

Abstract
Inherited thrombocytopenias are a heterogeneous group of disorders characterized by abnormally low platelet counts which can be associated with abnormal bleeding. Next-generation sequencing has previously been employed in these disorders for the confirmation of suspected genetic abnormalities, and more recently in the discovery of novel disease-causing genes. However its full potential has not yet been exploited. Over the past 6 years we have sequenced the exomes from 55 patients, including 37 index cases and 18 additional family members, all of whom were recruited to the UK Genotyping and Phenotyping of Platelets study. All patients had inherited or sustained thrombocytopenia of unknown etiology with platelet counts varying from 11×109/L to 186×109/L. Of the 51 patients phenotypically tested, 37 (73%), had an additional secondary qualitative platelet defect. Using whole exome sequencing analysis we have identified “pathogenic” or “likely pathogenic” variants in 46% (17/37) of our index patients with thrombocytopenia. In addition, we report variants of uncertain significance in 12 index cases, including novel candidate genetic variants in previously unreported genes in four index cases. These results demonstrate that whole exome sequencing is an efficient method for elucidating potential pathogenic genetic variants in inherited thrombocytopenia. Whole exome sequencing also has the added benefit of discovering potentially pathogenic genetic variants for further study in novel genes not previously implicated in inherited thrombocytopenia.
Introduction
Inherited thrombocytopenias (IT) are a heterogeneous group of disorders characterized by platelet counts of less than 150×109/L in whole blood. Platelet counts are considered normal when maintained at levels between 150×109/L and 450×109/L. This is achieved by homeostatic processes controlling platelet production (thrombopoiesis), platelet senescence and platelet consumption/destruction. Pathogenic mutations can result in a disruption of these balanced processes causing IT. However, the clinical manifestations are often dependent on both a decreased platelet count and a qualitative or acquired platelet defect and can vary dramatically from severe and potentially life-threatening bleeding to no symptoms. This variation is noted among individuals shown to have the same underlying genetic causes of disease, suggesting that bleeding risk and phenotype are complex traits.1
The average incidence of IT is estimated to be approximately 270 cases per 1×106 live births.2 To date there are 27 individual IT disorders with known causative mutations registered within the Online Mendelian Inheritance in Man (OMIM) catalog, although 33 disease-causing genes have been described.3
Genetic studies have played a major role in the diagnosis and progressive understanding of IT. The genes implicated in the disease encode proteins that vary widely in function and include transcription factors (ETV6, FLI1, GATA1, GFI1B and RUNX1) and proteins involved in cytoskeleton rearrangement and organization (ACTN1, FLNA, GP1BA, GP1BB, GP9, TUBB1 and WAS). However, some protein functions currently remain unknown (SLFN14 and GNE).4–9. Although our knowledge of the causes of IT continues to grow, presently a genetic diagnosis is only reported in approximately 50% of individuals.10–12
So far, genetic investigation into IT has focused on candidate gene sequencing and individual cases of whole exome sequencing (WES) when a causative gene is not obvious.9 With 50% of patients currently undiagnosed, a change in the way we approach genetic diagnosis is necessary. Here we present the first, large-scale, WES-only approach to patients with suspected IT. We demonstrate its application in determining possible genetic origins of IT including identification of variants in novel candidate causative genes. We combine this with an approach implemented by the Genotyping and Phenotyping of Platelets (GAPP) study, which combines WES analysis with extensive platelet phenotyping to create a complete method of diagnosis and gene discovery in this subset of patients.
Methods
Study approval
The UK-GAPP study was approved by the National Research Ethics Service Committee of West Midlands–Edgbaston (REC reference: 06/MRE07/36) and participants gave written informed consent in accordance with the Declaration of Helsinki. This study was registered at www.isrctn.org as #ISRCTN 77951167. The GAPP study is included in the National Institute of Health Research Non-Malignant Haematology study portfolio (ID9858).
Platelet counts, morphology and white blood cell counts
Results from patients’ samples were compared to ranges for healthy volunteers for the specific method of morphology used. Platelet counts for light transmission aggregometry and flow cytometry analysis as well as mean platelet volume in platelet-rich plasma were originally measured using the Beckman Coulter counter (n=44). Subsequently, platelet counts, morphology and white blood cell counts in whole blood were determined using the Sysmex XN-1000 (n=11). The PLT-F channel was used to measure platelet counts in whole blood and the immature platelet fraction. Mean platelet volume was determined from the impedance PLT-I channel. White blood cell counts were obtained using the Sysmex XN-DIFF channel. All samples were tested against a normal range which was established by measuring the counts for 40 healthy individuals using the Sysmex XN-1000.
Platelet preparation and platelet function testing
Platelet function was assessed by light transmission aggregometry, including lumiaggregometry, for samples with platelet counts in platelet-rich plasma of >1×108/mL (n=13). An in-house flow-cytometry assay was developed to assess platelet function in patients with platelet counts in platelet-rich plasma <1×108/mL (n=22). Platelets from individuals with borderline platelet counts in platelet-rich plasma, between 1.0 and 1.5×108/mL, were assessed using both assays (n=16).
Aggregometry was performed as previously described.13,14 For flow cytometry, resting surface levels of CD42b, CD41 and GPVI were assessed. The platelet-rich plasma was then stimulated with ADP (3 and 30 μM), CRP (0.3 and 3 μg/mL) and PAR-1 peptide (10 and 100 μM). Membrane expression of P-selectin (FITC-conjugated mouse anti-human CD62P antibody, BD Pharmingen), a marker of platelet alpha granule release, as well as fluorescent fibrinogen binding (a marker of integrin activation) was assessed by flow cytometry on an Accuri C6 flow cytometer. Incubation took place at 37°C for 2 min and was terminated by adding a 5-fold excess of ice-cold phosphate-buffered saline.
Whole exome sequencing
WES and bioinformatics analysis were performed as described previously8,15,16 (Figure 1).
Figure 1.
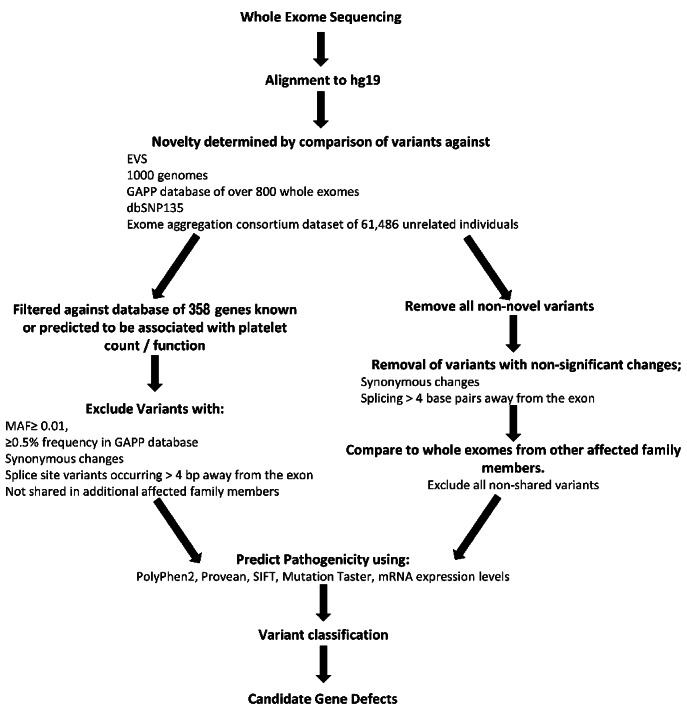
Bioinformatics pipeline analysis of whole exome sequencing data. Initial WES analysis focused on comparison with a panel of 358 genes (Online Supplementary Table S1), after which screening of exome variants focused on novel variants. Variants were classified using the ACMG consensus guidelines.
The pathogenicity of variants was determined and called using the consensus guidelines as set out by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG guidelines).17 Segregation was determined by Sanger sequencing of candidate variants in both affected and unaffected family members, when available, and the classification was adapted appropriately for the specific study and small sample size.
Sanger sequencing
To verify candidate mutations and examine their segregation among family members Sanger sequencing was performed using standard methods on an ABI 3730 automated sequencer, as described previously.8
Results
Recruitment of patients
To date, 55 patients with a suspected IT or sustained reduced platelet counts have been enrolled from 25 UK Haemophilia Care Centres and investigated as part of the GAPP study. Before enrollment in the study, all patients underwent clinical and genetic work-up to exclude known platelet disorders (including Bernard-Soulier syndrome and MYH9-related disorders, analyzed initially by blood film), idiopathic thrombocytopenic purpura and other non-platelet disorders including von Willebrand disease and inherited coagulation factor deficiencies. The patients’ bleeding phenotypes are displayed in Table 1. WES was performed on genomic DNA from all patients, including 37 index cases, all of whom met the study’s entry criteria. All patients, excluding F35.I and F35.II, were of white British or mixed British ethnicity. All results following platelet function testing and WES were reported back to the referring hematology consultants to aid in genetic counselling and disease management.
Table 1.
Platelet and bleeding phenotypes of 55 patients recruited to the UK-GAPP study.
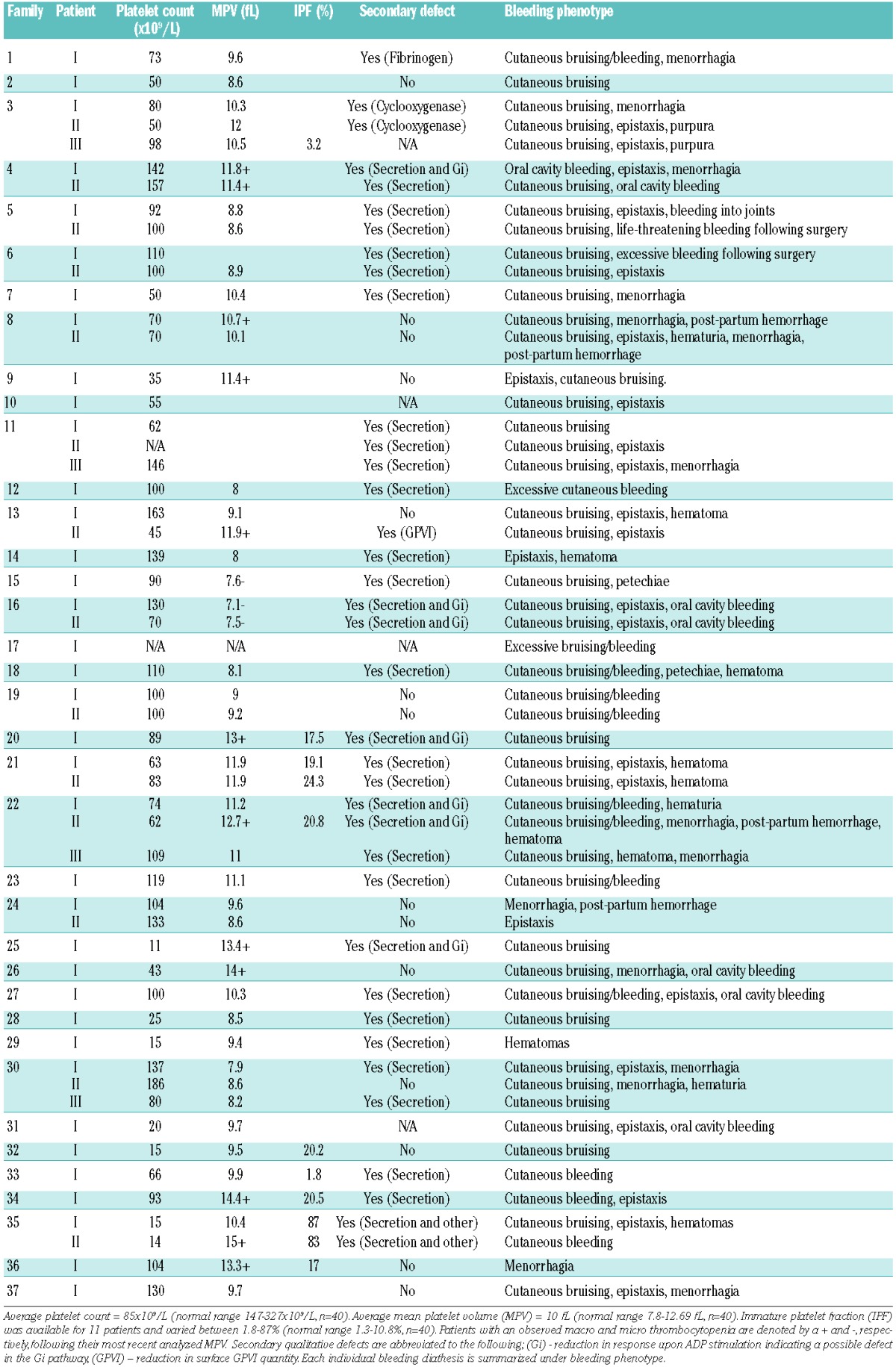
Platelet counts, morphology and function testing
Patients were recruited with a platelet count in whole blood, at the time of enrollment, of less than 150×109/L. Patients with platelet counts in the range of 150×109/L to 200×109/L remained enrolled in the study if they showed a similar phenotype to related affected family members and a platelet count below 150×109/L had been observed prior to enrollment (patients F4.II, F11.III, F13.I and F30.II). Platelet counts, mean platelet volume and immature platelet fraction are displayed in Table 1. Of the 55 recruited patients, 12 were deemed to have a macrothrombocytopenia and three a microthrombocytopenia (Table 1). White cell counts were within the normal range (3.78×109/L – 10.11×109/L, n=40) in all patients analyzed (n=11).
Platelet function studies revealed the presence of a secondary qualitative defect in addition to the low platelet count in 37/51 (73%) of the 55 patients whose DNA underwent WES and who were also available for platelet function testing (Table 1). Of the 37 patients with a secondary qualitative defect, 89% (33/37) displayed defects in both alpha and dense granule secretion. Five of these patients with an observed granule secretion defect were also suspected to have an additional Gi defect because of reduced responses to all concentrations of ADP. The remaining four patients without an observable granule secretion defect showed abnormalities in alternative pathways (integrin activation, cyclooxygenase pathway and GPVI surface levels) in addition to low platelet counts (Table 1).
Whole exome sequencing
WES was performed on genomic DNA from all 55 patients, including 37 index cases, following platelet function testing. An average fold-coverage of 111 was observed across all DNA samples analyzed by WES with an average of 91% of target sequences having >20× coverage. Areas of poor coverage were analyzed manually when occurring in previously IT-associated genes.
WES revealed between 24,000 and 25,000 variants (single nucleotide variants, small scale insertions/deletions, and splice site variations) in the DNA from each patient, with an average of 197 novel variants per exome. On average, per individual, 2401 variants with a mean allele frequency of <0.01 were observed, excluding synonymous variants. By evaluating the specificity of the pipeline in calling small variations, it was found that the sensitivity was over 99% and the false discovery rate was approximately 3%. The percentage of the genes with ≤20× coverage for a panel of 358 platelet-related genes is included within Online Supplementary Table S1.
Copy number variations were detected using ExomeDepth.18 The analysis revealed an average of 137 copy number variations per exome (n=32). No copy number variations were deemed potential candidates either because of a high allele frequency or a lack of expression or functional role of the gene within the megakaryocyte/platelet lineage.
Variants in known thrombocytopenia-causing genes
WES and downstream analysis identified variants within 33 known IT-causing genes in 25 index cases (68%). All variants exceeded 30× sequence coverage at the point of variation and have been confirmed by Sanger sequencing. Variants were selected from positive hits to genes within the panel of 358 IT-associated genes (Online Supplementary Table S1). On average, 37 variants per individual (range, 11–52) were noted in genes from the panel of 358 IT-associated genes, of which on average four (range, 0–7) variants were significant per exome analyzed.
In total 28 variants were noted in 14 genes previously known to cause IT (Table 2). Twenty-one index cases possessed a single variant in a gene previously known to cause IT. Four index cases possessed two variants in genes previously known to cause IT. One variant, RUNX1; c.270+1G>T, was noted in two index cases (F13.I and F14.I). Candidate variations were present within ACTN1, the 5′-UTR of ANKRD26, CYCS, FLI1, GFI1B, ITGB3, GP1BA (heterozygous), MYH9, NBEAL2, RUNX1, SLFN14, STIM1, TPM4 and TUBB1. All but six variants were novel and not present within the variant databases previously mentioned. Three variants, ANKRD26; c.-126T>G in F2.I, MYH9; c.3493C>T (rs80338829) in F9.I and RUNX1; c.530G>A in F19.I and F19.II have been previously associated with IT.1,19,20 The remaining three variants that have been previously observed occurred at frequencies of <0.005 (0.05%) in available databases. One of the databases scrutinized was that of the ExAC consortium (http://exac.broadinstitute.org) which may include data from individuals with low platelet counts who were either undiagnosed or recruited through an unrelated study (Table 2). Seven variants have previously been published as part of two separate publications from the UK-GAPP study group.8,15
Table 2.
Results of whole exome sequencing analysis of 55 patients with inherited thrombocytopenia showing variants in known thrombocytopenia-causing genes. 68% of individuals have a predicted genetic etiology in a previously IT-associated gene. When a variant has been previously observed it is annotated in the prevalence column with the database in which it is included. The ACMG consensus guideline results are also displayed in the final classification column.17
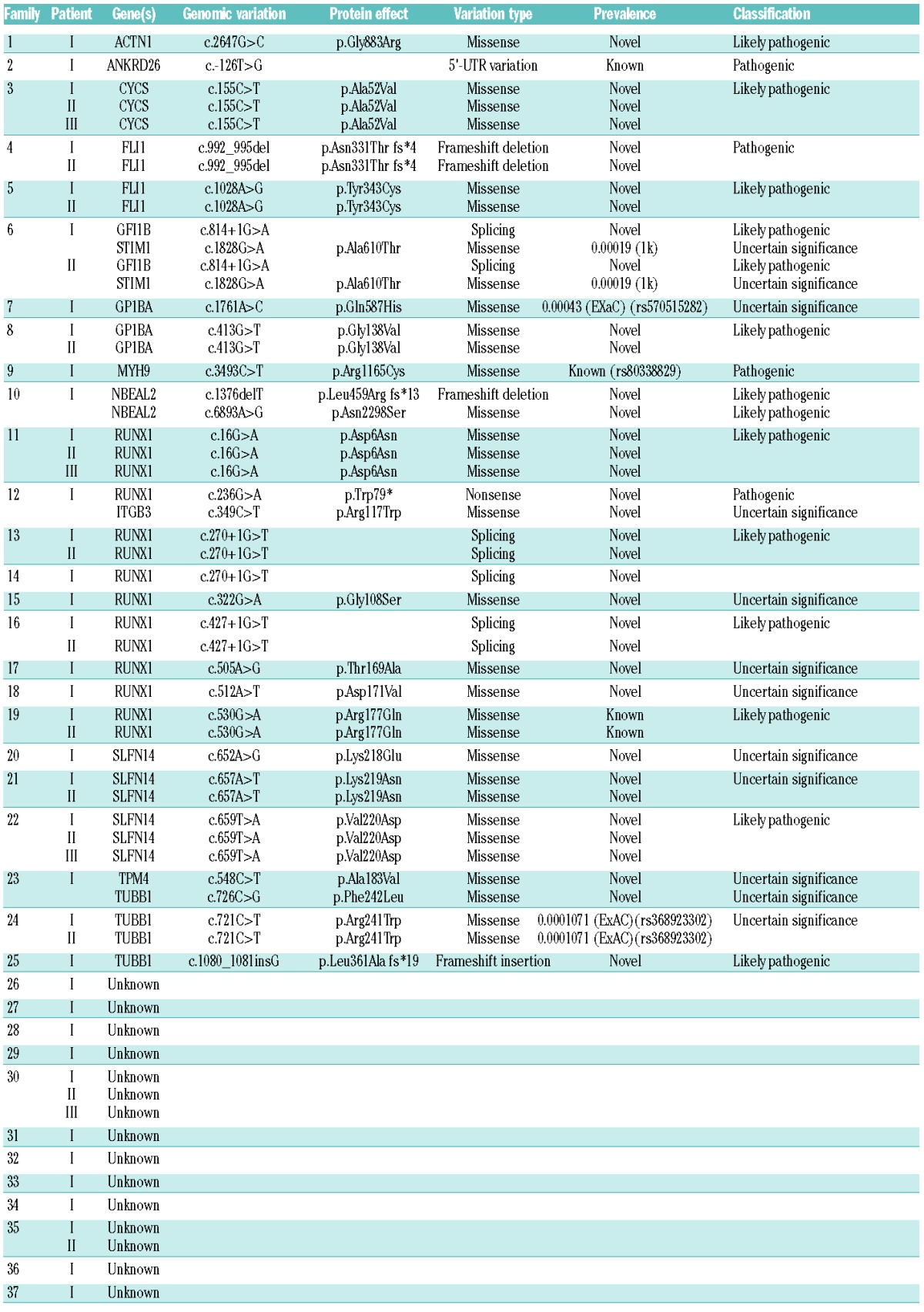
Classification of the 28 variants occurring within the known IT-related genes, following the interpretation guidelines set out by Richards et al.,17 revealed four variants to be “pathogenic”, 13 to be “likely pathogenic” and 11 to be of “uncertain significance”. Variants classified as “pathogenic” were either already known to be a genetic cause of IT; ANKRD26; c.-126T>G in F2.I and MYH9; p.Arg1165Cys in F9.I, or were predicted to be loss-of-function variants in genes for which a loss of function is known to cause disease; FLI1; p.Asn331Thr fs*4, in F4.I and F4.II and RUNX1; pTrp79* in P12.I.
On average, less than one novel variant was expected to be observed in the known IT-causing genes in which variants were observed. The number of variants occurring also exceeds the expected number when extending the analysis to cover variants with a mean allele frequency of <0.01.
Of the 37 index patients, four presented with two candidate variations in known disease-linked genes, which in one case were present in the same gene. These were as follows: F6.I (GFI1B; c.676+1G>A and STIM1; p.Ala610Thr), F10.I (NBEAL2; (p.Leu459Arg fs*13 and p.Asn2298Ser), F12.I (RUNX1; p.Trp79* and ITGB3 p.Arg117Trp) and F23.I (TPM4; p.Ala183Val and TUBB1; p.Phe242Leu).
Of the 25 index cases with variants in known disease-causing genes, nine were observed to have variants within the RUNT1-related transcription factor gene; RUNX1. One variant, RUNX1; p.Arg177Gln, observed in F19.I and F19.II has been previously reported as a causative germline mutation of a familial platelet disorder in two individuals from the same pedigree.20 The variations consisted of five missense variants, two splice-site variants and one nonsense variant. One splice-site variation, c.270+1G>T, was present within three affected individuals from two separate families (F13 and F14). All variants, with the exception of a missense substitution (p.D6N), lie within the genetic region encoding the RUNT homology domain (RHD) which mediates DNA binding and heterodimerization with CBFβ (Figure 2).21 Platelets from the majority of these patients (10/13) demonstrated a reduction in ATP secretion and, in keeping with previous reports, several of these patients displayed additional clinical features. Variations in RUNX1 are associated with a propensity to myelodysplastic syndrome and acute myeloid leukemia. To date, hematologic malignancies have not been reported in any patients; however, the brother of F16.1 did have a history of acute myeloid leukemia but was unavailable for testing.
Figure 2.
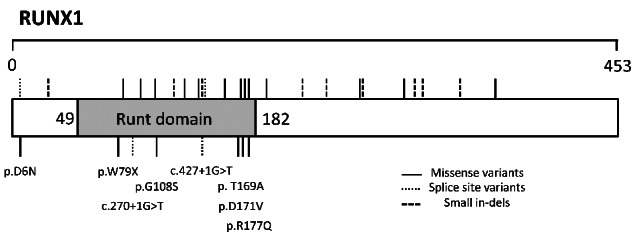
Spatial amino acid locations of all thrombocytopenia-causing variants present within RUNT transcription factor 1 (RUNX1) (RefSeq NP_001001890). Previously disease-causing variants found the HGMD (www.hgmd.cf.ac.uk) and ClinVar (www.ncbi.nlm.nih.gov/clinvar/) databases are denoted above. The eight variants found within RUNX1 in the GAPP cohort of 54 patients who have undergone whole exome sequencing are denoted below and the effect on the protein or predicted splice-site is shown.
Potentially damaging variants in novel candidate genes
After scrutinizing individuals for variants within the panel of 358 platelet-associated genes (Online Supplementary Table S1), individuals without a variant in a previously IT-associated gene were analyzed for variants in novel genes. WES analysis revealed potentially damaging candidate variants in three families with currently unknown genetic etiology (Table 3). All candidate variants are novel (excluding a previously annotated variant in MKL1; p.Val575Met, which occurs at a frequency of 0.007718 within the ExAC consortium), segregate with the disease status and have been confirmed by Sanger sequencing.
Table 3.
Potentially damaging variants in novel candidate genes.
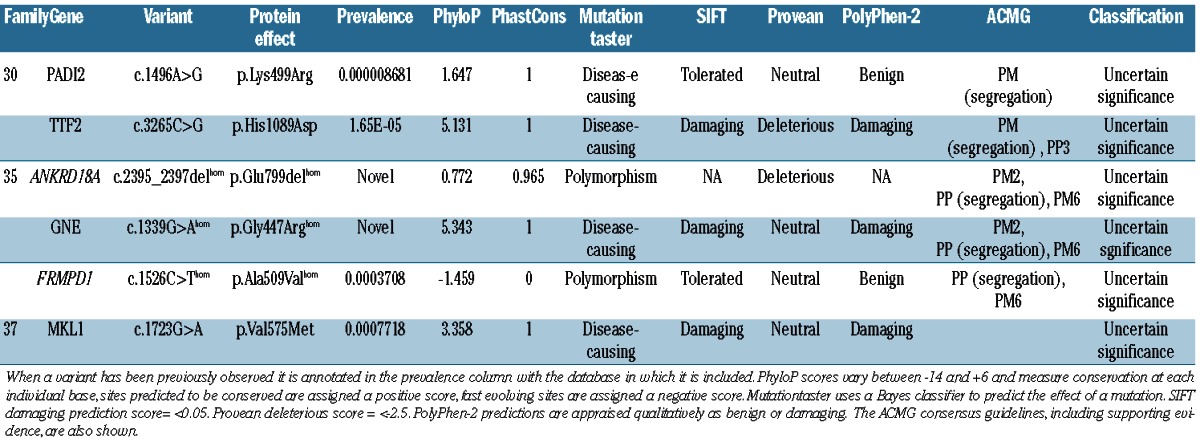
Variants within ANKRD18A, GNE and FRMPD1 in two related individuals from consanguineous relationships
WES analysis of two related patients (F35.I and F35.II) of South Asian ethnicity was approached differently to that of other patients in this study. Both patients displayed a similarly severe clinical phenotype with a significant reduction in circulating platelets (15×109/L). Platelet function testing revealed a reduction in P-selectin (CD62P) expression upon stimulation and variable fluorescent fibrinogen binding which was consistent across both affected individuals. The patients were cousins born from consanguineous relationships within a single consanguineous kindred so the analysis was focused on identification of a shared homozygous variant due to the recessive segregation of disease. Three variants occurring within ANKRD18A; p.Glu799del, GNE; p.Gly447Arg and FRMPD1; p.Ala509Val were present in both affected individuals and within a tightly linked region of homozygosity on chromosome 9p. The variations within ANKRD18A and GNE were novel according to the previously mentioned databases whereas the variant in FRMPD1 has been observed at a frequency of 0.0003708 including 39 times within the South Asian population (rs571037699). There is no ClinVar entry for this variant and all three variants are classified as variants of “uncertain significance”.
One missense variant in the recently proposed inherited thrombocytopenia-linked gene, MKL1
One individual was shown to harbor a rare (frequency <0.01) missense variant within the Megakaryoblastic Leukaemia (translocation) 1 gene; MKL1. The variant was the only variant occurring within a gene of hemostatic relevance within 109 significant novel variants. The variant; MKL1; c.1723G>A, p.Val575Met present in patient F37.I has been noted previously at a frequency of 0.0007718 (allele count of 6/7774 in the ExAC consortium). The patient has a mild reduction in platelet count (130×109/L) and no secondary qualitative defects in platelet function were observed. The variant is classified as of “unknown significance”.
Novel missense candidate variants in PADI2 and TTF2
Three affected individuals and four unaffected related individuals of a large kindred were recruited to the study. Mild thrombocytopenia was observed within the family with platelet counts ranging from 80×109/L to 186×109/L in the three affected individuals. All three affected individuals presented with a normal platelet size (7.9–8.6 fL) and a mild reduction in secretion was observed in F30.I and F30.III but not in F30.II. All affected individuals shared a similar bleeding phenotype, suffering from spontaneous epistaxis, excessive bruising and prolonged bleeding from minor wounds. WES analysis revealed 14 novel or rare (frequency <0.01) variants shared between the three affected individuals. Sanger sequencing of all 14 variants in four unaffected related individuals narrowed down candidates to only two missense variants; PADI2 (p.Lys499Arg) and TTF2 (p.His1089Asp). Both variants segregate with disease, not being present in the unaffected individuals. Both variants have been observed previously at a low frequency (<0.01) within the EXaC database (Table 3) and are currently classified as being of “uncertain significance”.
Discussion
Here we present the first, large-scale application of WES analysis to patients with inherited bleeding diatheses presenting with thrombocytopenia of unknown etiology.
Platelet counts and phenotypic presentations varied considerably among the patients studied, which is consistent with the variability observed in the spectrum of IT. However, the majority of patients (73%) were noted to have a secondary qualitative defect in platelet function which may explain the disproportionate bleeding when compared to the patients’ platelet counts. A lack of consistency was noted in families 13 and 30, which apparently included affected individuals both with and without defects in platelet function. Clinical complications are shared among the affected family members so this most likely represents limitations in the sensitivity of platelet function testing or intra-familial variability.
Overall, when considering pathogenicity WES analysis positively predicted pathogenicity in 46% of index cases (17/37) (results classified as “pathogenic” or “likely pathogenic” in a gene consistent with the patients’ phenotype and zygosity consistent with expected inheritance). Twenty-two percent of the index cases (8/37) had uncertain/possible pathogenicity (results classified as being of “uncertain significance” in known IT-causing genes). The remaining 32% of index cases (12/37) had a negative prediction of pathogenicity (no convincing variants identified in known IT-causing genes). WES is not without its limitations and, as with any genetic analysis, all variants must be functionally confirmed as deleterious to the coded protein. However, our positive variant discovery rate is comparable to or exceeds the rates in previous large-scale WES clinical multicenter studies of Mendelian disorders.22,23
Focusing our genetic analysis on patients with unknown etiology of disease with minor prior genetic testing has produced a spectrum of variants different from that from previous, large-scale, targeted genetic studies of IT. Patients were recruited to the study with clinically diagnosed bleeding disorders of unknown etiology. One caveat about this approach is the possible exclusion of individuals with known Bernard-Soulier syndrome or MYH9-related disorders as these two forms of IT are routinely tested for in many hematology centers in the UK. However, three index cases with variants in either GP1BA or MYH9 were noted in our analysis; these patients had atypical presentations of Bernard-Soulier syndrome or MYH9-related disorder and were, therefore, potentially falsely-negatively reported cases. The individuals with variants within GP1BA and MYH9 showed a slight increase in mean platelet volume; however, this was not at the magnitude of giant platelets normally attributed to this group of disorders and only patient F9.I showed any secondary syndromic symptoms with the individual suffering from congenital cataracts.
One attribute of excluding patients with known variants in GP1BA, GP1BB, MYH9 and potentially GP9 was the discovery of a relatively large percentage of individuals analyzed (24% of index cases) with variants in RUNX1 as a primary likely cause of disease. With the exception of one predicted loss-of-function variant, the variants present within RUNX1 are currently classified as either “likely pathogenic” or of “uncertain significance” and need functional confirmation to be considered the cause of disease. However, the presence of these variants in a large number of individuals with an often shared secondary functional defect in secretion does suggest that the prevalence of RUNX1 variants may be higher than previously thought. This raises the issue of whether they should be considered as clinically significant as Bernard-Soulier syndrome and MYH9-related disorders and be searched for in a primary genetic screening at the initial diagnosis of IT.
An advantage of using WES is the possibility of finding candidate variations in novel genes in subjects who do not possess variants in known IT-causing genes. The determination of whether these candidate variants are in fact pathogenic relies on functional confirmation of the deleterious effect of the variant. However, WES analysis, especially with combined segregation analysis by Sanger sequencing in extensive kindreds, can provide indications as to which variants may be of scientific and clinical relevance. This strategy has recently been utilized in the discovery of novel candidate variations in SLFN14 initially as part of the GAPP study.15,24
Family 35 is an interesting case of two affected related individuals born from consanguineous relationships. The molecular function of ANKRD18A is currently unknown, while FRMPD1 regulates the subcellular localization of activator of G-protein signaling 3 (AGS3).25 Both genes are expressed weakly in hematopoietic cells. However, GNE, coding for an enzyme in the sialic acid biosynthetic pathway, is expressed in all cells of the hematopoietic lineage. There are currently 88 registered mutations in GNE in the Human Genome Mutation Database (www.hgmd.cf.ac.uk). Mutations are known to be the genetic cause of sialuria (OMIM269921) and hereditary inclusion body myopathy (OMIM600737).26,27 Recently, two separate groups have reported patients with compound heterozygous variations in GNE, causing GNE-related myopathy with congenital thrombocytopenia.28,29 The platelet counts of the four reported affected individuals were below 45×109/L; platelet volume measurements were not recorded. None of the patients displayed signs of myopathy until mid-adolescence/early adulthood; F35.I and F35.II are currently aged 10 and 6, respectively. Without functional characterization of the effects of each variation, we cannot definitively conclude the genetic etiology of these two individuals’ severe thrombocytopenia. However, WES analysis has allowed us to focus our efforts on three potentially pathogenic variants in novel genes.
MKL1 was initially included in our panel of 358 genes for post-WES analysis due to its role in megakaryocyte maturation elucidated via its binding partner, serum response factor (SRF).30–32 Recently, the first case of a homozygous mutation in MKL1 in a patient with severe immunodeficiency and no hematologic malignancies was reported.33 One interesting phenotypic presentation of the affected individual was an intermittent mild thrombocytopenia with low platelet counts in whole blood of between 50×109/L and 150×109/L. Here we present a novel variant within MKL1, at a highly conserved genetic site. The missense variant observed in F37.I represents the only variant to occur in a gene with previous hematologic implications. One further variant in MKL1 was observed in addition to a “likely pathogenic” frameshift causing insertion within TUBB1 in patient F25.I. Due to the predicted loss of function of the frameshift causing the TUBB1 variant it is unlikely that the variant with MKL1 is additive to the phenotype of patient F25.I. However, the variant of uncertain significance in patient F37.I is an interesting candidate to take forward for functional studies.
WES and segregation determination using Sanger sequencing revealed candidate variants in PADI2 and TTF2 that segregate with disease in F30.I, F30.II and F30.III. The phenotypic presentations vary between the patients but clinical presentations are consistent, which may reflect limitations in the sensitivity of platelet function testing. Neither gene has previously been implicated in hematologic abnormalities: mutations in PADI2 have been associated with schizophrenia, breast cancer and rheumatoid arthritis, while mutations in TTF2 have been associated with thyroid dysgenesis.34–37 WES analysis has therefore provided us with the first steps for determining the impact of these two variants of uncertain significance and whether they have the propensity to be disease causing.
In summary, we show that WES can be applied to identify the underlying genetic cause in known IT-causing genes for patients with thrombocytopenia and unclear disease etiology. We show similar positive detection rates when compared to prior targeted studies and, with the addition of complementary functional studies, show an improved detection rate when compared to WES analysis of other developmental disorders. We also suggest the applicability of WES in providing preliminary insight into novel genes and their potential mechanism of action through candidate variations of unknown significance. This approach provides a foundation to enhance our current knowledge on megakaryopoiesis, platelet function and platelet senescence/death through subsequent functional studies.
Acknowledgments
We thank the families for providing samples and our clinical and laboratory colleagues for their help. This work was supported by the British Heart Foundation (RG/PG/13/36/30275; RG/09/007), an MRC Doctoral Training Partnership grant (BJ), a Wellcome Trust Combined Training Programme Fellowship (093994) (GCL), the Healing Foundation (PH) and the Platelet Charity. We thank the NIHR Haematology Specialty Group for their help in recruiting to the study, and all our clinical investigators and collaborators. The authors also acknowledge support from the Department of Health via the National Institute for Health Research (NIHR) Comprehensive Biomedical Research Centre Award to Guy’s & St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. We thank the Queen Elizabeth Hospital Charity for funding the Sysmex XN-1000.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/10/1170
References
- 1.Noris P, Perrotta S, Seri M, et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood. 2011;117(24):6673–6680. [DOI] [PubMed] [Google Scholar]
- 2.Balduini CL. Diagnosis and management of inherited thrombocytopenias. European Human Genetics Conference 2014; 2014; Milan, Italy. [Google Scholar]
- 3.Johnson B, Fletcher SF, Morgan NV. Inherited thrombocytopenia: novel insights into megakaryocyte maturation, proplatelet formation and platelet lifespan. Platelets. 2016:1–7. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canales ML, Mauer AM. Sex-linked hereditary thrombocytopenia as a variant of Wiskott-Aldrich syndrome. N Engl J Med. 1967;277(17):899–901. [DOI] [PubMed] [Google Scholar]
- 5.Kunishima S, Kobayashi R, Itoh TJ, Hamaguchi M, Saito H. Mutation of the beta1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood. 2009;113(2):458–461. [DOI] [PubMed] [Google Scholar]
- 6.Kunishima S, Okuno Y, Yoshida K, et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet. 2013;92(3):431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nichols KE, Crispino JD, Poncz M, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24(3): 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stockley J, Morgan NV, Bem D, et al. Enrichment of FLI1 and RUNX1 mutations in families with excessive bleeding and platelet dense granule secretion defects. Blood. 2013;122(25):4090–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balduini CL, Pecci A, Noris P. Inherited thrombocytopenias: the evolving spectrum. Hamostaseologie. 2012;32(4):259–270. [DOI] [PubMed] [Google Scholar]
- 11.Balduini CL, Savoia A. Genetics of familial forms of thrombocytopenia. Hum Genet. 2012;131(12):1821–1832. [DOI] [PubMed] [Google Scholar]
- 12.Savoia A. Molecular basis of inherited thrombocytopenias. Clin Genet. 2016;89(2): 154–162. [DOI] [PubMed] [Google Scholar]
- 13.Dawood BB, Wilde J, Watson SP. Reference curves for aggregation and ATP secretion to aid diagnose of platelet-based bleeding disorders: effect of inhibition of ADP and thromboxane A(2) pathways. Platelets. 2007;18(5):329–345. [DOI] [PubMed] [Google Scholar]
- 14.Dawood BB, Lowe GC, Lordkipanidze M, et al. Evaluation of participants with suspected heritable platelet function disorders including recommendation and validation of a streamlined agonist panel. Blood. 2012;120(25):5041–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fletcher SJ, Johnson B, Lowe GC, et al. SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J Clin Invest. 2015;125(9): 3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leo VC, Morgan NV, Bem D, et al. Use of next-generation sequencing and candidate gene analysis to identify underlying defects in patients with inherited platelet function disorders. J Thromb Haemost. 2015;13(4): 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plagnol V, Curtis J, Epstein M, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28(21):2747–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seri M, Cusano R, Gangarossa S, et al. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Hegglin/Fechtner Syndrome Consortium. Nature Genetics. 2000;26(1):103–105. [DOI] [PubMed] [Google Scholar]
- 20.Preudhomme C, Renneville A, Bourdon V, et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood. 2009;113(22):5583–5587. [DOI] [PubMed] [Google Scholar]
- 21.Kamachi Y, Ogawa E, Asano M, et al. Purification of a mouse nuclear factor that binds to both the A and B cores of the polyomavirus enhancer. J Virol. 1990;64(10): 4808–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chong JX, Buckingham KJ, Jhangiani SN, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015; 97(2):199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marconi C, Di Buduo CA, Barozzi S, et al. SLFN14-related thrombocytopenia: identification within a large series of patients with inherited thrombocytopenia. Thromb Haemost. 2016;115(5):1076–1079. [DOI] [PubMed] [Google Scholar]
- 25.An N, Blumer JB, Bernard ML, Lanier SM. The PDZ and band 4.1 containing protein Frmpd1 regulates the subcellular location of activator of G-protein signaling 3 and its interaction with G-proteins. J Biol Chem. 2008;283(36):24718–24728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seppala R, Lehto VP, Gahl WA. Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am J Hum Genet. 1999;64(6):1563–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisenberg I, Avidan N, Potikha T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29(1):83–87. [DOI] [PubMed] [Google Scholar]
- 28.Izumi R, Niihori T, Suzuki N, et al. GNE myopathy associated with congenital thrombocytopenia: a report of two siblings. Neuromuscul Disord. 2014;24(12):1068–1072. [DOI] [PubMed] [Google Scholar]
- 29.Zhen C, Guo F, Fang X, Liu Y, Wang X. A family with distal myopathy with rimmed vacuoles associated with thrombocytopenia. Neurol Sci. 2014;35(9):1479–1481. [DOI] [PubMed] [Google Scholar]
- 30.Cheng EC, Luo Q, Bruscia EM, et al. Role for MKL1 in megakaryocytic maturation. Blood. 2009;113(12):2826–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith EC, Thon JN, Devine MT, et al. MKL1 and MKL2 play redundant and crucial roles in megakaryocyte maturation and platelet formation. Blood. 2012;120(11):2317–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halene S, Gao Y, Hahn K, et al. Serum response factor is an essential transcription factor in megakaryocytic maturation. Blood. 2010;116(11):1942–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Record J, Malinova D, Zenner HL, et al. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood. 2015;126(13):1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe Y, Nunokawa A, Kaneko N, et al. A two-stage case-control association study of PADI2 with schizophrenia. J Hum Genet. 2009;54(7):430–432. [DOI] [PubMed] [Google Scholar]
- 35.McElwee JL, Mohanan S, Griffith OL, et al. Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer. 2012;12:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang X, Xia Y, Pan J, Meng Q, Zhao Y, Yan X. PADI2 is significantly associated with rheumatoid arthritis. PLoS One. 2013;8(12):e81259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castanet M, Polak M. Spectrum of human Foxe1/TTF2 mutations. Horm Res Paediatr. 2010;73(6):423–429. [DOI] [PubMed] [Google Scholar]