes
An abnormal increase in D1R protomer signaling produces LID [194]. Therefore, antagonizing over-activated D1R protomer signaling in various types of homo and heteroreceptor complexes could be a promising target for treating LID.
D2R-D3R heteroreceptor complexes
A detailed understanding of the function and potential dysfunction D2R-D4.2R and D2R-D4.4R heteroreceptor complexes in PD is lacking. However, due to the activity of antiparkinsonian drugs, such as L-DOPA and apomorphine, on D4Rs, they also act on D2R-D4.2R and D2R-D4.4R heteroreceptor complexes, which could open new doors by increasing the plasticity of responses to L-DOPA treatment [195].
A2AR-D2R heteroreceptor complexes
Fuxe and colleagues demonstrated antagonistic A2AR-D2R interactions at the level of D2R recognition in striatum from naive and hemiparkinson rats [196] and also at the level of the striatopallidal GABA pathway and its brain circuits in a naive rat model of PD [197]. These findings lead to the hypothesis that A2AR antagonists could be novel antiparkinsonian drugs by acting the A2AR homomer and protomer in the striatopallidal GABA neurons through antagonistic A2AR-D2R receptor-receptor interactions [198] resulting in antidyskinetic actions, which could help lessen the wearing off of the therapeutic effects of L-DOPA.
A2AR-D2R-mGluR5 heteroreceptor complexes
Due to the supremacy of co-activated A2AR and GluR5R receptor protomer signaling, chronic treatment with D2R agonists and L-DOPA is been often observed to produce a strong inhibition of D2R protomer signaling in A2AR-D2R-mGluR5 heteroreceptor complexes [199]. Therefore in the treatment of PD, we might need to aim A2AR and mGluR5 receptors located on striato-pallidal GABA neurons by co-antagonizing them via using heterobivalent compounds. These compounds will encompass A2A antagonistic and mGlu5 antagonist/negative allosteric modulator properties to eliminate the inhibition on D2R protomer signaling [195].
D2R-NMDAR (NR2B containing) heteroreceptor complexes
Liu and coworkers established that the NR2B subunit of the NMDAR directly binds with the D2R [200]. Based on this finding, it is plausible to consider aiming D2R-NMDAR (NR2B containing) heteroreceptor complexes by co-treatment of a NR2B-selective NMDA antagonist with an mGluR5 antagonist/negative allosteric mGluR5 modulator to treat PD.
A1R-D1R heteroreceptor complexes
Similar to antagonistic A2AR-D2R interactions, there is also an existence for antagonistic interactions between A1R-D1R in the modulation of GABA release in hemi-parkinsonian rat [198] and also in D1R agonist-stimulated motor effects in rodents [201]. Interestingly, it is worth to note that A1R agonists in rabbits can neutralize D1R agonist-stimulated oral dyskinesias [202]. Therefore with respect to the function of D1Rs in facilitating LIDs [203], these findings suggest the possibility that initial co-treatment with A1R agonists in A1RD1R heteroreceptor complexes can neutralize the development of LIDs. Hence, pharmacology of A1R should be investigated in cellular models with A1R-D1R and A1R-D1R-D3R heteroreceptor complexes as targets. Cumulatively the existing investigations on D1R and D2R heteroreceptor complexes in the basal ganglia propose a novel molecular mechanism for understanding the loss of efficacy of L-DOPA and DA receptor agonists and the development of dyskinesias by L-DOPA and DA receptor agonist.
In addition to the above mentioned heterereceptor complexes, growth hormone secretagogue receptor (GHSR1a) also is documented to form dimer with D1R. GHSR1a is a biological target of ghrelin peptide that is extensively disseminated throughout the brain. GHSR1a and D1R have been demonstrated to be co-expressed in areas including ventral tegmental areas, SN, and midbrain. Dimerization of D1R with GHSR1a amplifies cAMP signaling and subsequently D1R signaling. This suggests that dimerization is associated with mood, learning, and memory [204].
ATP13A2 as a PD target
The significance of ATP13A2 also known as PARK9 in PD has increased with the finding that mutations in this gene cause Kufor-Rakeb syndrome (KRS) which is an autosomal recessive, juvenile-onset form of parkinsonism [205]. With the knowledge that ATP13A2 is a disease-insinuating gene; a series of laboratory studies were initiated to unravel the molecular function and classify the pathophysiological mechanisms that result in the clinical phenotype. Early indications from various disease models suggested that ATP13A2 participates in Mn+2 and Zn+2 metabolism [206, 207], mitochondrial bioenergetics [208–210], and in the autophagy-lysosomal pathway [209, 211, 212]. Moreover, ATP13A2 has been exposed to control α-syn metabolism, one of the major components of LB [212]. Detecting this monogenic form of PD has spurred to a number of experiments examining the role of this gene in sporadic PD. Numerous solo heterozygous ATP13A2 mutations have been recognized with greater incidence in early-onset PD as compared to that in healthy controls, indicating that these mutations are an age-of-onset modifier or a risk factor for PD [213, 214]. Preliminary indications signifying the role of ATP13A2 in sporadic PD came from the observation that ATP13A2 mRNA levels increase in surviving dopaminergic SN neurons from brains of patients with sporadic PD [215]. Another distinct finding also confirmed a noticeable increase in the ATP13A2 protein in the SN and other brain regions of patients with sporadic PD [210]. Consequent studies consistently reported substantial changes in ATP13A2 levels in the brains of patients with sporadic PD [211, 216]. Additional evidence for a role of ATP13A2 in sporadic PD comes from the consequences of its absence. Functional ATP13A2 appears to be indispensable for breaking down hoarded or aggregated α-syn [212, 217]. Any slight deviation in its competence over a lifetime could contribute to the progression of synucleinopathy. Moreover, participation of ATP13A2 in externalization of α-syn through exosomes may be important in the development of PD, either by removing α-syn from the cytoplasm or by disseminating the protein into neighboring cells [207]. ATP13A2 may also unravel the relationship between α-syn metabolism and mitochondrial dysfunction in patients with sporadic PD. Mounting evidence puts mitochondrial dysfunction during neurodegeneration at the center of both familial [218, 219] and sporadic PD [220, 221]. Cell lines derived from a patient with KRS [208, 222] and mammalian ATP13A2-silenced cell lines [209, 210] had pathogenic changes in mitochondria function. The function of ATP13A2 in lysosomes and mitochondria appears to be directly connected to its role as a Zn+2 transporter and signifies the common fiber that connects these two apparently dissimilar organelle systems in PD [217, 222]. Dysregulation of zinc connected to insufficiency of ATP13A2 could clarify the established link between sporadic PD, higher brain zinc levels, and other tissues [223, 224]. Taken together these evidences back the participation of ATP13A2 in numerous overlapping pathogenic pathways intimately associated with PD. Additional examination on the expression levels of ATP13A2 as a target and their functional significances in these mechanisms, using suitable PD models, will be essential to widen the learning of ATP13A2 in PD pathogenesis.
Glutaredoxin as a PD target
Mutations in LRRK2 are related to autosomal dominant PD, and many of these mutations are observed to increase cellular ROS levels. Therefore, antioxidant proteins are essential to reinstate the redox balance and preserve cell viability. Studies in the past decade have begun to establish the prominence of redox proteins in facilitating neuroprotection in PD models [225]. Glutaredoxin (Grx) is small redox enzyme that precisely catalyzes the removal of glutathione from cysteine residues [226], thereby reinstating the function of proteins whose function is altered upon glutathionylation [227]. Grx1 is a well-recognized major deglutathionylating enzyme, exhibiting about 5000-fold greater catalytic competence for deglutathionylation compared to Trx1 [228]. The primary finding demonstarting that Grx1 participates in protecting dopaminergic neurons was observed when Grx1 mRNA and protein levels were elevated in mouse brain homogenate post-MPTP treatment [229], indicating a homeostatic upregulation of the chemical intoxication. Another study reported that female mice have higher Grx1 content than that of males and are more resistant to MPTP-induced dopaminergic cell death [230]. Supplementary experiments provided additional evidence that Grx1 facilitates neuronal protection. Treating SH-SY5Y cells with the pro-oxidant drug L-DOPA increased apoptosis. After examining the mechanism of drug-induced cell death, Grx1 was particularly inactivated compared to other redox enzymes [231]. This finding led to the suggestion that Grx1 plays an important role upholding neuronal cell viability. To check this concept, Grx1 was silenced in SH-SY5Y cells. Cells exposed to Grx1-siRNA presented an increased level of apoptosis, similar to control cells with non-targeting siRNA [231]. This result was also confirmed by Grx1 knockdown of Neuro-2a cells via shRNA, resulting in cell death [232]. Taken together, these results suggest that Grx1 plays a neuroprotective role in cultured cells. Although promising results have been seen in vitro, the neuroprotective role of Grx1 has not been investigated in vivo and its implications for PD in humans [233]. Johnson and colleagues examined the role of Grx1 in conferring protection to dopaminergic neurons in a C. elegans PD model. C. elegans worms overexpressing α-syn, TH, LRRK2-G2019S, and LRRK2-R1441C in dopaminergic neurons, simulating both familial and sporadic PD model, were crisscrossed with worms missing the Grx1 homolog called GLRX-10. Each of the hybrid worm lines missing the Grx1 homolog displayed a significantly more severe PD phenotype compared to the control worms with endogenous GLRX-10. Moreover, re-expression of wild-type GLRX-10, in the dopaminergic neurons of the GLRX-10−/−/LRRK2-R1441C worms salvaged the exacerbated PD-like phenotypes [225]. Johnson and coworkers also inspected Grx1 content in brain samples of patients with and without PD. Immunoblot analysis of midbrain homogenates showed an overall reduction in the Grx1 protein in the midbrains of patients with PD compared to control subjects. Additionally, midbrain tissue slices revealed that more dopaminergic neurons were devoid of Grx1 in patients with PD compared to those in control subjects. Overall, these data contributes to the in vivo evidence that Grx1 protects dopaminergic neurons in familial and sporadic PD. Also, it reveals that Grx1 protein content is reduced in PD brains, indicating that decreased Grx1 with aging precedes PD [225]. Furthermore, Johnson and colleagues found that loss of the Grx1 homolog in worms aggravated LRRK2-induced dopaminergic neuronal toxicity. Over production of DA or α-syn was observed to be responsible for the loss of Grx1 which further worsened PD phenotypes in models of sporadic PD. However, re-expression of the catalytically active site of the Grx1 homolog avoided the intensified toxicity. Largely these data noticeably implicates that reversible protein glutathionylation is the most likely mechanistic base for the catalytic role of Grx1 in facilitating protection to dopaminergic neuronal against the oxidative stress associated with overexpression of mutant α-syn or LRRK2. Thus, elimination of the glutathione modification from vital regulatory proteins by Grx1 is obligatory to reinstate their function and preserve cellular homeostasis and cell survival. Based on the available data and the need to combat oxidative stress in PD, Grx1 can serve as a novel therapeutic strategy for PD [225].
Metabotropic glutamate receptor as a PD target
Nickols and Conn have reported all pertinent preclinical indications that will lead to expanding mGlu4 receptor-positive allosteric modulators (PAMs) and mGlu5 receptor negative allosteric modulators (NAMs) as potential anti-parkinsonian drugs [234]. One major drawback in the existing treatment of PD is motor fluctuations and LIDs. No treatments are available for LIDs except weak NMDA channel blockers and amantadine. In a recent report by Xia and associates, it was demonstrated that the selective mGluR5 antagonist, 2-methyl-6- (phenylethynyl) pyridine in the presence of rotenone intoxication, mitigates DNA damage in MN9D dopaminergic neurons by decreasing intracellular calcium release which further decreases mitochondrial dysfunction and endoplasmic reticulum stress caused by ROS [235]. mGlu5 receptor NAMs have demonstrated in vivo activity by decreasing both LIDs and parkinsonian motor symptoms. These drugs may protect nigral dopaminergic neurons in rodents and non-human primates [236, 237]. Mavoglurant (AFQ056) is an mGlu5 receptor NAM that exhibits substantial anti-dyskinetic activity without restricting the therapeutic efficacy of L-DOPA in preliminary, Phase 2a, and Phase 2b clinical trials [238]. However, mavoglurant was discontinued, as it was not found active in further studies. Due to the phenotypic response by glutamate in manifesting motor behavior, it is plausible that mGlu5 receptor blockade is a reliable strategy to treat LIDs. Similarly, dipraglurant, another mGlu5 receptor NAM, showed good safety and acceptability profile with noteworthy efficacy against LIDs in a Phase 2a study [238]. Nevertheless, all of these studies were short-term and it remains unclear whether mGlu5 receptor NAMs can change synaptic plasticity underlying LIDs or whether they have symptomatic effects.
The mGlu4 receptor is another recognized target for treating PD, and several orthosteric agonists and mGlu4 receptor PAMs have presented symptomatic efficacy in experimental models of parkinsonism [234, 239]. Stimulating mGlu4 receptors could attenuate the degeneration of dopaminergic neurons in PD [240, 241]. Jeff Conn and colleagues discovered that mGlu4 receptors form homodimers at striatopallidal synapses, whereas they form inter-group heterodimers with mGlu2 receptors at corticostriatal synapses. PHCCC, an mGlu4 receptor PAM, selectively triggers mGlu4 receptor homodimers, whereas other PAMs, such as VU0155041 and Lu AF21934, also stimulate mGlu4/mGlu2 heteromers [242]. It will be exciting to associate the activity of the two dissimilar categories of PAMs in models of parkinsonism. mGlu3 receptor PAMs still have the potential as disease-modifying agents in PD since, mGlu3 receptors activate the production of neurotrophic factors such as GDNF and transforming growth factor-β [243].
Niacin receptor 1 as a PD target
Niacin receptor 1 (GPR109A) is a high-affinity niacin receptor that is overexpressed in the SN of patients with PD. Niacin is the primary source to synthesize NAD-NADH, which is required for DA production. Hence, niacin supplementation might assist three purposes. First, DA synthesis in the striatum could increase by supplying NADPH, and a higher NAD/NADH ratio would boost mitochondrial functions and decrease inflammation through GPR109A-related mechanisms [244]. The neuroprotective role of niacin in PD was suggested by an unreliable report [245]. Furthermore, various reports have documented that chronic use of levodopa decreases the level of niacin by interfering with tryptophan breakdown. Tryptophan metabolism itself is impaired in patients with untreated PD [246, 247]. Long-term niacin deficiency causes mitochondrial dysfunction, oxidative stress, and death of DA producing cells in the SN [248]. Therefore, the decrease in mitochondrial function may actually be an outcome of inflammation resulting from chronic niacin deficiency. Niacin is a very important factor for producing NADPH. An altered NAD-NADH ratio is implicated in ATP depletion and mitochondrial dysfunction, resulting in neuronal death in neurodegenerative diseases, such as PD [244]. NADPH is required to synthesize of tetrahydrofolate, which is crucial coenzyme required for DA synthesis. With decreased levels of niacin in PD, NADPH levels decrease and lessen striatal DA production. Therefore, it is reasonable to deliver a natural source for the coenzyme rather than supplying a ready-made coenzyme, which is niacin [244]. Wakade and colleagues suggested that niacin plays a significant role protecting dopaminergic neurons in PD via the GPR109A pathway, either by augmenting blood supply to hypoperfused areas in the brain or by increasing anti-inflammatory mechanisms. GPR109A is also called hydroxycarboxylic acid receptor 2 (HCAR2), HM74a in humans, and PUMA-G in mice. GPR109A is a G-protein-coupled high-affinity niacin receptor [249] and beta-hydroxyl butyrate is its physiological ligand [250]. GPR109A is expressed in different human tissues, including the brain [251]. However, immune cells express the highest amount of the GPR109A protein in humans [251]. GPR109A and its ligands have been recognized for their anti-inflammatory properties in a range of in vivo and in vitro experimental models, including the retina, skin, and gut. Although the anti-inflammatory effect of GPR109A has not been shown in the brain, one report has indicated that GPR109A agonists subdue LPS-induced inflammation via the NF-κB pathway in the gut [252]. Also, activation of GPR109A in mouse macrophages by interferon gamma substantiates the role of GPR109A in inflammation [253]. However knocking down GPR109A has no effect on NAD-NADH or DA production. NADH is required for DA synthesis and cultured PC12 cells show increased TH and DA levels [254] by augmenting the reuse of quinonoid dihydrobiopterin to tetrahydrobiopterin [255]. Additionally, systemic administration of NADH increases urinary excretion of homovanillinic acid, which is suggestive of increased endogenous L-DOPA synthesis [256]. Therefore, DA may be augmented directly by supplying NADH in vitro. Niacin is involved in many other signaling pathways, including increasing nitric oxide, arachidonic acid, and prostaglandin D2 and E2 levels [257, 258]. Niacin also stimulates adiponectin levels. Adiponectin may be neuroprotective by reducing oxidative damage [259, 260]. Therefore, GPR109A could be useful to battle acute or chronic inflammation in PD [244] and reduce PD progression.
Apolipoprotein E as a PD target
The Apolipoprotein E (APOE) gene is situated on chromosome 19 and encrypts a plasma glycoprotein composed of 299 amino acids, which is linked with low density lipoprotein (LDL), very low density lipoprotein (VLDL), and high-density lipoprotein (HDL) [261]. APOE is synthesized ubiquitously in the body, including the brain, liver, skin, and in macrophages [262]. Several major APOE isoforms have been described, such as E2, E3, and E4. Six different APOE phenotypes are detectable due to two single nucleotide polymorphisms at amino acid positions 112 and 158. These mutations could change the protein charge and stability; thus, inducing unique physiological functions. APOE decreases blood lipid and lipoprotein levels. APOE acts as a ligand for members of LDL receptor family and mediates removal of lipoproteins from the circulation for excretion via the liver. APOE forms VLDL and chylomicrons and changes the activity of other lipid metabolism-influenced proteins and enzymes, such as lipoprotein and lipase hepatic lipase. New data suggest that APOE and its isoform function beyond lipid metabolism to encompass maintenance of normal brain function [263]. Numerous APOE isoforms with structural differences have been discovered that serve functions in mitochondrial signaling, neuronal signaling, neuroinflammation, brain lipid transport, and glucose metabolism. Understanding the mutations in APOE, their isoforms, and their structural properties will help determine role in various diseases and to advance therapeutic strategies. Targeting APOE could help with risk assessment, diagnosis, prevention, and treatment of various neurodegenerative and cardiovascular diseases in humans. APOE is mostly manufactured by astrocytes or by neurons under certain pathological conditions [264, 265]. The human brain comprises up to 25% of the body's cholesterol, which is indispensable for myelin production, function, and its integrity. Cholesterol is a crucial constituent of axonal growth and synaptic formation, which are vital for learning and memory [266, 267]. Regulating cholesterol in the central nervous system (CNS) is independent of the peripheral system. Any dysfunction in CNS cholesterol could have influence on aging and the development of certain neurodegenerative diseases. APOE delivers cholesterol to neurons [268, 269], but the BBB limits the interchange of lipoproteins and APOE between the peripheral and CNS systems. One study reported that brain injury causes an increase in APOE protein content in the brain [270] but the mechanisms relating APOE to all of these biological processes have been elusive. Recently, association between APOE and PD has been demonstrated [271, 272]. Maximum studies were unsuccessful to testify any relation between APOE ε4 and vulnerability to PD and PD-associated cognitive dysfunction [273, 274]. Various experiments have shown that APOE ε4 is a risk factor for age of onset and reduced cognitive functioning associated with PD. Though, ε2 is considered a weak or inconsistent risk factor for PD [274–277], but a meta-analysis suggested that the ε2 allele is related with advanced risk for PD [72, 278]. However, another study reported that the ε4 allele is associated with PD development [271]. Until now, experiments concentrating on the role of APOE in PD remain largely inconclusive and more studies are needed to determine whether APOE is a PD target.
CONCLUSION AND PERSPECTIVE
Irrespective of the intensive research from the past few decades, all existing therapies for PD are focused on providing symptomatic relief and not towards achieving neuroprotective or disease-modifying strategies. Therefore, there is a serious need for an exemplar shift in discovering novel and effective targets that cures or stops the advancement of PD. However, discovering a definitive cure for PD is a highly difficult task, as many obstacles need to be handled for the development of effective disease-modifying or neuroprotective treatment. Novel targets can be discovered by the development of translationally accurate disease models, as current models of PD do not sufficiently simulate the advanced nature of the disease. Furthermore, a multi-target therapeutic approach (polypharmacology) might also combat the disease from various pathways, thus significantly improving the likelihood of disease modification. In our existing write-up and also as illustrated in Figure 1, we have summarized recent findings concerning preclinical investigations of various targets linked to PD. Current developments in understanding the molecular and cellular mechanisms of PD have facilitated us with a quick progress in identifying new PD targets. Additionally, multiple models can also be used to measure whether a target is capable of impacting different stages involved in PD pathogenesis. Quite a few targets have been examined in clinical trials designed to evaluate disease modification in PD, but all haven't been satisfactorily fruitful. Over the past 3 years, clinical trials exploring the possibility of coenzyme Q10, creatine, dopaminergic receptor, adeno-associated virus serotype 2 (AAV)-neuturin, and PPAR-γ receptors have reported negative outcomes. Regardless of these drawbacks, clinical trials presently are testing the potential of targets such as, calcium channel blocker, adenosine receptor, neuronal nicotinic receptor, glutathione, AAV2-GDNF, as well as active and passive immunization against α-syn as disease-modifying therapies. We believe that rather than achieving short term benefits for PD, setting long-terms goals by carrying out integrative research by merging or collaborating academic and industrial outcomes would manifest an actual therapy for PD.
Figure 1. Preclinical Targets for Parkinson's disease (PD).
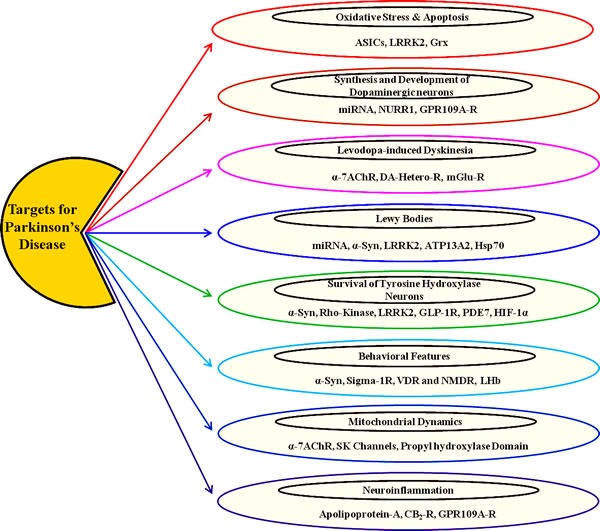
As illustrated, there are eight capsules, each signifying various targets acting on particular pathological process and/or outcome of PD.
Acknowledgments
This paper was supported by Konkuk University in 2016
Footnotes
CONFLICTS OF INTEREST
The authors state no conflict of interest.
REFERENCES
- 1.Gong H, Qian H, Ertl R, Astle CM, Wang GG, Harrison DE, Xu X. Histone modifications change with age, dietary restriction and rapamycin treatment in mouse brain. Oncotarget. 2015;6(18):15882–15890. doi: 10.18632/oncotarget.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tarazi FI, Sahli ZT, Wolny M, Mousa SA. Emerging therapies for Parkinson's disease: from bench to bedside. Pharmacol Ther. 2014;144(2):123–133. doi: 10.1016/j.pharmthera.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Schapira AH, Bezard E, Brotchie J, Calon F, Collingridge GL, Ferger B, Hengerer B, Hirsch E, Jenner P, Le Novere N, Obeso JA, Schwarzschild MA, Spampinato U, Davidai G. Novel pharmacological targets for the treatment of Parkinson's disease. Nat Rev Drug Discov. 2006;5(10):845–854. doi: 10.1038/nrd2087. [DOI] [PubMed] [Google Scholar]
- 4.Cookson MR, Bandmann O. Parkinson's disease: insights from pathways. Hum Mol Genet. 2010;19(R1):R21–27. doi: 10.1093/hmg/ddq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23(4):228–242. doi: 10.1177/0891988710383572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harbor perspectives in medicine. 2012;2(1):a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein C, Schlossmacher MG. The genetics of Parkinson disease: implications for neurological care. Nature clinical practice Neurology. 2006;2(3):136–146. doi: 10.1038/ncpneuro0126. [DOI] [PubMed] [Google Scholar]
- 8.Pan-Montojo F, Reichmann H. Considerations on the role of environmental toxins in idiopathic Parkinson's disease pathophysiology. Transl Neurodegener. 2014;3:10. doi: 10.1186/2047-9158-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu WY, Kang KH, Chen SL, Chiu SY, Yen AM, Fann JC, Su CW, Liu HC, Lee CZ, Fu WM, Chen HH, Liou HH. Hepatitis C virus infection: a risk factor for Parkinson's disease. J Viral Hepat. 2015;22:784–9. doi: 10.1111/jvh.12392. [DOI] [PubMed] [Google Scholar]
- 10.Hermanowicz N. Management of Parkinson's disease. Strategies, pitfalls, and future directions. Postgrad Med. 2001;110(6):15–18. doi: 10.3810/pgm.2001.12.1060. 21-13, 28. [DOI] [PubMed] [Google Scholar]
- 11.Bajaj N, Hauser RA, Grachev ID. Clinical utility of dopamine transporter single photon emission CT (DaT-SPECT) with (123I) ioflupane in diagnosis of parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 2013;84(11):1288–1295. doi: 10.1136/jnnp-2012-304436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de la Fuente-Fernandez R. Role of DaTSCAN and clinical diagnosis in Parkinson disease. Neurology. 2012;78(10):696–701. doi: 10.1212/WNL.0b013e318248e520. [DOI] [PubMed] [Google Scholar]
- 13.Michotte A. Recent developments in the neuropathological diagnosis of Parkinson's disease and parkinsonism. Acta Neurol Belg. 2003;103(3):155–158. [PubMed] [Google Scholar]
- 14.Olanow CW, Watts RL, Koller WC. An algorithm (decision tree) for the management of Parkinson's disease (2001): treatment guidelines. Neurology. 2001;56(11 Suppl 5):S1–S88. doi: 10.1212/wnl.56.suppl_5.s1. [DOI] [PubMed] [Google Scholar]
- 15.Huynh T. The Parkinson's disease market. Nat Rev Drug Discov. 2011;10(8):571–572. doi: 10.1038/nrd3515. [DOI] [PubMed] [Google Scholar]
- 16.Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162(6):1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5(10):821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Adelstein SJ, Kassis AI. Target discovery from data mining approaches. Drug Discov Today. 2009;14(3-4):147–154. doi: 10.1016/j.drudis.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9(10):768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- 20.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurosawa G, Akahori Y, Morita M, Sumitomo M, Sato N, Muramatsu C, Eguchi K, Matsuda K, Takasaki A, Tanaka M, Iba Y, Hamada-Tsutsumi S, Ukai Y, Shiraishi M, Suzuki K, Kurosawa M, et al. Comprehensive screening for antigens overexpressed on carcinomas via isolation of human mAbs that may be therapeutic. Proc Natl Acad Sci U S A. 2008;105(20):7287–7292. doi: 10.1073/pnas.0712202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging (Albany, NY) 2009;1(3):281–288. doi: 10.18632/aging.100034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henning SW BG. Loss-of-function strategies in drug target validation. Current Drug Discovery. 2002:17–21. [Google Scholar]
- 25.NP P. What constitutes target validation? Targets. 2003;2:125–127. [Google Scholar]
- 26.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zanders ED, Bailey DS, Dean PM. Probes for chemical genomics by design. Drug Discov Today. 2002;7(13):711–718. doi: 10.1016/s1359-6446(02)02325-5. [DOI] [PubMed] [Google Scholar]
- 28.Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, Obeso JA, Rascol O, Schapira A, Voon V, Weiner DM, Tison F, Bezard E. Priorities in Parkinson's disease research. Nat Rev Drug Discov. 2011;10(5):377–393. doi: 10.1038/nrd3430. [DOI] [PubMed] [Google Scholar]
- 29.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8(2):93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 30.Kamal MA, Mushtaq G, Greig NH. Current Update on Synopsis of miRNA Dysregulation in Neurological Disorders. CNS Neurol Disord Drug Targets. 2015;14(4):492–501. doi: 10.2174/1871527314666150225143637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper JM. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson's disease. Cell Death Dis. 2013;4:e545. doi: 10.1038/cddis.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winslow AR, Rubinsztein DC. The Parkinson disease protein alpha-synuclein inhibits autophagy. Autophagy. 2011;7(4):429–431. doi: 10.4161/auto.7.4.14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Decressac M, Bjorklund A. TFEB: Pathogenic role and therapeutic target in Parkinson disease. Autophagy. 2013;9(8):1244–1246. doi: 10.4161/auto.25044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maciotta S, Meregalli M, Torrente Y. The involvement of microRNAs in neurodegenerative diseases. Front Cell Neurosci. 2013;7:265. doi: 10.3389/fncel.2013.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi DC, Chae YJ, Kabaria S, Chaudhuri AD, Jain MR, Li H, Mouradian MM, Junn E. MicroRNA-7 protects against 1-methyl-4-phenylpyridinium-induced cell death by targeting RelA. J Neurosci. 2014;34(38):12725–12737. doi: 10.1523/JNEUROSCI.0985-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kabaria S, Choi DC, Chaudhuri AD, Jain MR, Li H, Junn E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radical Biology and Medicine. 2015;89:548–556. doi: 10.1016/j.freeradbiomed.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sotiriou S, Gibney G, Baxevanis AD, Nussbaum RL. A single nucleotide polymorphism in the 3′UTR of the SNCA gene encoding alpha-synuclein is a new potential susceptibility locus for Parkinson disease. Neurosci Lett. 2009;461(2):196–201. doi: 10.1016/j.neulet.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285(17):12726–12734. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106(31):13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margis R, Rieder CR. Identification of blood microRNAs associated to Parkinsonis disease. J Biotechnol. 2011;152(3):96–101. doi: 10.1016/j.jbiotec.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 42.Cardo LF, Coto E, de Mena L, Ribacoba R, Moris G, Menendez M, Alvarez V. Profile of microRNAs in the plasma of Parkinson's disease patients and healthy controls. J Neurol. 2013;260(5):1420–1422. doi: 10.1007/s00415-013-6900-8. [DOI] [PubMed] [Google Scholar]
- 43.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89(1):73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Millar NS, Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology. 2009;56(1):237–246. doi: 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 45.Quik M, Wonnacott S. alpha6beta2* and alpha4beta2* nicotinic acetylcholine receptors as drug targets for Parkinson's disease. Pharmacol Rev. 2011;63(4):938–966. doi: 10.1124/pr.110.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pauly JR, Stitzel JA, Marks MJ, Collins AC. An autoradiographic analysis of cholinergic receptors in mouse brain. Brain Res Bull. 1989;22(2):453–459. doi: 10.1016/0361-9230(89)90072-5. [DOI] [PubMed] [Google Scholar]
- 47.Elbaz A, Moisan F. Update in the epidemiology of Parkinson's disease. Curr Opin Neurol. 2008;21(4):454–460. doi: 10.1097/WCO.0b013e3283050461. [DOI] [PubMed] [Google Scholar]
- 48.Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur J Epidemiol. 2011;26(Suppl 1):S1–58. doi: 10.1007/s10654-011-9581-6. [DOI] [PubMed] [Google Scholar]
- 49.Lichtensteiger W, Hefti F, Felix D, Huwyler T, Melamed E, Schlumpf M. Stimulation of nigrostriatal dopamine neurones by nicotine. Neuropharmacology. 1982;21(10):963–968. doi: 10.1016/0028-3908(82)90107-1. [DOI] [PubMed] [Google Scholar]
- 50.Rapier C, Lunt GG, Wonnacott S. Nicotinic modulation of [3H]dopamine release from striatal synaptosomes: pharmacological characterisation. J Neurochem. 1990;54(3):937–945. doi: 10.1111/j.1471-4159.1990.tb02341.x. [DOI] [PubMed] [Google Scholar]
- 51.O'Neill MJ, Murray TK, Lakics V, Visanji NP, Duty S. The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2002;1(4):399–411. doi: 10.2174/1568007023339166. [DOI] [PubMed] [Google Scholar]
- 52.Quik M, O'Neill M, Perez XA. Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci. 2007;28(5):229–235. doi: 10.1016/j.tips.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Kawamata J, Suzuki S, Shimohama S. alpha7 nicotinic acetylcholine receptor mediated neuroprotection in Parkinson's disease. Curr Drug Targets. 2012;13(5):623–630. doi: 10.2174/138945012800399026. [DOI] [PubMed] [Google Scholar]
- 54.Janson AM, Hedlund PB, Hillefors M, von Euler G. Chronic nicotine treatment decreases dopamine D2 agonist binding in the rat basal ganglia. Neuroreport. 1992;3(12):1117–1120. doi: 10.1097/00001756-199212000-00021. [DOI] [PubMed] [Google Scholar]
- 55.Janson AM, Meana JJ, Goiny M, Herrera-Marschitz M. Chronic nicotine treatment counteracts the decrease in extracellular neostriatal dopamine induced by a unilateral transection at the mesodiencephalic junction in rats: a microdialysis study. Neurosci Lett. 1991;134(1):88–92. doi: 10.1016/0304-3940(91)90515-u. [DOI] [PubMed] [Google Scholar]
- 56.Soto-Otero R, Mendez-Alvarez E, Hermida-Ameijeiras A, Lopez-Real AM, Labandeira-Garcia JL. Effects of (−)-nicotine and (−)-cotinine on 6-hydroxydopamine-induced oxidative stress and neurotoxicity: relevance for Parkinson's disease. Biochem Pharmacol. 2002;64(1):125–135. doi: 10.1016/s0006-2952(02)01070-5. [DOI] [PubMed] [Google Scholar]
- 57.Sershen H, Hashim A, Wiener HL, Lajtha A. Effect of chronic oral nicotine on dopaminergic function in the MPTP-treated mouse. Neurosci Lett. 1988;93(2-3):270–274. doi: 10.1016/0304-3940(88)90094-8. [DOI] [PubMed] [Google Scholar]
- 58.Parain K, Marchand V, Dumery B, Hirsch E. Nicotine, but not cotinine, partially protects dopaminergic neurons against MPTP-induced degeneration in mice. Brain Res. 2001;890(2):347–350. doi: 10.1016/s0006-8993(00)03198-x. [DOI] [PubMed] [Google Scholar]
- 59.Quik M, Cox H, Parameswaran N, O'Leary K, Langston JW, Di Monte D. Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys. Ann Neurol. 2007;62(6):588–596. doi: 10.1002/ana.21203. [DOI] [PubMed] [Google Scholar]
- 60.Quik M, Chen L, Parameswaran N, Xie X, Langston JW, McCallum SE. Chronic oral nicotine normalizes dopaminergic function and synaptic plasticity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned primates. J Neurosci. 2006;26(17):4681–4689. doi: 10.1523/JNEUROSCI.0215-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quik M, Parameswaran N, McCallum SE, Bordia T, Bao S, McCormack A, Kim A, Tyndale RF, Langston JW, Di Monte DA. Chronic oral nicotine treatment protects against striatal degeneration in MPTP-treated primates. J Neurochem. 2006;98(6):1866–1875. doi: 10.1111/j.1471-4159.2006.04078.x. [DOI] [PubMed] [Google Scholar]
- 62.Bordia T, McGregor M, Papke RL, Decker MW, McIntosh JM, Quik M. The alpha7 nicotinic receptor agonist ABT-107 protects against nigrostriatal damage in rats with unilateral 6-hydroxydopamine lesions. Exp Neurol. 2015;263:277–284. doi: 10.1016/j.expneurol.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryan RE, Ross SA, Drago J, Loiacono RE. Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in alpha4 nicotinic receptor subunit knockout mice. Br J Pharmacol. 2001;132(8):1650–1656. doi: 10.1038/sj.bjp.0703989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Srinivasan R, Pantoja R, Moss FJ, Mackey ED, Son CD, Miwa J, Lester HA. Nicotine up-regulates alpha4beta2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J Gen Physiol. 2011;137(1):59–79. doi: 10.1085/jgp.201010532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Srinivasan R, Richards CI, Xiao C, Rhee D, Pantoja R, Dougherty DA, Miwa JM, Lester HA. Pharmacological chaperoning of nicotinic acetylcholine receptors reduces the endoplasmic reticulum stress response. Mol Pharmacol. 2012;81(6):759–769. doi: 10.1124/mol.112.077792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suzuki S, Kawamata J, Matsushita T, Matsumura A, Hisahara S, Takata K, Kitamura Y, Kem W, Shimohama S. 3-[(2,4-Dimethoxy)benzylidene]-anabaseine dihydrochloride protects against 6-hydroxydopamine-induced parkinsonian neurodegeneration through alpha7 nicotinic acetylcholine receptor stimulation in rats. J Neurosci Res. 2013;91(3):462–471. doi: 10.1002/jnr.23160. [DOI] [PubMed] [Google Scholar]
- 67.Yanagida T, Takeuchi H, Kitamura Y, Takata K, Minamino H, Shibaike T, Tsushima J, Kishimoto K, Yasui H, Taniguchi T, Shimohama S. Synergistic effect of galantamine on nicotine-induced neuroprotection in hemiparkinsonian rat model. Neurosci Res. 2008;62(4):254–261. doi: 10.1016/j.neures.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 68.Stuckenholz V, Bacher M, Balzer-Geldsetzer M, Alvarez-Fischer D, Oertel WH, Dodel RC, Noelker C. The alpha7 nAChR agonist PNU-282987 reduces inflammation and MPTP-induced nigral dopaminergic cell loss in mice. J Parkinsons Dis. 2013;3(2):161–172. doi: 10.3233/JPD-120157. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, Huang Z, Ellsworth K, Fan W. alpha7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J Neuroinflammation. 2012;9:98. doi: 10.1186/1742-2094-9-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Quik M, Zhang D, Perez XA, Bordia T. Role for the nicotinic cholinergic system in movement disorders; therapeutic implications. Pharmacol Ther. 2014;144(1):50–59. doi: 10.1016/j.pharmthera.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bordia T, Campos C, McIntosh JM, Quik M. Nicotinic receptor-mediated reduction in L-DOPA-induced dyskinesias may occur via desensitization. J Pharmacol Exp Ther. 2010;333(3):929–938. doi: 10.1124/jpet.109.162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang X, Chen PC, Poole C. APOE-[epsilon]2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology. 2004;62(12):2198–2202. doi: 10.1212/01.wnl.0000130159.28215.6a. [DOI] [PubMed] [Google Scholar]
- 73.Quik M, Mallela A, Chin M, McIntosh JM, Perez XA, Bordia T. Nicotine-mediated improvement in L-dopa-induced dyskinesias in MPTP-lesioned monkeys is dependent on dopamine nerve terminal function. Neurobiol Dis. 2013;50:30–41. doi: 10.1016/j.nbd.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quik M, Mallela A, Ly J, Zhang D. Nicotine reduces established levodopa-induced dyskinesias in a monkey model of Parkinson's disease. Mov Disord. 2013;28(10):1398–1406. doi: 10.1002/mds.25594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mihalak KB, Carroll FI, Luetje CW. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70(3):801–805. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- 76.Zhang D, Mallela A, Sohn D, Carroll FI, Bencherif M, Letchworth S, Quik M. Nicotinic receptor agonists reduce L-DOPA-induced dyskinesias in a monkey model of Parkinson's disease. J Pharmacol Exp Ther. 2013;347(1):225–234. doi: 10.1124/jpet.113.207639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang D, McGregor M, Decker MW, Quik M. The alpha7 nicotinic receptor agonist ABT-107 decreases L-Dopa-induced dyskinesias in parkinsonian monkeys. J Pharmacol Exp Ther. 2014;351(1):25–32. doi: 10.1124/jpet.114.216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Paolo T, Gregoire L, Feuerbach D, Elbast W, Weiss M, Gomez-Mancilla B. AQW051, a novel and selective nicotinic acetylcholine receptor alpha7 partial agonist, reduces l-Dopa-induced dyskinesias and extends the duration of l-Dopa effects in parkinsonian monkeys. Parkinsonism Relat Disord. 2014;20(11):1119–1123. doi: 10.1016/j.parkreldis.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 79.Shaw S, Bencherif M, Marrero MB. Janus kinase 2, an early target of alpha 7 nicotinic acetylcholine receptor-mediated neuroprotection against Abeta-(1-42) amyloid. J Biol Chem. 2002;277(47):44920–44924. doi: 10.1074/jbc.M204610200. [DOI] [PubMed] [Google Scholar]
- 80.Ren K, Puig V, Papke RL, Itoh Y, Hughes JA, Meyer EM. Multiple calcium channels and kinases mediate alpha7 nicotinic receptor neuroprotection in PC12 cells. J Neurochem. 2005;94(4):926–933. doi: 10.1111/j.1471-4159.2005.03223.x. [DOI] [PubMed] [Google Scholar]
- 81.Arredondo J, Chernyavsky AI, Jolkovsky DL, Pinkerton KE, Grando SA. Receptor-mediated tobacco toxicity: cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. Faseb J. 2006;20(12):2093–2101. doi: 10.1096/fj.06-6191com. [DOI] [PubMed] [Google Scholar]
- 82.Toborek M, Son KW, Pudelko A, King-Pospisil K, Wylegala E, Malecki A. ERK 1/2 signaling pathway is involved in nicotine-mediated neuroprotection in spinal cord neurons. J Cell Biochem. 2007;100(2):279–292. doi: 10.1002/jcb.21013. [DOI] [PubMed] [Google Scholar]
- 83.Parada E, Egea J, Romero A, del Barrio L, Garcia AG, Lopez MG. Poststress treatment with PNU282987 can rescue SH-SY5Y cells undergoing apoptosis via alpha7 nicotinic receptors linked to a Jak2/Akt/HO-1 signaling pathway. Free Radic Biol Med. 2010;49(11):1815–1821. doi: 10.1016/j.freeradbiomed.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 84.Rey P, Lopez-Real A, Sanchez-Iglesias S, Munoz A, Soto-Otero R, Labandeira-Garcia JL. Angiotensin type-1-receptor antagonists reduce 6-hydroxydopamine toxicity for dopaminergic neurons. Neurobiol Aging. 2007;28(4):555–567. doi: 10.1016/j.neurobiolaging.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 85.Tyagi E, Agrawal R, Nath C, Shukla R. Cholinergic protection via alpha7 nicotinic acetylcholine receptors and PI3K-Akt pathway in LPS-induced neuroinflammation. Neurochem Int. 2010;56(1):135–142. doi: 10.1016/j.neuint.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 86.Kaneko S, Maeda T, Kume T, Kochiyama H, Akaike A, Shimohama S, Kimura J. Nicotine protects cultured cortical neurons against glutamate-induced cytotoxicity via alpha7-neuronal receptors and neuronal CNS receptors. Brain Res. 1997;765(1):135–140. doi: 10.1016/s0006-8993(97)00556-8. [DOI] [PubMed] [Google Scholar]
- 87.Lawand NB, Saade NE, El-Agnaf OM, Safieh-Garabedian B. Targeting alpha-synuclein as a therapeutic strategy for Parkinson's disease. Expert Opin Ther Targets. 2015:1–10. doi: 10.1517/14728222.2015.1062877. [DOI] [PubMed] [Google Scholar]
- 88.Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci. 2002;22(8):3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alerte TN, Akinfolarin AA, Friedrich EE, Mader SA, Hong CS, Perez RG. Alpha-synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: lessons from viral transduction of knockout mice. Neurosci Lett. 2008;435(1):24–29. doi: 10.1016/j.neulet.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Spinelli KJ, Taylor JK, Osterberg VR, Churchill MJ, Pollock E, Moore C, Meshul CK, Unni VK. Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson's disease. J Neurosci. 2014;34(6):2037–2050. doi: 10.1523/JNEUROSCI.2581-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Swirski M, Miners JS, de Silva R, Lashley T, Ling H, Holton J, Revesz T, Love S. Evaluating the relationship between amyloid-beta and alpha-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson's disease. Alzheimers Res Ther. 2014;6(5-8):77. doi: 10.1186/s13195-014-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Khodr CE, Becerra A, Han Y, Bohn MC. Targeting alpha-synuclein with a microRNA-embedded silencing vector in the rat substantia nigra: positive and negative effects. Brain Res. 2014;1550:47–60. doi: 10.1016/j.brainres.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Khodr CE, Sapru MK, Pedapati J, Han Y, West NC, Kells AP, Bankiewicz KS, Bohn MC. An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson's disease, but displays toxicity in dopamine neurons. Brain Res. 2011;1395:94–107. doi: 10.1016/j.brainres.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Parnetti L, Castrioto A, Chiasserini D, Persichetti E, Tambasco N, El-Agnaf O, Calabresi P. Cerebrospinal fluid biomarkers in Parkinson disease. Nat Rev Neurol. 2013;9(3):131–140. doi: 10.1038/nrneurol.2013.10. [DOI] [PubMed] [Google Scholar]
- 95.Wang N, Gibbons CH, Lafo J, Freeman R. alpha-Synuclein in cutaneous autonomic nerves. Neurology. 2013;81(18):1604–1610. doi: 10.1212/WNL.0b013e3182a9f449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Christopher Gibbons NW, Roy Freeman. Alpha-Synuclein Within Cutaneous Autonomic Nerves In Parkinson Disease: A Biomarker Of Disease Severity And Autonomic Dysfunction. Neurology. 2014;82:10. [Google Scholar]
- 97.Ardah MT, Paleologou KE, Lv G, Menon SA, Abul Khair SB, Lu JH, Safieh-Garabedian B, Al-Hayani AA, Eliezer D, Li M, El-Agnaf OM. Ginsenoside Rb1 inhibits fibrillation and toxicity of alpha-synuclein and disaggregates preformed fibrils. Neurobiol Dis. 2015;74:89–101. doi: 10.1016/j.nbd.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Toth G, Gardai SJ, Zago W, Bertoncini CW, Cremades N, Roy SL, Tambe MA, Rochet JC, Galvagnion C, Skibinski G, Finkbeiner S, Bova M, Regnstrom K, Chiou SS, Johnston J, Callaway K, et al. Targeting the intrinsically disordered structural ensemble of alpha-synuclein by small molecules as a potential therapeutic strategy for Parkinson's disease. PLoS One. 2014;9(2):e87133. doi: 10.1371/journal.pone.0087133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dzamko N, Zhou J, Huang Y, Halliday GM. Parkinson's disease-implicated kinases in the brain; insights into disease pathogenesis. Front Mol Neurosci. 2014;7:57. doi: 10.3389/fnmol.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Braithwaite SP, Stock JB, Mouradian MM. alpha-Synuclein phosphorylation as a therapeutic target in Parkinson's disease. Rev Neurosci. 2012;23(2):191–198. doi: 10.1515/revneuro-2011-0067. [DOI] [PubMed] [Google Scholar]
- 101.Chu Y, Kordower JH. The prion hypothesis of Parkinson's disease. Curr Neurol Neurosci Rep. 2015;15(5):28. doi: 10.1007/s11910-015-0549-x. [DOI] [PubMed] [Google Scholar]
- 102.Dunning CJ, George S, Brundin P. What's to like about the prion-like hypothesis for the spreading of aggregated alpha-synuclein in Parkinson disease? Prion. 2013;7(1):92–97. doi: 10.4161/pri.23806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kovacs GG, Breydo L, Green R, Kis V, Puska G, Lorincz P, Perju-Dumbrava L, Giera R, Pirker W, Lutz M, Lachmann I, Budka H, Uversky VN, Molnar K, Laszlo L. Intracellular processing of disease-associated alpha-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol Dis. 2014;69:76–92. doi: 10.1016/j.nbd.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 104.Valera E, Masliah E. Immunotherapy for neurodegenerative diseases: focus on alpha-synucleinopathies. Pharmacol Ther. 2013;138(3):311–322. doi: 10.1016/j.pharmthera.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Romero-Ramos M, von Euler Chelpin M, Sanchez-Guajardo V. Vaccination strategies for Parkinson disease: induction of a swift attack or raising tolerance? Hum Vaccin Immunother. 2014;10(4):852–867. doi: 10.4161/hv.28578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106(31):13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brannstrom K, Lindhagen-Persson M, Gharibyan AL, Iakovleva I, Vestling M, Sellin ME, Brannstrom T, Morozova-Roche L, Forsgren L, Olofsson A. A generic method for design of oligomer-specific antibodies. PLoS One. 2014;9(3):e90857. doi: 10.1371/journal.pone.0090857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32(39):13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep. 2014;7(6):2054–2065. doi: 10.1016/j.celrep.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, Patrick C, Ubhi K, Nuber S, Sacayon P, Zago W, Seubert P, Barbour R, Schenk D, Masliah E. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson's disease-like models. J Neurosci. 2014;34(28):9441–9454. doi: 10.1523/JNEUROSCI.5314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yan J, Fu Q, Cheng L, Zhai M, Wu W, Huang L, Du G. Inflammatory response in Parkinson's disease (Review) Mol Med Rep. 2014;10(5):2223–2233. doi: 10.3892/mmr.2014.2563. [DOI] [PubMed] [Google Scholar]
- 112.Barkholt P, Sanchez-Guajardo V, Kirik D, Romero-Ramos M. Long-term polarization of microglia upon alpha-synuclein overexpression in nonhuman primates. Neuroscience. 2012;208:85–96. doi: 10.1016/j.neuroscience.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 113.Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chao Y, Wong SC, Tan EK. Evidence of inflammatory system involvement in Parkinson's disease. Biomed Res Int. 2014;2014:308654. doi: 10.1155/2014/308654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Macchi B, Di Paola R, Marino-Merlo F, Felice MR, Cuzzocrea S, Mastino A. Inflammatory and cell death pathways in brain and peripheral blood in Parkinson's disease. CNS Neurol Disord Drug Targets. 2015;14(3):313–324. doi: 10.2174/1871527314666150225124928. [DOI] [PubMed] [Google Scholar]
- 116.Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61(3):349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res. 2011;69(4):337–342. doi: 10.1016/j.neures.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 118.Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One. 2013;8(1):e55375. doi: 10.1371/journal.pone.0055375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Decressac M, Mattsson B, Bjorklund A. Comparison of the behavioural and histological characteristics of the 6-OHDA and alpha-synuclein rat models of Parkinson's disease. Exp Neurol. 2012;235(1):306–315. doi: 10.1016/j.expneurol.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 120.Sanchez-Guajardo V, Barnum CJ, Tansey MG, Romero-Ramos M. Neuroimmunological processes in Parkinson's disease and their relation to alpha-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro. 2013;5(2):113–139. doi: 10.1042/AN20120066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thome AD, Standaert DG, Harms AS. Fractalkine Signaling Regulates the Inflammatory Response in an α-Synuclein Model of Parkinson Disease. PLoS One. 2015;10(10):e0140566. doi: 10.1371/journal.pone.0140566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Labandeira-Garcia JL, Rodriguez-Perez AI, Villar-Cheda B, Borrajo A, Dominguez-Meijide A, Guerra MJ. Rho Kinase and Dopaminergic Degeneration: A Promising Therapeutic Target for Parkinson's Disease. Neuroscientist. 2015;21(6):616–29. doi: 10.1177/1073858414554954. [DOI] [PubMed] [Google Scholar]
- 123.Herskowitz JH, Feng Y, Mattheyses AL, Hales CM, Higginbotham LA, Duong DM, Montine TJ, Troncoso JC, Thambisetty M, Seyfried NT, Levey AI, Lah JJ. Pharmacologic inhibition of ROCK2 suppresses amyloid-beta production in an Alzheimer's disease mouse model. J Neurosci. 2013;33(49):19086–19098. doi: 10.1523/JNEUROSCI.2508-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Barcia C, Ros CM, Annese V, Carrillo-de Sauvage MA, Ros-Bernal F, Gomez A, Yuste JE, Campuzano CM, de Pablos V, Fernandez-Villalba E, Herrero MT. ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci Rep. 2012;2:809. doi: 10.1038/srep00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tonges L, Frank T, Tatenhorst L, Saal KA, Koch JC, Szego EM, Bahr M, Weishaupt JH, Lingor P. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson's disease. Brain. 2012;135(Pt 11):3355–3370. doi: 10.1093/brain/aws254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Villar-Cheda B, Dominguez-Meijide A, Joglar B, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL. Involvement of microglial RhoA/Rho-kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol Dis. 2012;47(2):268–279. doi: 10.1016/j.nbd.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 127.Borrajo A, Rodriguez-Perez AI, Villar-Cheda B, Guerra MJ, Labandeira-Garcia JL. Inhibition of the microglial response is essential for the neuroprotective effects of Rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology. 2014;85:1–8. doi: 10.1016/j.neuropharm.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 128.Mattson MP, Maudsley S. Live longer sans the AT1A receptor. Cell Metab. 2009;9(5):403–405. doi: 10.1016/j.cmet.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hiroki J, Shimokawa H, Higashi M, Morikawa K, Kandabashi T, Kawamura N, Kubota T, Ichiki T, Amano M, Kaibuchi K, Takeshita A. Inflammatory stimuli upregulate Rho-kinase in human coronary vascular smooth muscle cells. J Mol Cell Cardiol. 2004;37(2):537–546. doi: 10.1016/j.yjmcc.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 130.Sharma N, Nehru B. Apocyanin, a Microglial NADPH Oxidase Inhibitor Prevents Dopaminergic Neuronal Degeneration in Lipopolysaccharide-Induced Parkinson's Disease Model. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9267-2. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 131.Garrido-Gil P, Valenzuela R, Villar-Cheda B, Lanciego JL, Labandeira-Garcia JL. Expression of angiotensinogen and receptors for angiotensin and prorenin in the monkey and human substantia nigra: an intracellular renin-angiotensin system in the nigra. Brain Struct Funct. 2013;218(2):373–388. doi: 10.1007/s00429-012-0402-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rodriguez-Perez AI, Valenzuela R, Joglar B, Garrido-Gil P, Guerra MJ, Labandeira-Garcia JL. Renin angiotensin system and gender differences in dopaminergic degeneration. Mol Neurodegener. 2011;6(1):58. doi: 10.1186/1750-1326-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rodriguez-Perez AI, Valenzuela R, Villar-Cheda B, Guerra MJ, Labandeira-Garcia JL. Dopaminergic neuroprotection of hormonal replacement therapy in young and aged menopausal rats: role of the brain angiotensin system. Brain. 2012;135(Pt 1):124–138. doi: 10.1093/brain/awr320. [DOI] [PubMed] [Google Scholar]
- 134.Rodriguez-Perez AI, Valenzuela R, Villar-Cheda B, Guerra MJ, Lanciego JL, Labandeira-Garcia JL. Estrogen and angiotensin interaction in the substantia nigra. Relevance to postmenopausal Parkinson's disease. Exp Neurol. 2010;224(2):517–526. doi: 10.1016/j.expneurol.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 135.Rodriguez-Perez AI, Dominguez-Meijide A, Lanciego JL, Guerra MJ, Labandeira-Garcia JL. Inhibition of Rho kinase mediates the neuroprotective effects of estrogen in the MPTP model of Parkinson's disease. Neurobiol Dis. 2013;58:209–219. doi: 10.1016/j.nbd.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 136.He H, Yim M, Liu KH, Cody SC, Shulkes A, Baldwin GS. Involvement of G proteins of the Rho family in the regulation of Bcl-2-like protein expression and caspase 3 activation by Gastrins. Cell Signal. 2008;20(1):83–93. doi: 10.1016/j.cellsig.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 137.Wakita S, Izumi Y, Nakai T, Adachi K, Takada-Takatori Y, Kume T, Akaike A. Staurosporine induces dopaminergic neurite outgrowth through AMP-activated protein kinase/mammalian target of rapamycin signaling pathway. Neuropharmacology. 2014;77:39–48. doi: 10.1016/j.neuropharm.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 138.Tonges L, Gunther R, Suhr M, Jansen J, Balck A, Saal KA, Barski E, Nientied T, Gotz AA, Koch JC, Mueller BK, Weishaupt JH, Sereda MW, Hanisch UK, Bahr M, Lingor P. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia. 2014;62(2):217–232. doi: 10.1002/glia.22601. [DOI] [PubMed] [Google Scholar]
- 139.Gómez-Suaga P, Fdez E, Fernández B, Martínez-Salvador M, Blanca Ramírez M, Madero-Pérez J, Rivero-Ríos P, Fuentes JM, Hilfiker S. Novel insights into the neurobiology underlying LRRK2-linked Parkinson's disease. Neuropharmacology. 2014;85:45–56. doi: 10.1016/j.neuropharm.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 140.Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. Embo J. 2008;27(18):2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466(7306):637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Caesar M, Zach S, Carlson CB, Brockmann K, Gasser T, Gillardon F. Leucine-rich repeat kinase 2 functionally interacts with microtubules and kinase-dependently modulates cell migration. Neurobiol Dis. 2013;54:280–288. doi: 10.1016/j.nbd.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 143.Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, Delbroek L, Swerts J, Chavez-Gutierrez L, Esposito G, Daneels G, Karran E, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75(6):1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 144.Bravo-San Pedro JM, Niso-Santano M, Gomez-Sanchez R, Pizarro-Estrella E, Aiastui-Pujana A, Gorostidi A, Climent V, Lopez de Maturana R, Sanchez-Pernaute R, Lopez de Munain A, Fuentes JM, Gonzalez-Polo RA. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci. 2013;70(1):121–136. doi: 10.1007/s00018-012-1061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson's disease. Mov Disord. 2012;27(7):831–842. doi: 10.1002/mds.24962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rudenko IN, Chia R, Cookson MR. Is inhibition of kinase activity the only therapeutic strategy for LRRK2-associated Parkinson's disease? BMC Med. 2012;10:20. doi: 10.1186/1741-7015-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23(2):329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 148.Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL, Dawson TM. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med. 2010;16(9):998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res. 2010;316(4):649–656. doi: 10.1016/j.yexcr.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 150.Zhao J, Hermanson SB, Carlson CB, Riddle SM, Vogel KW, Bi K, Nichols RJ. Pharmacological inhibition of LRRK2 cellular phosphorylation sites provides insight into LRRK2 biology. Biochem Soc Trans. 2012;40(5):1158–1162. doi: 10.1042/BST20120137. [DOI] [PubMed] [Google Scholar]
- 151.Deng X, N SG. Pyrazolopyridines as inhibitors of the kinase LRRK2: a patent evaluation (WO2011141756) Expert Opin Ther Pat. 2012;22(6):709–713. doi: 10.1517/13543776.2012.691968. [DOI] [PubMed] [Google Scholar]
- 152.Gomez-Suaga P, Churchill GC, Patel S, Hilfiker S. A link between LRRK2, autophagy and NAADP-mediated endolysosomal calcium signalling. Biochem Soc Trans. 2012;40(5):1140–1146. doi: 10.1042/BST20120138. [DOI] [PubMed] [Google Scholar]
- 153.Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314(10):2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 154.Ness D, Ren Z, Gardai S, Sharpnack D, Johnson VJ, Brennan RJ, Brigham EF, Olaharski AJ. Leucine-rich repeat kinase 2 (LRRK2)-deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS One. 2013;8(6):e66164. doi: 10.1371/journal.pone.0066164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Baker KD, Shewchuk LM, Kozlova T, Makishima M, Hassell A, Wisely B, Caravella JA, Lambert MH, Reinking JL, Krause H, Thummel CS, Willson TM, Mangelsdorf DJ. The Drosophila orphan nuclear receptor DHR38 mediates an atypical ecdysteroid signaling pathway. Cell. 2003;113(6):731–742. doi: 10.1016/s0092-8674(03)00420-3. [DOI] [PubMed] [Google Scholar]
- 156.Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, Xu H, Walker NP, Perlmann T. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423(6939):555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- 157.Alavian KN, Jeddi S, Naghipour SI, Nabili P, Licznerski P, Tierney TS. The lifelong maintenance of mesencephalic dopaminergic neurons by Nurr1 and engrailed. J Biomed Sci. 2014;21:27. doi: 10.1186/1423-0127-21-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kadkhodaei B, Alvarsson A, Schintu N, Ramskold D, Volakakis N, Joodmardi E, Yoshitake T, Kehr J, Decressac M, Bjorklund A, Sandberg R, Svenningsson P, Perlmann T. Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. Proc Natl Acad Sci U S A. 2013;110(6):2360–2365. doi: 10.1073/pnas.1221077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Heng X, Jin G, Zhang X, Yang D, Zhu M, Fu S, Li X, Le W. Nurr1 regulates Top IIbeta and functions in axon genesis of mesencephalic dopaminergic neurons. Mol Neurodegener. 2012;7:4. doi: 10.1186/1750-1326-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jacobs FM, van der Linden AJ, Wang Y, von Oerthel L, Sul HS, Burbach JP, Smidt MP. Identification of Dlk1, Ptpru and Klhl1 as novel Nurr1 target genes in meso-diencephalic dopamine neurons. Development. 2009;136(14):2363–2373. doi: 10.1242/dev.037556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Joseph B, Wallen-Mackenzie A, Benoit G, Murata T, Joodmardi E, Okret S, Perlmann T. p57(Kip2) cooperates with Nurr1 in developing dopamine cells. Proc Natl Acad Sci U S A. 2003;100(26):15619–15624. doi: 10.1073/pnas.2635658100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24(1):78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 163.Volakakis N, Kadkhodaei B, Joodmardi E, Wallis K, Panman L, Silvaggi J, Spiegelman BM, Perlmann T. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc Natl Acad Sci U S A. 2010;107(27):12317–12322. doi: 10.1073/pnas.1007088107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Barneda-Zahonero B, Servitja JM, Badiola N, Minano-Molina AJ, Fado R, Saura CA, Rodriguez-Alvarez J. Nurr1 protein is required for N-methyl-D-aspartic acid (NMDA) receptor-mediated neuronal survival. J Biol Chem. 2012;287(14):11351–11362. doi: 10.1074/jbc.M111.272427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kim CH, Han BS, Moon J, Kim DJ, Shin J, Rajan S, Nguyen QT, Sohn M, Kim WG, Han M, Jeong I, Kim KS, Lee EH, Tu Y, Naffin-Olivos JL, Park CH, et al. Nuclear receptor Nurr1 agonists enhance its dual functions and improve behavioral deficits in an animal model of Parkinson's disease. Proc Natl Acad Sci U S A. 2015;112(28):8756–8761. doi: 10.1073/pnas.1509742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137(1):47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Smith GA, Rocha EM, Rooney T, Barneoud P, McLean JR, Beagan J, Osborn T, Coimbra M, Luo Y, Hallett PJ, Isacson O. A Nurr1 agonist causes neuroprotection in a Parkinson's disease lesion model primed with the toll-like receptor 3 dsRNA inflammatory stimulant poly(I:C) PLoS One. 2015;10(3):e0121072. doi: 10.1371/journal.pone.0121072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Decressac M, Ulusoy A, Mattsson B, Georgievska B, Romero-Ramos M, Kirik D, Bjorklund A. GDNF fails to exert neuroprotection in a rat alpha-synuclein model of Parkinson's disease. Brain. 2011;134(Pt 8):2302–2311. doi: 10.1093/brain/awr149. [DOI] [PubMed] [Google Scholar]
- 169.Decressac M, Volakakis N, Bjorklund A, Perlmann T. NURR1 in Parkinson disease--from pathogenesis to therapeutic potential. Nat Rev Neurol. 2013;9(11):629–636. doi: 10.1038/nrneurol.2013.209. [DOI] [PubMed] [Google Scholar]
- 170.Yu XW, Hu ZL, Ni M, Fang P, Zhang PW, Shu Q, Fan H, Zhou HY, Ni L, Zhu LQ, Chen JG, Wang F. Acid-sensing ion channels promote the inflammation and migration of cultured rat microglia. Glia. 2015;63(3):483–496. doi: 10.1002/glia.22766. [DOI] [PubMed] [Google Scholar]
- 171.Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci. 2011;31(6):2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15(20):3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- 173.Santiago JA, Potashkin JA. System-based approaches to decode the molecular links in Parkinson's disease and diabetes. Neurobiol Dis. 2014;72(Pt A):84–91. doi: 10.1016/j.nbd.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 174.Mattson MP. Interventions that improve body and brain bioenergetics for Parkinson's disease risk reduction and therapy. J Parkinsons Dis. 2014;4(1):1–13. doi: 10.3233/JPD-130335. [DOI] [PubMed] [Google Scholar]
- 175.Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T. Exenatide and the treatment of patients with Parkinson's disease. J Clin Invest. 2013;123(6):2730–2736. doi: 10.1172/JCI68295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson's disease. J Parkinsons Dis. 2014;4(3):337–344. doi: 10.3233/JPD-140364. [DOI] [PubMed] [Google Scholar]
- 177.Cai HY, Holscher C, Yue XH, Zhang SX, Wang XH, Qiao F, Yang W, Qi JS. Lixisenatide rescues spatial memory and synaptic plasticity from amyloid beta protein-induced impairments in rats. Neuroscience. 2014;277:6–13. doi: 10.1016/j.neuroscience.2014.02.022. [DOI] [PubMed] [Google Scholar]
- 178.Han WN, Holscher C, Yuan L, Yang W, Wang XH, Wu MN, Qi JS. Liraglutide protects against amyloid-beta protein-induced impairment of spatial learning and memory in rats. Neurobiol Aging. 2013;34(2):576–588. doi: 10.1016/j.neurobiolaging.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 179.McClean PL, Holscher C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer's disease. Neuropharmacology. 2014;86:241–258. doi: 10.1016/j.neuropharm.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 180.Sharma MK, Jalewa J, Holscher C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J Neurochem. 2014;128(3):459–471. doi: 10.1111/jnc.12469. [DOI] [PubMed] [Google Scholar]
- 181.Liu S, Cheng XY, Wang F, Liu CF. Acid-sensing ion channels: potential therapeutic targets for neurologic diseases. Transl Neurodegener. 2015;4:10. doi: 10.1186/s40035-015-0031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Xiong ZG, Pignataro G, Li M, Chang SY, Simon RP. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr Opin Pharmacol. 2008;8(1):25–32. doi: 10.1016/j.coph.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013;14(7):461–471. doi: 10.1038/nrn3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wemmie JA, Price MP, Welsh MJ. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 2006;29(10):578–586. doi: 10.1016/j.tins.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 185.Benarroch EE. Acid-sensing cation channels: structure, function, and pathophysiologic implications. Neurology. 2014;82(7):628–635. doi: 10.1212/WNL.0000000000000134. [DOI] [PubMed] [Google Scholar]
- 186.Chu XP, Miesch J, Johnson M, Root L, Zhu XM, Chen D, Simon RP, Xiong ZG. Proton-gated channels in PC12 cells. J Neurophysiol. 2002;87(5):2555–2561. doi: 10.1152/jn.00741.2001. [DOI] [PubMed] [Google Scholar]
- 187.Wang YZ, Xu TL. Acidosis, acid-sensing ion channels, and neuronal cell death. Mol Neurobiol. 2011;44(3):350–358. doi: 10.1007/s12035-011-8204-2. [DOI] [PubMed] [Google Scholar]
- 188.Wang YZ, Zeng WZ, Xiao X, Huang Y, Song XL, Yu Z, Tang D, Dong XP, Zhu MX, Xu TL. Intracellular ASIC1a regulates mitochondrial permeability transition-dependent neuronal death. Cell Death Differ. 2013;20(10):1359–1369. doi: 10.1038/cdd.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Arias RL, Sung ML, Vasylyev D, Zhang MY, Albinson K, Kubek K, Kagan N, Beyer C, Lin Q, Dwyer JM, Zaleska MM, Bowlby MR, Dunlop J, Monaghan M. Amiloride is neuroprotective in an MPTP model of Parkinson's disease. Neurobiol Dis. 2008;31(3):334–341. doi: 10.1016/j.nbd.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 190.Joch M, Ase AR, Chen CX, MacDonald PA, Kontogiannea M, Corera AT, Brice A, Seguela P, Fon EA. Parkin-mediated monoubiquitination of the PDZ protein PICK1 regulates the activity of acid-sensing ion channels. Mol Biol Cell. 2007;18(8):3105–3118. doi: 10.1091/mbc.E05-11-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Borroto-Escuela DO, Romero-Fernandez W, Garriga P, Ciruela F, Narvaez M, Tarakanov AO, Palkovits M, Agnati LF, Fuxe K. G protein-coupled receptor heterodimerization in the brain. Methods Enzymol. 2013;521:281–294. doi: 10.1016/B978-0-12-391862-8.00015-6. [DOI] [PubMed] [Google Scholar]
- 192.Perreault ML, Hasbi A, O'Dowd BF, George SR. Heteromeric dopamine receptor signaling complexes: emerging neurobiology and disease relevance. Neuropsychopharmacology. 2014;39(1):156–168. doi: 10.1038/npp.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, O'Dowd BF, George SR. D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci U S A. 2007;104(2):654–659. doi: 10.1073/pnas.0604049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fiorentini C, Savoia P, Savoldi D, Missale C. Receptor heteromers in Parkinson's disease and L-DOPA-induced dyskinesia. CNS Neurol Disord Drug Targets. 2013;12(8):1101–1113. [PubMed] [Google Scholar]
- 195.Fuxe K, Guidolin D, Agnati LF, Borroto-Escuela DO. Dopamine heteroreceptor complexes as therapeutic targets in Parkinson's disease. Expert Opin Ther Targets. 2015;19(3):377–398. doi: 10.1517/14728222.2014.981529. [DOI] [PubMed] [Google Scholar]
- 196.Fuxe K, Agnati LF, von Euler G, Tanganelli S, O'Connor WT, Ferre S, Hedlund P, Zoli M. Neuropeptides, excitatory amino acid and adenosine A2 receptors regulate D2 receptors via intramembrane receptor-receptor interactions. Relevance for Parkinson's disease and schizophrenia. Neurochem Int. 1992;20(Suppl):215S–224S. doi: 10.1016/0197-0186(92)90242-j. [DOI] [PubMed] [Google Scholar]
- 197.Ferraro L, Beggiato S, Tomasini MC, Fuxe K, Antonelli T, Tanganelli S. A(2A)/D(2) receptor heteromerization in a model of Parkinson's disease. Focus on striatal aminoacidergic signaling. Brain Res. 2012;1476:96–107. doi: 10.1016/j.brainres.2012.01.032. [DOI] [PubMed] [Google Scholar]
- 198.Fuxe K, Ferre S, Zoli M, Agnati LF. Integrated events in central dopamine transmission as analyzed at multiple levels. Evidence for intramembrane adenosine A2A/dopamine D2 and adenosine A1/dopamine D1 receptor interactions in the basal ganglia. Brain Res Brain Res Rev. 1998;26(2-3):258–273. doi: 10.1016/s0165-0173(97)00049-0. [DOI] [PubMed] [Google Scholar]
- 199.Antonelli T, Fuxe K, Agnati L, Mazzoni E, Tanganelli S, Tomasini MC, Ferraro L. Experimental studies and theoretical aspects on A2A/D2 receptor interactions in a model of Parkinson's disease. Relevance for L-dopa induced dyskinesias. J Neurol Sci. 2006;248(1-2):16–22. doi: 10.1016/j.jns.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 200.Liu XY, Chu XP, Mao LM, Wang M, Lan HX, Li MH, Zhang GC, Parelkar NK, Fibuch EE, Haines M, Neve KA, Liu F, Xiong ZG, Wang JQ. Modulation of D2R-NR2B interactions in response to cocaine. Neuron. 2006;52(5):897–909. doi: 10.1016/j.neuron.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 201.Rimondini R, Ferre S, Gimenez-Llort L, Ogren SO, Fuxe K. Differential effects of selective adenosine A1 and A2A receptor agonists on dopamine receptor agonist-induced behavioural responses in rats. European Journal of Pharmacology. 1998;347(2-3):153–158. doi: 10.1016/s0014-2999(98)00107-1. [DOI] [PubMed] [Google Scholar]
- 202.Ferre S, O'Connor WT, Snaprud P, Ungerstedt U, Fuxe K. Antagonistic interaction between adenosine A2A receptors and dopamine D2 receptors in the ventral striopallidal system. Implications for the treatment of schizophrenia. Neuroscience. 1994;63(3):765–773. doi: 10.1016/0306-4522(94)90521-5. [DOI] [PubMed] [Google Scholar]
- 203.Westin JE, Vercammen L, Strome EM, Konradi C, Cenci MA. Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry. 2007;62(7):800–810. doi: 10.1016/j.biopsych.2006.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wellman M, Abizaid A. Growth Hormone Secretagogue Receptor Dimers: A New Pharmacological Target. eneuro. 2015;2(2) doi: 10.1523/ENEURO.0053-14.2015. ENEURO. 0053-0014.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Park JS, Blair NF, Sue CM. The role of ATP13A2 in Parkinson's disease: Clinical phenotypes and molecular mechanisms. Mov Disord. 2015;30(6):770–779. doi: 10.1002/mds.26243. [DOI] [PubMed] [Google Scholar]
- 206.Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, Lindquist S. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41(3):308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA. Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum Mol Genet. 2014;23(11):2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- 208.Grunewald A, Arns B, Seibler P, Rakovic A, Munchau A, Ramirez A, Sue CM, Klein C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. 2012;33(8):e1841–1847. doi: 10.1016/j.neurobiolaging.2011.12.035. 1843. [DOI] [PubMed] [Google Scholar]
- 209.Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012;45(3):962–972. doi: 10.1016/j.nbd.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E, Pletnikova O, Troncoso JC, Glauser L, Moore DJ. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum Mol Genet. 2012;21(8):1725–1743. doi: 10.1093/hmg/ddr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109(24):9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32(12):4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, Marini P, Squitieri F, Horstink MW, Montagna P, Libera AD, Stocchi F, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68(19):1557–1562. doi: 10.1212/01.wnl.0000260963.08711.08. [DOI] [PubMed] [Google Scholar]
- 214.Lin CH, Tan EK, Chen ML, Tan LC, Lim HQ, Chen GS, Wu RM. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology. 2008;71(21):1727–1732. doi: 10.1212/01.wnl.0000335167.72412.68. [DOI] [PubMed] [Google Scholar]
- 215.Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38(10):1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 216.Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun. 2013;1:11. doi: 10.1186/2051-5960-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tsunemi T, Krainc D. Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23(11):2791–2801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26(19):5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson's disease. J Neurochem. 1990;55(6):2142–2145. doi: 10.1111/j.1471-4159.1990.tb05809.x. [DOI] [PubMed] [Google Scholar]
- 220.Hao LY, Giasson BI, Bonini NM. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci U S A. 2010;107(21):9747–9752. doi: 10.1073/pnas.0911175107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75(22):2017–2020. doi: 10.1212/WNL.0b013e3181ff9685. [DOI] [PubMed] [Google Scholar]
- 222.Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM. Parkinson's disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet. 2014;23(11):2802–2815. doi: 10.1093/hmg/ddt623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bocca B, Alimonti A, Senofonte O, Pino A, Violante N, Petrucci F, Sancesario G, Forte G. Metal changes in CSF and peripheral compartments of parkinsonian patients. J Neurol Sci. 2006;248(1-2):23–30. doi: 10.1016/j.jns.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 224.Hozumi I, Hasegawa T, Honda A, Ozawa K, Hayashi Y, Hashimoto K, Yamada M, Koumura A, Sakurai T, Kimura A, Tanaka Y, Satoh M, Inuzuka T. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J Neurol Sci. 2011;303(1-2):95–99. doi: 10.1016/j.jns.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 225.Johnson WM, Wilson-Delfosse AL, Chen SG, Mieyal JJ. The roles of redox enzymes in Parkinson's disease: Focus on glutaredoxin. Ther Targets Neurol Dis. 2015;2(2) [PMC free article] [PubMed] [Google Scholar]
- 226.Gravina SA, Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32(13):3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 227.Allen EM, Mieyal JJ. Protein-thiol oxidation and cell death: regulatory role of glutaredoxins. Antioxid Redox Signal. 2012;17(12):1748–1763. doi: 10.1089/ars.2012.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chrestensen CA, Starke DW, Mieyal JJ. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J Biol Chem. 2000;275(34):26556–26565. doi: 10.1074/jbc.M004097200. [DOI] [PubMed] [Google Scholar]
- 229.Kenchappa RS, Ravindranath V. Glutaredoxin is essential for maintenance of brain mitochondrial complex I: studies with MPTP. Faseb J. 2003;17(6):717–719. doi: 10.1096/fj.02-0771fje. [DOI] [PubMed] [Google Scholar]
- 230.Kenchappa RS, Diwakar L, Annepu J, Ravindranath V. Estrogen and neuroprotection: higher constitutive expression of glutaredoxin in female mice offers protection against MPTP-mediated neurodegeneration. Faseb J. 2004;18(10):1102–1104. doi: 10.1096/fj.03-1075fje. [DOI] [PubMed] [Google Scholar]
- 231.Sabens EA, Distler AM, Mieyal JJ. Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: implications for therapy of Parkinson's disease. Biochemistry. 2010;49(12):2715–2724. doi: 10.1021/bi9018658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Saeed U, Ray A, Valli RK, Kumar AM, Ravindranath V. DJ-1 loss by glutaredoxin but not glutathione depletion triggers Daxx translocation and cell death. Antioxid Redox Signal. 2010;13(2):127–144. doi: 10.1089/ars.2009.2832. [DOI] [PubMed] [Google Scholar]
- 233.Johnson WM, Yao C, Siedlak SL, Wang W, Zhu X, Caldwell GA, Wilson-Delfosse AL, Mieyal JJ, Chen SG. Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson's disease. Hum Mol Genet. 2015;24(5):1322–1335. doi: 10.1093/hmg/ddu542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Nickols HH, Conn PJ. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol Dis. 2014;61:55–71. doi: 10.1016/j.nbd.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Xia N, Zhang Q, Wang ST, Gu L, Yang HM, Liu L, Bakshi R, Yang H, Zhang H. Blockade of metabotropic glutamate receptor 5 protects against DNA damage in a rotenone-induced Parkinson's disease model. Free Radical Biology and Medicine. 2015;89:567–580. doi: 10.1016/j.freeradbiomed.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 236.Battaglia G, Busceti CL, Molinaro G, Biagioni F, Storto M, Fornai F, Nicoletti F, Bruno V. Endogenous activation of mGlu5 metabotropic glutamate receptors contributes to the development of nigro-striatal damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J Neurosci. 2004;24(4):828–835. doi: 10.1523/JNEUROSCI.3831-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Masilamoni GJ, Bogenpohl JW, Alagille D, Delevich K, Tamagnan G, Votaw JR, Wichmann T, Smith Y. Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain. 2011;134(Pt 7):2057–2073. doi: 10.1093/brain/awr137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Rascol O, Fox S, Gasparini F, Kenney C, Di Paolo T, Gomez-Mancilla B. Use of metabotropic glutamate 5-receptor antagonists for treatment of levodopa-induced dyskinesias. Parkinsonism Relat Disord. 2014;20(9):947–956. doi: 10.1016/j.parkreldis.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 239.Amalric M, Lopez S, Goudet C, Fisone G, Battaglia G, Nicoletti F, Pin JP, Acher FC. Group III and subtype 4 metabotropic glutamate receptor agonists: discovery and pathophysiological applications in Parkinson's disease. Neuropharmacology. 2013;66:53–64. doi: 10.1016/j.neuropharm.2012.05.026. [DOI] [PubMed] [Google Scholar]
- 240.Battaglia G, Busceti CL, Molinaro G, Biagioni F, Traficante A, Nicoletti F, Bruno V. Pharmacological activation of mGlu4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurosci. 2006;26(27):7222–7229. doi: 10.1523/JNEUROSCI.1595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Williams CJ, Dexter DT. Neuroprotective and symptomatic effects of targeting group III mGlu receptors in neurodegenerative disease. J Neurochem. 2014;129(1):4–20. doi: 10.1111/jnc.12608. [DOI] [PubMed] [Google Scholar]
- 242.Yin S, Noetzel MJ, Johnson KA, Zamorano R, Jalan-Sakrikar N, Gregory KJ, Conn PJ, Niswender CM. Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J Neurosci. 2014;34(1):79–94. doi: 10.1523/JNEUROSCI.1129-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wroblewski JT, Pin JP. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011;60(7-8):1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wakade C, Chong R. A novel treatment target for Parkinson's disease. J Neurol Sci. 2014;347(1-2):34–38. doi: 10.1016/j.jns.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 245.Fall PA, Fredrikson M, Axelson O, Granerus AK. Nutritional and occupational factors influencing the risk of Parkinson's disease: a case-control study in southeastern Sweden. Mov Disord. 1999;14(1):28–37. doi: 10.1002/1531-8257(199901)14:1<28::aid-mds1007>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 246.Karobath M, Díaz J-L, Huttunen MO. The effect of L-dopa on the concentrations of tryptophan, tyrosine and serotonin in rat brain. European Journal of Pharmacology. 1971;14(4):393–396. doi: 10.1016/0014-2999(71)90195-6. [DOI] [PubMed] [Google Scholar]
- 247.Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, Saso S. Kynurenine pathway abnormalities in Parkinson's disease. Neurology. 1992;42(9):1702–1706. doi: 10.1212/wnl.42.9.1702. [DOI] [PubMed] [Google Scholar]
- 248.Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, Saso S. Kynurenine pathway abnormalities in Parkinson's disease. Neurology. 1992;42(9):1702–1706. doi: 10.1212/wnl.42.9.1702. [DOI] [PubMed] [Google Scholar]
- 249.Offermanns S. The nicotinic acid receptor GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends Pharmacol Sci. 2006;27(7):384–390. doi: 10.1016/j.tips.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 250.Gharbaoui T, Skinner PJ, Shin Y-J, Averbuj C, Jung J-K, Johnson BR, Duong T, Decaire M, Uy J, Cherrier MC, Webb PJ, Tamura SY, Zou N, Rodriguez N, Boatman PD, Sage CR, et al. Agonist lead identification for the high affinity niacin receptor GPR109a. Bioorganic & Medicinal Chemistry Letters. 2007;17(17):4914–4919. doi: 10.1016/j.bmcl.2007.06.028. [DOI] [PubMed] [Google Scholar]
- 251.Maciejewski-Lenoir D, Richman JG, Hakak Y, Gaidarov I, Behan DP, Connolly DT. Langerhans Cells Release Prostaglandin D2 in Response to Nicotinic Acid. J Invest Dermatol. 2006;126(12):2637–2646. doi: 10.1038/sj.jid.5700586. [DOI] [PubMed] [Google Scholar]
- 252.Digby JE, Martinez F, Jefferson A, Ruparelia N, Chai J, Wamil M, Greaves DR, Choudhury RP. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler Thromb Vasc Biol. 2012;32(3):669–676. doi: 10.1161/ATVBAHA.111.241836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Benyo Z, Gille A, Kero J, Csiky M, Suchankova MC, Nusing RM, Moers A, Pfeffer K, Offermanns S. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest. 2005;115(12):3634–3640. doi: 10.1172/JCI23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Vrecko K, Birkmayer JGD, Krainz J. Stimulation of dopamine biosynthesis in cultured PC 12 phaeochromocytoma cells by the coenzyme nicotinamide adeninedinucleotide (NADH) J Neural Transm Gen Sect. 1993;5(2):147–156. doi: 10.1007/BF02251205. [DOI] [PubMed] [Google Scholar]
- 255.Vrecko K, Storga D, Birkmayer JGD, Reinhard M, Tafeit E, Horejsi R, Reibnegger G. NADH stimulates endogenous dopamine biosynthesis by enhancing the recycling of tetrahydrobiopterin in rat phaeochromocytoma cells. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1997;1361(1):59–65. doi: 10.1016/s0925-4439(97)00016-1. [DOI] [PubMed] [Google Scholar]
- 256.Birkmayer W, Birkmayer GJD. Nicotinamidadenindinucleotide (NADH): The new approach in the therapy of Parkinson's disease. Annals of Clinical and Laboratory Science. 1989;19(1):38–43. [PubMed] [Google Scholar]
- 257.Chang PC, Chien LY, Yeo JF, Wang YP, Chung MC, Chong LY, Kuo MY, Chen CH, Chiang HC, Ng BN, Lee QQ, Phay YK, Ng JR, Erk KY. Progression of periodontal destruction and the roles of advanced glycation end products in experimental diabetes. J Periodontol. 2013;84(3):379–388. doi: 10.1902/jop.2012.120076. [DOI] [PubMed] [Google Scholar]
- 258.Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. International Journal of Clinical Practice. 2009;63(9):1369–1377. doi: 10.1111/j.1742-1241.2009.02099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Plaisance EP, Lukasova M, Offermanns S, Zhang Y, Cao G, Judd RL. Niacin stimulates adiponectin secretion through the GPR109A receptor. American Journal of Physiology - Endocrinology and Metabolism. 2009;296(3):E549–E558. doi: 10.1152/ajpendo.91004.2008. [DOI] [PubMed] [Google Scholar]
- 260.Song W, Huo T, Guo F, Wang H, Wei H, Yang Q, Dong H, Wang Q, Xiong L. Globular adiponectin elicits neuroprotection by inhibiting NADPH oxidase-mediated oxidative damage in ischemic stroke. Neuroscience. 2013;248:136–144. doi: 10.1016/j.neuroscience.2013.05.063. [DOI] [PubMed] [Google Scholar]
- 261.Rall SC, Jr, Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. 1982;257(8):4171–4178. [PubMed] [Google Scholar]
- 262.Mahley RW, Huang Y. Apolipoprotein E: from atherosclerosis to Alzheimer's disease and beyond. Curr Opin Lipidol. 1999;10(3):207–217. doi: 10.1097/00041433-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 263.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 264.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240(4852):622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 265.Zhang H, Wu LM, Wu J. Cross-talk between apolipoprotein E and cytokines. Mediators Inflamm. 2011;2011:949072. doi: 10.1155/2011/949072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45(8):1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 267.Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8(4):468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- 268.Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294(5545):1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 269.Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60(6):1158–1171. doi: 10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res Mol Brain Res. 1991;11(2):97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- 271.Li YJ, Hauser MA, Scott WK, Martin ER, Booze MW, Qin XJ, Walter JW, Nance MA, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Goetz CG, Small GW, et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology. 2004;62(11):2005–2009. doi: 10.1212/01.wnl.0000128089.53030.ac. [DOI] [PubMed] [Google Scholar]
- 272.Pulkes T, Papsing C, Mahasirimongkol S, Busabaratana M, Kulkantrakorn K, Tiamkao S. Association between apolipoprotein E genotypes and Parkinson's disease. J Clin Neurosci. 2011;18(10):1333–1335. doi: 10.1016/j.jocn.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 273.Ezquerra M, Campdelacreu J, Gaig C, Compta Y, Munoz E, Marti MJ, Valldeoriola F, Tolosa E. Lack of association of APOE and tau polymorphisms with dementia in Parkinson's disease. Neurosci Lett. 2008;448(1):20–23. doi: 10.1016/j.neulet.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 274.Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB. A large study reveals no association between APOE and Parkinson's disease. Neurobiol Dis. 2012;46(2):389–392. doi: 10.1016/j.nbd.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Buchanan DD, Silburn PA, Prince JA, Mellick GD. Association of APOE with Parkinson disease age-at-onset in women. Neurosci Lett. 2007;411(3):185–188. doi: 10.1016/j.neulet.2006.07.080. [DOI] [PubMed] [Google Scholar]
- 276.Kurz MW, Dekomien G, Nilsen OB, Larsen JP, Aarsland D, Alves G. APOE alleles in Parkinson disease and their relationship to cognitive decline: a population-based, longitudinal study. J Geriatr Psychiatry Neurol. 2009;22(3):166–170. doi: 10.1177/0891988709332945. [DOI] [PubMed] [Google Scholar]
- 277.Ryu HG, Kwon OD. Apolipoprotein E epsilon 4 allele is not associated with age at onset or MMSE of Parkinson's disease in a Korean study. Parkinsonism Relat Disord. 2010;16(9):615–617. doi: 10.1016/j.parkreldis.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 278.Harhangi BS, de Rijk MC, van Duijn CM, Van Broeckhoven C, Hofman A, Breteler MM. APOE and the risk of PD with or without dementia in a population-based study. Neurology. 2000;54(6):1272–1276. doi: 10.1212/wnl.54.6.1272. [DOI] [PubMed] [Google Scholar]
- 279.Kermer P, Kohn A, Schnieder M, Lingor P, Bahr M, Liman J, Dohm CP. BAG1 is neuroprotective in in vivo and in vitro models of Parkinson's disease. J Mol Neurosci. 2015;55(3):587–595. doi: 10.1007/s12031-014-0396-2. [DOI] [PubMed] [Google Scholar]
- 280.Kermer P, Köhn A, Schnieder M, Lingor P, Bähr M, Liman J, Dohm CP. BAG1 is neuroprotective in in vivo and in vitro models of Parkinson's disease. Journal of Molecular Neuroscience. 2015;55(3):587–595. doi: 10.1007/s12031-014-0396-2. [DOI] [PubMed] [Google Scholar]
- 281.Uchida S-i, Soshiroda K, Okita E, Kawai-Uchida M, Mori A, Jenner P, Kanda T. The adenosine A 2A receptor antagonist, istradefylline enhances anti-parkinsonian activity induced by combined treatment with low doses of L-DOPA and dopamine agonists in MPTP-treated common marmosets. European Journal of Pharmacology. 2015;766:25–30. doi: 10.1016/j.ejphar.2015.09.028. [DOI] [PubMed] [Google Scholar]
- 282.Mizuno Y, Kondo T. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson's disease. Movement Disorders. 2013;28(8):1138–1141. doi: 10.1002/mds.25418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Litim N, Bourque M, Al Sweidi S, Morissette M, Di Paolo T. The 5α-reductase inhibitor Dutasteride but not Finasteride protects dopamine neurons in the MPTP mouse model of Parkinson's disease. Neuropharmacology. 2015;97:86–94. doi: 10.1016/j.neuropharm.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 284.Bortolato M, Cannas A, Solla P, Bini V, Puligheddu M, Marrosu F. Finasteride attenuates pathological gambling in patients with Parkinson disease. Journal of clinical psychopharmacology. 2012;32(3):424–425. doi: 10.1097/JCP.0b013e3182549c2a. [DOI] [PubMed] [Google Scholar]
- 285.Gómez-Gálvez Y, Palomo-Garo C, Fernández-Ruiz J, García C. Potential of the cannabinoid CB2 receptor as a pharmacological target against inflammation in Parkinson's disease. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2016;64:200–208. doi: 10.1016/j.pnpbp.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 286.Ternianov A, Pérez-Ortiz JM, Solesio ME, García-Gutiérrez MS, Ortega-Álvaro A, Navarrete F, Leiva C, Galindo MF, Manzanares J. Overexpression of CB2 cannabinoid receptors results in neuroprotection against behavioral and neurochemical alterations induced by intracaudate administration of 6-hydroxydopamine. Neurobiol Aging. 2012;33(2):421. doi: 10.1016/j.neurobiolaging.2010.09.012. e421-421. e416. [DOI] [PubMed] [Google Scholar]
- 287.Alquezar C, Barrio E, Esteras N, de la Encarnacion A, Bartolome F, Molina JA, Martin-Requero A. Targeting cyclin D3/CDK6 activity for treatment of Parkinson's disease. J Neurochem. 2015;133(6):886–897. doi: 10.1111/jnc.13070. [DOI] [PubMed] [Google Scholar]
- 288.Morales-Garcia JA, Alonso-Gil S, Gil C, Martinez A, Santos A, Perez-Castillo A. Phosphodiesterase 7 inhibition induces dopaminergic neurogenesis in hemiparkinsonian rats. Stem Cells Transl Med. 2015;4(6):564–575. doi: 10.5966/sctm.2014-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Morales-Garcia JA, Redondo M, Alonso-Gil S, Gil C, Perez C, Martinez A, Santos A, Perez-Castillo A. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS One. 2011;6(2):e17240. doi: 10.1371/journal.pone.0017240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Chen X, Wales P, Quinti L, Zuo F, Moniot S, Herisson F, Rauf NA, Wang H, Silverman RB, Ayata C, Maxwell MM, Steegborn C, Schwarzschild MA, Outeiro TF, Kazantsev AG. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson's disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS One. 2015;10(1):e0116919. doi: 10.1371/journal.pone.0116919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Patel VP, Chu CT. Decreased SIRT2 activity leads to altered microtubule dynamics in oxidatively-stressed neuronal cells: Implications for Parkinson's disease. Exp Neurol. 2014;257:170–181. doi: 10.1016/j.expneurol.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Aime P, Sun X, Zareen N, Rao A, Berman Z, Volpicelli-Daley L, Bernd P, Crary JF, Levy OA, Greene LA. Trib3 Is Elevated in Parkinson's Disease and Mediates Death in Parkinson's Disease Models. J Neurosci. 2015;35(30):10731–10749. doi: 10.1523/JNEUROSCI.0614-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Na SJ, DiLella AG, Lis EV, Jones K, Levine DM, Stone DJ, Hess J. Molecular profiling of a 6-hydroxydopamine model of Parkinson's disease. Neurochemical research. 2010;35(5):761–772. doi: 10.1007/s11064-010-0133-3. [DOI] [PubMed] [Google Scholar]
- 294.Dolga AM, de Andrade A, Meissner L, Knaus HG, Hollerhage M, Christophersen P, Zischka H, Plesnila N, Hoglinger GU, Culmsee C. Subcellular expression and neuroprotective effects of SK channels in human dopaminergic neurons. Cell Death Dis. 2014;5:e999. doi: 10.1038/cddis.2013.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Chen L, Deltheil T, Turle-Lorenzo N, Liberge M, Rosier C, Watabe I, Sreng L, Amalric M, Mourre C. SK channel blockade reverses cognitive and motor deficits induced by nigrostriatal dopamine lesions in rats. International Journal of Neuropsychopharmacology. 2014;17(8):1295–1306. doi: 10.1017/S1461145714000236. [DOI] [PubMed] [Google Scholar]
- 296.Francardo V, Bez F, Wieloch T, Nissbrandt H, Ruscher K, Cenci MA. Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain. 2014;137(Pt 7):1998–2014. doi: 10.1093/brain/awu107. [DOI] [PubMed] [Google Scholar]
- 297.Francardo V. Sigma-1 receptor: a potential new target for Parkinson's disease? Neural regeneration research. 2014;9(21):1882. doi: 10.4103/1673-5374.145351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, Stankowski J, Kim MS, Zhong J, Kumar M, Andrabi SA, Xiong Y, Dickson DW, Wszolek ZK, Pandey A, Dawson TM, et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson's disease. Cell. 2014;157(2):472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Valadas JS, Vos M, Verstreken P. Therapeutic strategies in Parkinson's disease: what we have learned from animal models. Annals of the New York Academy of Sciences. 2015;1338(1):16–37. doi: 10.1111/nyas.12577. [DOI] [PubMed] [Google Scholar]
- 300.Rajagopalan S, Chinta SJ, Andersen JK. Pharmacological prolyl hydroxylase domain inhibition as a therapeutic target for Parkinson's disease. CNS Neurol Disord Drug Targets. 2014;13(1):120–125. doi: 10.2174/18715273113126660131. [DOI] [PubMed] [Google Scholar]
- 301.Johansen JL, Sager TN, Lotharius J, Witten L, Mørk A, Egebjerg J, Thirstrup K. HIF prolyl hydroxylase inhibition increases cell viability and potentiates dopamine release in dopaminergic cells. J Neurochem. 2010;115(1):209–219. doi: 10.1111/j.1471-4159.2010.06917.x. [DOI] [PubMed] [Google Scholar]
- 302.Ogundele OM, Nanakumo ET, Ishola AO, Obende OM, Enye LA, Balogun WG, Cobham AE, Abdulbasit A. NMDA R/+VDR pharmacological phenotype as a novel therapeutic target in relieving motor-cognitive impairments in Parkinsonism. Drug Chem Toxicol. 2015:1–13. doi: 10.3109/01480545.2014.975355. [DOI] [PubMed] [Google Scholar]
- 303.Butler MW, Burt A, Edwards TL, Zuchner S, Scott WK, Martin ER, Vance JM, Wang L. Vitamin D receptor gene as a candidate gene for Parkinson disease. Annals of human genetics. 2011;75(2):201–210. doi: 10.1111/j.1469-1809.2010.00631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Han B, Jin HJ, Song MY, Wang T, Zhao H. A potential target for the treatment of Parkinson's disease: effect of lateral habenula lesions. Parkinsonism Relat Disord. 2014;20(11):1191–1195. doi: 10.1016/j.parkreldis.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 305.Sourani D, Eitan R, Gordon N, Goelman G. The habenula couples the dopaminergic and the serotonergic systems: application to depression in Parkinson's disease. European Journal of Neuroscience. 2012;36(6):2822–2829. doi: 10.1111/j.1460-9568.2012.08200.x. [DOI] [PubMed] [Google Scholar]
- 306.Feng Y, Liu T, Li XQ, Liu Y, Zhu XY, Jankovic J, Pan TH, Wu YC. Neuroprotection by Orexin-A via HIF-1alpha induction in a cellular model of Parkinson's disease. Neurosci Lett. 2014;579:35–40. doi: 10.1016/j.neulet.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 307.Wu Y, Li X, Xie W, Jankovic J, Le W, Pan T. Neuroprotection of deferoxamine on rotenone-induced injury via accumulation of HIF-1α and induction of autophagy in SH-SY5Y cells. Neurochem Int. 2010;57(3):198–205. doi: 10.1016/j.neuint.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 308.Sun X, Liu J, Crary JF, Malagelada C, Sulzer D, Greene LA, Levy OA. ATF4 protects against neuronal death in cellular Parkinson's disease models by maintaining levels of parkin. J Neurosci. 2013;33(6):2398–2407. doi: 10.1523/JNEUROSCI.2292-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Bouman L, Schlierf A, Lutz A, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death & Differentiation. 2011;18(5):769–782. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Kilpatrick K, Novoa JA, Hancock T, Guerriero CJ, Wipf P, Brodsky JL, Segatori L. Chemical induction of Hsp70 reduces alpha-synuclein aggregation in neuroglioma cells. ACS Chem Biol. 2013;8(7):1460–1468. doi: 10.1021/cb400017h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Witt SN. Hsp70 molecular chaperones and Parkinson's disease. Biopolymers. 2010;93(3):218–228. doi: 10.1002/bip.21302. [DOI] [PubMed] [Google Scholar]
- 312.Xilouri M, Brekk OR, Kirik D, Stefanis L. LAMP2A as a therapeutic target in Parkinson disease. Autophagy. 2013;9(12):2166–2168. doi: 10.4161/auto.26451. [DOI] [PubMed] [Google Scholar]
- 313.Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper J. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson's disease. Cell Death Dis. 2013;4(3):e545. doi: 10.1038/cddis.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. The Journal of Neuroscience. 2010;30(37):12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kolosova NG, Vitovtov AO, Muraleva NA, Akulov AE, Stefanova NA, Blagosklonny MV. Rapamycin suppresses brain aging in senescence-accelerated OXYS rats. Aging (Albany NY) 2013;5(6):474–484. doi: 10.18632/aging.100573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Mudo G, Makela J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Malkia A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D. Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci. 2012;69(7):1153–1165. doi: 10.1007/s00018-011-0850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA. PGC-1α, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. 2010;2(52):52ra73–52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Tai CH, Yang YC, Pan MK, Huang CS, Kuo CC. Modulation of subthalamic T-type Ca(2+) channels remedies locomotor deficits in a rat model of Parkinson disease. J Clin Invest. 2011;121(8):3289–3305. doi: 10.1172/JCI46482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Miwa H, Koh J, Kajimoto Y, Kondo T. Effects of T-type calcium channel blockers on a parkinsonian tremor model in rats. Pharmacology Biochemistry and Behavior. 2011;97(4):656–659. doi: 10.1016/j.pbb.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 320.Su M, Shi JJ, Yang YP, Li J, Zhang YL, Chen J, Hu LF, Liu CF. HDAC6 regulates aggresome-autophagy degradation pathway of alpha-synuclein in response to MPP+-induced stress. J Neurochem. 2011;117(1):112–120. doi: 10.1111/j.1471-4159.2011.07180.x. [DOI] [PubMed] [Google Scholar]
- 321.Simões-Pires C, Zwick V, Nurisso A, Schenker E, Carrupt P-A, Cuendet M. HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs. Mol Neurodegener. 2013;8(7):22. doi: 10.1186/1750-1326-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Qin R, Li X, Li G, Tao L, Li Y, Sun J, Kang X, Chen J. Protection by tetrahydroxystilbene glucoside against neurotoxicity induced by MPP+: The involvement of PI3K/Akt pathway activation. Toxicology Letters. 2011;202(1):1–7. doi: 10.1016/j.toxlet.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 323.Jha SK, Jha NK, Kar R, Ambasta RK, Kumar P. p38 MAPK and PI3K/AKT Signalling Cascades inParkinson's Disease. International journal of molecular and cellular medicine. 2015;4(2):67. [PMC free article] [PubMed] [Google Scholar]