

Graphical abstract

1. Introduction
The pyrimidine biosynthesis pathway is an attractive target for antibacterial drug design, as cells rely on nucleotide synthesis for survival and proliferation, and pyrimidines are essential building blocks of RNA and DNA. Mammals have a large, multifunctional dihydroorotate synthetase (CAD) enzyme for the first three steps of the de novo pyrimidine biosynthesis pathway, while prokaryotes use three separate monofunctional enzymes for each.1, 2 Dihydroorotase (DHOase), the third enzyme in the de novo pathway, is responsible for the reversible cyclization of carbamyl-asparate (Ca-asp) to dihydroorotate (DHO) (Fig. 1A). Historically, DHOase was divided into two evolutionary classes, with very low sequence identity (< 30%) between classes. Class I DHOases are found in gram-positive bacteria, mold, and insects, while Class II DHOases are found in gram-negative bacteria and fungi. There are two distinct differences between these two classes of DHOases. First, the main differences are structural, with Class II being slightly smaller at ~38 kDa, and with a long flexible catalytic loop, compared to ~45 kDa Class I enzymes with either no or a very short corresponding loop. Second, DHOases contain four conserved histidines and one aspartate that coordinate the active site Zn2+ ions. Two active site Zn2+ ions in Class I are bridged by an aspartate, whereas a carboxylated lysine serves the same role in Class II. More recently, a third class has been suggested for human and Porphyromonas gingivalis DHOases because they have higher sequence similarity to Class I, but have a long catalytic loop and the active site Zn2+ ions are bridged by a carboxylated lysine, distinctive to Class II (Fig. 1B).3
Figure 1. The mechanism of DHOase.

(A) The enzyme catalyzes the reversible cyclization of Ca-asp to DHO. (B) Ca-asp is stabilized by the Zn2+ ions coordinated by a carboxylated K102, while D250 abstracts the proton from the amide nitrogen of Ca-asp. (PDB: 1XGE). The exocyclic carboxyl group of Ca-asp is stabilized by N44, R20, and H254, in addition to the T109 and T110 from the ‘in’ formation of the catalytic loop. (C) Comparison of the three classes of DHOase with the catalytic loop highlighted in pink.
So far, the Class II DHOase from Escherichia coli has been most extensively studied, producing many apo and complex structures. The structure revealed DHOase to be a homodimer zinc metalloenzyme that belongs to the ‘amidohydrolase superfamily’, a classification proposed for a group of functionally diverse metal-dependent hydrolase enzymes with a central (β/α)8 barrel fold in the catalytic domain.4,5 The mechanism of DHOase was structurally determined as well, and is a cyclization reaction where the Ca-asp is stabilized by the active site Zn2+ and several active site residues (Asn44, Arg20, His254), while the carboxylate of Asp250 abstracts a proton from the amide nitrogen of Ca-asp, allowing for a nucleophilic attack (Fig. 1C).2 The proposed chemical mechanism coincides with previously observed effects of pH on DHOase activity, where the forward enzymatic reaction is favored at lower pH, while the reverse reaction is favored at higher pH.6,7
Crystal structures of ligand-bound DHOase show that the catalytic loop extends toward the active site when Ca-asp is bound, whereas it is in an ‘out’ formation when DHO is the substrate.8–10 The ligand and inhibitor complexes of E. coli DHOase and mutagenesis studies identified the importance of two threonine residues (T109, T110) on the flexible loop of Class II DHOase to stabilize the transition-state of Ca-asp, with T110 mutations resulting in decreased activity and T109 mutations demonstrating the essentiality of the hydrogen bond interaction from the hydroxyl group for converting Ca-asp to DHO (Fig. 1C).8–12 However, Class I DHOase has no distinct loop, and there are no structures of substrate or inhibitor bound Class I enzymes available to date to elucidate the interactions that would replace the role of these important threonines.
Currently, no inhibitors for gram-positive bacterial DHOase have been identified. Bacillus anthracis is the Gram-positive etiologic agent of anthrax, and DHOase has been identified as critical for its survival in human serum.13 Human anthrax infections can be developed through exposure via skin, ingestion of infected animals, inhalation of spores, or more recently, by injection.14 Early diagnosis and treatment is critical, but can be difficult if the person is unaware of being exposed to the pathogen as the initial clinical presentation is consistent with flu-like symptoms.15 Even with treatment, the mortality rates of gastrointestinal and inhalation anthrax are >40%.16, 17 Current treatment is focused on post-exposure prophylaxis (PEP), using a combination of antibiotics and antitoxins. Unfortunately, the long term therapy recommended with antimicrobials, such as β-lactams, introduce the risk of developing resistance. Therefore, developing novel antibiotics against B. anthracis would increase treatment options and make anthrax bioterrorism attacks less feasible.
Here, we present the first crystal structure of substrate-bound B. anthracis DHOase, which provides further insight into the differences in the catalytic loop between Class I and Class II DHOase and the role it may play in inhibitor or substrate binding and recognition. We previously screened inhibitors of Class II DHOase and DHO analogs against B. anthracis DHOase, and observed little to no inhibition.18 To identify potential inhibitors, we screened ~28,000 compounds and fragments against B. anthracis DHOase using a high throughput enzymatic colorimetric assay18, 19 and a thermal shift binding assay20–23, followed by an “orthogonal” surface plasmon resonance analysis.24
2. Results and discussion
2.1 Structure of substrate-bound DHOase from B. anthracis
Class II DHOase from E. coli has been extensively studied with many reported crystal structures of apo wild-type and mutants and complexes with substrates or inhibitors.8–10, 12 However, only apo structures have been solved for the two Gram positive bacteria DHOases from B. anthracis and Staphylococcus aureus. Here, we report the first substrate-bound crystal structure of a bacterial Class I DHOase. (PDB: 4YIW) The Ca-asp bound B. anthracis DHOase structure was solved and deposited as a homodimer at 2.45 Å, with chains A and B in one asymmetric unit. The substrate, Ca-asp, was found in the active site of each monomer, and both substrates have relatively weak electron density due to low substrate occupancy (Fig. 2A). The data processing and structure refinement statistics are summarized in Table 1. The overall structure of B. anthracis DHOase in complex with the Ca-asp looks similar to the apo structure (PDB: 3MPG) with an overall RMSD of 0.359 Å, indicating that binding of the Ca-asp did not cause much structural change. An overlay of Ca-asp-bound E. coli DHOase (PDB:1XGE) and our B. anthracis DHOase complex structure reveal a nice overlap in the catalytic aspartic acid (D250 and D304 in Fig. 2B). Another aspartate (D151) of B. anthracis DHOase aligns well with the carboxylated lysine (K102) of E. coli DHOase, serving the same purpose of bridging two active site Zn2+ ions. These two Zn2+ ions hold the substrate Ca-asp in position along with the active site residues, Arg60, Asn93, and His308, which also overlap well, suggesting the substrate is stabilized by the same hydrogen bond interactions. However, a major structural difference between the Class I and Class II DHOase is the longer catalytic loop of the Class II counterparts (shown in brown color in Fig. 2C). In the case of E. coli DHOase, previous studies have shown that the hydrogen bond interaction between Ca-asp and the two threonines (T109 and T110) on the catalytic loop play a crucial role.10 However, neither of the threonine residues is present in B. anthracis DHOase, so it has been previously hypothesized that the glycine (G152) in the shorter Class I loop may serve this function.3 According to our complex structure, this could be the case since the peptide backbone hydrogen of glycine is located at a distance of 3.75 Å from Ca-asp, which is within weak H-bond interaction range. (shown in blue dotted line in Fig. 2C). Flexible catalytic loops from the three DHOase classes are aligned for comparison (Fig. 2D and Fig. 2E). In the case of both Class II and III, long catalytic loops showed a distinct shift (dotted arrows) in the substrate-bound structures, indicating the regulatory role of the loops. On the other hand, the corresponding loop of B. anthracis DHOase (Class I) is very short and remained in the same location in the presence and absence of the substrate Ca-asp, and whether this loop plays the same stabilizing role or not is yet to be determined.
Figure 2. Comparison of Class I and Class II DHOase.

(A) Electron density of Ca-asp in the active site. The ligand density is countered at 2Fo − Fc = 0.51 rmsd. (B) Overlay of substrate bound B. anthracis DHOase (PDB:4YIW, cyan) and E. coli DHOase (PDB:1XGE, tan). (C) Catalytic loop of E. coli DHOase (PDB:1XGE) highlighted in blue, and H-bond interaction of substrate with the peptide backbone of glycine in B. anthracis DHOase. (PDB:4YIW). (D) Overlay of the catalytic loop. (B. anthracis apo PDB:3MPG; B. anthracis + Ca-asp PDB:4YIW; E. coli apo PDB:2EG6; E. coli + Ca-asp PDB:1XGE; human apo PDB:4BYE; human + Ca-asp PDB:4C6I). (E) Sequence alignment of catalytic loop highlighted in yellow.
Table 1.
X-ray crystallography statistics for the refinement of 4YIW.
| 4YIW | |
|---|---|
| Data collection | |
| Space group | P2(1) (No. 4) |
| Cell constants (a, b, c, β) | 50.18Å 81.68Å 104.53Å 100.29° |
| Resolution (Å) | 19.72 (2.45) Å |
| Total No. of reflections | 211917 |
| No. of averaged reflections (unique reflections) | 32367 |
| Redundancy | 6.5 |
| Rmerge (%) | 14.0 (99.1)† |
| <I/σ(I)> | 5.84 (1.08)† |
| Completeness (%) | 71.0 (99.6)† |
| Refinement | |
| Resolution range (Å) | 19.72-2.45 |
| No. of reflections | 28978 |
| No. of reflections in test set | 1521 |
| Completeness (%) | 99.33 |
| Rcryst (%) | 20.6 |
| Rfree (%) | 26.5 |
| Wilson B factor (Å2) | 36.9 |
| Mean B factor | 42.0 |
| No. of protein molecules in asymmetric unit | 2 |
| R.m.s.d.s from ideal geometry | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 1.023 |
| Ramachandran plot | |
| Favoured (%) | 93.85 |
| Allowed (%) | 5.56 |
| Disallowed (%) | 0.59 |
| No. of solvent molecules | 310 |
| Ligand | Zn |
| NCD | |
| Mean B factor NCD | 67.9 |
| Mean B factor H2O | 34.1 |
Highest resolution shell shown in parentheses
2.2 Mutagenesis Studies
Based on our substrate bound crystal structure, three active site residues (Arg63, Asn93, and Asp304) appear to stabilize the substrate and Zn2+ ion, indicating that these three residues could affect enzyme activity of B. anthracis DHOase. To further elucidate substrate-enzyme interactions, the three residues were sequentially mutated to an alanine. The affinities (KM) of wild-type B. anthracis DHOase for Ca-asp and DHO are essentially identical, at 112 ± 24 µM and 114 ± 15 µM, respectively (Table 2). In contrast, the affinities of E. coli DHOase for Ca-asp and DHO differ significantly, with KM values of 1700 µM and 80 µM, respectively.2 The catalytic activities of B. anthracis DHOase and E. coli DHOase also differ. For instance, the E. coli DHOase kcat values for Ca-asp and DHO are 160 ± 8 s−1 and 100 ± 1.6 s−1, respectively, compared to 2.1 ± 0.1 s−1 and 1.9 ± 0.1 s−1 for B. anthracis DHOase. This finding is similar to the catalytic kinetics of S. aureus DHOase, another active Class I DHOase.25 Interestingly, the catalytic efficiency (kcat/KM) of B. anthracis DHOase with Ca-asp as the substrate is similar to that of E. coli DHOase when Thr110 is mutated to Serine, and the essential Thr109 is maintained. The serine could still make the hydrogen bond interaction between the loop and Ca-asp, but this mutation caused a 6-fold decrease in kcat/KM compared to the wild-type, making its activity comparable to that of B. anthracis and S. aureus DHOases. Previously, it has been hypothesized that either the glycine in the shorter loop of Class I DHOase or the ATCase domain plays the critical role of the threonines in Class I DHOase, explaining why some Class I DHOase are active, while others, such as A. aeolicus DHOase is inactive unless it associates with the ATCase domain. The comparison of our kinetic parameters suggests that although the peptide backbone of glycine in Class I DHOase may contribute H-bond interactions for enzyme activity, an association with the ATCase domain may increase the enzyme activity. Despite the ~6-fold difference in kcat/KM values, the crystal structures show that Ca-asp binds in a similar manner in both classes of DHOase and has the same physicochemical interactions with the active site residues.
Table 2.
Comparison of kinetic values of wild-type and mutants of B. anthracis DHOases.
| Enzyme | Substrate | KM (µM) | kcat (s−1) | Kcat/KM (M−1s−1) |
|---|---|---|---|---|
| WT-DHOase | Ca-asp | 112 ± 24 | 2.1 ± 0.1 | 1.9 × 104 |
| WT-DHOase | DHO | 114 ± 16 | 1.9 ± 0.1 | 1.7 × 104 |
| R63A-DHOase | Ca-asp | na | na | na |
| R63A-DHOase | DHO | na | na | na |
| N93A-DHOase | Ca-asp | 1183 ± 251 | 1.4 ± 0.1 | 1.2 × 103 |
| N93A-DHOase | DHO | na | na | na |
| D304A-DHOase | Ca-asp | na | na | na |
| D304A-DHOase | DHO | na | na | na |
na : no activity (not measurable)
The E. coli DHOase Asp250 stabilizes a Zn2+ ion and initiates the enzyme reaction, and this aspartic acid residue is required for catalytic activity. This is also the case for Class I B. anthracis DHOase, as mutagenesis of D304 to alanine completely abolished activity in B. anthracis DHOase, with minimal recovery of activity after supplementing with zinc sulfate up to 2.5 mM. The R63A mutant also showed no enzyme activity with either DHO or Ca-asp. This is because Arg63 stabilizes the exocyclic carboxylic acid of both the substrate and product in the active site. On the other hand, Asn93 has an electrostatic interaction with the carboxylic group of Ca-asp, which undergoes nucleophilic attack by the amide nitrogen to form DHO. The N93A mutant has minor activity, even with almost 10-fold higher concentrations of Ca-asp compared to wild-type DHOase, most likely due to the inability to stabilize Ca-asp in the active site. When DHO is the substrate, no enzyme activity is observed, presumably because DHO lacks the carboxylic terminal to interact with Asn93 (Table 2).
2.3 High-throughput Screening and hit validation
Previously, inhibitors of E. coli and P. falciparum DHOase, such as 5-fluoroorotate (FOA), 5-aminoorotate (AOA), and three analogs of DHO showed no inhibition of B. anthracis DHOase.18 Therefore, to identify inhibitors for B. anthracis DHOase, a high-throughput screen (HTS) of 3,352 fragments from the Chembridge fragment and Zenobia fragment libraries were performed using a colorimetric enzymatic assay. A second thermal shift binding assay was performed to identify those that bind to the target enzyme. The enzymatic colorimetric assay uses a DAMO-TSC color mix to detect the ureido moiety of Ca-asp, as previously described.18 Compounds that inhibit DHOase should prevent the reverse cyclization process from DHO to Ca-asp. Therefore, absence or lowered concentrations of Ca-asp will exhibit little to no color change. For our studies, any fragment that showed < 50% of color development (red square in Fig. 3A) compared to the control suggested enzyme activity inhibition and were considered to be initial hits. HTS using this method resulted in 142 hits. The thermal shift binding assay uses a fluorescence dye to monitor unfolding and exposure of hydrophobic regions of an enzyme while gradually increasing the temperature.26 If a fragment binds to DHOase, it should stabilize the enzyme, resulting in a shift in melting temperature (Tm) to a higher temperature (Fig. 3C). Fragments that showed > 2 °C shift in temperature were identified as hits (Fig. 3B). This second method of HTS resulted in 160 hits, with 10 fragments overlapping with the enzymatic screen. One of the disadvantages of the fluorescence thermal shift assay is that fragments which saturate the target may not show any temperature shift by displaying a broad fluorescence curve. For that reason, we did not want to disregard any promising hits from our enzymatic assay. After testing the enzymatic hits for compound interference on color development, we selected 14 fragments to perform a secondary confirmation assay. Surface plasmon resonance (SPR) was used to eliminate non-specific binders and to determine the binding affinity (dissociation equilibrium constant, KD). Promiscuous binders were identified as those that did not show a trend toward a saturable curve (Fig. S5). Sensorgrams of compound 4 are shown at eight different concentrations in Fig. 3D as an example, and the determined KD values of 8 final fragment hits varied between 67 µM and 334 µM (Fig. 4).
Figure 3. High-throughput screening (HTS) of DHOase.

(A) Replicate plot of colorimetric enzymatic assay with fragments showing >50% inhibition in red. (B) Replicate plot of thermal shift binding assay with fragments showing >2 °C shift in red. (C) Melting curve of DHOase without substrate (black) and DHOase with Compound 4 (red). Compound 4 shows a shift in melting temperature (Tm) as indicated by the arrow. (D) Sensorgram of Compound 4 at increasing concentrations.
Figure 4. High-throughput screening hits.

Bar graphs of the dissociation equilibrium constants (KD) of 8 initial hit compounds determined by Surface Plasmon Resonance (SPR). KD values were compared in the presence (green) and in the absence (black) of the substrate Ca-asp as a competitor. Bars that reached the top of the graph represent KD values of over 1000 µM (no binding).
2.4 Identification of a lead compound and Structure-Activity Relationship (SAR)
To determine if the inhibitors bind to the active site, the KD was determined for each compound in the absence and presence of substrate Ca-asp (Fig. 4). If the compounds bind to the active site, the expectation is that the compound KD should be affected by the presence of high concentration of substrate. Overall, the binding affinity did not change in the presence of 1 mM Ca-asp, suggesting the hits may not be competitive inhibitors, with the exception of compound 7 (Fig. S7). These results suggest that the compounds may bind to an allosteric site, or possibly in the active site but not in a position to compete with substrate. Our crystal structure shows an open active site with a large chemical space which could accommodate two smaller fragments, such as our hit compounds and the natural substrate. This possibility could be observed when comparing compounds 3 and 4. Compounds 3 and 4 are similar in structure with similar binding affinities of 157 ± 2 µM and 170 ± 4 µM, respectively. However, with 1 mM Ca-asp, the KD of compound 3 nearly doubled, to 281 ± 19 µM, indicating that Ca-asp and 3 may be competing at least in part for the active site, whereas that of compound 4 remained the same. Previously, it has been shown that DHOase is highly specific for its substrate, and the substitution positions played a critical role, in that 5-substituted analogs of the substrate were more effective inhibitors and more competitive for the position in the active site that is normally occupied by either DHO or Ca-asp.27 Additionally, this may explain why compound 3 exhibited competition with the substrate, while compound 4 did not. Compounds 6–8 have a similar and much more rigid 3-ring scaffold, with compound 7 having the highest affinity for DHOase with a KD value of 67 ± 7 µM. Interestingly, in the presence of substrate, the binding affinity weakened significantly enough that KD was unable to be determined, confirming that 7 is a competitive inhibitor.
A few commercially available fragment analogs of compounds 6 and 7 were obtained and tested by SPR, and a preliminary Structure-Activity-Relationships (SAR) is summarized in Table 3. When the tetrahydro-2H-pyran ring of compound 6 was removed from R2 position, the KD increased ~1.5-fold, going from 380 µM to 588 µM and was not affected by the addition of substrate. When the methoxy group of 6a in the R1 position was replaced by a methyl group (6b), the compound interaction with DHOase was completely abolished. Similarly, when the methoxy group in the R1 was retained, but the tetrahydropyridine of R2 was removed (6c), the compound also showed no interaction with DHOase, indicating the interactions of the methoxy group and pyridine ring are necessary for active site binding. When the R1 position was kept as the same methoxy group and the pyran ring in R1 position was substituted by thiomethyl and cyanamide (7a), the binding affinity increased 18-fold from 380 µM to 21 µM, most likely due to the interaction between cyanamide and the Zn ions in the active site. When the methoxy group replaced the fluorine (7) in R1 position and kept R2 position same, the affinity decreased slightly to 84 µM. When the tetrahydropyridine ring was tethered with acetyl on the nitrogen (8) in the R2 position, interaction became weaker. These results together suggest that fluorine is the most favored, and methoxy is tolerated in the R1 position. A tetrahydropyridine in R2 is required, and being tethered with thiomethyl and cyanamide is the most favored, while tetrahydropyridine alone, fused with pyran, or tethered with acetyl on the nitrogen is tolerated (Fig. 5). Based on this preliminary Structure-Activity Relationship (SAR) map, our lead compound, 7, can be further optimized through synthesis.
Table 3.
Preliminary Structure-Activity Relationship (SAR) of HTS hit analogs.
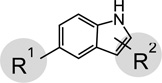 | |||||
|---|---|---|---|---|---|
| Compound | MW | R1 | R2 | KDa (µM) | KD (µM) with 1 mM Ca-asp |
| 6 | 308.8 | -OCH3 | 380 ± 1 | 314 ± 34 | |
| 6a | 202.3 | -OCH3 | 588 ± 38 | 675 ± 12 | |
| 6b | 186.3 | -CH3 | NBb | NBb | |
| 6c | 176.2 | -OCH3 | -2-CH2NH2 | NBb | NTc |
| 7 | 276.3 | -F | 67 ± 7 | >1 mM | |
| 7a | 288.4 | -OCH3 | 119 ± 13 | 144 ± 69 | |
| 8 | 244.3 | -OCH3 | 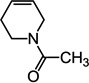 |
205 ± 8 | 215 ± 2 |
KD: Equilibrium dissociation constant determined by SPR; the standard deviation are from three independent experiments.
NB: Non-specific binding pattern
: NT: Not tested
Figure 5. Preliminary Structure-Activity Relationship (SAR) map of HTS hit analogs.

The KD of nine similarly structured fragment compounds were analyzed to generate this map.
3. Conclusions
We were able to solve the crystal structure of B. anthracis DHOase with Ca-asp in the active site (PDB: 4YIW). The structure confirms that the key interactions between the substrate and active site residues are similar between Class I and Class II DHOase enzymes, except for the lack of substrate stabilization by the catalytic loop in Class I DHOase. The crystal structure of E. coli (PDB:1XGE) shows two threonine residues that stabilize the substrate, which may also play a role in stabilizing small inhibitors. In contrast, the shorter loop in B. anthracis DHOase interaction with the substrate is minimal, and our structure confirms the previously hypothesized role of glycine as the hydrogen bond contributor. To further understand the importance of Ca-asp stabilization by hydrogen bonding, the enzyme kinetics will be compared to G152S and G152T in ongoing studies. The lack of loop stabilization may be one of the reasons why inhibitors of Class II DHOase have not been effective in B. anthracis DHOase. In order to identify potential inhibitors, we performed high-throughput screening of B. anthracis DHOase against several libraries using an enzymatic assay and an orthogonal binding assay. The binding affinity constants for eight inhibitors were determined by SPR, as well as by competition analysis with Ca-asp to determine if the inhibitors bound in the active site. The few hit compounds share a similar scaffold, and analogs were selected for further analysis based on binding affinities, with a preliminary SAR map. Our results provide more insight into the dihydroorotase family and identified several compounds that can be further optimized as potential antimicrobials against B. anthracis.
4. Experimental section
4.1 Preparation and purification of DHOase
The DHOase gene from Bacillus anthracis (Sterne strain) was cloned into a pET15b vector (Invitrogen) and purified as previously described.18, 28 In brief, the recombinant plasmid was inserted into BL21(DE3) Gold cells (Invitrogen) and grown in Terrific Broth (TB) media (Fisher Scientific) at 37° C while shaking at 220 rpm until the OD600 reached 0.6, when it was induced with 1 mM isopropyl B-D-1-thiogalactopyranoside (IPTG) (Sigma) and incubated for an additional 4 hours before harvesting. The cell pellet was resuspended in 50 mM Tris, pH 8.0, 500 mM NaCl, 20 mM imidazole, and 5 mM β-mercaptoethanol (β-MCE), 1 mg/mL lysozyme, 0.01% Triton X-100, and 0.025 mg/mL DNase I, and lysed by sonication. A HisTrap HP column (GE Healthcare) was used to purify the histidine-tagged protein using a stepwise gradient of elution buffer (50 mM Tris, pH 8.0, 500 mM NaCl, 500 mM imidazole, and 5 mM β-MCE) with either an AKTA purifier or AKTAxpress FPLC system. The eluted protein was dialyzed in 50 mM Tris, pH 7.5, and 100 mM NaCl overnight to remove imidazole. The histidine tag was removed by incubating the dialyzed protein with 1.5 units of thrombin per mg protein at room temperature for 1 hour, followed by 45 minutes at 37 °C. The digested protein was reloaded onto a HisTrap HP column stacked with a HiTrap Benzamidine column that was equilibrated with 50 mM Tris, pH 8.0, 500 mM NaCl, and 5 mM β-MCE. The histidine-tag cleaved DHOase was collected in the flow-through and loaded onto a HiLoad 16/60 Superdex 75 PG gel filtration column that was equilibrated with 25 mM Tris, pH 7.5 and 100 mM NaCl. Protein samples were analyzed by SDS-PAGE and final purity was above 90%.
4.2 Site-directed mutagenesis
R63A, N93A, and D304A mutations on DHOase were generated by site-directed mutagenesis PCR using the NEB Q5 mutagenesis Kit and wild-type pET15b plasmid containing B. anthracis DHOase as a template. The primers used are as follows: R63A, 5’-GGATTAGTAGATGTACACGTACACCT GCAGAACCAGGTGGTGAACATAAAG-3’; N93A, 5’-CACTACAATTTGCGCAATGCCAGCAACACGCCCAGTACCAG-3’; D304A, 5’-GGAACAATCGATATGATCGCAACTGCACATGCACCGCATACAGC-3’. The PCR reaction contained 0.3 µM each of forward and reverse primers, 0.5 µL template, 12.5 µL Q5 Hot Start High-Fidelity 2X Mix, and sterile water adjusted to 50 µL total volume for each forward and reverse reaction. The PCR product was incubated with the provided Kinase-Ligase-DpnI enzyme mix for 30 minutes at room temperature. The final PCR product was transformed into DH5α cells via heat shock for 30 seconds at 42° C and plated onto ampicillin treated plates (ampicillin 100 µg/mL). Transformants with the correct mutation were used to extract the plasmid and retransform into a protein expression cell line, BL21(DE3) Gold cells, via heat shock at 42° C for 30 seconds. Each mutant was expressed and purified by the same way as the wild-type DHOase.
4.3 Enzyme kinetics determination
The Michaelis-Menten constant (KM) was determined as described previously.18, 25 In brief, the Km values were determined by monitoring the UV absorbance at 230 nm. The reverse reaction of 100 nM final concentration wild-type DHOase or its mutants (R63A, N93A, or D304A-DHOase) were performed in 50 mM Tris, pH 8.3 with concentrations of substrate DHO ranging from 0 – 500 µM at 2-fold serial dilution in a 96-well UV plate (Corning). The forward reaction was performed in the same manner, but with a different assay buffer consisting of 50 mM MES, pH 5.8 at 100 nM final concentration wild-type DHOase or its mutants. The substrate Ca-asp concentrations for wild-type and two mutants (R63A and D304A) DHOase reaction ranged from 0 – 650 µM, while a much higher concentration range (0 – 5 mM) was used for N93A-DHOase. The enzyme reaction was continuously monitored using a SPECTRAmax Plus (Molecular Devices). The KM value and maximum activity (Vmax) were calculated using Sigmaplot 11.0 to fit the data to the hyperbolic equation (1), where y is the initial velocity and x is the concentration of the substrate.
4.4 Crystallization of substrate-bound BaDHOase
The pooled histidine-tag cleaved wild-type DHOase was concentrated to 10 mg/mL. It was then incubated with 1.5 mM of either DHO or Ca-asp overnight on ice. DHOase was crystallized using hanging-drop vapor diffusion with the crystallization condition of 0.1 M Bis-Tris, pH 5.5, 0.2 M NaCl, and 20% PEG 3350. The crystals diffracted at λ= 0.979800 Å at 100K using a MARMOSAIC CCD 300 mm detector at an oscillation angle of 1.0° for 360 frames at the Advanced Photon Source LS-cat beamline, station 21-ID-D at the Argonne National Laboratory. The data was indexed, integrated, and scaled by XDS.29 The molecular replacement was completed using Phaser30 in CCP4 and the structural refinement was completed with Refmac5.531 and Coot32.
4.5 Primary high-throughput screening
High-throughput screening of 3,352 fragments from the Chembridge fragment and Zenobia fragment libraries were performed using the Tecan Freedom EVO 200 liquid handling robot and the DAMO-TSC assay which has previously been optimized for this target.18 All assays were done in duplicate in transparent 384-well plates (Grenier). 30 µL of 133 nM his-tagged DHOase in HTS buffer (50 mM Tris, pH 8.3, 0.01% Triton X-100, 0.1 mg/mL BSA, 4 mM TCEP) was dispensed to plates, and final concentrations of 400 µM of each fragment or 25 µM of each compound was added and incubated for 10 minutes at room temperature. Then 10 µL of 320 µM substrate DHO in the same assay buffer was added prior to incubation for another 30 minutes after 30 seconds of shaking. To quench the reaction, 64 µL of DAMO-TSC acid color mix was added. The assay plates were sealed and incubated in the dark for 16 hours at room temperature prior to measuring the absorbance at 540 nm. Each plate contained 32 positive and 32 negative controls.
4.6 Fluorescence thermal shift assay
A fluorescence thermal shift (FTS) binding assay was performed on 3,352 fragments. FTS assay plates were prepared by the Tecan Freedom EVO 200 liquid handling robot, and melting curves were monitored using a Viia7 Real-Time PCR (Applied Biosystems). 5 µL of histidine-tagged DHOase (5 µM final concentration) in FTS buffer consisting of 50 mM MES, pH 6.5, 0.01% CHAPS, 150 mM NaCl, and 2 mM TCEP was dispensed in white 384-well PCR plates (BioRad). 0.1 µL of each fragment (400 µM final concentration) was added to the plate using a 384-pin stainless steel pin tool (V&P Scientific) with a 100-nL capillary capacity and incubated for 10 minutes at room temperature. Then 5 µL of 10X SYPRO Orange dye, diluted from 5000X stock, was added prior to shaking for 30 seconds. The assay plates were gradually heated from 25 °C to 95 °C at 0.07 °C/min while the fluorescence intensity was continuously measured at excitation/emission 470/623 nm. A Protein Thermal Shift™ Software (Applied Biosystems) was used to calculate the Tm shifts, and any fragments with Tm > 2° C were identified as positive hits.
4.7 Hit validation with surface plasmon resonance
Purified histidine-tag cleaved DHOase was buffer exchanged to PBS-P (10 mM phosphate, pH 7.4, 2.7 mM KCl, 137 mM NaCl, and 0.05% surfactant P-20). Surface Plasmon Resonance was performed at 25 °C using a Biacore T200 instrument and a CM5 sensor chip. The flow channels were activated by a 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrocholoride (EDC)/N-hydroxy succinimide (NHS) mixture. DHOase was diluted in 10 mM sodium acetate (pH 4.5) and immobilized to flow channels 2, 3, and 4 at levels of 12,974 RU, 9,853 RU, and 12,150 RU, respectively. Flow channel 1 was blocked by ethanolamine (pH 8.5) as a control. Compound solutions at increasing concentrations (0–200 µM at 2-fold dilutions) in binding buffer were applied to all four channels at a 10 µL/min flow rate. The SPR binding buffer was PBS-P, 2% DMSO and 0.5 mM TCEP. 1 mM Ca-asp was added to the binding buffer for competition SPR. The sensorgrams were analyzed using Biaevaluation software 2.0.3. All data was reference with the blank channel response unit signal prior to the fitting. The response unit difference at each concentration was measured during the binding equilibration phase and KD values were determined using the steady-state affinity fitting equation embedded in the Biaevaluation software.
Supplementary Material
Acknowledgments
AJR was supported during a portion of this work by NIDCR T32-DE018381, UIC College of Dentistry, MOST Program. We thank Dr. Kiira Ratia for compound library preparation and preparation of TSA plates, and Dr. Hyunwoo Lee for the B. anthracis ΔANR strain. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. 2004;279:33035–33038. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 2.Porter TN, Li Y, Raushel FM. Mechanism of the dihydroorotase reaction. Biochemistry. 2004;43:16285–16292. doi: 10.1021/bi048308g. [DOI] [PubMed] [Google Scholar]
- 3.Grande-García A, Lallous N, Díaz-Tejada C, Ramón-Maiques S. Structure, functional characterization, and evolution of the dihydroorotase domain of human CAD. Structure. 2014;22:185–198. doi: 10.1016/j.str.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Holm L, Sander C. An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins. 1997;28:72–82. [PubMed] [Google Scholar]
- 5.Liu A, Huo L. Amidohydrolase Superfamily. eLS, John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 6.Sander EG, Heeb MJ. Purification and properties of dihydroorotase from Escherichia coli B. Biochim Biophys Acta. 1971;227:442–452. doi: 10.1016/0005-2744(71)90075-1. [DOI] [PubMed] [Google Scholar]
- 7.Wang CC, Tsau HW, Chen WT, Huang CY. Identification and characterization of a putative dihydroorotase, KPN01074, from Klebsiella pneumoniae. Protein J. 2010;29:445–452. doi: 10.1007/s10930-010-9272-2. [DOI] [PubMed] [Google Scholar]
- 8.Lee M, Chan CW, Mitchell Guss J, Christopherson RI, Maher MJ. Dihydroorotase from Escherichia coli: loop movement and cooperativity between subunits. J Mol Biol. 2005;348:523–533. doi: 10.1016/j.jmb.2005.01.067. [DOI] [PubMed] [Google Scholar]
- 9.Lee M, Chan CW, Graham SC, Christopherson RI, Guss JM, Maher MJ. Structures of ligand-free and inhibitor complexes of dihydroorotase from Escherichia coli: implications for loop movement in inhibitor design. J Mol Biol. 2007;370:812–825. doi: 10.1016/j.jmb.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 10.Lee M, Maher MJ, Christopherson RI, Guss JM. Kinetic and structural analysis of mutant Escherichia coli dihydroorotases: a flexible loop stabilizes the transition state. Biochemistry. 2007;46:10538–10550. doi: 10.1021/bi701098e. [DOI] [PubMed] [Google Scholar]
- 11.Lee M, Maher MJ, Guss JM. Structure of the T109S mutant of Escherichia coli dihydroorotase complexed with the inhibitor 5-fluoroorotate: catalytic activity is reflected by the crystal form. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:154–161. doi: 10.1107/S1744309107004009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thoden JB, Phillips GN, Jr, Neal TM, Raushel FM, Holden HM. Molecular structure of dihydroorotase: a paradigm for catalysis through the use of a binuclear metal center. Biochemistry. 2001;40:6989–6997. doi: 10.1021/bi010682i. [DOI] [PubMed] [Google Scholar]
- 13.Samant S, Lee H, Ghassemi M, Chen J, Cook JL, Mankin AS, Neyfakh AA. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008;4:e37. doi: 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger T, Kassirer M, Aran AA. Injectional anthrax - new presentation of an old disease. Euro Surveill. 2014;19 doi: 10.2807/1560-7917.es2014.19.32.20877. [DOI] [PubMed] [Google Scholar]
- 15.Sweeney DA, Hicks CW, Cui X, Li Y, Eichacker PQ. Anthrax infection. Am J Respir Crit Care Med. 2011;184:1333–1341. doi: 10.1164/rccm.201102-0209CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.W.M. Hendricks KA, Shadomy SV, Bradley JS, Morrow MG, Pavia AT, et al. Centers for Disease Control and Prevention expert panel meetings on prevention and treatment of anthrax in adults. Emerg Infect Dis [Internet] 2014 doi: 10.3201/eid2002.130687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owen JL, Yang T, Mohamadzadeh M. New insights into gastrointestinal anthrax infection. Trends Mol Med. 2015;21:154–163. doi: 10.1016/j.molmed.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rice AJ, Truong L, Johnson ME, Lee H. A colorimetric assay optimization for high-throughput screening of dihydroorotase by detecting ureido groups. Anal Biochem. 2013;441:87–94. doi: 10.1016/j.ab.2013.05.035. [DOI] [PubMed] [Google Scholar]
- 19.Knipp M, Vasak M. A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline. Analytical Biochemistry. 2000;286:257–264. doi: 10.1006/abio.2000.4805. [DOI] [PubMed] [Google Scholar]
- 20.Sorrell FJ, Greenwood GK, Birchall K, Chen B. Development of a differential scanning fluorimetry based high throughput screening assay for the discovery of affinity binders against an anthrax protein. J Pharm Biomed Anal. 2010;52:802–808. doi: 10.1016/j.jpba.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 22.Layton CJ, Hellinga HW. Thermodynamic analysis of ligand-induced changes in protein thermal unfolding applied to high-throughput determination of ligand affinities with extrinsic fluorescent dyes. Biochemistry. 2010;49:10831–10841. doi: 10.1021/bi101414z. [DOI] [PubMed] [Google Scholar]
- 23.Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 24.Karlsson R. Real-time competitive kinetic analysis of interactions between low-molecular-weight ligands in solution and surface-immobilized receptors. Anal Biochem. 1994;221:142–151. doi: 10.1006/abio.1994.1390. [DOI] [PubMed] [Google Scholar]
- 25.Truong L, Hevener KE, Rice AJ, Patel K, Johnson ME, Lee H. High-level expression, purification, and characterization of Staphylococcus aureus dihydroorotase (PyrC) as a cleavable His-SUMO fusion. Protein Expr Purif. 2013;88:98–106. doi: 10.1016/j.pep.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andreotti G, Monticelli M, Cubellis MV. Looking for protein stabilizing drugs with thermal shift assay. Drug Test Anal. 2015 doi: 10.1002/dta.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christopherson RI, Jones ME. The effects of pH and inhibitors upon the catalytic activity of the dihydroorotase of multienzymatic protein pyr1–3 from mouse Ehrlich ascites carcinoma. J Biol Chem. 1980;255:3358–3370. [PubMed] [Google Scholar]
- 28.Mehboob S, Mulhearn DC, Truong K, Johnson ME, Santarsiero BD. Structure of dihydroorotase from Bacillus anthracis at 2.6 Å resolution. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:1432–1435. doi: 10.1107/S1744309110037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.