

Abstract
Recombination is an important source of metabolic innovation, especially in prokaryotes, which have evolved the ability to survive on many different sources of chemical elements and energy. Metabolic systems have a well-understood genotype–phenotype relationship, which permits a quantitative and biochemically principled understanding of how recombination creates novel phenotypes. Here, we investigate the power of recombination to create genome-scale metabolic reaction networks that enable an organism to survive in new chemical environments. To this end, we use flux balance analysis, an experimentally validated computational method that can predict metabolic phenotypes from metabolic genotypes. We show that recombination is much more likely to create novel metabolic abilities than random changes in chemical reactions of a metabolic network. We also find that phenotypic innovation is more likely when recombination occurs between parents that are genetically closely related, phenotypically highly diverse, and viable on few rather than many carbon sources. Survival on a new carbon source preferentially involves reactions that are superessential, that is, essential in many metabolic networks. We validate our observations with data from 61 reconstructed prokaryotic metabolic networks. Our systematic and quantitative analysis of metabolic systems helps understand how recombination creates innovation.
Keywords: genome-scale metabolic networks, recombination, metabolic innovation, superessential reactions, metabolic genotype, metabolic phenotype
1. Introduction
Organisms that reproduce sexually and recombine their DNA bear high evolutionary costs, among them the disruption of well-adapted phenotypes caused by recombination. Nonetheless, recombination is common in nature. This paradox has spurred many efforts to resolve it [1–3].
From a genetic perspective, the major benefit and costs of recombination are similar in kind, because recombination can create well-adapted phenotypes just as it can destroy them. Recombination has the ability to join beneficial mutations from two organisms or molecules [4,5], and thus speed up adaptive evolution [6]. In recent years, experimental evidence obtained through DNA shuffling experiments has made clear how great this benefit of recombination can be in creating proteins with novel phenotypes [7]. Many experimental studies have confirmed the power of recombination in generating genes, pathways, and genomes with novel features [8–11].
Because recombination can involve large-scale genotypic change, its power to disrupt existing well-adapted phenotypes may also be large. However, recent directed evolution experiments and computational studies based on model transcriptional gene regulatory circuits and lattice proteins suggest that random mutations are more likely to disrupt existing phenotypes than recombination [12–14].
To understand the relative costs and benefits of recombination, one needs to understand how genotypic change causes phenotypic change, but this is difficult even for well-studied systems like proteins. For example, measuring the phenotypic effects of genetic changes engineered into proteins is laborious, and our ability to predict altered protein functions from altered amino acid sequences computationally is very limited. However, in one class of biological systems, the complex and genetically encoded chemical reaction network of metabolism, we understand genotype–phenotype relationships better [15–18]. The reason is that computational tools such as flux balance analysis (FBA) provide a means to predict metabolic phenotypes—the ability of an organism to survive on specific nutrients—from information about metabolic genotypes, i.e. the collection of chemical reactions that a metabolic reaction network is able to catalyse [19]. What is more, FBA-based qualitative predictions of metabolic phenotypes are in good agreement with experimental data [20]. We here use FBA to quantify and understand the disruptive and creative effects of recombination on the biochemistry of metabolic systems. We will focus especially on metabolic innovation, the ability of recombination to create metabolic networks that are able to survive on new sources of carbon and energy. In doing so, we represent metabolic genotypes not as DNA sequences but as sets of metabolic reactions that can be altered through recombination, a common approach in metabolic systems biology [15–18,21–25].
Metabolism is an ideal system to study innovation, especially for microorganisms, because the prokaryotic world is rife with examples of metabolic innovations. For instance, microorganisms have acquired the capability to extract energy from a bewildering variety of non-natural and even toxic substances [26–29]. Microbial isolates from pristine soils have not only acquired resistance to a wide range of antibiotics, but they can even use some of these molecules as food [30]. And halophilic bacteria can tolerate high salt concentration by synthesizing novel molecules such as ectoine or glycine betaine [31].
In eukaryotes, recombination occurs during meiosis and is thus linked to reproduction. It involves parents that are usually genetically similar and belong to the same population and species. By contrast, prokaryotic recombination is not usually linked to reproduction. It occurs via horizontal gene transfer [32], whose incidence is large and greater than that of point mutations [33–35]. It changes the organization and gene content of genomes on short evolutionary time scales [32,36,37], and can involve very distantly related organisms [38,39]. Although horizontal gene transfer adds genes from a donor to a recipient, incorporating such genes into the recipient genome relies on DNA rearrangements that can also delete resident genes [40]. More generally, the majority of newly acquired genes obtained via horizontal gene transfer reside in the genome only for short amounts of time [41], and prokaryotic genomes show a bias towards DNA deletions [42]. Motivated by these observations, we here model prokaryotic recombination as a process where the transfer of biochemical reactions from a donor to a recipient is accompanied by concurrent deletion of reactions from the recipient.
Our work builds on an approach that we developed previously to study typical properties of a metabolic network with a given phenotype—the ability to survive on a given set of carbon and energy sources. These are properties that are independent of any one organism such as Escherichia coli [18,23–25,43,44]. The method explores a vast space of possible metabolic genotypes to create a random sample of metabolic networks that are viable on specific carbon sources such as glucose. We here use this approach to create pairs of ‘parental’ metabolic networks with well-defined genotypes and phenotypes. We ask how likely it is that recombination between these parents (i) disrupts their metabolic phenotypes and (ii) creates novel, innovative metabolic networks that can survive on at least one novel source of carbon and energy, among 50 different such sources we consider. We validate our observations with data from 61 prokaryotic genome-scale metabolic networks.
Our observations show that recombination creates more metabolic innovations than an equivalent amount of random change in a metabolic network's reaction complement—our model's representation of random mutation. At the same time, recombination is no more disruptive than random change. Importantly, the innovative power of recombination increases with the phenotypic diversity of the parents. By contrast, it decreases with their genotypic diversity and with their phenotypic complexity (the number of carbon sources on which they are viable). Moreover, we find that a class of metabolic reactions that we refer to as superessential plays an important role in metabolic innovation [45].
2. Material and methods
(a). Genome-scale metabolic networks and their phenotypic representations
Metabolism is a network of enzyme-catalysed biochemical reactions. Each such metabolic network contains a subset of the ‘reaction universe’ of all biochemical reactions that take place in the biosphere. We have curated a representation of this universe, which comprises 5 906 reactions and is based on current metabolic knowledge (for more details, see electronic supplementary material, text S1a and S1b) [18,46–49]. We represent an organism's metabolic genotype as a binary vector of length 5 906. Each entry of this vector corresponds to a given reaction in the universe, and is equal to one if the corresponding reaction is present in the network and zero otherwise. Thus, each genotype can be thought of as a single member of a vast space of all possible metabolic networks, which contains 25 906 distinct genotypes. We define the phenotype of a given metabolic genotype based on its viability on 50 distinct minimal environments that differ only in the carbon source (electronic supplementary material, text S1c). We consider a genotype viable on a given carbon source, if it can produce all the essential biomass precursors from the given carbon source, and we use FBA (see electronic supplementary material, text S1d) to determine viability [19].
(b). Generation of random metabolic networks
We here employ a previously described in silico process which relies on Markov Chain Monte Carlo (MCMC) random walks to generate metabolic networks comprising random sets of reactions that are viable on a given carbon source (electronic supplementary material, text S1e) [18,23]. This procedure can produce metabolic networks that are sampled uniformly from the set of all metabolic networks viable on a given carbon source.
(c). Generation of parental metabolic network pairs
Our analyses required us to recombine pairs of ‘parental’ metabolic networks with particular features, such as (i) their genotypic distance (D), defined as the number of reactions differing between the parents, (ii) their phenotypic complexity (||P||), that is, the number of carbon sources on which they are viable, (iii) their phenotypic distance (ΔP), that is, the number of carbon sources on which only one but not the other member of a parental pair is viable, and (iv) their genotypic complexity (||G||, or metabolic network size), defined as the number of reactions in each metabolic network. We used simultaneous genotype-converging MCMC random walks to generate pairs of metabolic networks with the features described above (see electronic supplementary material, texts S1f and S2).
(d). Modelling recombination and mutation in metabolic networks
To implement recombination for each parental metabolic network pair, we generated 1 000 recombiant offspring by (i) adding to the recipient metabolic network a given number n/2 of randomly chosen reactions that were present in the donor and absent in the recipient, followed by (ii) deleting n/2 reactions randomly chosen from the recipient. Thus, the total number of reactions changed by a recombination event in the recipient is equal to n (for more details, see electronic supplementary material texts S1g, S3, and figures S1, S2, and S3). We then quantify the incidence of metabolic innovation as the fraction (finnov) of offspring retaining viability on parental carbon sources that also gain viability on at least one additional carbon source.
To implement an amount of random change—our model's equivalent of ‘mutation’— in a metabolic network comparable with the same amount of recombinational change for a given n, we created a network's ‘mutational’ offspring by adding n/2 randomly chosen reactions from the reaction universe, and deleting n/2 randomly chosen reactions from the network. Note that the n/2 reactions added to recombinant offspring are chosen randomly from another viable network (the donor), whereas in mutation they are taken from the whole reaction universe.
To validate our model's results, we also analysed the metabolic networks of 61 prokaryotes obtained from the Biochemical Genetic and Genomic (BiGG) database [50], using the R package Sybil [51]. In these networks, we incorporated information about the linkage of the genes encoding metabolic reactions. To this end, we used the gene–reaction association rules defined in the BiGG database for each organism (in .mat files, grRules) [50], and ordered the genes in each organism based on their genomic position, as obtained from the RefSeq microbial genome database [52] (for more details, see electronic supplementary material, text S1h).
3. Results
(a). Recombination causes more metabolic innovations than random change
To quantify the power of recombination to create novel phenotypes in metabolic systems, we created 1 000 donor–recipient pairs of random viable metabolic networks with a fixed metabolic genotypic distance of D = 100 reactions. (Genomic data show that bacteria at this or greater metabolic divergence often recombine successfully; see electronic supplementary material, text S4 and figure S4.) For each of these pairs, we generated 1 000 recombinant offspring by recombining a given number n of reactions (see Material and methods). We quantified the incidence of metabolic innovation as the fraction (finnov) of offspring retaining viability on glucose that also gain viability on at least one additional carbon source. Moreover, we compared the effects of recombination on innovation with those of an equivalent amount of random change (‘mutation’, Material and methods). That is, we computed the fraction of innovative offspring (finnov) for metabolic networks with a number of random reaction deletions or additions equivalent to that caused by recombination.
Figure 1a shows the mean and standard error of finnov as a function of n. Two patterns are germane. First, the fraction of innovative offspring (finnov) is consistently greater for recombination (figure 1a, red) than for random change (black). For example, for recombination events involving n = 10 reactions, 5% of the viable recombinant offspring gain viability on at least one novel carbon source on average. By contrast, for random change involving n = 10 reactions, this fraction is more than four times smaller and below 1.3%. Second, finnov increases with the number of reactions exchanged through recombination. Our observations are robust to an alternative approach to generating metabolic network pairs with a fixed D = 100, and to the choice of an alternative primary carbon source (see Material and methods; electronic supplementary material, figures S3a–c). In sum, recombination produces a higher incidence of innovation. The diversity of metabolic innovations it produces is similar to those produced by random change (electronic supplementary material, figure S5).
Figure 1.

Cost–benefit relationship of recombination as compared with random reaction change (mutation). (a) Mean (bars) and standard error (vertical lines) of the fraction of innovative offspring (finnov) among all offspring retaining viability on glucose that are generated by recombination (red) and random reaction change (black), as a function of the number of reaction changes (n, x-axis). (b) Recombinational robustness (red), that is, the fraction of recombinant offspring retaining viablity on glucose, and mutational robustness (black), that is, the fraction of mutant offspring that can retain viability on glucose, as a function of the number of reaction changes (n, x-axis). Boxes span the 25% to 75% percentile, and whiskers indicate maxima and minima.
Next, we quantified the cost of recombination, i.e. its power to disrupt existing phenotypes. To this end, we measured recombinational robustness, the fraction of a parental pair's recombinant offspring that retains viability on glucose. Figure 1b (red) shows the recombinational robustness of the 1 000 donor–recipient pairs as a function of the number n of exchanged reactions. We also wanted to compare this recombinational robustness with our model's equivalent of mutational robustness, i.e. robustness to a comparable amount of random additions and deletions of reactions from a metabolic network. Figure 1b shows that recombinational robustness is not lower than robustness to random reaction change, regardless of the number (n) of altered reactions. Again, an alternative approach to generating metabolic network pairs with a fixed D = 100, or an alternative primary carbon source yield similar observations (see Material and methods; electronic supplementary material, figures S3d–f). We also note that innovative offspring grow even faster than parental metabolic networks on the parental carbon source (electronic supplementary material, figure S6).
Therefore, recombination can cause more innovation than random change, but it does not incur higher costs.
(b). Superessential reactions play an important role in metabolic innovation
To understand the specific reaction changes associated with metabolic innovation, we next analysed all 8 171 recombinant offspring with metabolic innovations that our analysis had identified. In most of them, only one of the multiple reactions added in a recombination event was responsible for gaining viability on a novel carbon source. For example, among three recombinant offspring that had independently gained viability on acetate, all three had gained the phosphoglycerate kinase reaction. Likewise, the five recombinant offspring that had independently gained viability on pyruvate achieved this gain through addition of the ribulose 5-phosphate 3-epimerase reaction in the pentose phosphate pathway. More generally in 98.91% of innovative offspring, a single reaction accounted for the innovation. An example of the 89 instances (1.09%) where multiple reaction additions are responsible for an innovation is the newly acquired viability on the carbon source trehalose. It was caused by the simultaneous addition of reactions catalysed by trehalose 6-phosphate phosphorylase and 2-dehydro-3-deoxy-phosphogluconate aldolase.
The reactions that cause viability on new carbon sources come from a relatively small subset of the ‘universe’ of 5 906 reactions (see Material and methods). Specifically, only 19 reactions among the 5 906 reactions are responsible for gaining viability on new carbon sources in the majority (53%, 4 430) of the 8 171 innovative offspring. The remaining 47% of innovations are caused by only 147 other reactions. What is more, these reactions tend to share a property that we refer to as their superessentiality [45]. The superessentiality index (ISE) of a metabolic reaction denotes the fraction of metabolic networks in which this reaction is essential for viability on carbon source C [45]. It can be computed from randomly sampled metabolic networks viable on that carbon source. The greater a reaction's ISE, the larger is also the number of bacterial genomes encoding this reaction [45]. Reactions with ISE > 0.5 are essential for viability in the majority of metabolic networks where they occur.
Reactions that cause viability on a new carbon source tend to have a higher ISE than those which rarely or never cause viability on new carbon sources (electronic supplementary material, figure S7a). Examples include the ribulose-5-phosphate 3-epimerase reaction, which causes viability on new carbon sources in 731 innovative offspring, and has a superessentiality index of ISE = 0.9714. They also include ribose-5-phosphate isomerase (ISE = 0.9530), which causes metabolic innovation in 677 offspring. More generally, we observed (i) a significant correlation between the ISE and the number of innovations a reaction causes (electronic supplementary material, figure S7b) and (ii) that the fraction of innovative offspring (finnov) of metabolic network pairs increases with fsuper, i.e. with the fraction of reactions with ISE > 0.5 (electronic supplementary material, figure S7c; Pearson's r = 0.18, p < 10−8).
(c). Genotypically more similar parental metabolic networks are more likely to generate metabolically innovative offspring
Thus far, we have examined the effects of recombination on metabolic innovation and robustness among parental metabolic networks with a fixed genotypic distance D. This distance, however, could have an influence on the effect of recombination. For example, recombination among more distant parents could lead to a smaller incidence of metabolic innovation, because fewer reactions ‘imported’ from a distant metabolic network may integrate productively into the resident metabolic network. To find out whether this is the case, we varied the distance among recombining parents between D = 100 and D = 1 500. We did not examine greater distances, because according to available data on the rate of successful recombination among prokaryotic species, this rate becomes negligible at such large distances (electronic supplementary material, text S4). However, as a reference point for including parents with the maximal genotype distance (Dmax), we analysed random metabolic changes, where new reactions are added not from another parental metabolic network but from the (maximally diverse) reaction universe. Specifically, for each value of D, we created 1 000 random pairs of parental metabolic networks, and from each pair we formed 1 000 recombinant offspring by recombining a fixed number n of randomly chosen reactions (see Material and methods).
Figure 2a shows that for any given number n of recombining reactions, the fraction of innovative offspring (finnov) decreases with increasing parental genotype distance. In other words, the more distant two recombining parents are, the smaller the likelihood that their offspring can survive on novel carbon sources. Although this relationship could be the result of an increasing fraction of inviable offspring when distantly related parents recombine, electronic supplementary material, figure S8 shows that this is not the case. These observations still hold if we require parental metabolic networks to be viable on acetate instead of glucose (electronic supplementary material, figures S9a,b).
Figure 2.

Effect of parental genotypic and phenotypic features on the incidence of innovative offspring. The vertical axes show mean (bar) and standard deviation (vertical line) of the fraction of innovative offspring (finnov), generated by recombination between parental metabolic networks with (a) genotypic distance (D), (c) phenotypic distance (ΔP), and (e) phenotypic complexity (||P||), where D, ΔP, and ||P|| are colour-coded according to their corresponding legend. The horizontal axis shows the number of recombined reactions (n). In (b), (d), and (f), the vertical axes show the fraction of reactions with a superessentiality index exceeding 0.5 (fsuper, x-axis) among reactions that can potentially be transferred from the parental donor to the recipient metabolic network. The horizontal axes show (b) genotypic distance (D), (d) phenotypic distance (ΔP), and (f) phenotypic complexity (||P||). Boxes span the 25%–75% percentile, and whiskers indicate maxima and minima.
We also examined how the fraction of reactions with ISE > 0.5 (fsuper) that can potentially be transferred from the donor to the recipient metabolic network changes with genotypic distance. Both the median and the variance of fsuper decreases with increasing D (figure 2b; electronic supplementary material, figure S9c). This observation further supports the importance of highly superessential reactions for metabolic innovation.
Moreover, we showed that parental metabolic networks with higher phenotypic diversity have greater potential to create innovative offspring (electronic supplementary material, texts S5 and S6 and figures S10 and S11; figure 2c,d). In addition, we also found that parental metabolic networks with more reactions (higher genotypic complexity), and those viable on fewer carbon sources (lower phenotypic complexity) are more likely to generate innovative offspring (electronic supplementary material, texts S7 and S8, S12–S14; figure 2e,f).
(d). Recombination in prokaryotic metabolic networks has similar innovation potential as in randomly sampled metabolic networks
While sampling viable metabolic networks from a metabolic genotype space permitted us to control parameters such as phenotypic and genotypic diversity, this analysis also has limitations. For example, it neglects the potential influence of gene linkage on metabolic innovation by recombination because it is based on the exchange of biochemical reactions rather than genes. We thus wanted to validate our observations with genome-scale metabolic networks of prokaryotic organisms. To this end, we used genome-scale metabolic networks of 61 bacterial species from the BiGG database [50], which differ in both their genotypes and phenotypes, i.e. their viability on 137 different carbon sources (see Material and methods). We first determined the innovation potential of each of the 3 660 possible pairs that can be formed from these metabolic networks, by asking whether the union of a pair's reaction sets confers viability on a carbon source on which neither member of the pair is viable. This was the case for 1 126 pairs (30.77%).
For each of these 1 126 metabolic network pairs, we generated 1 000 recombinant offspring through a procedure we refer to as ‘linkage-based’ recombination, which maps reactions onto genes, and recombines random stretches of DNA whose length is chosen such that a given number of n reactions is altered in the offspring's metabolic network (see Material and methods). For the purpose of comparison, we also used a complementary ‘free recombination’ procedure, which disregards linkage and creates recombinant offspring by altering a given number n of reactions in the recipient, just as we had done for randomly sampled viable networks. When analysing robustness to recombination, we found that linkage-based recombination is much more likely to preserve viability than free recombination (figures 3a; electronic supplementary material, figure S15a). In other words, the linear organization of metabolic genes in the genome facilitates robustness to recombination.
Figure 3.
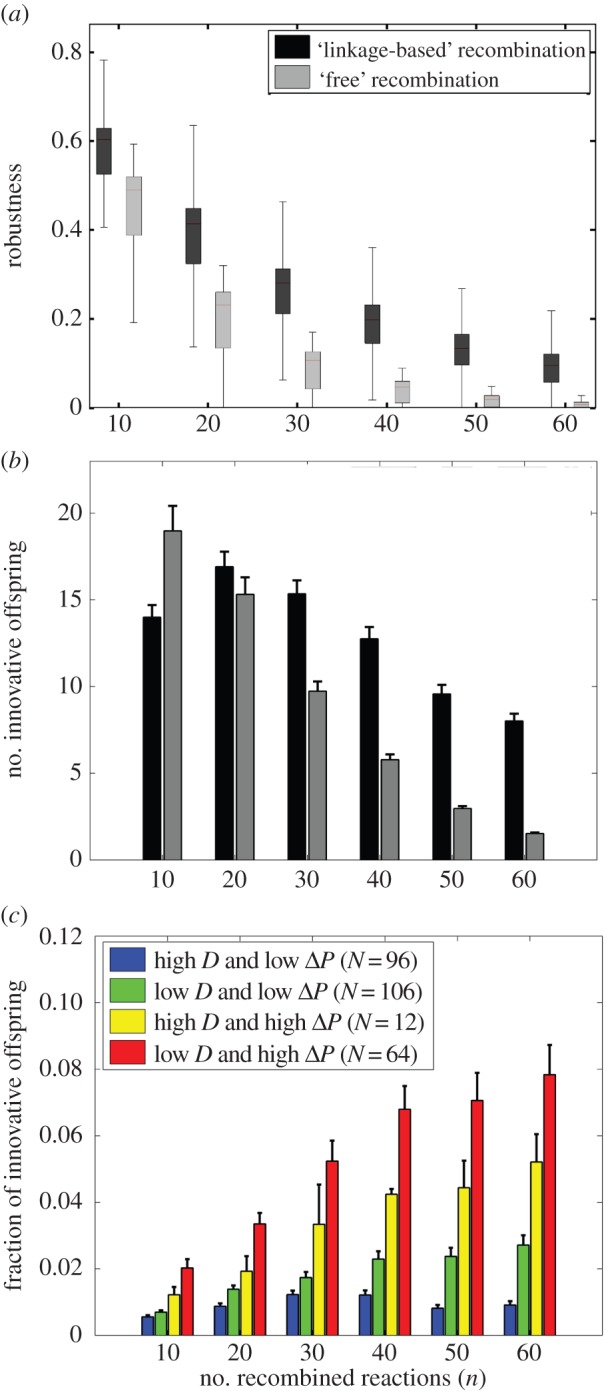
Recombination between prokaryotic metabolic networks shows the same innovation potential as in randomly sampled metabolic networks. (a) Robustness to recombination, i.e. the fraction of recombinant offspring retaining viability on at least one parental carbon source, and (b) mean (bar) and standard error (vertical line) of the number of innovative offspring among 1 000 recombinant offspring (y-axis), as a function of the number of recombined reactions (x-axis). Offspring were generated by (i) linkage-based recombination between prokaryotic metabolic networks (black), and (ii) free recombination between prokaryotic metabolic networks (grey). (c) The vertical axis shows the mean (bar) and standard error (vertical line) of the fraction of innovative offspring (finnov) generated by linkage-based recombination between prokaryotic parental metabolic networks with (i) high genotypic distance (D > 40), low phenotypic distance (ΔP < 30) and high phenotypic complexity (||P|| > 60) (blue, N = 96 parental pairs), (ii) low genotypic distance (D < 30), low phenotypic distance (ΔP < 30) and high phenotypic complexity (||P|| > 60) (green, N = 106 parental pairs), (iii) high genotypic distance (D > 40), high phenotypic distance (ΔP > 40) and low phenotypic complexity (||P||<40) (yellow, N = 12 parental pairs), and (iv) low genotypic distance (D < 30), high phenotypic distance (ΔP > 40) and low phenotypic complexity (||P||<40) (red, N = 64 parental pairs).
The fraction of innovative offspring is somewhat lower under linkage-based recombination (electronic supplementary material, figure S15b). However, a higher overall fraction of viable offspring (figure 3a) results in a substantially higher total number of innovative offspring under linkage-based recombination than free recombination, (especially for higher numbers of recombined reactions (n), (figure 3b)). Therefore, higher robustness to recombination in prokaryotic metabolic networks results in a higher potential for metabolic innovation.
In a majority of the 1 126 prokaryotic metabolic network pairs where metabolic innovation is possible (854 of 1 126, or 75.84%), the addition of a single reaction from the donor to the recipient was sufficient to gain viability on a new carbon source, just as we had observed for randomly sampled metabolic networks. Also, only a small fraction (106) of the 3 404 reactions that occurred in these 61 metabolic networks caused metabolic innovation. And just as in randomly sampled metabolic networks, reactions that cause innovation are more often essential (higher ISE [45]) than those that do not cause innovation (electronic supplementary material, figure S15c). In addition, the fraction of innovative offspring (finnov) increases with the fraction of reactions with superessentiality index ISE larger than 0.5 (electronic supplementary material, figure S15d; Pearson's r = 0.13, p < 10−5).
Finally, we showed that parental prokaryotic metabolic networks with low genotypic distance, high phenotypic distance, and low phenotypic complexity are more likely to generate innovative offspring (figure 3c, and electronic supplementary material, text S9 and figures S16 and S17a), just as they did in randomly sampled viable metabolic networks. Moreover, genotypic distance, phenotypic complexity, and phenotypic diversity do not strongly influence recombinational robustness, just as they did not for randomly sampled metabolic networks (electronic supplementary material, text S9 and figures S16, S17b,c).
In sum, our observations show that recombination in prokaryotic metabolic networks resembles those in randomly sampled metabolic networks in its innovation potential, and in the mechanisms by which it causes metabolic innovation.
3. Discussion
Recombination is a major force behind many innovations in biological systems [12–14,53,54]. Here, we studied its innovation potential in metabolic systems, where innovations enable organisms to survive on novel sources of energy and chemical elements. To do so, we computationally recombined biochemical reactions among thousands of metabolic network pairs that are viable on specific carbon sources. We sampled most of these from a vast space of such networks with a MCMC technique, but also validated our observations by analysing 61 prokaryotic metabolic networks.
We found that recombination provides greater benefit—a greater number of new metabolic abilities—at no greater cost in terms of viability loss than an equivalent amount of random mutation, modelled as random alterations in a metabolic network's complement of chemical reactions. Metabolic innovation is more likely when recombination occurs between parents that are genetically closely related, phenotypically highly diverse, and viable on few rather than many carbon sources. Survival on a new carbon source preferentially involves reactions that are highly superessential, that is, they are required in most metabolic networks viable on this carbon source.
One well-studied facilitator of evolutionary adaptation and innovation is the robustness of a biological system to genetic change [55,56]. Robustness can facilitate a population's exploration of a genotype space, and thus accelerate the origin of novel phenotypes [57]. Our observations support this positive role of robustness. Specifically, the frequently higher robustness of prokaryotic metabolic networks under linkage-based recombination results in a higher total number of innovative offspring (figure 3a,b). Moreover, larger metabolic networks are more robust to recombination, and they are more likely to create metabolic innovations (electronic supplementary material, figure S14). We also showed, however, that robustness is not the only factor affecting innovation by recombination. For example, parental genotypic distance, phenotypic diversity and complexity impact innovation without influencing robustness, so they modulate the incidence of innovation independently of robustness. The ability to study these factors in isolation is a key advantage of our computational approach.
Systemic properties like robustness and parental diversity are not the only factors influencing innovation by recombination. One property of individual reactions—superessentiality—is at least as important. Multiple reactions may be transferred in a recombination event, but in the vast majority of such events, only the addition of a single reaction causes innovation, and this reaction is often highly superessential. What is more, reaction superessentiality can help explain multiple systemic patterns in our data, e.g. that phenotypically diverse parents have greater innovation potential (electronic supplementary material, text S10 and figure S18). That being said, exceptions to the importance of superessentiality exist, where innovations are caused by reactions that are rarely essential. An extreme example is adenyl cyclase, which catalyses the conversion of adenosine triphosphate (ATP) to 3′,5′-cyclic adenosine monophosphate (AMP) and diphosphate. It is not essential for viability on any of the 10 000 randomly sampled metabolic networks (ISE = 0), yet it is responsible for metabolic innovation in 10 out of 81 71 innovative offspring. Relatedly, we found three examples of reactions that were blocked (i.e. inactive, with zero flux) in the donor metabolic network that caused innovation after being added to a recipient metabolic network. However, the innovation potential of such inactive reactions is small compared with active or highly superessential reactions (electronic supplementary material, figure S2).
Our approach of using randomly sampled viable metabolic networks has several advantages, most notably that we can arrive at general conclusions that go beyond any one organism, and that we can control important quantities such as parental genotypic diversity. However, it also has several limitations. First, our computational analysis is based on FBA, which neglects regulatory constraints that can arise through suboptimal enzyme expression. However, as discussed in more detail in the electronic supplementary material, text S1d, this limitation is not likely to affect our main observations.
Second, our approach ignores the linkage of related metabolic genes on chromosomes, for example, in operons [58]. Although on long evolutionary time scales operons often break up and reform [59,60], functionally related genes tend to be linked. Randomly sampled metabolic networks may contain combinations of reactions that are not found in any known organism, so that we cannot meaningfully assign linkage patterns to them. Third, our specification of a metabolic genotype represents this genotype on the level of the reaction rather than that of a gene and considers only the presence or absence of metabolic reactions. Although widely used [15–18,21–25], this representation neglects potentially important pieces of information, among them a myriad of mechanistic details of DNA recombination. Perhaps even more importantly, it also neglects that some reactions are catalysed by multiple enzymes [61], and that some enzymes catalyse multiple reactions [62–64].
We were able to mitigate the last two limitations by comparing our observations with those obtained from 61 curated prokaryotic metabolic networks, where gene–reaction maps and linkage information is available for metabolic genes. The incidence of metabolic innovation among hundreds of pairs of these prokaryotic metabolic networks shows the same patterns as our randomly sampled viable metabolic networks. In addition, these metabolic networks revealed an additional intriguing pattern, their increased metabolic robustness to recombination. Essential genes in general are known to be clustered in prokaryotic genomes [65,66], which may increase a genome's robustness to large-scale gene deletions. However, the evolutionary forces shaping the organization of metabolic genes are not well known, and call for further research.
Recombination and DNA mutations, such as point mutations and gene duplications, play complementary roles in creating metabolic innovation. In an evolving population, recombination cannot be effective unless mutation has created diversity beforehand. Mutations introduce novel parts (enzymes, reactions), and recombination creates novel combinations of these parts (metabolic pathways). Our results demonstrate the power of this combinatorial principle for metabolic innovation. One source of this power is that recombination shuffles system parts that have been ‘pre-tested’ in evolution, because they form part of a viable metabolic network. This is also why superessential reactions are important for innovation, and why robustness facilitates innovation.
Supplementary Material
Data accessibility
This work uses only publicly accessible data (see Material and methods section).
Authors' contributions
S.-R.H. and A.W. designed the study and wrote the manuscript. S.-R.H. and O.C.M. provided computational tools and suggested algorithms. S.-R.H. performed the simulations and computational analyses. S.-R.H. and A.W. analysed and interpreted the results. All authors read and approved the final manuscript.
Competing interests
We declare that we have no competing interests.
Funding
We acknowledge support through Swiss National Science Foundation grant no. 31003A_146137, as well as through the University Priority Research Program in Evolutionary Biology at the University of Zurich.
References
- 1.Smith JM. 1978. The evolution of sex. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 2.Otto SP, Lenormand T. 2002. Resolving the paradox of sex and recombination. Nat. Rev. Genet. 3, 252–261. ( 10.1038/nrg761) [DOI] [PubMed] [Google Scholar]
- 3.Hartfield M, Keightley PD. 2012. Current hypotheses for the evolution of sex and recombination. Integr. Zool. 7, 192–209. ( 10.1111/j.1749-4877.2012.00284.x) [DOI] [PubMed] [Google Scholar]
- 4.Fisher RA. 1930. The genetical theory of natural selection: a complete variorum edition. Oxford, UK: Oxford University Press. [Google Scholar]
- 5.Muller HJ. 1932. Some genetic aspects of sex. Am. Nat. 66, 118–138. ( 10.1086/280418) [DOI] [Google Scholar]
- 6.Keightley PD, Otto SP. 2006. Interference among deleterious mutations favours sex and recombination in finite populations. Nature 443, 89–92. ( 10.1038/nature05049) [DOI] [PubMed] [Google Scholar]
- 7.Stemmer WP. 1994. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc. Natl Acad. Sci. USA 91, 10 747–10 751. ( 10.1073/pnas.91.22.10747) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y-X, Perry K, Vinci VA, Powell K, Stemmer WPC, del Cardayré SB. 2002. Genome shuffling leads to rapid phenotypic improvement in bacteria. Nature 415, 644–646. ( 10.1038/415644a) [DOI] [PubMed] [Google Scholar]
- 9.Crameri A, Dawes G, Rodriguez E, Silver S, Stemmer WP. 1997. Molecular evolution of an arsenate detoxification pathway by DNA shuffling. Nat. Biotechnol. 15, 436–438. ( 10.1038/nbt0597-436) [DOI] [PubMed] [Google Scholar]
- 10.Chang CC, Chen TT, Cox BW, Dawes GN, Stemmer WP, Punnonen J, Patten PA. 1999. Evolution of a cytokine using DNA family shuffling. Nat. Biotechnol. 17, 793–797. ( 10.1038/70147) [DOI] [PubMed] [Google Scholar]
- 11.Ness JE, Welch M, Giver L, Bueno M, Cherry JR, Borchert TV, Stemmer WP, Minshull J. 1999. DNA shuffling of subgenomic sequences of subtilisin. Nat. Biotechnol. 17, 893–896. ( 10.1038/12884) [DOI] [PubMed] [Google Scholar]
- 12.Martin OC, Wagner A. 2009. Effects of recombination on complex regulatory circuits. Genetics 183, 673–684, 1SI–8SI ( 10.1534/genetics.109.104174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui Y, Wong WH, Bornberg-Bauer E, Chan HS. 2002. Recombinatoric exploration of novel folded structures: a heteropolymer-based model of protein evolutionary landscapes. Proc. Natl Acad. Sci. USA 99, 809–814. ( 10.1073/pnas.022240299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drummond DA, Silberg JJ, Meyer MM, Wilke CO, Arnold FH. 2005. On the conservative nature of intragenic recombination. Proc. Natl Acad. Sci. USA 102, 5380–5385. ( 10.1073/pnas.0500729102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feist AM, Palsson BØ. 2008. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat. Biotechnol. 26, 659–667. ( 10.1038/nbt1401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCloskey D, Palsson BØ, Feist AM. 2013. Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol. Syst. Biol. 9, 661 ( 10.1038/msb.2013.18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis NE, Nagarajan H, Palsson BO. 2012. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 10, 291–305. ( 10.1038/nrmicro2737) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matias Rodrigues JF, Wagner A. 2009. Evolutionary plasticity and innovations in complex metabolic reaction networks. PLoS Comput. Biol. 5, e1000613 ( 10.1371/journal.pcbi.1000613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orth JD, Thiele I, Palsson BØ. 2010. What is flux balance analysis? Nat. Biotechnol. 28, 245–248. ( 10.1038/nbt.1614) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards JS, Ibarra RU, Palsson BO. 2001. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19, 125–130. ( 10.1038/84379) [DOI] [PubMed] [Google Scholar]
- 21.Edwards JS, Palsson BO. 2000. The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc. Natl Acad. Sci. USA 97, 5528–5533. ( 10.1073/pnas.97.10.5528) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards JS, Palsson BO. 1999. Systems properties of the Haemophilus influenzae Rd metabolic genotype. J. Biol. Chem. 274, 17 410–17 416. ( 10.1074/jbc.274.25.17410) [DOI] [PubMed] [Google Scholar]
- 23.Samal A, Matias Rodrigues JF, Jost J, Martin OC, Wagner A. 2010. Genotype networks in metabolic reaction spaces. BMC Syst. Biol. 4, 30 ( 10.1186/1752-0509-4-30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barve A, Hosseini S-R, Martin OC, Wagner A. 2014. Historical contingency and the gradual evolution of metabolic properties in central carbon and genome-scale metabolisms. BMC Syst. Biol. 8, 48 ( 10.1186/1752-0509-8-48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosseini SR, Barve A, Wagner A. 2015. Exhaustive analysis of a genotype space comprising 1015 central carbon metabolisms reveals an organization conducive to metabolic innovation. PLoS Comput. Biol. 11, e1004329 ( 10.1371/journal.pcbi.1004329) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Copley SD. 2000. Evolution of a metabolic pathway for degradation of a toxic xenobiotic: the patchwork approach. Trends Biochem. Sci. 25, 261–265. ( 10.1016/S0968-0004(00)01562-0) [DOI] [PubMed] [Google Scholar]
- 27.Rehmann L, Daugulis AJ. 2008. Enhancement of PCB degradation by Burkholderia xenovorans LB400 in biphasic systems by manipulating culture conditions. Biotechnol. Bioeng. 99, 521–528. ( 10.1002/bit.21610) [DOI] [PubMed] [Google Scholar]
- 28.Van der Meer JR, Werlen C, Nishino S, Spain J. 1998. Evolution of a pathway for chlorobenzene metabolism leads to natural attenuation in contaminated groundwater. Appl. Environ. Microbiol. 64, 4185–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cline RE, Hill RH, Phillips DL, Needham LL. 1989. Pentachlorophenol measurements in body fluids of people in log homes and workplaces. Arch. Environ. Contam. Toxicol. 18, 475–481. ( 10.1007/BF01055012) [DOI] [PubMed] [Google Scholar]
- 30.Dantas G, Sommer MOA, Oluwasegun RD, Church GM. 2008. Bacteria subsisting on antibiotics. Science 320, 100–103. ( 10.1126/science.1155157) [DOI] [PubMed] [Google Scholar]
- 31.Detkova EN, Boltyanskaya YV. 2007. Osmoadaptation of haloalkaliphilic bacteria: role of osmoregulators and their possible practical application. Microbiology 76, 511–522. ( 10.1134/S0026261707050013) [DOI] [PubMed] [Google Scholar]
- 32.Thomas CM, Nielsen KM. 2005. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721. ( 10.1038/nrmicro1234) [DOI] [PubMed] [Google Scholar]
- 33.Guttman DS, Dykhuizen DE. 1994. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science 266, 1380–1383. ( 10.1126/science.7973728) [DOI] [PubMed] [Google Scholar]
- 34.Feil EJ, et al. 2001. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc. Natl Acad. Sci. USA 98, 182–187. ( 10.1073/pnas.98.1.182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitaker RJ, Grogan DW, Taylor JW. 2005. Recombination shapes the natural population structure of the hyperthermophilic archaeon Sulfolobus islandicus. Mol. Biol. Evol. 22, 2354–2361. ( 10.1093/molbev/msi233) [DOI] [PubMed] [Google Scholar]
- 36.Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. ( 10.1038/35012500) [DOI] [PubMed] [Google Scholar]
- 37.Pál C, Papp B, Lercher MJ. 2005. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 37, 1372–1375. ( 10.1038/ng1686) [DOI] [PubMed] [Google Scholar]
- 38.Fraser C, Hanage WP, Spratt BG. 2007. Recombination and the nature of bacterial speciation. Science 315, 476–480. ( 10.1126/science.1127573) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majewski J, Zawadzki P, Pickerill P, Cohan FM, Dowson CG. 2000. Barriers to genetic exchange between bacterial species: Streptococcus pneumoniae transformation. J. Bacteriol. 182, 1016–1023. ( 10.1128/JB.182.4.1016-1023.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 58, 401–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo C-H, Ochman H. 2009. The fate of new bacterial genes. FEMS Microbiol. Rev. 33, 38–43. ( 10.1111/j.1574-6976.2008.00140.x) [DOI] [PubMed] [Google Scholar]
- 42.Mira A, Ochman H, Moran NA. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17, 589–596. ( 10.1016/S0168-9525(01)02447-7) [DOI] [PubMed] [Google Scholar]
- 43.Matias Rodrigues JF, Wagner A. 2011. Genotype networks, innovation, and robustness in sulfur metabolism. BMC Syst. Biol. 5, 39 ( 10.1186/1752-0509-5-39) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagner A, Andriasyan V, Barve A. 2014. The organization of metabolic genotype space facilitates adaptive evolution in nitrogen metabolism. J. Mol. Biochem. 3, 2–13. ( 10.5167/uzh-107400) [DOI] [Google Scholar]
- 45.Barve A, Rodrigues JFM, Wagner A. 2012. Superessential reactions in metabolic networks. Proc. Natl Acad. Sci. USA 109, E1121–E1130. ( 10.1073/pnas.1113065109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goto S, Nishioka T, Kanehisa M. 2000. LIGAND: chemical database of enzyme reactions. Nucleic Acids Res. 28, 380–382. ( 10.1093/nar/28.1.380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. 2010. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 38, D355–D360. ( 10.1093/nar/gkp896) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. 2006. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–D357. ( 10.1093/nar/gkj102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BØ. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3, 121 ( 10.1038/msb4100155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.King ZA, Lu J, Dräger A, Miller P, Federowicz S, Lerman JA, Ebrahim A, Palsson BO, Lewis NE. 2015. BiGG models: a platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 44, D515–D522. ( 10.1093/nar/gkv1049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gelius-Dietrich G, Desouki AA, Fritzemeier CJ, Lercher MJ. 2013. Sybil-efficient constraint-based modelling in R. BMC Syst. Biol. 7, 125 ( 10.1186/1752-0509-7-125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. 2014. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 42, D553–D559. ( 10.1093/nar/gkt1274) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trudeau DL, Smith MA, Arnold FH. 2013. Innovation by homologous recombination. Curr. Opin. Chem. Biol. 17, 902–909. ( 10.1016/j.cbpa.2013.10.007) [DOI] [PubMed] [Google Scholar]
- 54.Apic G, Russell RB. 2010. Domain recombination: a workhorse for evolutionary innovation. Sci. Signal. 3, e30. ( 10.1126/scisignal.3139pe30) [DOI] [PubMed] [Google Scholar]
- 55.Wagner A. 2012. The role of robustness in phenotypic adaptation and innovation. Proc. R. Soc. B 279, 1249–1258. ( 10.1098/rspb.2011.2293) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fares MA. 2015. Survival and innovation: the role of mutational robustness in evolution. Biochimie 119, 254–261. ( 10.1016/j.biochi.2014.10.019) [DOI] [PubMed] [Google Scholar]
- 57.Wagner A. 2011. The molecular origins of evolutionary innovations. Trends Genet. 27, 397–410. ( 10.1016/j.tig.2011.06.002) [DOI] [PubMed] [Google Scholar]
- 58.Jacob F, Monod J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3, 318–356. ( 10.1016/S0022-2836(61)80072-7) [DOI] [PubMed] [Google Scholar]
- 59.Itoh T, Takemoto K, Mori H, Gojobori T. 1999. Evolutionary instability of operon structures disclosed by sequence comparisons of complete microbial genomes. Mol. Biol. Evol. 16, 332–346. ( 10.1093/oxfordjournals.molbev.a026114) [DOI] [PubMed] [Google Scholar]
- 60.Price MN, Arkin AP, Alm EJ. 2006. The life-cycle of operons. PLoS Genet. 2, e96 ( 10.1371/journal.pgen.0020096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hunter RL, Markert CL. 1957. Histochemical demonstration of enzymes separated by zone electrophoresis in starch gels. Science 125, 1294–1295. ( 10.1126/science.125.3261.1294-a) [DOI] [PubMed] [Google Scholar]
- 62.Khersonsky O, Tawfik DS. 2010. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 79, 471–505. ( 10.1146/annurev-biochem-030409-143718) [DOI] [PubMed] [Google Scholar]
- 63.Kim J, Kershner JP, Novikov Y, Shoemaker RK, Copley SD. 2010. Three serendipitous pathways in E. coli can bypass a block in pyridoxal-5′-phosphate synthesis. Mol. Syst. Biol. 6, 436 ( 10.1038/msb.2010.88) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nam H, Lewis NE, Lerman JA, Lee D-H, Chang RL, Kim D, Palsson BO. 2012. Network context and selection in the evolution to enzyme specificity. Science 337, 1101–1104. ( 10.1126/science.1216861) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fang G, Rocha EPC, Danchin A. 2008. Persistence drives gene clustering in bacterial genomes. BMC Genomics 9, 4 ( 10.1186/1471-2164-9-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fang G, Rocha E, Danchin A. 2005. How essential are nonessential genes? Mol. Biol. Evol. 22, 2147–2156. ( 10.1093/molbev/msi211) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This work uses only publicly accessible data (see Material and methods section).