

Abstract
The kidney is a vital organ for the elimination of therapeutic drugs and their metabolites. Renal drug transporters, which are primarily located in the renal proximal tubules, play an important role in tubular secretion and reabsorption of drug molecules in the kidney. Tubular secretion is characterized by high clearance capacities, broad substrate specificities, and distinct charge selectivity for organic cations and anions. In the past two decades, substantial progress has been made in understanding the roles of transporters in drug disposition, efficacy, toxicity and drug–drug interactions (DDIs). In the kidney, several transporters are involved in renal handling of organic cation (OC) and organic anion (OA) drugs. These transporters are increasingly recognized as the target for clinically significant DDIs. This review focuses on the functional characteristics of major human renal drug transporters and their involvement in clinically significant DDIs.
Abbreviations: ABC, ATP-binding cassette; ATP, adenosine triphosphate; AUC, area under the plasma concentration curve; BBB, blood–brain barrier; Cmax, maximum plasma concentration; CHO, Chinese hamster ovary; CL, plasma clearance; CLR, renal clearance; DDIs, drug–drug interactions; fe, fraction of the absorbed dose excreted unchanged in urine; FDA, U.S. Food and Drug Administration; GSH, glutathione; HEK, human embryonic kidney; IC50, half maximal inhibitory concentration; ITC, International Transporter Consortium; Ki, inhibitory constant; MATE, multidrug and toxin extrusion protein; MPP+, 1-methyl-4-phenylpyridimium; MRP, multidrug resistance-associated protein; MSD, membrane-spanning domain; MW, molecular weight; NBD, nucleotide-binding domain; NME, new molecular entity; NSAID, non-steroidal anti-inflammatory drugs; OA, organic anion; OAT or Oat, organic anion transporters; OATP or Oatp, organic anion-transporting peptide; OC, organic cation; OCT or Oct, organic cation transporter; OCTN, Organic zwitterions/cation transporters; PAH, p-aminohippurate; P-gp, P-glycoprotein; SLC, solute carrier; SNP, single-nucleotide polymorphism; TMD, transmembrane domain; TEA, tetraethylammonium; URAT, urate transporter
KEY WORDS: Renal drug transporters, Drug–drug interactions, Organic cations, Organic anions, Nephrotoxicity
Graphical abstract
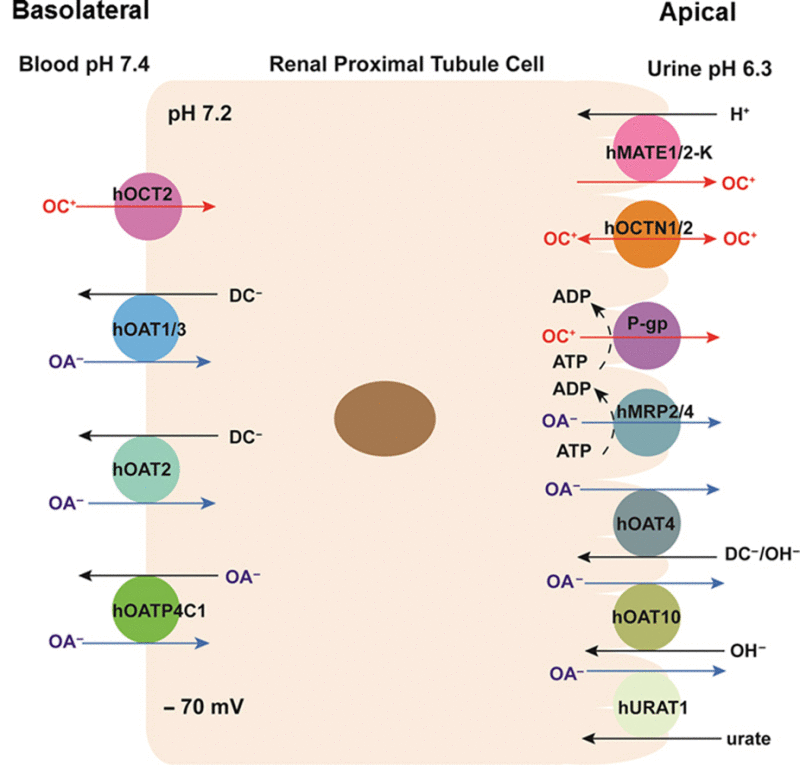
Renal drug transporters, expressed in the basolateral and apical membrane of renal proximal tubules, play an important role in tubular secretion and reabsorption of drug molecules in the kidney. These transporters are increasingly recognized as the target for clinically significant drug–drug interactions.
1. Introduction
Renal clearance is a major pathway of drug elimination. About 32% of the top 200 prescribed drugs in the U.S. in 2010 are renally eliminated with more than 25% of the absorbed dose excreted unchanged in urine1. Renal elimination is the result of three concurrent processes occurring in the nephron, which include glomerular filtration, tubular secretion, and tubular reabsorption. Glomerular filtration is a passive process while tubular secretion, and sometimes reabsorption, involves a variety of transporters located on the basolateral and luminal membranes of the tubular epithelium. These transporters are predominantly expressed in the proximal tubule and they work in tandem to eliminate drugs from the blood circulation to the urine1, 2, 3. Both basolateral and apical transporters tend to be charge selective for anionic and cationic drugs, although recent study suggests that there is some degree of overlap3, 4. In humans, major transporters involved in tubular secretion of cationic drugs include organic cation transporter 2 (hOCT2) on the basolateral membrane and the multidrug and toxin extrusion proteins 1 and 2-K (hMATE1 and hMATE2-K) on the apical membrane1, 3. P-glycoprotein (P-gp) is also expressed in the apical member to facilitate the excretion of larger and more hydrophobic cations. The major transporters engaged in secretion of anionic drugs include organic anion transporters 1 and 3 (hOAT1 and hOAT3) on the basolateral membrane and multidrug resistance-associated proteins 2 and 4 (hMRP2 and hMRP4) on the apical membrane1, 3. In addition, several closely related transporters are present in the proximal tubules and they may also contribute to renal handling of drugs and metabolic wastes.
Transporter-mediated drug–drug interactions (DDIs) are increasingly recognized as an important modifier of the pharmacokinetics and pharmacodynamics of drugs2, 3, 5. Drugs inhibiting renal drug transporters may cause marked changes in the pharmacokinetics of the affected drug, resulting in clinically significant DDIs1, 2, 5. Furthermore, expression and inhibition of renal drug transporters may result in abnormal drug accumulation in renal tubular cells, leading to drug-induced nephrotoxicity. This review focuses on renal drug transporters and their significance in DDIs and drug-induced nephrotoxicity. We first briefly summarize the current knowledge on major renal drug transporters including their expression, cellular localization, transport mechanisms, and substrate specificities. We then review the basic principles underlying renal DDIs and highlight the importance of renal drug transporters in clinically significant DDIs. The relevant consequences on pharmacokinetics, pharmacodynamics, and drug-induced nephrotoxicity are illustrated using several well-studied clinical DDI examples. Lastly, a brief summary along with current challenges in the field is presented.
2. Major drug transporters in human kidney
More than 400 membrane transporters are encoded by the human genome, and generally fall into the following two superfamilies: the adenosine triphosphate (ATP)-binding cassette (ABC) and the solute carrier (SLC)1, 3. ABC transporters are primary active transporters that can transport substrates against their electrochemical gradients, utilizing energy generated from ATP hydrolysis. SLC transporters have diverse modes of transport. Facilitative SLC transporters transport substrates down their electrochemical gradients without coupling to an energy input. On the other hand, active SLC transporters can mediate uphill transport of a substrate against its electrochemical gradient by coupling to a co-transported ion (e.g., Na+ and H+) or solute1. The major drug transporters involved in OC and OA transport in the human kidney are shown in Fig. 1. The molecular and functional characteristics of these transporters are described below.
Figure 1.

Major drug transporters expressed in human renal proximal tubule cells. ADP, adenosine diphosphate; ATP, adenosine triphosphate; DC, dicarboxylate; OA, organic anion; and OC, organic cation.
2.1. Cationic drug transporters
2.1.1. hOCTs (SLC22A)
hOCTs belong to the SLC22 family6. Following the first cloning of rat OCT1 (rOCT1) in 19947, 16 additional OCTs were cloned from different species6. In human, three OCT isoforms (hOCT1, 2, and 3) have been identified. hOCT2 is about 70% identical to hOCT18, and hOCT3 is about 50% identical to hOCT1 and hOCT29. hOCTs are membrane proteins with 553–556 amino acid residues8, 9 and are predicted to have 12 transmembrane domains (TMDs)6. In humans, hOCT2 is the major OCT isoform expressed in the kidney6, 8. hOCT1, on the other hand, is predominantly expressed in the liver; and hOCT3 is broadly expressed in many tissues including the skeletal muscle, heart, placenta, and salivary glands6, 9, 10. hOCT1-3 are polyspecific transporters with a large overlap in substrate specificity6. They typically translocate relatively small, hydrophilic, and structurally diverse organic cations2, 6. In the kidney, hOCT2 is located in the basolateral membrane of renal proximal tubule cells1. It mediates the first step in OC secretion in the kidney by translocating drug molecules from systemic circulation into the renal tubule cells2, 6, 11. Transport by hOCT2 is electrogenic and Na+-independent, and facilitated by the inside-negative membrane potential existing in the kidney tubular cells8. Common substrates for hOCT2 include model cations tetraethylammonium (TEA) and 1-methyl-4-phenylpyridimium (MPP+), endogenous monoamines, the antidiabetic drug metformin, the antihypertensive drug atenolol, the antiviral drug lamivudine, and the cytostatic drug oxaliplatin1, 2, 12, 13. Most hOCT2 inhibitors are larger, more hydrophobic cations that may or may not be transported by the transporter1, 2, 6. Several clinically used drugs, including cimetidine, quinidine and dolutegravir, are known hOCT2 inhibitors2, 14. The mRNA of hOCT3 is also detectable in the kidney but at a much lower level15, 16. The membrane localization of hOCT3 in human kidney is unclear. Further investigation is needed to elucidate the role of hOCT3 in renal excretion of drug molecules.
2.1.2. hMATEs (SLC47A)
hMATEs belong to SLC47 family. Two human orthologues of the bacterial MATE proteins, MATE1 and MATE2 were first cloned in 200517. Soon after, two splice variants of hMATE2 were isolated from kidney and brain separately and were designated as hMATE2-K and hMATE2-B, respectively18. hMATE1 and hMATE2 are 47.5% identical17. hMATE1, hMATE2 and hMATE2-K are proteins of 570, 602 and 566 amino acids17, 18, respectively, and are currently predicted to have 13 TMDs19, 20. hMATE2-B is a truncated protein of 220 amino acids and is not functional with respect to transport18. hMATE1 has the highest expression level in the kidney and is also strongly expressed in other tissues including the liver, skeleton muscle and adrenal gland17, 18. Immunohistochemistry of human tissue revealed that in the kidney, hMATE1 is localized to the apical membrane of renal proximal tubule cells and distal convoluted tubules; and in the liver, it is expressed in bile canaliculi17. The full-length hMATE2 and the kidney-specific splice variant hMATE2-K are predominantly expressed in the kidney17, 18, 21. Immunostaining showed both of them are expressed in the renal proximal tubule and hMATE2-K is localized to the luminal membrane of the tubule cells18, 21. Different from hMATE1/2-K, hMATE2 was localized in intracellular vesicular structures upon expression in human embryonic kidney (HEK) 293 cells and only showed transport activity when reconstituted into liposomes21. hMATE1 and hMATE2-K are OC/proton exchangers and need an oppositely oriented proton gradient to drive the transport17, 18, 21. In the nephron, the tubular lumen is more acidic (~pH 6.3) than the cytosol, providing an inwardly directed proton gradient across the apical membrane of proximal tubule epithelial cells. hMATE-mediated influx of protons is coupled with the efflux of OCs into the urine. hMATE1/2-K share a broad spectrum of substrates and inhibitors with the hOCT222. In the kidney, hMATE1/2-K mainly coordinate with hOCT2 to mediate OC secretion. However, hMATE1/2-K can also transport several anionic compounds and zwitterions22, which suggests that they may also partner with hOATs for renal excretion of anionic and zwitterionic drugs.
2.1.3. hOCTN (SLC22A)
Organic zwitterions/cation transporters (OCTNs) belong to the same SLC22 gene subfamily as OCTs. There are three OCTN isomers (OCTN1–3) in rodents, but humans only have OCTN1 and OCTN26. The first human OCTN, hOCTN1, was cloned in 1997 from human fetal liver23. Soon after, hOCTN2 was cloned by screening a human kidney cDNA library24. hOCTN1 and hOCTN2 have 75.8% identity and both have high expression level in the kidney23, 24, where they are located in the apical membrane of renal proximal tubule cells6, 25, 26. Both hOCTN1 and hOCTN2 can transport OC and zwitterions, but the transport mechanisms are substrate-dependent and quite different for each transporter. hOCTN1 has a high affinity for the zwitterionic antioxidant ergothioneine, the uptake of which is stimulated by extracellular sodium27. hOCTN1 also appears to transport OCs such as TEA by an OC/H+ exchange mechanism23, 28. The exact role of hOCTN1 in the renal proximal tubules is unclear. It may participate in Na+-dependent reabsorption of ergothioneine from the filtrate; alternatively, it may contribute to tubular secretion by mediating OC efflux at the apical membrane driven by the acidic pH in the lumen23, 27, 28. hOCTN2 has a high affinity for l-carnitine and functions as a Na+--l-carnitine cotransporter24. In addition, hOCTN2 can also transport OCs in Na+-independent manner29. Similar to hOCTN1, hOCTN2 may participate in either renal reabsorption of zwitterions (e.g., l-carnitine) or secretion of xenobiotic OCs depending on its mode of transport. While the proton/OC antiporters hMATE1/2-K are apparently the most important extrusion transporters for OC efflux at the luminal membrane30, hOCTN1/2 have different substrate selectivity and may contribute to the secretion of certain OC or zwitterion drugs. Interestingly, a recent pharmacogenomics study suggested that hOCTN1 is involved in active tubular secretion of gabapentin, an anticonvulsant widely prescribed for epilepsy and other neuropathic disorders31.
2.1.4. P-gp (ABCB1)
P-glycoprotein (P-gp) is probably the most well studied ABC transporter to date. It was first identified in 1976 as a cell surface glycoprotein from Chinese hamster ovary (CHO) cells resistant to colchicine32. Overexpressed in many cancer cells, P-gp decreases drug accumulation in multidrug-resistant cells and mediates the development of resistance to anticancer drugs32. As a typical ABC transporter, it has two membrane-spanning domains (MSDs) and two cytoplasmic nucleotide-binding domains (NBDs). Using energy generated from ATP hydrolysis, P-gp actively transports its substrates out of cells against their concentration gradients. A vast number of therapeutic drugs, such as anticancer drugs, HIV protease inhibitors, immunosuppressants, cardioactive drugs and antifungals, interact with P-gp33, 34, 35. Typical P-gp substrates are lipophilic or amphipathic large molecules (molecular weight > 400 Da) carrying a positive charge at pH 7.4. However, neutral drugs with bulky ring structures (steroids and cyclic peptides) are also transported by P-gp. Interestingly, many of drugs transported by P-gp are also substrate of drug-metabolizing cytochrome P450 (CYP) enzymes, especially CYP3A4/533.
Besides cancer cells, P-gp is broadly expressed in many normal tissues including excretory organs and tissue barriers important for drug disposition. The transporter has been localized to the luminal membrane of brain endothelial cells forming the blood–brain barrier (BBB), canalicular membrane of hepatocytes, apical surface of intestinal columnar epithelial cells, the apical membrane of kidney proximal tubule cells, and the apical membrane of placental syncytiotrophoblast cells2, 34, 36. The expression of P-gp in organs important for drug elimination and distribution is consistent with a protective role of P-gp in promoting drug elimination from the body and preventing drug entry into critical organs such as the brain and the developing fetus33, 37, 38, 39. In the human intestine, P-gp and CYP3A are co-localized to the mucosal epithelial cells36, 40. It was suggested that P-gp and CYP3A work together to synergistically limit oral bioavailability of many drugs33. Both P-gp and CYP3A are inducible by pregnane X receptor ligands (e.g., rifampin)41, 42. In the kidney, P-gp has been identified in the apical membrane of human proximal tubule cells by immunostaining, consistent with a role in facilitating renal drug excretion34. There is also evidence that expression of P-gp is increased after ischemic reperfusion injury in kidney43.
2.2. Anionic drug transporters
2.2.1. hOATs (SLC22A)
Despite transporting a largely different group of anionic substrates, OATs belong to the same SLC22 family that also encodes the OCTs. OAT was first discovered in 1997 with the cloning of rat and flounder Oat144, 45, 46. The cloned OAT/Oats are proteins of 536–556 amino acids and are predicted to have 12 TMDs47, 48, 49. In human, 10 OAT isoforms have been identified, including hOAT1–8, hOAT10, and the urate transporter 1 (hURAT1)47. Among them, hOAT1–4, hOAT7, hOAT10 and hURAT1 have been functionally characterized47, 50. hOAT1, the first cloned human OAT51, has 4 splice variants, hOAT1-1, hOAT1-2, hOAT1-3 and hOAT1-452. hOAT1-1 and hOAT1-2 are longer and showed similar transport activity while hOAT1-3 and hOAT1-4 are shorter and lack of transport activity52. Most hOATs have expression in the renal proximal tubule, except hOAT7, which is restrictedly expressed in the liver47, 53. In the kidney, hOAT1–3 are located on the basolateral membrane of renal tubule cells whereas hOAT4, hOAT10 and hURAT1 are expressed on the luminal membrane47. Basally-expressed hOAT1–3 function as organic anion (OA)/dicarboxylate exchangers which mediate the first step of OA renal excretion by transporting OAs into renal tubule cells utilizing the outward dicarboxylate (e.g., α-ketoglutarate for hOAT1/3, succinate for hOAT2) gradient established by the Na+–dicarboxylate cotransporter47. hOAT1 and hOAT3 have substantial overlap in their substrate specificities, accepting relatively small and hydrophilic OAs2, 50. hOAT3 appears to be more tolerant in size and charge of its substrates than hOAT1 and can transport bulkier (e.g., estrone sulfate) and even positively charged (e.g., cimetidine) compounds2, 50. Numerous drugs have been shown to be substrates of hOAT1/3, including antibiotics, antivirals, antihypertensive drugs, diuretics, cytostatics, H2-antagonists, non-steroidal anti-inflammatory drugs (NSAIDs), statins and uricosurics1, 54. The role of hOAT2 in renal handling of drugs is less clear. Reported substrates of hOAT2 include some endogenous compounds, such as glutamate, nucleobases, nucleosides and nucleotides, and some drug molecules, such as salicylate, bumetanide and erythromycin50.
Apically-expressed hOATs and hURAT1 may have multiple transport mechanisms. hOAT4 can transport in both influx and efflux modes55. As an influx transporter, it can take up estrone sulfate and urate through OA/dicarboxylate or OA/OH− exchange mode55, 56. As an efflux transporter, it can release PAH into the tubule lumen via PAH/Cl− exchange55. hOAT10 is an antiporter, taking up p-aminohippurate (PAH), urate and nicotinate possibly by OA/OH– exchange57. Although hOAT4 and hOAT10 have both been implicated in drug transport in the kidney, their roles in tubular drug secretion and/or reabsorption still need to be clarified. hURAT1 is known to play an important role in urate homeostasis. It reabsorbs urate from lumen of renal tubule by exchanging extracellular urate with intracellular OAs such as lactate and nicotinate58.
2.2.2. hMRPs (ABCC)
MRPs are ATP-dependent efflux transporters. They use energy generated from ATP hydrolysis to export molecules out of cells. They are part of the C branch of ABC family, which can be further divided into two subfamilies, “long” (MRP1, 2, 3, 6, and 7) and “short” (MRP4, 5, 8, 9, and 10)59. The short MRPs have the typical ABC transporter structure with two MSDs and two cytoplasmic NBDs, while the long MRPs have an additional MSD59. Among the 10 identified hMRP genes, 8 (hMRP1–8) have been confirmed to encode functional proteins59. Several hMRP isoforms are expressed in the kidney, including hMRP1, hMRP2, hMRP3, and hMRP460, 61, 62, 63, 64. In particular, hMRP2 and hMRP4 are located in the apical membrane domain of renal proximal tubule cells, suggesting their role in efflux of molecules into the tubule lumen60, 61. In mouse kidney, MRP1 was found in the basolateral membrane of the distal and collecting tubule cells, but not in proximal tubule cells65. Similarly, in human kidney, hMRP3 is located in the basolateral membrane of distal convoluted tubules66. The role of hMRP1 and hMRP3 in the kidney remains unclear. The typical substrates of hMRPs are the smaller unconjugated organic anions, such as PAH, and the larger conjugated organic anions, including glutathione (GSH) conjugates and glucuronides2. hMRP2/4 have some substrate overlap with hOAT1/3. Accordingly, hMRP2 and hMRP4 may coordinate with hOAT1/3 to mediate renal excretion of certain anionic drugs.
2.2.3. hOATPs (SLCO)
Organic anion-transporting peptides (OATPs) are SLC carriers predicted to have 12 TMDs67. The first OATP was cloned from rat in 199468. One year later, the first human OATP, OATP1A2, was isolated from human liver69. Today, OATP superfamily consists of more than 300 members from over 40 species, which form 6 families, OATP1–670. In human, 11 members have been identified, which are hOATP1A2, hOATP1B1, hOATP1B3, hOATP1C1, hOATP2A1, hOATP2B1, hOATP3A1, hOATP4A1, hOATP4C1, hOATP5A1 and hOATP6A170. OATPs can transport anionic and amphipathic molecules that are relatively large (>450) and have a high degree of albumin binding under physiological conditions71. The transport by OATPs is Na+-independent, but the exact transport mechanisms are unclear70. They are believed to act as an OA/OA exchanger, coupling cellular uptake of organic compounds with efflux of intracellular bicarbonate, GSH and GSH conjugates70. In addition, uptake by some OATPs is pH-sensitive and appears to have higher uptake rate at lower extracellular pH70. Among the 11 hOATPs, hOATP1B1 and 1B3 are considered to be liver-specific72, while hOATP4C1 was predicted to be kidney-specific73. hOATP4C1 can transport cardiac glycoside (digoxin and ouabain) and thyroid hormone (tri-iodothyronine) with high affinities73. Its rat counterpart OATP4C1 is localized to the basolateral membrane of rat kidney proximal tubule cells, suggesting that hOATP4C1 might mediate the first step in renal excretion of digoxin and other compounds73.
3. Renal transporter-mediated drug interactions
In the human kidney, elimination of drugs consists of passive glomerular filtration, active tubular secretion and passive or active reabsorption. For xenobiotics, reabsorption is believed to occur mainly through a passive process74. DDIs due to inhibition of tubular secretion thus represent the most common type of drug interactions at the renal level. Inhibition at a tubular secretion site decreases renal secretion clearance, which may result in increased drug concentrations in the plasma, altered pharmacological and toxicological responses. Furthermore, renal DDIs may change drug accumulation in proximal tubule cells, leading to drug-induced nephrotoxicity and kidney injury1, 2. Although renal DDIs are often unwanted as they may lead to adverse drug reactions, occasionally, coadministration of an inhibitor (e.g., probenecid) is used deliberately to either alter renal clearance or reduce nephrotoxicity of another drug75, 76. Recognizing the important roles of transporters in drug disposition and interactions, the International Transporter Consortium (ITC) and the U.S. Food and Drug Administration (FDA) have recently published a series of papers and recommendations for assessing DDI potentials between a new molecular entity (NME) and clinically important transporters including the renal hOCT2 and hOAT1/377, 78, 79, 80.
Historically, numerous clinically significant DDIs in the kidney have been reported and attributed to the inhibition of renal organic cation and anion secretion systems1, 2, 5. Cimetidine has been historically used as the classic inhibitor for the OC system whereas probenecid is considered as the prototypical inhibitor of the OA system1, 2, 5. Inhibitors of the renal OC and OA secretion systems are often non-specific and interact with both apical and basolateral transporters. While inhibition of a basolateral or an apical transporter both decreases tubular secretion, the impact on intrarenal drug accumulation and toxicity is completely different. As illustrated in Fig. 2, inhibition of a basolateral uptake transporter reduces drug accumulation within renal tubular cells, thus is nephron-protective. In contrast, inhibition of apical efflux transporters diminishes drug exit from renal tubular cells, which can lead to increased drug accumulation and nephrotoxicity. Such scenarios are demonstrated in the clinical DDI examples later. Therefore, knowing the precise site of interaction (i.e., apical vs. basolateral) is critical to predict whether an inhibitor has a nephron-toxic or a nephron-protective effect in vivo.
Figure 2.

Hypothesized effects of transporter inhibition on tubular drug secretion and intracellular accumulation. When a basolateral uptake transporter such as hOCT2 is the main inhibition site, both renal secretion and intracellular drug accumulation are decreased. In contrast, when an apical efflux transporter such as hMATE1 is the primary inhibition site, tubular secretion is decreased but the intracellular drug level is increased.
Clinically, several pharmacokinetic conditions must be satisfied for significant DDIs to occur at the level of renal transporters. First, the affected drug must be actively secreted in the kidney and transporter-mediated renal clearance must account for a significant portion of its total clearance. Second, clinical unbound concentrations of the interacting drug (i.e., the inhibitor) must be high enough in order to produce a pronounced effect. When plasma concentrations of the inhibitor are much less than the inhibitory constant (Ki), the potential for significant drug interactions is small. However, for drugs with a narrow therapeutic window, even small changes in their pharmacokinetic profiles may be clinically relevant. In the following section, we highlight the importance of renal OC and OA drug transporters in mediating clinically significant DDIs. The relevant consequences on pharmacokinetics, pharmacodynamics, and drug-induced nephrotoxicity are illustrated using several well-studied clinical DDI examples as summarized in Table 114, 76, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90.
Table 1.
Examples of clinically observed DDIs involving renal drug transporters.
| Implicated transporters | Victim drug | Perpetrator drug | AUC fold increase | CLR decrease (%) | References |
|---|---|---|---|---|---|
| hOCT2, hMATE1, and hMATE2-K | Metformin | Cimetidine | 1.5 | 28 | 81 |
| Metformin | Cimetidine | 1.5 | 45 | 82 | |
| Metformin | Pyrimethamine | 1.4 | 35 | 83 | |
| Metformin | Dolutegravir | 2.5 | N.D. | 14 | |
| hOAT1 and hOAT3 | Furosemide | Probenecid | 2.7 | 66 | 84 |
| Furosemide | Probenecid | 3.1 | 80 | 85 | |
| Cidofovir | Probenecid | 1.8 | 52 | 76 | |
| Fexofenadine | Probenecid | 1.5 | 73 | 86 | |
| Fexofenadine | Probenecid | 1.5 | 70 | 87 | |
| P-gp | Digoxin | Quinidine | N.D. | 56 | 88 |
| Digoxin | Quinidine | N.D. | 33 | 89 | |
| Digoxin | Quinidine | N.D. | 34 | 90 |
N.D.: not determined.
3.1. Interactions involving hOCT2 and hMATE1/2-K
hOCT2 and hMATE1/2-K form a major pathway for renal elimination of small hydrophilic drugs carrying a positive charge. Inhibition of either hOCT2 or hMATE1/2-K has been implicated in many interactions involving cationic drugs1, 2, 5. In the current ITC and FDA recommendations, metformin is suggested as the in vivo probe for assessing the inhibition potential of a NME towards hOCT2 and hMATE1/2-K3, 77, 78. Metformin is the first-line treatment for type 2 diabetes. The drug is minimally metabolized in vivo and exclusively eliminated unchanged by the kidney91, 92. Its reported renal clearance (CLR) is about 454 mL/min, which is much larger than its glomerular filtration clearance92. hOCT2-hMATE1/2-K–mediated active secretion plays an important role in metformin renal elimination. To date, some of the well-established DDIs involving renal OC transport system were observed with metformin. Besides DDIs, hOCT2-mediated drug uptake and accumulation in renal proximal tubule cells is known to contribute to drug-induced kidney injury as demonstrated in the case of cisplatin nephrotoxicity.
3.1.1. Cimetidine–metformin interaction
Cimetidine, a histamine H2-receptor antagonist, is a classic inhibitor of renal OC secretion. Cimetidine is 20% protein bound in the plasma and the reported unbound maximum plasma concentration (Cmax) after a typical 400 mg oral dose is around 8 µmon/L93, 94. There have been several reports of cimetidine–metformin interaction81, 82. The largest observed area under the plasma concentration curve (AUC) increase and renal clearance (CLR) decrease is 1.5-fold and 45%, respectively82. Metformin is a substrate of both hOCT2 and hMATE1/2-K83, and is eliminated predominantly unchanged by the kidney. Historically, inhibition of basolateral hOCT2-mediated metformin uptake was thought to be the mechanism underlying the observed interaction2, 3. In addition, the inhibitory effect of cimetidine on metformin renal clearance has been reported to depend on a genetic polymorphism of hOCT2 in a cohort of Chinese subjects82. However, Ito et al.95 recently demonstrated that cimetidine has much greater in vitro inhibition potencies towards the apical hMATE1/2-K (Ki=1.1–6.9 µmol/L) than for the basolateral hOCT2 (Ki=95–146 μmol/L). These data suggest that cimetidine inhibition of apical hMATE1/2-K, but not basolateral hOCT2, is the likely mechanism underlying clinically observed cimetidine–metformin DDIs95. However, cimetidine is a substrate of hOCT2 and hMATE1/2-K, and it has been proposed that cimetidine interferes with hMATE1/2-K through an intracellular binding site96, 97. Therefore, hOCT2-mediated uptake into kidney cells could have an impact on cimetidine׳s inhibitory effect towards hMATE1/2-K, which may explain the hOCT2 genotype-dependent effect on cimetidine–metformin interaction82.
3.1.2. Pyrimethamine–metformin interaction
Pyrimethamine is an antiparasitic commonly used for malarial infection. Co-administration of pyrimethamine and metformin has been reported to result in clinically significant DDIs, leading to a 1.4-fold increase of AUC and a 35% decrease of CLR of metformin83. Pyrimethamine is a selective inhibitor of hMATE1/2-K, and its potency toward hMATE1/2-K is about 100-fold higher than that of hOCT283. Thus inhibition of apical hMATE1/2-K has been proposed to be the underlying mechanism of pyrimethamine–metformin interaction83. However, pyrimethamine is highly protein bound, the unbound concentration of the drug in the plasma is low at clinically used doses. This may explain the relative small magnitudes of changes in metformin AUC and CLR when co-administrated with pyrimethamine83. Whether pyrimethamine is actively transported into renal tubule cells is still unknown, but its lipophilic nature (logP = 2.7) and small molecular weight (MW=248.7) may allow passive diffusion into the renal cells, leading to significant inhibition of the apical hMATE1/2-K.
3.1.3. Dolutegravir–metformin interaction
Dolutegravir is a newly approved anti-HIV drug and also an inhibitor of hOCT2 and hMATE1/2-K. In vitro, dolutegravir is a more potent inhibitor for hOCT2 (half maximal inhibitory concentration (IC50) is ~1.9 µmol/L) than for hMATE1/2-K (IC50 ~6.3–25 µmol/L)14. Co-administration of dolutegravir increased metformin AUC by 2.5-fold14, a magnitude well exceeded what has been observed for cimetidine and pyrimethamine. The observed metformin AUC change in the presence of dolutegravir is higher than anticipated. Based on its IC50 values and its unbound Cmax, dolutegravir is predicted to be an irrelevant in vivo inhibitor of hMATE1/2-K but a moderate in vivo inhibitor of hOCT214, 98. Therefore, inhibition of hOCT2 only partially explains the observed AUC change of metformin. Evaluation of the effect of dolutegravir on putative transporters involved in absorption and distribution of metformin also showed negative results14, 99, 100. Thus, it is possible that other unidentified mechanism(s) may be involved in dolutegravir–metformin interaction. Nevertheless, based on the significant metformin AUC change caused by dolutegravir, it is recommended that dose adjustments of metformin be considered when patients are starting or stopping dolutegravir while on metformin therapy.
3.1.4. Cisplatin nephrotoxicity
Cisplatin is a chemotherapeutic agent used in the treatment of lung, bladder, colon, testis, and brain cancer101, 102, 103. However, nephrotoxicity, primarily in proximal tubules, is a major dose limiting toxicity of cisplatin104, 105. In vitro, cisplatin is an excellent OCT2 substrate; however, it is a poor substrate of either MATE1 or MATE2-K106, 107, 108. In animal studies, Oct1/Oct2-deficient mice exhibited impaired urinary excretion of cisplatin and were protected from severe cisplatin-induced renal tubular necrosis109, 110. In addition, a nonsynonymous single-nucleotide polymorphism (SNP) 808 G>T in hOCT2 gene was associated with reduced cisplatin-induced nephrotoxicity in cancer patients109. All these evidence supports a significant role of hOCT2 in renal handling and nephrotoxicity of cisplatin. The discovery of the critical role of OCT2 in cisplatin toxicity provided a rationale for using OCT2-selective inhibitors to mitigate the debilitating side effect of cisplatin109, 111, 112. In fact, co-administration of cisplatin and high dose cimetidine has been reported to lead to partial protection against cisplatin-induced nephrotoxicity113. These findings collectively support future exploration of hOCT2 inhibitors as potential therapeutic agents to prevent cisplatin-induced nephrotoxicity. However, as stated earlier, many OCT inhibitors also inhibit MATEs, which may increase intracellular cisplatin accumulation and toxicity. In needed, selective inhibition of MATE transporters with pyrimethamine or ondansetron was shown to increase the nephrotoxicity of cisplatin in mice114, 115. Therefore the risk of using chemical inhibitors as a cisplatin nephroprotectant should be carefully addressed given the opposing effect of hOCT2 and hMATEs in cisplatin intrarenal accumulation and toxicity (Fig. 2).
3.2. Interactions involving hOATs
Probenecid is the prototype inhibitor for the renal organic anion secretion system2, 3, 77. During World War II, probenecid was first developed as a penicillin-sparing agent to prevent the rapid urinary loss of the antibiotic. Numerous interactions between probenecid and penicillin-derivatives, or other anionic drugs, have been reported1, 2, 5. Clinically, inhibition of renal anion secretion by probenecid has also been employed to produce beneficial drug interactions to either enhance activity of antibiotics or reduce renal accumulation and nephrotoxicity of certain antiviral drugs1, 2. Probenecid exhibits similar inhibition potencies towards hOAT1 and hOAT3 with Ki values around 4–12 µmol/L1, 2. Less inhibitory effects were reported with apical hMRP2, hMRP4 and hOAT4 (Ki of 44.6, 2300, and 54.9 µmol/L, respectively)2, 116, 117, 118. At typical oral doses (e.g., 0.5–2 g), probenecid produces unbound plasma concentrations in the range of 3–50 µmol/L119, suggesting that both hOAT1 and hOAT3 are likely to be the site of drug interactions with probenecid in vivo. Nevertheless, as probenecid at higher doses also inhibits other transporters and some phase II drug metabolizing enzymes, cautions should be taken when interpreting in vivo DDI data with probenecid.
3.2.1. Probenecid–furosemide interaction
Furosemide is a loop diuretic, which exerts its pharmacological effects by inhibiting Na+-K+-2Cl– cotransporter located in the luminal membrane of loop of Henle120. Renal excretion is the major elimination pathway for furosemide with fraction of the absorbed dose excreted unchanged in urine (fe) 71%121. Due to high protein binding, glomerular filtration of furosemide is very limited121. Thus, active tubular secretion may represent the major route for both furosemide renal elimination and delivery of the diuretic to its effective site. In vitro, furosemide has been shown to be a substrate of hOAT1 and hOAT3120. Oat1-knockout mice also showed impaired furosemide renal excretion and diuretic responsiveness122, further supporting involvement of OATs in furosemide renal excretion. In humans, probenecid markedly reduces furosemide CLR and urinary excretion while increases its system exposure and half-life84, 85 (Table 1). Intriguingly, mixed results were reported regarding the effect of probenecid on the diuretic effect of furosemide85, 123, 124, 125. In some studies, pretreatment with probenecid even increased the overall response to furosemide124, 125. A detailed analysis of the time-course of the increased diuresis and natriuresis showed that probenecid decreased the response for the first 60–90 min after furosemide but increased the subsequent response sufficiently to result in a greater overall effect124. Thus, the effect of probenecid on the pharmacodynamics of furosemide in humans is complex and may not be simply predicted from changes in plasma or urinary drug levels.
3.2.2. Probenecid–cidofovir interaction
Cidofovir is an acyclic nucleotide analog used in the treatment of cytomegalovirus infection of the eye. Cidofovir is eliminated largely through renal excretion with approximately 90% of intravenous dose recovered in urine unchanged76. Nephrotoxicity, due to excessive drug accumulation in renal proximal tubule cells, is the dose-limiting toxicity for cidofovir126. Cidofovir is an hOAT1 substrate and hOAT1-mediate cytotoxicity was markedly reduced with probenecid treatment75, 127. Co-administration of high-dose probenecid with cidofovir in HIV patients reduced cidofovir CLR to a level approaching glomerular filtration, supporting the clinical use of probenecid as a nephroprotectant during cidofovir therapy76. Nowadays, co-administration of probenecid with cidofovir is required by FDA to protect patients against cidofovir-induced nephrotoxicity1.
3.2.3. Probenecid–fexofenadine interaction
Fexofenadine, an active metabolite of terfenadine, is a selective histamine H1 receptor antagonist used for the treatment of allergic rhinitis and chronic idiopathic urticaria. After oral administration, fexofenadine is mainly eliminated through biliary excretion, but renal clearance also makes a significant contribution to its total body clearance128. Several reports showed that probenecid could increase fexofenadine AUC by 1.5-fold and decrease its CLR by approximately 70%86, 87. Although fexofenadine is a known substrate of P-gp and OATPs, probenecid appears to be a weak inhibitor for these transporters. In vitro, fexofenadine showed significant accumulation in hOAT3-expressing HEK cells but not in hOAT1- and hOAT2-expressing HEK cells128. Probenecid also showed high inhibition potency toward fexofenadine uptake in hOAT3 cells with Ki value of 1.3 µmol/L128, which is much lower than the maximum unbound concentration of probenecid at typical clinical dosages119. It is likely that inhibition of hOAT3-mediated renal uptake of fexofenadine contributes to the observed probenecid–fexofenadine interactions.
3.3. Interaction involving P-gp
As an efflux pump with broad substrate specificity, P-gp plays an important role in drug disposition1. In the kidney, P-gp is located in the apical membrane of proximal tubule cells where it can actively export hydrophobic drug molecules into the urine36. There have been many reports of P-gp-mediated DDIs, but the most well studied interaction is probably P-gp-mediated interaction with digoxin2, 3, a well-established P-gp substrate. Digoxin, a commonly used cardiac glycoside, is metabolically stable and primarily eliminated through renal excretion129. Because digoxin has a narrow therapeutic window, even small changes in serum levels of digoxin may lead to clinically significant toxicities that can affect multiple organ systems130. Thus cautions must be taken when using other co-medications with digoxin.
Quinidine is a substrate and inhibitor of P-gp131. There have been several reports of quinidine–digoxin interactions with the largest reported plasma clearance (CL) decrease of digoxin being 64%132. Serum digoxin levels can reach dangerously high concentrations when co-administered with quinidine. In Caco-2 monolayers, basal-to-apical transport of digoxin was strongly inhibited by quinidine133. In addition, quinidine at same in vivo concentration markedly increased digoxin plasma concentration in wild-type mice, but not in P-gp knockout mice133. Both in vitro and in vivo data strong support that inhibition of P-gp--mediated digoxin efflux is the major underlying mechanism of quinidine–digoxin interaction. Similar digoxin–drug interactions with reduced renal clearance and have also been observed with other P-gp inhibitors such as verapamil and clarithromycin134, 135.
4. Conclusions
In conclusion, renal drug transporters play an important role in drug disposition, efficacy and toxicity. Like drug-metabolizing enzymes, they are also the target sites for DDIs. Despite the significant progresses made in our understanding on drug transporters, our knowledge of renal drug transporters and our comprehension of their roles in the kidney and the mechanisms of renal transporter-mediated DDIs are still limited. There are still significant challenges to predict and understand DDIs mediated by renal drug transporters. For example, it is still difficult to precisely locate the actual sites (apical vs. basal membranes) of renal DDIs in vivo. While the plasma concentrations of the inhibitor drug are used for DDI prediction, the actual concentrations of inhibitor that the transporter encounters at the site of inhibition may be significantly different and difficult to measure. Lastly, substrate-dependent and time-dependent inhibitions have been recently reported136, 137, 138, 139, which further complicates the assessment and in vitro–to–in vivo prediction of DDIs. Nevertheless, the field of drug transporters is rapidly evolving. With the conceptual and technological advancements in drug transport research, we are now at the forefront to gain a better understanding of renal drug transporters, predict and ameliorate adverse renal DDIs, and design beneficial DDIs to improve drug efficacy and minimize drug toxicity.
Acknowledgements
This study was supported by the U. S. National Institutes of Health National Institute of General Medical Sciences (Grant R01 GM066233) and the National Center for Advancing Translational Sciences (Grant TL1 TR000422). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Morrissey K.M., Stocker S.L., Wittwer M.B., Xu L., Giacomini K.M. Renal transporters in drug development. Annu Rev Pharmacol Toxicol. 2012;53:503–529. doi: 10.1146/annurev-pharmtox-011112-140317. [DOI] [PubMed] [Google Scholar]
- 2.Li M., Anderson G.D., Wang J. Drug–drug interactions involving membrane transporters in the human kidney. Expert Opin Drug Metab Toxicol. 2006;2:505–532. doi: 10.1517/17425255.2.4.505. [DOI] [PubMed] [Google Scholar]
- 3.International Transporter Consortium, Giacomini K.M., Huang S.M., Tweedie D.J., Benet L.Z., Brouwer K.L. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahn S.Y., Eraly S.A., Tsigelny I., Nigam S.K. Interaction of organic cations with organic anion transporters. J Biol Chem. 2009;284:31422–31430. doi: 10.1074/jbc.M109.024489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masereeuw R., Russel F.G. Mechanisms and clinical implications of renal drug excretion. Drug Metab Rev. 2001;33:299–351. doi: 10.1081/dmr-120000654. [DOI] [PubMed] [Google Scholar]
- 6.Koepsell H., Lips K., Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24:1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- 7.Gründemann D., Gorboulev V., Gambaryan S., Veyhl M., Koepsell H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature. 1994;372:549–552. doi: 10.1038/372549a0. [DOI] [PubMed] [Google Scholar]
- 8.Gorboulev V., Ulzheimer J.C., Akhoundova A., Ulzheimer-Teuber I., Karbach U., Quester S. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16:871–881. doi: 10.1089/dna.1997.16.871. [DOI] [PubMed] [Google Scholar]
- 9.Gründemann D., Schechinger B., Rappold G.A., Schömig E. Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nat Neurosci. 1998;1:349–351. doi: 10.1038/1557. [DOI] [PubMed] [Google Scholar]
- 10.Lee N., Duan H., Hebert M.F., Liang C.J., Rice K.M., Wang J. Taste of a pill: organic cation transporter-3 (OCT3) mediates metformin accumulation and secretion in salivary glands. J Biol Chem. 2014;289:27055–27064. doi: 10.1074/jbc.M114.570564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujita T., Urban T.J., Leabman M.K., Fujita K., Giacomini K.M. Transport of drugs in the kidney by the human organic cation transporter, OCT2 and its genetic variants. J Pharm Sci. 2006;95:25–36. doi: 10.1002/jps.20536. [DOI] [PubMed] [Google Scholar]
- 12.Nies A.T., Koepsell H., Damme K., Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011;2011:105–167. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- 13.Yin J., Duan H., Shirasaka Y., Prasad B., Wang J. Atenolol renal secretion is mediated by human organic cation transporter 2 and multidrug and toxin extrusion proteins. Drug Metab Dispos. 2015;43:1872–1881. doi: 10.1124/dmd.115.066175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song I.H., Zong J., Borland J., Jerva F., Wynne B., Zamek-Gliszczynski M.J. The effect of dolutegravir on the pharmacokinetics of metformin in healthy subjects. J Acquir Immune Defic Syndr. 2016;72:400–407. doi: 10.1097/QAI.0000000000000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright S.H., Dantzler W.H. Molecular and cellular physiology of renal organic cation and anion transport. Physiol Rev. 2004;84:987–1049. doi: 10.1152/physrev.00040.2003. [DOI] [PubMed] [Google Scholar]
- 16.Motohashi H., Sakurai Y., Saito H., Masuda S., Urakami Y., Goto M. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol. 2002;13:866–874. doi: 10.1681/ASN.V134866. [DOI] [PubMed] [Google Scholar]
- 17.Otsuka M., Matsumoto T., Morimoto R., Arioka S., Omote H., Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci U S A. 2005;102:17923–17928. doi: 10.1073/pnas.0506483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masuda S., Terada T., Yonezawa A., Tanihara Y., Kishimoto K., Katsura T. Identification and functional characterization of a new human kidney–specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol. 2006;17:2127–2135. doi: 10.1681/ASN.2006030205. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X., Cherrington N.J., Wright S.H. Molecular identification and functional characterization of rabbit MATE1 and MATE2-K. Am J Physiol Ren Physiol. 2007;293:F360–F370. doi: 10.1152/ajprenal.00102.2007. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X., Wright S.H. MATE1 has an external COOH terminus, consistent with a 13-helix topology. Am J Physiol Ren Physiol. 2009;297:F263–F271. doi: 10.1152/ajprenal.00123.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komatsu T., Hiasa M., Miyaji T., Kanamoto T., Matsumoto T., Otsuka M. Characterization of the human MATE2 proton-coupled polyspecific organic cation exporter. Int J Biochem Cell Biol. 2011;43:913–918. doi: 10.1016/j.biocel.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Tanihara Y., Masuda S., Sato T., Katsura T., Ogawa O., Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem Pharmacol. 2007;74:359–371. doi: 10.1016/j.bcp.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Tamai I., Yabuuchi H., Nezu J., Sai Y., Oku A., Shimane M. Cloning and characterization of a novel human pH-dependent organic cation transporter OCTN1. FEBS Lett. 1997;419:107–111. doi: 10.1016/s0014-5793(97)01441-5. [DOI] [PubMed] [Google Scholar]
- 24.Tamai I., Ohashi R., Nezu J., Yabuuchi H., Oku A., Shimane M. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998;273:20378–20382. doi: 10.1074/jbc.273.32.20378. [DOI] [PubMed] [Google Scholar]
- 25.Tamai I., Nakanishi T., Kobayashi D., China K., Kosugi Y., Nezu J. Involvement of OCTN1 (SLC22A4) in pH-dependent transport of organic cations. Mol Pharm. 2004;1:57–66. doi: 10.1021/mp0340082. [DOI] [PubMed] [Google Scholar]
- 26.Tamai I., China K., Sai Y., Kobayashi D., Nezu J., Kawahara E. Na+-coupled transport of l-carnitine via high-affinity carnitine transporter OCTN2 and its subcellular localization in kidney. Biochim Biophys Acta. 2001;1512:273–284. doi: 10.1016/s0005-2736(01)00328-5. [DOI] [PubMed] [Google Scholar]
- 27.Gründemann D., Harlfinger S., Golz S., Geerts A., Lazar A., Berkels R. Discovery of the ergothioneine transporter. Proc Natl Acad Sci U S A. 2005;102:5256–5261. doi: 10.1073/pnas.0408624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yabuuchi H., Tamai I., Nezu J., Sakamoto K., Oku A., Shimane M. Novel membrane transporter OCTN1 mediates multispecific, bidirectional, and pH-dependent transport of organic cations. J Pharmacol Exp Ther. 1999;289:768–773. [PubMed] [Google Scholar]
- 29.Wu X., Huang W., Prasad P.D., Seth P., Rajan D.P., Leibach F.H. Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J Pharmacol Exp Ther. 1999;290:1482–1492. [PubMed] [Google Scholar]
- 30.Terada T., Inui K. Physiological and pharmacokinetic roles of H+/organic cation antiporters (MATE/SLC47A) Biochem Pharmacol. 2008;75:1689–1696. doi: 10.1016/j.bcp.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Urban T.J., Brown C., Castro R.A., Shah N., Mercer R., Huang Y. Effects of genetic variation in the novel organic cation transporter, OCTN1, on the renal clearance of gabapentin. Clin Pharmacol Ther. 2008;83:416–421. doi: 10.1038/sj.clpt.6100271. [DOI] [PubMed] [Google Scholar]
- 32.Juliano R.L., Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 33.Cummins C.L., Jacobsen W., Benet L.Z. Unmasking the dynamic interplay between intestinal P-glycoprotein and CYP3A4. J Pharmacol Exp Ther. 2002;300:1036–1045. doi: 10.1124/jpet.300.3.1036. [DOI] [PubMed] [Google Scholar]
- 34.Schinkel A.H., Jonker J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 35.Yu J., Ritchie T.K., Zhou Z., Ragueneau-Majlessi I. Key findings from preclinical and clinical drug interaction studies presented in new drug and biological license applications approved by the food and drug administration in 2014. Drug Metab Dispos. 2016;44:83–101. doi: 10.1124/dmd.115.066720. [DOI] [PubMed] [Google Scholar]
- 36.Thiebaut F., Tsuruo T., Hamada H., Gottesman M.M., Pastan I., Willingham M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gramatté T., Oertel R., Terhaag B., Kirch W. Direct demonstration of small intestinal secretion and site-dependent absorption of the β-blocker talinolol in humans. Clin Pharmacol Ther. 1996;59:541–549. doi: 10.1016/S0009-9236(96)90182-4. [DOI] [PubMed] [Google Scholar]
- 38.Chandra P., Brouwer K.L. The complexities of hepatic drug transport: current knowledge and emerging concepts. Pharm Res. 2004;21:719–735. doi: 10.1023/b:pham.0000026420.79421.8f. [DOI] [PubMed] [Google Scholar]
- 39.Chen C., Liu X., Smith B.J. Utility of Mdr1-gene deficient mice in assessing the impact of P-glycoprotein on pharmacokinetics and pharmacodynamics in drug discovery and development. Curr Drug Metab. 2003;4:272–291. doi: 10.2174/1389200033489415. [DOI] [PubMed] [Google Scholar]
- 40.Kolars J.C., Lown K.S., Schmiedlin-Ren P., Ghosh M., Fang C., Wrighton S.A. CYP3A gene expression in human gut epithelium. Pharmacogenetics. 1994;4:247–259. doi: 10.1097/00008571-199410000-00003. [DOI] [PubMed] [Google Scholar]
- 41.Kolars J.C., Schmiedlin-Ren P., Schuetz J.D., Fang C., Watkins P.B. Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J Clin Invest. 1992;90:1871–1878. doi: 10.1172/JCI116064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greiner B., Eichelbaum M., Fritz P., Kreichgauer H.P., von Richter O., Zundler J. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104:147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huls M., van den Heuvel J.J., Dijkman H.B., Russel F.G., Masereeuw R. ABC transporter expression profiling after ischemic reperfusion injury in mouse kidney. Kidney Int. 2006;69:2186–2193. doi: 10.1038/sj.ki.5000407. [DOI] [PubMed] [Google Scholar]
- 44.Sekine T., Watanabe N., Hosoyamada M., Kanai Y., Endou H. Expression cloning and characterization of a novel multispecific organic anion transporter. J Biol Chem. 1997;272:18526–18529. doi: 10.1074/jbc.272.30.18526. [DOI] [PubMed] [Google Scholar]
- 45.Sweet D.H., Wolff N.A., Pritchard J.B. Expression cloning and characterization of ROAT1. The basolateral organic anion transporter in rat kidney. J Biol Chem. 1997;272:30088–30095. doi: 10.1074/jbc.272.48.30088. [DOI] [PubMed] [Google Scholar]
- 46.Wolff N.A., Werner A., Burkhardt S., Burckhardt G. Expression cloning and characterization of a renal organic anion transporter from winter flounder. FEBS Lett. 1997;417:287–291. doi: 10.1016/s0014-5793(97)01304-5. [DOI] [PubMed] [Google Scholar]
- 47.Burckhardt G. Drug transport by organic anion transporters (OATs) Pharmacol Ther. 2012;136:106–130. doi: 10.1016/j.pharmthera.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 48.Perry J.L., Dembla-Rajpal N., Hall L.A., Pritchard J.B. A three-dimensional model of human organic anion transporter 1: aromatic amino acids required for substrate transport. J Biol Chem. 2006;281:38071–38079. doi: 10.1074/jbc.M608834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srimaroeng C., Perry J.L. Pritchard JB. Physiology, structure, and regulation of the cloned organic anion transporters. Xenobiotica. 2008;38:889–935. doi: 10.1080/00498250801927435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koepsell H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Asp Med. 2013;34:413–435. doi: 10.1016/j.mam.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 51.Reid G., Wolff N.A., Dautzenberg F.M., Burckhardt G. Cloning of a human renal p-aminohippurate transporter, hROAT1. Kidney Blood Press Res. 1998;21:233–237. doi: 10.1159/000025863. [DOI] [PubMed] [Google Scholar]
- 52.Bahn A., Ebbinghaus C., Ebbinghaus D., Ponimaskin E.G., Fuzesï L., Burckhardt G. Expression studies and functional characterization of renal human organic anion transporter 1 isoforms. Drug Metab Dispos. 2004;32:424–430. doi: 10.1124/dmd.32.4.424. [DOI] [PubMed] [Google Scholar]
- 53.Shin H.J., Anzai N., Enomoto A., He X., Kim D.K., Endou H. Novel liver-specific organic anion transporter OAT7 that operates the exchange of sulfate conjugates for short chain fatty acid butyrate. Hepatology. 2007;45:1046–1055. doi: 10.1002/hep.21596. [DOI] [PubMed] [Google Scholar]
- 54.Rizwan A.N., Burckhardt G. Organic anion transporters of the SLC22 family: biopharmaceutical, physiological, and pathological roles. Pharm Res. 2007;24:450–470. doi: 10.1007/s11095-006-9181-4. [DOI] [PubMed] [Google Scholar]
- 55.Hagos Y., Stein D., Ugele B., Burckhardt G., Bahn A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J Am Soc Nephrol. 2007;18:430–439. doi: 10.1681/ASN.2006040415. [DOI] [PubMed] [Google Scholar]
- 56.Ekaratanawong S., Anzai N., Jutabha P., Miyazaki H., Noshiro R., Takeda M. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J Pharmacol Sci. 2004;94:297–304. doi: 10.1254/jphs.94.297. [DOI] [PubMed] [Google Scholar]
- 57.Bahn A., Hagos Y., Reuter S., Balen D., Brzica H., Krick W. Identification of a new urate and high affinity nicotinate transporter, hOAT10 (SLC22A13) J Biol Chem. 2008;283:16332–16341. doi: 10.1074/jbc.M800737200. [DOI] [PubMed] [Google Scholar]
- 58.Enomoto A., Kimura H., Chairoungdua A., Shigeta Y., Jutabha P., Cha S.H. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 59.Deeley R.G., Westlake C., Cole S.P. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev. 2006;86:849–899. doi: 10.1152/physrev.00035.2005. [DOI] [PubMed] [Google Scholar]
- 60.Schaub T.P., Kartenbeck J., König J., Spring H., Dörsam J., Staehler G. Expression of the MRP2 gene-encoded conjugate export pump in human kidney proximal tubules and in renal cell carcinoma. J Am Soc Nephrol. 1999;10:1159–1169. doi: 10.1681/ASN.V1061159. [DOI] [PubMed] [Google Scholar]
- 61.van Aubel R.A., Smeets P.H., Peters J.G., Bindels R.J., Russel F.G. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: putative efflux pump for urinary cAMP and cGMP. J Am Soc Nephrol. 2002;13:595–603. doi: 10.1681/ASN.V133595. [DOI] [PubMed] [Google Scholar]
- 62.Belinsky M.G., Bain L.J., Balsara B.B., Testa J.R., Kruh G.D. Characterization of MOAT-C and MOAT-D, new members of the MRP/cMOAT subfamily of transporter proteins. J Natl Cancer Inst. 1998;90:1735–1741. doi: 10.1093/jnci/90.22.1735. [DOI] [PubMed] [Google Scholar]
- 63.Kiuchi Y., Suzuki H., Hirohashi T., Tyson C.A., Sugiyama Y. cDNA cloning and inducible expression of human multidrug resistance associated protein 3 (MRP3) FEBS Lett. 1998;433:149–152. doi: 10.1016/s0014-5793(98)00899-0. [DOI] [PubMed] [Google Scholar]
- 64.Flens M.J., Zaman G.J., van der Valk P., Izquierdo M.A., Schroeijers A.B., Scheffer G.L. Tissue distribution of the multidrug resistance protein. Am J Pathol. 1996;148:1237–1247. [PMC free article] [PubMed] [Google Scholar]
- 65.Peng K.C., Cluzeaud F., Bens M., Duong Van Huyen J.P., Wioland M.A., Lacave R. Tissue and cell distribution of the multidrug resistance-associated protein (MRP) in mouse intestine and kidney. J Histochem Cytochem. 1999;47:757–768. doi: 10.1177/002215549904700605. [DOI] [PubMed] [Google Scholar]
- 66.Scheffer G.L., Kool M., de Haas M., de Vree J.M., Pijnenborg A.C., Bosman D.K. Tissue distribution and induction of human multidrug resistant protein 3. Lab Invest. 2002;82:193–201. doi: 10.1038/labinvest.3780411. [DOI] [PubMed] [Google Scholar]
- 67.Hagenbuch B., Meier P.J. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003;1609:1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- 68.Jacquemin E., Hagenbuch B., Stieger B., Wolkoff A.W., Meier P.J. Expression cloning of a rat liver Na+-independent organic anion transporter. Proc Natl Acad Sci U S A. 1994;91:133–137. doi: 10.1073/pnas.91.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kullak-Ublick G.A., Hagenbuch B., Stieger B., Schteingart C.D., Hofmann A.F., Wolkoff A.W. Molecular and functional characterization of an organic anion transporting polypeptide cloned from human liver. Gastroenterology. 1995;109:1274–1282. doi: 10.1016/0016-5085(95)90588-x. [DOI] [PubMed] [Google Scholar]
- 70.Hagenbuch B., Stieger B. The SLCO (former SLC21) superfamily of transporters. Mol Asp Med. 2013;34:396–412. doi: 10.1016/j.mam.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hagenbuch B., Meier P.J. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 72.Roth M., Obaidat A., Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 2012;165:1260–1287. doi: 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mikkaichi T., Suzuki T., Onogawa T., Tanemoto M., Mizutamari H., Okada M. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci U S A. 2004;101:3569–3574. doi: 10.1073/pnas.0304987101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rowland M., Tozer T.N. Clinical Pharmacokinetics and pharmacodynamics: Concepts and Applications. 4th ed. LWW; Baltimore: 2011. [Google Scholar]
- 75.Cihlar T., Lin D.C., Pritchard J.B., Fuller M.D., Mendel D.B., Sweet D.H. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Mol Pharmacol. 1999;56:570–580. doi: 10.1124/mol.56.3.570. [DOI] [PubMed] [Google Scholar]
- 76.Cundy K.C., Petty B.G., Flaherty J., Fisher P.E., Polis M.A., Wachsman M. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus–infected patients. Antimicrob Agents Chemother. 1995;39:1247–1252. doi: 10.1128/aac.39.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.U.S. Food and Drug Administration. Guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling. Available from: 〈http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf〉, 2012.
- 78.Hillgren K.M., Keppler D., Zur A.A., Giacomini K.M., Stieger B., Cass C.E. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther. 2013;94:52–63. doi: 10.1038/clpt.2013.74. [DOI] [PubMed] [Google Scholar]
- 79.Brouwer K.L., Keppler D., Hoffmaster K.A., Bow D.A., Cheng Y., Lai Y. In vitro methods to support transporter evaluation in drug discovery and development. Clin Pharmacol Ther. 2013;94:95–112. doi: 10.1038/clpt.2013.81. [DOI] [PubMed] [Google Scholar]
- 80.Zhang L., Huang S.M., Lesko L.J. Transporter-mediated drug–drug interactions. Clin Pharmacol Ther. 2011;89:481–484. doi: 10.1038/clpt.2010.359. [DOI] [PubMed] [Google Scholar]
- 81.Somogyi A., Stockley C., Keal J., Rolan P., Bochner F. Reduction of metformin renal tubular secretion by cimetidine in man. Br J Clin Pharmacol. 1987;23:545–551. doi: 10.1111/j.1365-2125.1987.tb03090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Z.J., Yin O.Q., Tomlinson B., Chow M.S. OCT2 polymorphisms and in-vivo renal functional consequence: studies with metformin and cimetidine. Pharmacogenet Genom. 2008;18:637–645. doi: 10.1097/FPC.0b013e328302cd41. [DOI] [PubMed] [Google Scholar]
- 83.Kusuhara H., Ito S., Kumagai Y., Jiang M., Shiroshita T., Moriyama Y. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther. 2011;89:837–844. doi: 10.1038/clpt.2011.36. [DOI] [PubMed] [Google Scholar]
- 84.Vree T.B., van den Biggelaar-Martea M., Verwey-van Wissen C.P. Probenecid inhibits the renal clearance of frusemide and its acyl glucuronide. Br J Clin Pharmacol. 1995;39:692–695. [PMC free article] [PubMed] [Google Scholar]
- 85.Smith D.E., Gee W.L., Brater D.C., Lin E.T., Benet L.Z. Preliminary evaluation of furosemide-probenecid interaction in humans. J Pharm Sci. 1980;69:571–575. doi: 10.1002/jps.2600690526. [DOI] [PubMed] [Google Scholar]
- 86.Yasui-Furukori N., Uno T., Sugawara K., Tateishi T. Different effects of three transporting inhibitors, verapamil, cimetidine, and probenecid, on fexofenadine pharmacokinetics. Clin Pharmacol Ther. 2005;77:17–23. doi: 10.1016/j.clpt.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 87.Liu S., Beringer P.M., Hidayat L., Rao A.P., Louie S., Burckart G.J. Probenecid, but not cystic fibrosis, alters the total and renal clearance of fexofenadine. J Clin Pharmacol. 2008;48:957–965. doi: 10.1177/0091270008319707. [DOI] [PubMed] [Google Scholar]
- 88.Schenck-Gustafsson K., Dahlqvist R. Pharmacokinetics of digoxin in patients subjected to the quinidine--digoxin interaction. Br J Clin Pharmacol. 1981;11:181–186. doi: 10.1111/j.1365-2125.1981.tb01122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fenster P.E., Hager W.D., Goodman M.M. Digoxin–quinidine–spironolactone interaction. Clin Pharmacol Ther. 1984;36:70–73. doi: 10.1038/clpt.1984.141. [DOI] [PubMed] [Google Scholar]
- 90.Hager W.D., Fenster P., Mayersohn M., Perrier D., Graves P., Marcus F.I. Digoxin–quinidine interaction pharmacokinetic evaluation. N Engl J Med. 1979;300:1238–1241. doi: 10.1056/NEJM197905313002202. [DOI] [PubMed] [Google Scholar]
- 91.Scheen A.J. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 1996;30:359–371. doi: 10.2165/00003088-199630050-00003. [DOI] [PubMed] [Google Scholar]
- 92.Pentikainen P.J., Neuvonen P.J., Penttila A. Pharmacokinetics of metformin after intravenous and oral administration to man. Eur J Clin Pharmacol. 1979;16:195–202. doi: 10.1007/BF00562061. [DOI] [PubMed] [Google Scholar]
- 93.Somogyi A.A., Hovens C.M., Muirhead M.R., Bochner F. Renal tubular secretion of amiloride and its inhibition by cimetidine in humans and in an animal model. Drug Metab Dispos. 1989;17:190–196. [PubMed] [Google Scholar]
- 94.Somogyi A., Rohner H.G., Gugler R. Pharmacokinetics and bioavailability of cimetidine in gastric and duodenal ulcer patients. Clin Pharmacokinet. 1980;5:84–94. doi: 10.2165/00003088-198005010-00003. [DOI] [PubMed] [Google Scholar]
- 95.Ito S., Kusuhara H., Yokochi M., Toyoshima J., Inoue K., Yuasa H. Competitive inhibition of the luminal efflux by multidrug and toxin extrusions, but not basolateral uptake by organic cation transporter 2, is the likely mechanism underlying the pharmacokinetic drug–drug interactions caused by cimetidine in the kidney. J Pharmacol Exp Ther. 2012;340:393–403. doi: 10.1124/jpet.111.184986. [DOI] [PubMed] [Google Scholar]
- 96.Ito S., Kusuhara H., Kuroiwa Y., Wu C., Moriyama Y., Inoue K. Potent and specific inhibition of mMate1-mediated efflux of type I organic cations in the liver and kidney by pyrimethamine. J Pharmacol Exp Ther. 2010;333:341–350. doi: 10.1124/jpet.109.163642. [DOI] [PubMed] [Google Scholar]
- 97.Tahara H., Kusuhara H., Endou H., Koepsell H., Imaoka T., Fuse E. A species difference in the transport activities of H2 receptor antagonists by rat and human renal organic anion and cation transporters. J Pharmacol Exp Ther. 2005;315:337–345. doi: 10.1124/jpet.105.088104. [DOI] [PubMed] [Google Scholar]
- 98.Zamek-Gliszczynski M.J., Lee C.A., Poirier A., Bentz J., Chu X., Ellens H. ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport-mediated PK and DDIs in humans. Clin Pharmacol Ther. 2013;94:64–79. doi: 10.1038/clpt.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Graham G.G., Punt J., Arora M., Day R.O., Doogue M.P., Duong J.K. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50:81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 100.Reese M.J., Savina P.M., Generaux G.T., Tracey H., Humphreys J.E., Kanaoka E. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab Dispos. 2013;41:353–361. doi: 10.1124/dmd.112.048918. [DOI] [PubMed] [Google Scholar]
- 101.Rosenberg B., van Camp L., Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 102.Wang D., Lippard S.J. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 103.Cohen S.M., Lippard S.J. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol. 2001;67:93–130. doi: 10.1016/s0079-6603(01)67026-0. [DOI] [PubMed] [Google Scholar]
- 104.Arany I., Safirstein R.L. Cisplatin nephrotoxicity. Semin Nephrol. 2003;23:460–464. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 105.Pabla N., Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 106.Filipski K.K., Loos W.J., Verweij J., Sparreboom A. Interaction of cisplatin with the human organic cation transporter 2. Clin Cancer Res. 2008;14:3875–3880. doi: 10.1158/1078-0432.CCR-07-4793. [DOI] [PubMed] [Google Scholar]
- 107.Yonezawa A., Masuda S., Yokoo S., Katsura T., Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family) J Pharmacol Exp Ther. 2006;319:879–886. doi: 10.1124/jpet.106.110346. [DOI] [PubMed] [Google Scholar]
- 108.Tanihara Y., Masuda S., Katsura T., Inui K. Protective effect of concomitant administration of imatinib on cisplatin-induced nephrotoxicity focusing on renal organic cation transporter OCT2. Biochem Pharmacol. 2009;78:1263–1271. doi: 10.1016/j.bcp.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 109.Filipski K.K., Mathijssen R.H., Mikkelsen T.S., Schinkel A.H., Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86:396–402. doi: 10.1038/clpt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ciarimboli G., Deuster D., Knief A., Sperling M., Holtkamp M., Edemir B. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am J Pathol. 2010;176:1169–1180. doi: 10.2353/ajpath.2010.090610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pabla N., Gibson A.A., Buege M., Ong S.S., Li L., Hu S. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci U S A. 2015;112:5231–5236. doi: 10.1073/pnas.1424313112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sprowl J.A., van Doorn L., Hu S., van Gerven L., de Bruijn P., Li L. Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: influence on antitumor efficacy and systemic clearance. Clin Pharmacol Ther. 2013;94:585–592. doi: 10.1038/clpt.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sleijfer D.T., Offerman J.J., Mulder N.H., Verweij M., van der Hem G.K., Schraffordt Koops H.S. The protective potential of the combination of verapamil and cimetidine on cisplatin-induced nephrotoxicity in man. Cancer. 1987;60:2823–2828. doi: 10.1002/1097-0142(19871201)60:11<2823::aid-cncr2820601138>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 114.Li Q., Guo D., Dong Z., Zhang W., Zhang L., Huang S.M. Ondansetron can enhance cisplatin-induced nephrotoxicity via inhibition of multiple toxin and extrusion proteins (MATEs) Toxicol Appl Pharmacol. 2013;273:100–109. doi: 10.1016/j.taap.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nakamura T., Yonezawa A., Hashimoto S., Katsura T., Inui K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem Pharmacol. 2010;80:1762–1767. doi: 10.1016/j.bcp.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 116.Reid G., Wielinga P., Zelcer N., De Haas M., Van Deemter L., Wijnholds J. Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol Pharmacol. 2003;63:1094–1103. doi: 10.1124/mol.63.5.1094. [DOI] [PubMed] [Google Scholar]
- 117.Horikawa M., Kato Y., Tyson C.A., Sugiyama Y. The potential for an interaction between MRP2 (ABCC2) and various therapeutic agents: probenecid as a candidate inhibitor of the biliary excretion of irinotecan metabolites. Drug Metab Pharmacokinet. 2002;17:23–33. doi: 10.2133/dmpk.17.23. [DOI] [PubMed] [Google Scholar]
- 118.Enomoto A., Takeda M., Shimoda M., Narikawa S., Kobayashi Y., Kobayashi Y. Interaction of human organic anion transporters 2 and 4 with organic anion transport inhibitors. J Pharmacol Exp Ther. 2002;301:797–802. doi: 10.1124/jpet.301.3.797. [DOI] [PubMed] [Google Scholar]
- 119.Emanuelsson B.M., Beermann B., Paalzow L.K. Non-linear elimination and protein binding of probenecid. Eur J Clin Pharmacol. 1987;32:395–401. doi: 10.1007/BF00543976. [DOI] [PubMed] [Google Scholar]
- 120.Hasannejad H., Takeda M., Taki K., Shin H.J., Babu E., Jutabha P. Interactions of human organic anion transporters with diuretics. J Pharmacol Exp Ther. 2004;308:1021–1029. doi: 10.1124/jpet.103.059139. [DOI] [PubMed] [Google Scholar]
- 121.Brunton L.L., Chabner B.A., Knollman B.C. Goodman & Gilman׳s the Pharmacological Basis of Therapeutics. 12th ed. McGraw-Hill Medical; New York: 2011. [Google Scholar]
- 122.Eraly S.A., Vallon V., Vaughn D.A., Gangoiti J.A., Richter K., Nagle M. Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock-out mice. J Biol Chem. 2006;281:5072–5083. doi: 10.1074/jbc.M508050200. [DOI] [PubMed] [Google Scholar]
- 123.Homeida M., Roberts C., Branch R.A. Influence of probenecid and spironolactone on furosemide kinetics and dynamics in man. Clin Pharmacol Ther. 1977;22:402–409. doi: 10.1002/cpt1977224402. [DOI] [PubMed] [Google Scholar]
- 124.Brater D.C. Effects of probenecid on furosemide response. Clin Pharmacol Ther. 1978;24:548–554. doi: 10.1002/cpt1978245548. [DOI] [PubMed] [Google Scholar]
- 125.Sommers D.K., Meyer E.C., Moncrieff J. The influence of co-administered organic acids on the kinetics and dynamics of frusemide. Br J Clin Pharmacol. 1991;32:489–493. doi: 10.1111/j.1365-2125.1991.tb03936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bischofberger N., Hitchcock M.J., Chen M.S., Barkhimer D.B., Cundy K.C., Kent K.M. 1-((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl] cytosine, an intracellular prodrug for (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine with improved therapeutic index in vivo. Antimicrob Agents Chemother. 1994;38:2387–2391. doi: 10.1128/aac.38.10.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ho E.S., Lin D.C., Mendel D.B., Cihlar T. Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1. J Am Soc Nephrol. 2000;11:383–393. doi: 10.1681/ASN.V113383. [DOI] [PubMed] [Google Scholar]
- 128.Tahara H., Kusuhara H., Maeda K., Koepsell H., Fuse E., Sugiyama Y. Inhibition of OAT3-mediated renal uptake as a mechanism for drug–drug interaction between fexofenadine and probenecid. Drug Metab Dispos. 2006;34:743–747. doi: 10.1124/dmd.105.008375. [DOI] [PubMed] [Google Scholar]
- 129.Hinderling P.H., Hartmann D. Pharmacokinetics of digoxin and main metabolites/derivatives in healthy humans. Ther Drug Monit. 1991;13:381–401. doi: 10.1097/00007691-199109000-00001. [DOI] [PubMed] [Google Scholar]
- 130.Bauman J.L., Didomenico R.J., Galanter W.L. Mechanisms, manifestations, and management of digoxin toxicity in the modern era. Am J Cardiovasc Drugs. 2006;6:77–86. doi: 10.2165/00129784-200606020-00002. [DOI] [PubMed] [Google Scholar]
- 131.Feng B., Mills J.B., Davidson R.E., Mireles R.J., Janiszewski J.S., Troutman M.D. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos. 2008;36:268–275. doi: 10.1124/dmd.107.017434. [DOI] [PubMed] [Google Scholar]
- 132.Ochs H.R., Bodem G., Greenblatt D.J. Impairment of digoxin clearance by coadministration of quinidine. J Clin Pharmacol. 1981;21:396–400. doi: 10.1002/j.1552-4604.1981.tb01739.x. [DOI] [PubMed] [Google Scholar]
- 133.Fromm M.F., Kim R.B., Stein C.M., Wilkinson G.R., Roden D.M. Inhibition of P-glycoprotein–mediated drug transport: a unifying mechanism to explain the interaction between digoxin and quinidine. Circulation. 1999;99:552–557. doi: 10.1161/01.cir.99.4.552. [DOI] [PubMed] [Google Scholar]
- 134.Pedersen K.E., Dorph-Pedersen A., Hvidt S., Klitgaard N.A., Nielsen-Kudsk F. Digoxin–verapamil interaction. Clin Pharmacol Ther. 1981;30:311–316. doi: 10.1038/clpt.1981.165. [DOI] [PubMed] [Google Scholar]
- 135.Rengelshausen J., Göggelmann C., Burhenne J., Riedel K.D., Ludwig J., Weiss J. Contribution of increased oral bioavailability and reduced nonglomerular renal clearance of digoxin to the digoxin–clarithromycin interaction. Br J Clin Pharmacol. 2003;56:32–38. doi: 10.1046/j.1365-2125.2003.01824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hacker K., Maas R., Kornhuber J., Fromm M.F., Zolk O. Substrate-dependent inhibition of the human organic cation transporter OCT2: a comparison of metformin with experimental substrates. PLoS One. 2015;10:e0136451. doi: 10.1371/journal.pone.0136451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Belzer M., Morales M., Jagadish B., Mash E.A., Wright S.H. Substrate-dependent ligand inhibition of the human organic cation transporter OCT2. J Pharmacol Exp Ther. 2013;346:300–310. doi: 10.1124/jpet.113.203257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Martínez-Guerrero L.J., Wright S.H. Substrate-dependent inhibition of human MATE1 by cationic ionic liquids. J Pharmacol Exp Ther. 2013;346:495–503. doi: 10.1124/jpet.113.204206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ma L., Qin Y., Shen Z., Hu H., Zhou H., Yu L. Time-dependent inhibition of hOAT1 and hOAT3 by anthraquinones. Biol Pharm Bull. 2015;38:992–995. doi: 10.1248/bpb.b15-00217. [DOI] [PubMed] [Google Scholar]