

Full and partial deletion of the DEK1 Linker implicate DEK1 in the three major developmental phases of the moss Physcomitrella patens.
Abstract
The DEFECTIVE KERNEL1 (DEK1) calpain is a conserved 240-kD key regulator of three-dimensional body patterning in land plants acting via mitotic cell plane positioning. The activity of the cytosolic C-terminal calpain protease is regulated by the membrane-anchored DEK1 MEM, which is connected to the calpain via the 600-amino acid residue Linker. Similar to the calpain and MEM domains, the Linker is highly conserved in the land plant lineage, the similarity dropping sharply compared with orthologous charophyte sequences. Using site-directed mutagenesis, we studied the effect on Physcomitrella patens development by deleting the Linker and two conserved Linker motifs. The results show that removal of the Linker has nearly the same effect as removal of the entire DEK1 gene. In contrast, deletion of the conserved Laminin_G3 (LG3) domain had a milder effect, perturbing leafy gametophore patterning and archegonia development. The LG3 domain from Marchantia polymorpha is fully functional in P. patens, whereas angiosperm sequences are not functional. Deletion of a C-terminal Linker subsegment containing a potential calpain autolytic site severely disturbs gametophore development. Finally, changing one of the three calpain active-site amino acid residues results in the same phenotype as deleting the entire DEK1 gene. Based on the conserved nature of animal and DEK1 calpains, we propose that the DEK1 MEM-Linker complex inactivates the calpain by forcing apart the two calpain subunits carrying the three amino acids of the active site.
The three-dimensional (3D) architecture of land plant bodies is determined by the orientation of cell walls deposited between dividing nuclei in developing organs. The ability to determine cell wall orientation in various planes was a novel feature that evolved in the transition from filamentous charophyte algae to the first land plants (Pires and Dolan, 2012). In contrast to land plants, tip expansion and fixed division planes of the cells drove a two-dimensional (2D) growth pattern of charophyte members represented by filamentous or discoid forms. The membrane-anchored DEFECTIVE KERNEL1 (DEK1) calpain is a candidate protein involved in position-dependent cell wall deposition in the plant lineage (Perroud et al., 2014). Evidence for a conserved function in land plants comes from the observation that DEK1 is essential for cell wall orientation in land plants ranging from angiosperms to mosses, an evolutionary time span of 450 million years (Olsen et al., 2015). Support for a central function of DEK1 also comes from a recent review in which analyses of meristematic shoot tip transcriptomes of widely divergent extant vascular plant lineage members suggest that different genes were recruited to regulate similar meristematic processes during evolution. Exceptions identified include PIN-FORMED, DEK1, and LONELY GUY1 (Frank et al., 2015) proteins that represent potential genetic homologs in meristem function (Harrison, 2015).
The DEK1 calpain derives its name from the maize (Zea mays) mutant dek1, which lacks aleurone cells in the endosperm (Lid et al., 2002). The DEK1 protease core domain, CysPc, and the additional C-terminal C2L domain are connected to DEK1 MEM, a 23-transmembrane (TM) regulatory domain interrupted by a Loop segment via a Linker (previously referred to as Arm). Phylogenetic analysis concluded that four ancestral calpain architectures existed approximately 1.5 billion years ago (Zhao et al., 2012). Of these, TML-calpains, to which DEK1 belongs, consisted of a TM domain with at least 15 TM segments linked to the CysPc-C2L domains. The common ancestor of the Chlorophyceae and Charophyceae algae is inferred to have contained both cytosolic and TML-calpains (Demko et al., 2014). The Chlorophyceae group appears to have lost TML-calpains, whereas the Charophyceae algae, the closest ancestors to land plants, retained both cytosolic and TML-calpain variants. In the transition to land plants, cytosolic calpains were lost, leaving DEK1 as the single calpain of land plants (Demko et al., 2014). We have hypothesized that DEK1 evolved a novel function in the development of the 3D architecture of land plants during the transition from charophyte land plant ancestors to land plants (Demko et al., 2014). Our working hypothesis is that the calpain activity of DEK1 is regulated by the MEM segment via an unknown ligand or stimulus or the lack of such (release of inhibition) and that the Linker transmits the signaling from MEM, thereby activating the catalytic calpain domain. The substrate(s) of the DEK1 calpain is unknown.
We are pursuing systematic genetic studies of the function and evolutionary conservation of DEK1 and its component domains (Fig. 1). In the first set of experiments, we focused on CysPc-C2L by exploiting the observation that the Arabidopsis (Arabidopsis thaliana) dek1-3 mutant phenotype is complemented by Arabidopsis CysPc-C2L under the control of the RIBOSOMAL PROTEIN5A promoter (Johnson et al., 2008). Our results support the conserved nature of DEK1 function in these experiments; the Physcomitrella patens DEK1 CysPc-C2L domains fully complement the Arabidopsis dek1-3 mutant phenotype (Liang et al., 2013). The CysPc domain of conventional mammalian calpains (CAPN1 and CAPN2, also called µCL/calpain-1 and mCL/calpain-2, respectively) consists of two subdomains, PC1 and PC2, which are connected via a hinge region; PC1 carries the active-site residue Cys, whereas PC2 carries the two remaining active-site residues His and Asn (Strobl et al., 2000). The two subdomains also each harbor a conserved Ca2+-binding site (Moldoveanu et al., 2002). Regulation of the CysPc domain of CAPN1 occurs via an electrostatic switch mechanism operated via Ca2+ binding, causing the two CysPc subdomains to swing into position around the hinge, thus aligning the three active-site amino acid residues into an active configuration (Moldoveanu et al., 2002). Additional levels of regulation of conventional calpains also exist that depend on structures that are absent from DEK1. One example is calpastatin, a multiheaded inhibitor that binds to the catalytic CysPc domain, blocking access to the active site (Wendt et al., 2004). Three conserved features between DEK1 and animal CysPc suggest a similar activation mechanism in plants and animals. First, the three active-site residues of the CysPc domain, as well as their configuration with Cys located on PC1 and His and Asn located on PC2, are conserved in DEK1 (Liang et al., 2013). Substituting the active-site residue Cys with the amino acid Ser inactivates the DEK1 calpain in vivo (Johnson et al., 2008) and in vitro (Wang et al., 2003). Second, residues corresponding to the hinge also are conserved in the DEK1 CysPc domain (Wang et al., 2003; Liang et al., 2013), suggesting that a similar swinging motion may be involved in the activation of DEK1 calpain. Third, the PC1 and PC2 amino acids involved in Ca2+ binding in conventional mammalian CysPc domains (Moldoveanu et al., 2002) are structurally and functional conserved in DEK1 (Liang et al., 2013). DEK1 CysPc harboring substitution of these amino acids to Ala fails to complement the Arabidopsis dek1-3 mutant phenotype (Liang et al., 2013), suggesting that DEK1 calpain activity may be Ca2+ dependent. This conclusion is potentially in conflict with the observation that the recombinant protein of maize CysPc-C2L displays a low, but significant, in vitro proteolytic activity in the presence of EDTA (Wang et al., 2003). Although sharing structural features, chimeric calpains (the CysPc domain of animals fused to the C2L domain of DEK1) fail to complement the Arabidopsis dek1-3 mutant, and coevolution analysis also identified differences in the interactions between the CysPc-C2L residues of DEK1 and classical calpains (Liang et al., 2013).
Figure 1.

P. patens DEK1 protein domain structure and DEK1 Linker sequence conservation. A, Schematic representation of DEK1 domain structure. DEK1 MEM is composed of two parts, with multiple TM segments disrupted by the Loop. The DEK1 Linker is divided into the three subsegments, N, LG3, and C. DEK1 calpain consists of CysPc, the catalytic domain, and C2L. Amino acid numbering is given above the DEK1 outline. B, Heat map showing the degree of DEK1 Linker sequence conservation among 73 land plant species. Percentage sequence identities are indicated in different colors, from blue (0%; insertions/deletions) to red (100%; absolute conserved positions). Amino acid numbering for the different subsegments of the P. patens DEK1 Linker is given above the top part. Percentages above the heat map give the sequence identities between the Arabidopsis and P. patens DEK1 Linker subsegments. C, Predicted structure of the P. patens DEK1 LG3 domain. I, Structural alignment of the predicted I-TASSER (blue), RaptorX (magnenta), and Phyre2 (green) structures showing that the LG3 domain adopts a β-sandwich fold and that individual servers predicted highly similar structures. II, Structural alignment between the Phyre2 model and the Protein Data Base (PDB) structure 3FLP (yellow) of native heptameric serum amyloid P component (SAP)-like pentraxin from Limulus polyphemus.
In the next series of studies of DEK1 function and evolution, we used the moss P. patens. Moss represents one of the, if not the, earliest extant relatives of the first land plants, permitting studies of ancestral DEK1 function. Second, mosses form a branched system of multicellular filaments called protonemata, a gametophyte stage in which DEK1 is nonessential (Perroud et al., 2014), allowing tissues to be cultivated and transformed, utilizing the potential of homologous recombination for site-directed mutagenesis and gene replacement. This is in contrast to other land plants, in which dek1 null mutations are lethal (Becraft et al., 2002; Lid et al., 2005), severely restricting the ability to study DEK1 function. Third, DEK1 studies in mosses are relevant for higher plants, since its role in cell wall orientation is conserved in land plants (Olsen et al., 2015). When P. patens spores germinate, the protonema, consisting of cells extending by tip growth and dividing in one plane, represent a 2D growth form reminiscent of the charophyte algae predecessors of land plants. Shortly thereafter, buds are initiated from the protonema, which develop into gametophores with phyllids (leaf-like structures) and rhizoids (rooting structures), representing 3D organ forms typical of land plants. At maturity, the gametophore produces archegonia and antheridia in a cluster at the apex of the stem, which, upon fertilization, develop into diploid sporophytes containing the spores (Prigge and Bezanilla, 2010). During its life cycle, P. patens develops several (eight) meristematic cells that promote growth in the gametophyte and sporophyte generations (Kofuji and Hasebe, 2014).
In our first approach using P. patens to study DEK1 function, we deleted the entire DEK1 gene (Perroud et al., 2014). In the resulting Δdek1 mutant, protonema growth appears normal, but the mutant displays an over-budding phenotype, producing four times more buds than the wild type. Of the four AP2-type transcription factors (APB1–APB4, for AINTEGUMENTA, PLETHORA, and BABY BOOM) shown to be indispensable for the formation of the bud initial cell in P. patens (Aoyama et al., 2012), APB2 and APB3 were found to be up-regulated in the Δdek1 mutant (Demko et al., 2014). In the Δdek1 mutant, abnormal cell divisions in the emerging bud result in developmental arrest and the absence of gametophores.
Next, we investigated the function of the DEK1 Loop, an approximately 300-amino acid segment interrupting DEK1 MEM, by creating the Loop deletion mutant dek1Δloop. The dek1Δloop mutant forms twice as many buds as the wild type (Demko et al., 2014). The division plane in the dek1Δloop bud apical cell is perpendicular, similar to that in the wild type, although with a slightly bent cell wall. In contrast to the Δdek1 mutant, the dek1Δloop bud continues to divide, producing a stem-like structure without emerging phyllids. Gametangia do not form. The liverwort DEK1 Loop segment, sharing approximately 43% amino acid identity with the P. patens DEK1 Loop, is fully functional in the moss. However, the DEK1 Loop sequences from Arabidopsis and maize only partly complement the dek1Δloop phenotype; the numbers of buds, and the positioning of the first division of the bud apical cell, occur as in the wild type. However, gametophores develop narrow phyllids and sporophyte is not formed (Demko et al., 2014).
In this study, we focus on the DEK1 Linker, which we propose functions to transmit the activation of DEK1 MEM by positional signaling into calpain activation. Here, we first show that the DEK1 Linker is highly conserved among land plants but divergent from orthologous charophyte algae sequences. Bioinformatic analyses identify functional elements, including a Laminin_G3 (LG3) domain and a potential autolysis site. To assess the importance of the Linker segment, the LG3 domain, and the putative autolysis site, we create partial P. patens DEK1 deletion mutants. We demonstrate the conserved nature of the LG3 domain between mosses and liverworts by genetic complementation assay. Finally, we show that mutating one of the three calpain active-site amino acid residues generates the Δdek1 phenotype. We use these data to discuss possible models for the regulation of DEK1 calpain activation.
RESULTS
The Land Plant DEK1 Linker Is Highly Conserved and Contains a Predicted β-Sandwich Fold and a Putative Autolytic Cleavage Site
The DEK1 Linker is approximately 600 amino acids long with a predicted molecular mass of 69 kD (Fig. 1A). Conserved domain database searches with DEK1 Linker as query identify a single domain, LG3, of approximately 165 amino acids, in the Linker segment at positions 1,443 to 1,610 in the P. patens DEK1 sequence. Based on the identification of this domain, we divide the Linker into the three subsegments N, LG3, and C (Fig. 1A). Sequence alignment of 73 land plant DEK1 Linker proteins and four orthologous sequences from charophyte algae (Supplemental Table S1; Supplemental Data S1) show that land plant DEK1 Linker sequences are highly conserved (Fig. 1B), with sequence identities ranging from 59% to 98% (Supplemental Table S2; Supplemental Data S2). In contrast, the identity to the orthologous charophyte sequences is moderate to low (less than 36%), and these also are highly divergent from each other (Supplemental Table S2). Overall, sequence conservation between, for example, Arabidopsis and P. patens is highest in the N segment (60% identity and 73% similarity), followed by the LG3 domain (55% identity and 68% similarity), and lowest in the C segment (34% identity and 60% similarity; Fig. 1B). In the Linker N segment, for which no conserved domain has been identified, several long stretches (up to 16 amino acids) of 100% identity between the moss P. patens and the angiosperm Arabidopsis sequences are present. For segment C, we identify one highly conserved stretch of 18 amino acids that are not present in motif databases. The position of this sequence coincides with a proposed autolytic cleavage site for DEK1 (Johnson et al., 2008).
In BLAST searches, the land plant DEK1 LG3 domains align, although with low sequence similarities (less than 25%) and insignificant E values, to metazoan Sushi, von Willebrand factor type A, EGF, and pentraxin (PTX) domain-containing protein 1 and to prokaryotic Laminin G (LG) domain-containing proteins, domains belonging to the concanavalin A-like lectin/glucanases superfamily (SCOPe version 2.06; http://scop.berkeley.edu/). Protein domains of this superfamily adopt a sandwich structure of 12 to 14 β-strands arranged in two sheets with a complex topology, referred to as the legume lectin fold or β-sandwich fold. To investigate whether the LG3 domain of DEK1 has the potential to adopt a similar fold, we predicted the structure of the P. patens DEK1 LG3 domain using three different threading servers: I-TASSER, RaptorX, and Phyre2. All gave highly similar structures, all of which adopted a typical β-sandwich fold composed of eight antiparallel β-strands arranged in two sheets with loops connecting the β-strands on both edges (Fig. 1C, I). All three servers modeled the fold with high confidence levels; for example, an I-TASSER C score of −0.1 was obtained, indicating that the model was predicted with correct global topology. In addition, an average normalized Z score of greater than 2 shows good threading alignment between the template and the query sequence. Structure search of the model against the Research Collaboratory for Structural Bioinformatics Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) using PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm/) also gave similar top hits, belonging to the PTX family and the β-sandwich structure fold (Fig. 1C, II).
Deleting P. patens DEK1 Linker Yields a Phenotype Similar to Δdek1
To assess the importance of the Linker for DEK1 function, we created a P. patens dek1Δlinker mutant, in which a genomic sequence corresponding to 564 amino acids (amino acid residues 1,121–1,684; XP_001774206.1) of the DEK1 Linker was removed (Fig. 2A). First, we transformed wild-type protoplasts with the pBHRF-ΔLinkerG1 vector (Supplemental Fig. S1A). This vector, containing 5′ and 3′ targeting sequences (TGSs) flanking the genomic region encoding the DEK1 Linker, was designed to insert the Hygromycin phosphotransferase resistance cassette (HRC) flanked by loxP sites to intron 17. One of the resulting hygromycin-resistant lines, dek1ΔlinkerG1#20, gave PCR signals for targeted integration, loss of DEK1 Linker, and a single insertion event at the locus, as demonstrated by Southern-blot analysis (Supplemental Fig. S1B). The dek1ΔlinkerG1#20 mutant displayed the Δdek1 phenotype lacking gametophores (Perroud et al., 2014). Since insertion of the HRC into an intron potentially can interfere with posttranscriptional processing of the DEK1 transcript, the HRC was removed by Cre/lox-mediated excision, resulting in the mutant dek1ΔlinkerG1/cre. PCR genotyping and sequencing of the DEK1 locus from intron 12 to intron 26 confirmed removal of the HRC, leaving the loxP site in the fused intron between exons 17 and 24 of the DEK1ΔLinkerG1/cre locus. However, RT-PCR and subsequent sequencing of the cDNA product revealed that the DEK1 transcript was not properly spliced, leaving the fused intron in the processed mRNA, creating a premature stop codon 5′ to the CysPc coding sequence (Supplemental Fig. S1C). In order to remove the fused intron from the mutant, dek1ΔlinkerG1/cre protoplasts were transformed with the pArrow-ΔLinkerG3 construct (Supplemental Fig. S2A), containing the full exon 17 sequence fused in-frame with the 3′ end of exon 24, designed to insert the Neomycin phosphotransferase resistance cassette after the DEK1 stop codon. One of the resulting mutants, dek1ΔlinkerG3#2, was subjected to Cre/lox-mediated excision to obtain the dek1Δlinker/cre#123 mutant line (hereafter referred to as the dek1Δlinker mutant). The dek1Δlinker mutant was first characterized by PCR genotyping and Southern-blot analysis (Supplemental Fig. S2B), confirming elimination of the DEK1 Linker and showing no evidence of off-locus integrations. Second, RT-PCR using forward (Ex7-F) and reverse (Ex30-R) primers annealing to DEK1 exons 7 and 30, respectively (Fig. 2A), showed amplification of a single PCR product of the expected sizes of 4.6 and 2.9 kb for the wild type and dek1Δlinker, respectively (Fig. 2B). Subsequent sequencing of the 2.9-kb RT-PCR product confirmed proper splicing of the truncated DEK1ΔLinker cDNA with exon 17 fused in-frame to the 3′ end of exon 24 (Supplemental Fig. S2C). Quantitative RT-PCR was performed, revealing no significant changes in the steady-state DEK1 transcript level in 6- and 14-d-old dek1Δlinker mutant tissue compared with wild-type plants of the same age (Fig. 2C), supporting the conclusion that deletion of the Linker did not affect the stability of the truncated DEK1 mRNA.
Figure 2.

DEK1 cDNA structure and expression analyses in wild-type, dek1Δlinker, dek1Δlg3, dek1Δc-linker, and dek1° lines. A, Schematic representation of the DEK1 coding sequence architecture in wild-type and dek1 mutant plants. The truncated DEK1 variants are missing either the Linker or the Linker subsegments LG3 and C, while DEK10 harbors two point mutations leading to a single amino acid replacement from Cys to Ser in the CysPc coding sequence of the active site. Numbering identifies deleted nucleotides in the individual truncated DEK1 variants. The codon TGC starting at the wild-type sequence in nucleotide position 5,341 (black star) is mutated to TCT (white star) in DEK10. The positions of the PCR primers (Ex7-F and Ex30-R) used in B are indicated by arrows, and expected reverse transcription (RT)-PCR product sizes using this primer set are given. For comparison, wild-type DEK1 also is shown. B, RT-PCR analysis of DEK1 transcripts in wild-type (WT) and dek1 mutant plants using primers Ex7-F and Ex30-R showing PCR amplicons of the expected sizes. M, Marker; NC, negative control. C, Real-time quantitative RT-PCR analysis of relative DEK1 transcript levels in dek1 mutant plants at two time points (6 and 14 d). The graph shows mean fold change values in the dek1 mutants relative to the wild type, which was set to 1, normalized to the average transcript level of RIBOSOMAL PROTEIN S9 (RSB; Pp3c18_1350V3.1) used as an internal reference. Error bars indicate se of two biological replicates.
In this section, we describe phenotypic characteristics of the dek1Δlinker mutant compared with wild type plants, a mutant lacking the entire DEK1 gene (Δdek1; Perroud et al., 2014), and a partial dek1 deletion mutant harboring a deletion within the DEK1 MEM segment (dek1Δloop; Demko et al., 2014). The phenotypes of wild-type, Δdek1, and dek1Δloop plants are shown in Figure 3, A to C, respectively. In the wild type, one bud is typically initiated per filament of 15 cells (Fig. 3A1). The bud initial cell divides by oblique division (Fig. 3A2, arrowheads), giving rise to the basal cell and the apical cell. The next division plane in the apical cell is perpendicular to the first oblique division (Fig. 3A2, arrow). This division gives rise to the bud apical initial (Fig. 3A2, blue dot) and the first apical lateral cell (Fig. 3A2, yellow dot). Successive oriented divisions then lead to the formation of the gametophore apical cell (tetrahedral stem cell) and primordial lateral domains (Fig. 3A3). A wild-type young gametophore with primordial phyllids is shown in Figure Fig. 3A4. Primordial phyllids are shaped by mitotic activity of the phyllid apical cell (Fig. 3A4, green dot) that divides by asymmetric oblique divisions. Further mitotic activity that takes place within the sectors of developing phyllid contributes to phyllid lateral and longitudinal expansion. Wild-type gametophores (Fig. 3A5) form phyllids with different morphologies depending on their position along the apical-basal axis of the gametophore, a phenomenon known as heteroblasty. A typical wild-type phyllid from the middle part of the gametophore is shown in Figure Fig. 3A6. It has expanded lamina with pointed apex, serrulated margins, and central midrib. Archegonia, the female reproductive organs, are formed on gametophore apices. During the final stages of their development, the neck apical cells become vacuolated and swollen, and the apex opens (Fig. 3A7). After fertilization of the egg cell enclosed in the archegonium, a sporophyte is formed (Fig. 3A8).
Figure 3.

Wild-type and dek1 mutant phenotypes. A, Wild-type (WT) plants. B to G, dek1 mutants. The phenotypes at different developmental stages are shown in columns 1 to 8. Column 1 shows the number of buds per 15-cell filament (n = 100). Column 2 shows buds at the four-cell stage. Arrowheads indicate the first oblique division of the bud initial. Arrows indicate the plane of the first division in the bud apical cell. Blue dots mark the bud apical initial. Yellow dots mark apical lateral cells. Brown dots mark basal lateral cells. Column 3 shows bud development at the six-cell or more stage. Arrowheads indicate the position of the initial oblique division. Blue dots mark bud apical cells. Yellow dots mark apical lateral cells (forming a hair in A and E). Green dots in E mark a phyllid initial cell. Brown dots mark basal lateral cells. Column 4 details young gametophores with primordial phyllids in A and E. Green dots mark phyllid initials. Yellow dots mark a hair. Note that the latter structures are not formed in B and G. Column 5 shows leafy gametophores in A and E and reduced stems lacking phyllids in C and F. These structures are not formed in B, D, and G. Column 6 displays the morphology of a phyllid isolated from the middle part of a gametophore. Column 7 represents the morphology of archegonia at later stages of development. The archegonium apex is opened, and a neck canal is marked by brown pigmentation in A. The neck remains closed and is distorted in E. Column 8 shows a mature sporophyte. Bars = 50 μm (columns 2 and 3, A7, and E7), 100 μm (column 4), 1 mm (A5 and E5), 500 μm (C5, F5, and A8), and 200 μm (A6 and E6).
In contrast to the wild type, the Δdek1 mutant produces four buds per 15-cell filament (Fig. 3B1) while the dek1Δloop mutant produces two (Fig. 3C1). The first oblique division of the bud initial cell appears normal in both Δdek1 (Fig. 3B2) and dek1Δloop (M). In Δdek1, the division of the bud apical cell does not orient as in the wild type (Fig. 3B2), and the cell divides in variable random planes (Perroud et al., 2014). Moreover, the division of the resulting apical cell also is misoriented, while the apical lateral cell often does not continue to divide. Consequently, bud development is arrested early after a few divisions (Fig. 3B3). In dek1Δloop, the division plane in the bud apical cell orients perpendicularly to the first oblique division; however, the cell wall tends to bend (Fig. 3C2). As the bud continues to grow, the pattern of cell divisions becomes irregular (Fig. 3C3). Phyllid initiation is abortive in dek1Δloop (Fig. 3C4). In the dek1Δloop mutant, expanded phyllids are never formed; instead, filamentous protrusions emerge from a stem (Fig. 3C5).
The dek1Δlinker mutant produces two buds per 15-cell filament compared with one bud in the wild type and four buds in the Δdek1 mutant (Figs. 3, A1–D1, and 4). Similar to the Δdek1 mutant, the first oblique division of the bud initial cell appears normal. However, the cell wall of the divided apical cell points toward the lower ventral side of the bud, and its intersection relative to the first oblique division also varies compared with the wild type (Fig. 3D2). Resulting daughter cells then do not expand as in the wild type, and their successive divisions are misoriented, which leads to disorganized buds with abortive growth (Fig. 3D3). In contrast to the Δdek1 mutant, where buds are arrested early after a few divisions, a proportion of dek1Δlinker buds continue to divide, giving rise to larger structures of variable size (Fig. 3D4). No phyllid initiation was observed. From these results, we infer that removal of the Linker segment leaves a minimal calpain activity sufficient to support cell proliferation but lacking an organized division pattern.
Figure 4.
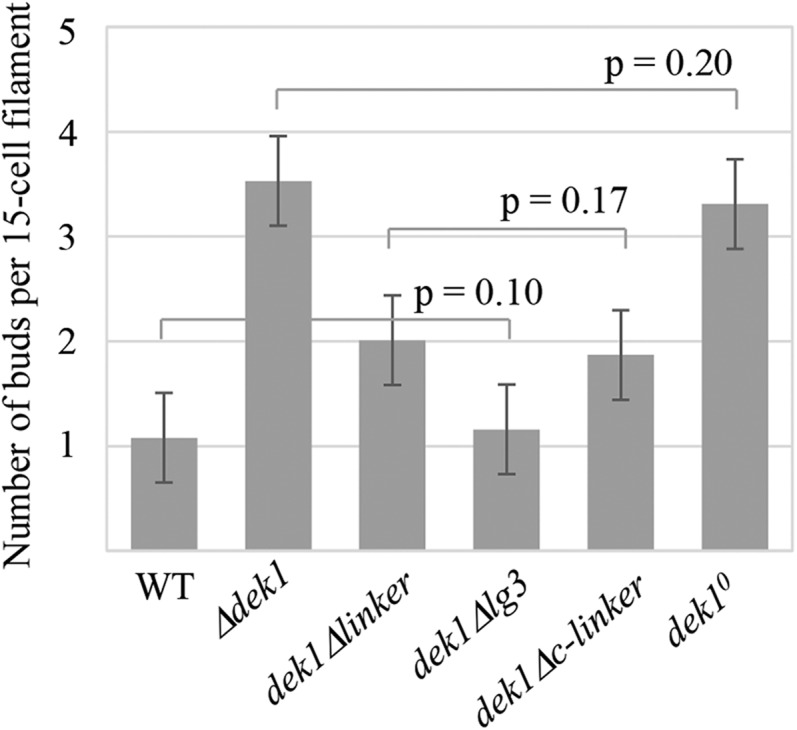
Number of buds initiated in P. patens wild-type (WT) and dek1 mutant plants. The graph shows the average number of buds per 15-cell filament (n = 100) in wild-type, Δdek1, dek1Δlinker, dek1Δlg3, dek1Δc-linker, and dek1° lines. Lines above the bars indicate no significant differences by paired Student’s t test. Error bars indicate se.
Finally, in a control experiment, we retargeted the DEK1 Linker sequence to the DEK1ΔLinker locus as described in Supplemental Fig. S3A. PCR genotyping (Supplemental Fig. S3B) and sequencing of the DEK1 cDNA (data not shown) confirmed correct insertion of the Linker sequence. Two independent lines were obtained, both forming normal gametophores with fully expanded phyllids, as shown for one of these lines (Supplemental Fig. S3C).
Deletion of the DEK1 LG3 Domain Perturbs Leafy Gametophore Patterning and Archegonia Development
We next studied the effect of deleting the LG3 domain in the dek1Δlg3 mutant. This mutant was generated by transforming the pB-ΔLG3 construct (Supplemental Fig. S4A) into dek1ΔlinkerG1#20 protoplasts and harbors a deletion corresponding to 188 amino acids (amino acids 1,426–1,613; XP_001774206.1) that spans the LG3 domain (Fig. 2A). PCR genotyping and Southern-blot analysis confirmed deletion of the LG3 sequence and no evidence of off-locus integrations of the construct (Supplemental Fig. S4B). To verify in-frame removal of the LG3 coding sequence in the mutant, a 4-kb RT-PCR product spanning from exon 7 to 30 was amplified (Fig. 2B) and subsequently sequenced, confirming proper splicing of the truncated DEK1 transcript with the 5′ end of exon 22 fused in-frame with the 3′ end of exon 23 (Supplemental Fig. S4C). No significant changes in the steady-state level of the DEK1 transcript were observed between 6- and 14-d-old wild-type and dek1Δlg3 tissue (Fig. 2C).
Removal of the DEK1 LG3 domain causes developmental changes in the gametophyte that differ distinctly from Δdek1, dek1Δloop, and dek1Δlinker mutants (Fig. 3). The bud initiation rate is similar to that of the wild type (Figs. 3, A1 and E1, and 4). Early cell divisions in the dek1Δlg3 buds follow the wild-type pattern. After the first oblique division of the bud initial, the division plane in the apical cell orients perpendicularly (Fig. 3E2), and the resultant daughter cells continue to divide correctly to form tetrahedral stem cell (Fig. 3E3, blue dot) and lateral segments of the young gametophore (Fig. 3E3). The effects of the LG3 deletion become apparent later during young gametophore development. In particular, the mitotic activity of the phyllid apical cell is reduced, affecting phyllid primordia development (Fig. 3E4). As a result, the dek1Δlg3 mutant forms reduced gametophores with narrow phyllids lacking several features of the wild-type phyllids, including the midrib (Fig. 3, E5 and E6). Archegonium development also is affected in the mutant. The archegonium neck is deformed and does not open at the apex (Fig. 3E7). Consequently, fertilization is prevented and no sporophytes are formed in the mutant.
Altered phyllid and archegonium morphology in the dek1Δlg3 mutant is shown in more detail in Figure 5. An example of heteroblastic phyllids typically seen in wild-type gametophores is shown in Figure 5A. Basal phyllids lack the midrib, and their marginal cells do not acquire the highly elongated pointed shape as the phyllids that develop later, also called upper phyllids. Mature upper phyllids form a midrib that passes through from the base to the tip in the most expanded phyllids. Proximal cells of the phyllid lamina are considerably larger than more distal cells. Phyllid edges are formed of elongated pointed cells forming marginal serration. The juvenile apical phyllids also have serrulated edges and possess a midrib. In contrast to mature fully expanded phyllids, the proximal cells in the apical phyllids are not yet elongated and continue to divide. In dek1Δlg3, basal phyllids consist of two or three files of cells with irregular shape (Fig. 5B). Mature upper phyllids lack several characteristics of the wild type, including a midrib, marginal serration, and acute tip. They do not expand as in the wild type, and their narrow lamina lack the typical proximal-distal distribution of cells of variable size. The same is true for the mutant young apical phyllids. In the wild type, phyllids continue to expand by longitudinal and transverse cell divisions that take place in sectors within the phyllid. From the phyllid morphologies in dek1Δlg3, we infer that both phyllid apical cell activity and sectorial cell divisions within the phyllid are impaired in the mutant.
Figure 5.

Phyllid and reproductive organ morphology in the wild type (WT) and dek1Δlg3. A, Wild-type phyllids isolated from the basal, middle, and apical parts of the gametophore. The positions of the isolated phyllids on gametophores are marked by arrowheads in the insets. B, dek1Δlg3 phyllids isolated from the basal, middle, and apical parts of the gametophore. The positions of the isolated phyllids on gametophores are marked by arrowheads in the insets. C, Young wild-type archegonium with highlighted cell walls in the neck. D, Examples of wild-type archegonia at the later stages of development. Note the open apices and brown pigmentation marking the neck canal. E, Wild-type gametophore with sporophyte. Antheridium is shown in the inset. F, Young dek1Δlg3 archegonium with highlighted cell walls in the neck. G, dek1Δlg3 archegonia at the later stages of development. Note the closed and deformed necks and patchy distribution of the brown pigmentation that reflects defects in the neck canal differentiation. H, dek1Δlg3 gametophores cultivated under the sporogenesis conditions. Sporophytes are not formed. Antheridium is shown in the inset. Bars = 500 μm (A and B), 50 μm (C, D, F, and G), and 1 mm (E and H).
Closer examination of the gametangia revealed that archegonium development was affected in the dek1Δlg3 mutant. In the wild type, reproductive organ formation is highly organized, as described in detail previously (Landberg et al., 2013). In the first stages of development, the primordial archegonium develops through a series of mostly anticlinal cell divisions forming a layer of outer cells and inner cells (Fig. 5C). Upper inner cells elongate and give rise to the archegonium canal. Basal inner cells divide asymmetrically and give rise to the egg cell. Upper outer cells also divide anticlinally and elongate to shape the archegonium neck. Basal outer cells divide both anticlinally and periclinally to form the basal part of the archegonium with an egg cavity. During the final stages of archegonium development, inner canal cells lyse and degrade, and the resulting channel acquires yellow-brown pigmentation (Fig. 5D). The content of the neck outer cells also degrades. The neck apical cells become swollen, and the apex is fully opened (Fig. 5D). In the dek1Δlg3 mutant, the development of the archegonium appears irregular from the early stages, with altered morphology and organization of cells forming the neck (Fig. 5F). Defects become more apparent in the later stages of mutant archegonium development. The canal fails to form properly, which is marked by dislocated patchy brown pigmentation (Fig. 5G). The neck apical cells do not swell, and the apex fails to open. The egg and the egg cavity appear to form in the mutant (Fig. 5G). These defects prevent fertilization, and the sporophyte is not formed in the mutant (Fig. 5H). No major morphological defects were detected in the mutant antheridia (Fig. 5H), although we do not exclude further anatomical/ultrastructural differences.
In our interpretation, the LG3 domain is needed for regulated activation of the calpain during certain stages of gametophore development, including gametophore apical cell activity and cell divisions in phyllids and archegonia. On the other hand, truncated DEK1 lacking the LG3 domain is fully efficient to promote normal early bud and antheridia development.
The Marchantia polymorpha DEK1 LG3 Domain Complements the dek1Δlg3 Mutant of P. patens
To investigate whether DEK1 LG3 from angiosperms, liverwort, and bryophytes are functionally conserved, we introduced the M. polymorpha and Arabidopsis DEK1 LG3 sequences into the DEK1 locus of the P. patens dek1Δlg3 mutant. First, to verify the complementation assay, we retargeted the wild-type LG3 sequence into the dek1Δlg3 locus by transforming dek1Δlg3 protoplasts with the pCR-PpLG3 construct (Supplemental Fig. S5A). Three independent lines were obtained, all of which showed reversion to the wild-type phenotype, forming gametophores with fully expanded phyllids (Fig. 6). PCR genotyping confirmed insertion of the wild-type LG3 sequence (Supplemental Fig. S5B). Next, we targeted the M. polymorpha DEK1 LG3 sequence to the dek1Δlg3 locus using the pCR-MpLG3 construct (Supplemental Fig. S5A). Regenerating plants were screened by visual inspection, resulting in the identification of three independent lines, all of which developed wild-type gametophores, as shown for the line named MpLG3 in Figure 6. PCR genotyping (Supplemental Fig. S5B) and sequencing of the heterologous DEK1 cDNA (data not shown) confirmed in-frame insertion of the M. polymorpha DEK1 LG3 sequence at the DEK1 locus. These results show that the heterologous DEK1 LG3 domain from a liverwort, sharing approximately 67% amino acid identity with the P. patens DEK1 LG3 domain, is functional in moss gametophores, demonstrating the functional conservation of DEK1 LG3 within the groups of early-diverging land plants.
Figure 6.

DEK1 LG3 complementation. A, Wild-type (WT) gametophore. B, dek1Δlg3 gametophore. C, Gametophore of the dek1Δlg3 strain after retargeting of the P. patens DEK1-LG3 sequence. D, dek1Mplg3 gametophore: dek1Δlg3 after introduction of the M. polymorpha DEK1 LG3 sequence. Bars = 1 mm.
Despite extensive testing, we were unable to identify regenerating plants exhibiting gametophores with a different phenotype than that of dekΔlg3 after transforming mutant protoplasts with pCR-AtLG3 (Supplemental Fig. S5A). This strongly suggests that the Arabidopsis DEK1 LG3 domain does not complement the mutant phenotype or, alternatively, that no targeted lines were obtained in this experiment. To explore this further, we chose an alternative strategy using the construct pBHRF-AtLG3 designed to insert the Arabidopsis LG3 sequence together with HRC into the dek1Δlg3 locus, as described in Supplemental Figure S5C. Seventy-four hygromycin-resistant lines were obtained in this experiment, and PCR genotyping was performed, identifying one line with proper 5′ and 3′ targeting signals. After cre/lox-mediated removal of the HRC in this line, the resulting PCR genotyping-positive lines all displayed the Δdek1 phenotype. To investigate DEK1 transcript processing, DEK1 cDNA was amplified from one of the after-cre lines and subsequently sequenced, revealing that the Arabidopsis DEK1 intron 21 was not spliced in the P. patens DEK1 AtLG3 transcript, thereby creating a premature stop codon in the mRNA (data not shown). Therefore, more studies are needed to firmly conclude whether DEK1 LG3 function is conserved between mosses and angiosperms.
Deleting the Potential Autolysis Site Severely Disturbs Gametophore Development
We also created the dek1Δc-linker mutant, in which a genomic region corresponding to 71 amino acids (amino acids residues 1,614–1,684; XP_001774206.1) directly upstream of the DEK1 calpain domain was removed (Supplemental Fig. S6A). The rationale for making this mutant was a previous study proposing an autolytic cleavage site in close proximity to the start of the Arabidopsis DEK1 calpain domain. The dek1Δc-linker mutant (Fig. 3) was verified using PCR genotyping, Southern-blot analysis (Supplemental Fig. S6B), quantitative RT-PCR analysis (Fig. 2C), and sequencing of the 4-kb DEK1ΔC-Linker RT-PCR product (Fig. 2B; Supplemental Fig. S6C).
The resulting mutant initiates approximately two buds per 15-cell filament (Figs. 3F1 and 4). Early cell divisions of the dek1Δc-linker mutant bud showed a similar pattern to the dek1Δlinker mutant. The cell wall dividing the bud apical cell is leaning toward the lower ventral side of the bud (Fig. 3F2). The resulting cells then continue to divide in a disorganized manner, forming distorted buds of various sizes (Fig. 3F3). In contrast to Δdek1 and dek1Δlinker, dek1Δc-linker bud cells continue to proliferate and form severely deformed gametophore-like structures (Fig. 3F4). In a number of cases, such gametophores form stems with filamentous protrusions instead of phyllids similar to dek1Δloop (Fig. 3F5). The remaining DEK1 calpain activity is sufficient to enhance cell proliferation in the buds; however, the structures lack organized patterns, and such residual activity is insufficient to initiate normal phyllid formation.
P. patens DEK1 Function Is Dependent on the Cys Active-Site Residue in the CysPc Protease Core Domain
Finally, we created the P. patens dek1° single-codon replacement mutant in which Cys-1782 is replaced by a Ser (Supplemental Fig. S7A). Molecular analyses of the dek1° mutant using PCR genotyping, Southern-blot analysis (Supplemental Fig. S7B), RT-PCR (Fig. 2B), and subsequent sequencing of the DEK1° cDNA showed targeted integration, no evidence of off-locus integration, and the Cys-to-Ser substitution, respectively. Quantitative RT-PCR analysis confirmed that the steady-state level of the DEK1° transcript was comparable to the DEK1 transcript level in wild-type plants (Fig. 2C).
The dek1° mutant produces, on average, three buds per 15-cell filament, not significantly different from the average bud number in the Δdek1 mutant (Figs. 3G1 and 4). The mutant does not form leafy gametophores, and bud development is affected at its early stage (Fig. 3), mimicking the Δdek1 mutant phenotype (Perroud et al., 2014). After the first oblique division of the bud initial, the apical cell fails to expand and divide as in the wild type (Fig. 3G2). Bud growth stops after a few divisions without establishing the tetrahedral stem cell and lateral domains (Fig. 3G3). These results show, first, that an intact catalytic triad of the CysPc protease core domain is essential for DEK1 function and, second, that the Cys-to-Ser substitution gives the same effect as deleting the DEK1 gene. The P. patens dek1° mutant phenotype thus confirms previous mutant studies in Arabidopsis (Johnson et al., 2008; Liang et al., 2013) and in vitro studies of maize DEK1 (Wang et al., 2003) and animal calpains (Arthur et al., 1995) showing that calpain activity is dependent on the active-site residue Cys.
DISCUSSION
The DEK1 protein of land plants forms a separate phylogenetic clade in the streptophyte lineage, with the highest degree of conservation in the calpain-specific catalytic domain CysPc, showing more than 80% amino acid identity between the member species (Liang et al., 2013). As shown here, replacement of the CysPc active-site residue Cys with Ser in the P. patens dek1° mutant generates the Δdek1 mutant phenotype (Fig. 3), confirming that an intact catalytic triad is necessary for DEK1 activity in P. patens and underscoring the calpain-dependent activity of DEK1. A high degree of sequence conservation also exists for DEK1 MEM, with 54% overall amino acid identity between Arabidopsis and P. patens. Here, we show that the high degree of sequence conservation in the DEK1 land plant clade also extends to the approximately 600-amino acid residue 69-kD Linker, the identity between land plant members ranging from 59% to 98%. The identity between P. patens and Arabidopsis is 60%. Similar to DEK1 MEM and CysPc-C2L (Liang et al., 2013), DEK1 Linker sequence identity is reduced dramatically when comparing land plants with charophytes, the algal predecessors of land plants, dropping to below 35%. The charophyte sequences also are highly divergent from each other, suggesting a conserved functional role for the DEK1 Linker in land plants that may differ from the function in charophyte algae. This accords well with the proposal that a major change in DEK1 function occurred during the evolutionary transition from charophyte algae to land plants (Demko et al., 2014). The phenotype of the dek1Δlinker mutant confirms an essential role for the Linker in regulating DEK1 activity. Notably, the effect of deleting the Linker is slightly less severe than deleting the entire DEK1 gene, suggesting that a DEK1 protein missing the Linker retains some residual calpain activity. If so, this activity is sufficient to limit the increase in bud number to two buds per 15-cell filament, compared with four in Δdek1, as well as to support cell proliferation beyond that of the Δdek1 mutant (Fig. 3).
Within the Linker, the highest overall identity between P. patens and Arabidopsis is in the 340-amino acid N segment, 60%, including several stretches of up to 16 amino acids with 100% identity between P. patens and Arabidopsis (Fig. 1B). In spite of the high amino acid sequence conservation, no known structural element that may hint at the function of this part of the Linker can be identified.
The identification of a conserved LG3 domain in DEK1 is supported by structural predictions, the domain adopting a typical β-sandwich fold similar to the protein domains belonging to the concanavalin A-like lectin/glucanase superfamily of which LG3 is a member (SCOPe v2.06). However, the number of predicted β-strands in DEK1 LG3 is lower, eight, compared with 12 to 14 β-strands in other family members. The β-sandwich fold, also referred to as the lectin fold, is a widely occurring fold present in 25 distinct protein families (SCOPe v.2.06) of very diverse functions. The predominant function of these proteins appears to be carbohydrate binding (Chandra et al., 2001). However, despite the highly conserved structural fold, sequence similarity among the different members is very low, identity in some cases being only 2% (Chandra et al., 2001), excluding the prediction of structure-function relationships (Rudenko et al., 2001). The land plant DEK1 LG3 domains display more than 59% sequence identity and only insignificant sequence similarities to other protein domains in the National Center for Biotechnology Information nonredundant database, suggesting that DEK1 LG3 may represent a new family member of the concanavalin A-like lectin/glucanase superfamily. A number of the templates selected by the servers to build the models for DEK1 LG3 included Protein Data Bank structures for PTX, such as serum amyloid P component (SAP) from L. polyphemus and human C-reactive protein. Interestingly, these proteins are characterized by Ca2+-dependent binding to their ligands, including carbohydrates, and function as soluble pattern-recognition molecules (Du Clos, 2013). Whether the DEK1 LG3 domain binds carbohydrates in a Ca2+-dependent process has yet to be determined. However, it is notable that the DEK1 Cys protease has been identified as a potential candidate involved in the initial proteolysis and turnover of CELLULOSE SYNTHASE A (CesA) in cotton (Gossypium hirsutum) rosettes (Jacob-Wilk et al., 2006) responsible for cellulose biosynthesis in primary and secondary cell walls (Festucci-Buselli et al., 2007). Both calcium signaling and CesA have been implicated in moss bud development (Saunders and Hepler, 1982; Goss et al., 2012). Cytokinin treatment induces bud formation, which coincides with elevated intracellular Ca2+. A similar effect can be achieved after treatment with an ionophore (Saunders and Hepler, 1982). We have shown previously that the Δdek1 mutant responds to cytokinin-induced bud formation (Perroud et al., 2014), and DEK1 is involved in the spatial control of bud formation via the negative regulation of APB transcription factors (Demko et al., 2014). Disorganized buds are formed instead of leafy gametophores in P. patens cesa5 mutants (Goss et al., 2012). Whether DEK1 senses cell wall properties or is involved in the regulation of cell wall remodeling needs to be further investigated.
Deletion of the LG3 domain in the P. patens dek1Δlg3 mutant is much less severe than deletion of the Linker in dek1Δlinker, bud and leaf apical stem cell specification being unaffected (Fig. 3). The effect of deleting the LG3 domain, however, does become apparent during later developmental stages, as both phyllid and archegonia development are perturbed in the dek1Δlg3 mutant (Fig. 3), indicating that distinct DEK1 activities are required throughout 3D development of the gametophyte. This observation suggests that the LG3 domain is involved in fine-tuning of calpain activity and that the dek1Δlg3 mutant regulates calpain activity close to the wild-type level, sufficient to allow wild-type bud development but insufficient to maintain wild-type phyllid and archegonia patterning. Moreover, general reduction of dek1Δlg3 gametophore growth indicates that the activity of the gametophore stem cell also is reduced in the mutant. Thus, the DEK1 LG3 domain may play its role depending on cellular context. Similar to what was demonstrated for the DEK1 Loop (Demko et al., 2014), the M. polymorpha DEK1 LG3 domain complements the dek1Δlg3 mutant of P. patens. The DEK1 LG3 domain from liverwort shares 67% amino acid identity with the P. patens DEK1 LG3 domain, demonstrating functional conservation both in MEM and in the Linker. Due to missplicing of the Arabidopsis DEK1 transcript in the P. patens dek1Atlg3 mutant, we were not able to conclude whether DEK1 LG3 function is conserved between angiosperms and mosses.
Compared with N and LG3, the 71-amino acid C segment is the least conserved part of the Linker, with an overall identity of 34% and a similarity of 60% between land plants (Fig. 1B). Interestingly, a highly conserved stretch of 18 amino acid residues is located approximately 36 amino acids upstream of CysPc, corresponding approximately to the location of a proposed autolytic cleavage site of the DEK1 calpain (Johnson et al., 2008). The consensus sequence of this amino acid stretch is YDRDDVDWDGQYSSGRKR, the residues underlined being 100% conserved in land plant DEK1 proteins included in this study. The consensus sequence is not recognized as a conserved motif in current databases. The signal for animal calpain cleavage and the structural clues for substrate recognition are largely unknown (Sorimachi et al., 2012). However, in a recent study, both primary and higher order structures were identified as determinants for CAPN1 and CAPN2 substrate specificity (Shinkai-Ouchi et al., 2016). In an effort to determine the amino acid preference around the cleavage site, Tompa et al. (2004) analyzed 106 CAPN1 and CAPN2 cleavage sites in 49 substrates compiled from the literature. The preferred residues were identified as Leu, Val, and Thr in the P2 position and Tyr, Arg, and Lys in P1. Small hydrophilic residues, Ser and to a lesser extent Thr and Ala, were found to occur more often in P1′. In addition, a high preference for Pro in positions P3 and P2′ to P4′ was identified. This sequence pattern is not present in the consensus sequence of the conserved 18-amino acid residue C-segment site. Thus, if this sequence is indeed a DEK1 autolytic cleavage site, the DEK1 calpain has different cleavage site preferences than conventional animal CAPN1 and CAPN2 proteins.
The functional significance of the Linker C region is clearly demonstrated by the phenotype of the dek1Δc-linker mutant, resembling dek1Δlinker in severity during early stages of bud development (Fig. 3). In both mutants, the tetrahedral apical cell is not formed; however, mitotic activity is present, allowing cell proliferation beyond that of the Δdek1 mutant, resulting in buds of variable sizes. In contrast to the dek1Δlinker, dek1Δc-linker proliferates to form stem-like structures, similar to dek1Δloop (Demko et al., 2014; Fig. 3). These observations suggest that the dek1Δc-linker and dek1Δloop mutants regulate DEK1 calpain activity to similar levels. Further analyses are required to determine whether the conserved 18-amino acid residue site alone causes the dek1Δc-linker phenotype.
The multiple phenotypes obtained when deleting different regions of DEK1 point to different functions of the different parts of the Linker throughout P. patens development. A fuller understanding of DEK1 function has to await the 3D structure of DEK1. Meanwhile, our working hypothesis for DEK1 function is that the DEK1 MEM and Linker segments regulate the activity of CysPc-C2L. The observation that deletion of the Loop leads to a severe phenotype (Demko et al., 2014; Fig. 3) supports the role of DEK1 MEM in regulating DEK1 calpain activity. The exact role of the Loop in positional sensing, if any, needs further studies. Upon the activation of DEK1 MEM, our working model for DEK1 regulation proposes that a conformational change of DEK1 MEM activates the CysPc-C2L moiety in a process that involves the Linker. The activated protease then cleaves the Linker and is thereby released in its active form, ready to cleave its unknown substrate(s). Although the mode of activation is unknown, the structural similarities between DEK1 CysPc and CAPN2 CysPc make it tempting to speculate that DEK1 calpain activation occurs by a similar mechanism. If so, a change in the conformation of the activated MEM-Linker allows the CysPc subdomains PC1 and PC2 to swing close together, thereby forming an active site. In CAPN2, the inactive form represents the ground state of the protein, in which electrostatic forces in an acidic loop within C2L are partly responsible for the separation of PC1 and PC2 (Strobl et al., 2000). Activation by Ca2+ occurs in two steps, first by releasing constraints imposed by the circular arrangement of the domains, then by cooperative binding of Ca2+ to PC1 and PC2 (Khorchid and Ikura, 2002). We propose that the DEK1 MEM-Linker acts via a similar mechanism, although it remains to be determined whether the ground state of the DEK1 calpain is an active state or not. It is possible that both endogenous DEK1 and ectopically expressed CysPc-C2L that is capable of complementing the Arabidopsis dek1-3 mutant phenotype require the binding of Ca2+ to be active. Indeed, the observation that disruption of the conserved amino acids coordinating Ca2+-binding sites in conventional calpain makes the Arabidopsis CysPc-C2L incapable of complementing the dek1-3 mutant phenotype (Liang et al., 2013) supports this interpretation. The observation that deletion of the Linker does not lead to DEK1 activation suggests that the Linker alone does not function to inactivate CysPc but, rather, that MEM and Linker acts together. This conclusion also is supported by the observation that ectopically expressed Linker-CysPc-C2L is capable of complementing the Arabidopsis dek1-3 mutant phenotype (Johnson et al., 2008). The structure of DEK1 MEM-Linker is unknown. However, it is interesting that part of the MEM shows similarity to members of the major facilitator superfamily of proteins of bacterial origin (Demko et al., 2014). Members of this family have evolved from transporters, via dual-function molecules referred to as transceptors, to receptors (Yan, 2013). Given the long evolutionary history of TML-calpains and the DEK1 clade of TML-calpains, it is an interesting possibility that a similar functional change took place with DEK1.
CONCLUSION
In contrast to higher plants, where loss-of-function alleles of DEK1 are lethal, the protonemata stage of mosses is not critically affected by the loss of DEK1. Interestingly, the unique moss protonemata stage represents an image of the transition from charophyte algae to land plants. Initially, it grows in a 2D mode typical of the algae predecessors of land plants. It then switches to the 3D growth form of land plants, initially in the form of the gametophore bud. DEK1 plays a role in two major processes related to 3D growth: first, by affecting the number of buds via APB transcription factors; and second, by its essential function in positioning of the cell wall resulting from the first division of the apical bud cell. This study completes a series of investigations into the functional effect of deleting specific domains of DEK1, including the entire gene and the DEK1 Loop. Here, we first show that the Linker performs an essential function in the regulation of DEK1 calpain activity; loss of the Linker has nearly the same effect as loss of the entire DEK1 gene. In addition, our analysis also identifies an important function in the Linker C segment containing a highly conserved 18-amino acid sequence, the deletion of which also has a severe effect on moss development. One potential function of this sequence is to serve as a substrate for the DEK1 calpain protease, although the sequence does not represent a typical target sequence for conventional mammalian calpains. The second conserved Linker subsequence is LG3, the deletion of which leads to less dramatic perturbation of moss development. Earlier studies showed that the DEK1 Loop sequence of M. polymorpha is functional in moss but sequences from angiosperm are not. Similarly, the LG3 sequence of M. polymorpha functions in moss but not in angiosperms LG3 sequences. Earlier studies have shown the essential nature of the Cys residue of the active site of CysPc. Here, we confirm the essential nature of the P. patens Cys-1782 residue to DEK1 function. Finally, we propose that DEK1 MEM-Linker in an inactive state inhibits calpain activity by forcing apart the CysPc PC1 and PC2 subdomains. In an active state, this inhibition is abolished and the two subunits swing closer, bringing the three active-site amino acid residues together into its active-site configuration.
MATERIALS AND METHODS
Plant Material and Growth Conditions
The Physcomitrella patens Gransden strain was used throughout this study. Tissue maintenance and production were performed on BCDA medium as described by Cove et al. (2009). P. patens tissue and protoplasts were grown under long-day conditions (16 h of light [70–80 µmol m−2 s−1]/8 h of dark) at 25°C. Growth medium was supplemented with 20 µg L−1 hygromycin B or 30 µg L−1 G418 for selection of transformed cells. Tissue for phenotypic characterizations was grown on BCD medium unless stated otherwise. Cultures for bud counting were established as follows. A BCD-containing petri dish was inoculated with 16 equally spaced spot inoculums consisting of 10 µL of protonemal tissue suspension. After 14 d of growth, buds were counted on 100 filaments using a dissecting microscope, and the average number of buds per filament was calculated. Buds were counted on 15 cells from the tip of the filaments. The 100 bud-containing filaments were randomly chosen from different areas of the plate. Tissue for sporophyte production was grown on sterile Jiffy7 soil blocks placed in glass jars under short-day conditions (8 h of light [70–80 µmol m−2 s−1]/16 h of dark) at 15°C and manipulated as described (Perroud et al., 2011). Tissue for real-time quantitative RT-PCR was grown and harvested as follows. Protonemal tissue grown on BCDA under long-day conditions was collected, homogenized in sterile water, and inoculated on BCD medium overlaid with cellophane discs. The tissue was collected after 6 and 14 d of growth, snap frozen in liquid nitrogen, and stored at −80°C until further processing.
In Silico Analyses
The orthologous DEK1 sequences used in this study are listed in Supplemental Table S1. To identify and extract the amino acid sequences corresponding to the DEK1 Linker, defined as the segment between the TM-spanning domain (MEM) and the CysPc domain (Fig. 1A), full-length DEK1 sequences were submitted to the SMART server (http://smart.embl-heidelberg.de). The extracted DEK1 Linker sequences were further submitted to Pfam (pfam.xfam.org) and the Conserved Domain Database (ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) to identify putative conserved domains. Pfam domain models were extracted using SMART. Multiple sequence alignment of DEK1 Linker sequences was performed with the L-INS-I algorithm using MAFFT version 7 (http://mafft.cbrc.jp/alignment/server/index.html; Katoh and Standley, 2013). Pairwise comparison of each pair of sequences in the multiple sequence alignment was calculated as the percentage of identical residues in alignment positions between the two sequences using the software CLC Genomic Workbench version 7. To predict the 3D structure of the P. patens DEK1 LG3 domain, the amino acid sequence corresponding to LG3 (residues 1,426–1,613; XP_001774206.1) was submitted to three different fold recognition Web servers: RaptorX (http://raptorx.uchicago.edu/), I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/), and Phyre2 (http://www.sbg.bio.ic.ac.uk/∼phyre2/html/page.cgi?id=index). The SAVES metaserver (http://services.mbi.ucla.edu/SAVES/) was used for structural analysis and validation of the generated models to verify their stereochemical quality and overall structural geometry.
Construction of Plasmids for Gene Targeting
Oligonucleotides used for vector construction are listed in Supplemental Table S3. The schematics for the gene-deletion and knock-in complementation constructs are shown in Supplemental Figures S1 to S7. Plasmids were generated using the In-Fusion HD Cloning kit (Clontech Laboratories) according to the manufacturer’s instructions, unless stated otherwise. The PCR-generated fragments used in the In-Fusion reactions were amplified using Thermo Scientific Phusion High-Fidelity DNA Polymerase. To verify the constructed vectors, restriction digestion and DNA sequencing analyses were performed. Nucleotide numberings are relative to the ATG start site of the P. patens DEK1 sequence (Pp3c17_17550; www.phytozome.net).
Two plasmid vectors, named pBHRF-ΔlinkerG1 and pArrow-ΔlinkerG3, were used to generate the P. patens dek1Δlinker mutant. GenScript performed fragment synthesis and cloning of the pBHRF-ΔlinkerG1 vector. In short, the 5′ TGS, P. patens DEK1 nucleotides 7,249 to 8,346 flanked by 5′ HindIII and 3′ NruI recognition sequences, and the 3′ TGS, P. patens DEK1 nucleotides 11,545 to 12,546 flanked by 5′ SpeI and 3′ NsiI recognition sequences, were synthetized de novo. The 5′ and 3′ TGSs were cloned sequentially into pBHRF (Schaefer et al., 2010) using HindIII/NruI and SpeI/NsiI restriction enzyme pairs, respectively, creating construct pBHRF-ΔLinkerG1 (Supplemental Fig. S1A). Prior to P. patens transformation, pBHRF-ΔlinkerG1 was digested with BmrI and PacI. The pTDek1-3′ plasmid (Perroud et al., 2014), which contains a 3′ TGS of 1,003 bp 3′ to the DEK1 termination code, was used to build the pArrow-ΔlinkerG3 construct. The 5′ TGS of the latter construct, designed to contain a mutagenized P. patens DEK1 fragment spanning from nucleotides 7,250 to 14,569 with a 3,398-bp deletion from nucleotides 8,321 to 11,718, was amplified using two PCR-generated fragments (fragments A and B) in an overlapping PCR reaction. Fragments A and B were first separately PCR amplified using primer pairs SP-A/ASP-A and SP-B/ASP-B, respectively, with genomic P. patens DNA as template. The 5′ TGS was then amplified with primer set SP-A and ASP-B in a second round of PCR using fragments A and B as template. The SP-A and ASP-B primers were designed to add 15-bp 5′ and 3′ extensions to the resulting PCR product, facilitating insertion of the 5′ TGS into SmaI-digested pTDek1-3′ using In-Fusion reaction. The resulting construct, pArrow-ΔLinkerG3 (Supplemental Fig. S2A), was digested with BamHI prior to P. patens protoplast transformation.
In order to generate the P. patens dek1Δlg3 mutant, construct pB-ΔLG3 was used. This plasmid, containing a mutagenized P. patens DEK1 fragment spanning from nucleotides 7,249 to 12,546 harboring a 673-bp deletion from nucleotides 10,567 to 11,239 spanning the DEK1 LG3 sequence, was made in several steps. First, we PCR amplified an In-Fusion fragment containing P. patens DEK1 nucleotides 8,347 to 11,544 with primers SP_PpLinker and ASP_PpLinker and genomic P. patens DNA as template. This insert was mixed with linearized vector pBHRF-ΔlinkerG1, produced by PCR using primers SP_Inf1 and ASP_Inf1 to exclude the sequence between the 5′ and 3′ TGSs in this vector (e.g. the HRC). The insert and linearized vector were ligated in an In-Fusion reaction, resulting in plasmid pB_Linker, containing a full P. patens DEK1 fragment spanning from nucleotides 7,249 to 12,546. Finally, a second PCR amplification was performed to delete nucleotides 10,567 to 11,239, using pB_Linker as template and primers SP_Inf2 and ASP_Inf2, resulting in the generation of the pB-ΔLG3 construct (Supplemental Fig. S4A). The construct was digested with RsrII and PacI prior to P. patens protoplast transformation.
To generate the P. patens dek1Δc-linker mutant, the construct pBHRF-ΔC-2G was used. The 3′ TGS of this construct, spanning from nucleotides 8,402 to 12,893, contains a 479-bp deletion (nucleotides 11,240–1,1718) corresponding to the Linker C segment (Fig. 1A). Construct pBHRF-ΔC-2G was built from the pBHRF-ΔlinkerG2 plasmid. The latter plasmid was generated by GenScript as follows. In short, the 5′ TGS, P. patens DEK1 nucleotides 7,249 to 8,404 flanked by 5′ HindIII and 3′ NruI recognition sequences, and a mutagenized 3′ TGS, P. patens DEK1 nucleotides 8,402 to 12,893 flanked by 5′ SpeI and 3′ NsiI recognition sequences, were synthetized de novo. The 3′ TGS was designed to contain the following sequence (from 5′ to 3′): the 3′ end of intron 17 to the 5′ end of exon 18 (nucleotides 8,402–8,461) fused in frame with the 3′ end of exon 24 to the start of intron 26 (nucleotides 11,719–12,893). The 5′ and 3′ TGSs were cloned sequentially into pBHRF (Schaefer et al., 2010) using the HindIII/NruI and SpeI/NsiI restriction enzyme pairs, respectively, creating plasmid pBHRF-ΔlinkerG2. We then PCR amplified, from genomic P. patens DNA, an In-Fusion fragment containing P. patens DEK1 nucleotides 8,462 to 11,718 with primers SP_2G_E and ASP_2G_E, designed to add 15-bp 5′ and 3′ extensions complementary to the ends of the linearized vector. The insert was mixed with linearized (at P. patens DEK1 nucleotide position 8,461) pBHRF-ΔlinkerG2 DNA, produced by PCR using primers SP_2G_F and ASP_2G_F, then ligation using an In-Fusion reaction was performed, resulting in plasmid pBHR-LinkerG2. Finally, a second PCR amplification was performed to delete nucleotides 11,240 to 11,718, using pBHR-LinkerG2 and primers SP_2GDC_Inf and ASP_2GDC_Inf, followed by an In-Fusion reaction, resulting in the generation of the pBHRF-ΔC-2G construct (Supplemental Fig. S6A). The construct was digested with BmrI and PacI prior to P. patens protoplast transformation.
To generate the P. patens dek1° mutant, the construct pArrow-DEK1° was used. The 5′ TGS in this construct contains a mutagenized PpDEK1 fragment spanning from nucleotides 11,763 to 14,569 containing the two substitutions G to C and C to T at nucleotide positions 1,186 and 1,187, respectively. The wild-type PpDEK1 fragment (nucleotides 11,763–14,569) was first amplified from genomic P. patens DNA using primers SP-PpCysPc and ASP-PpC2L designed to insert 5′ SmaI and 3′ BsiWI recognition sequences and then cloned into the Zero Blunt PCR cloning vector (Invitrogen), resulting in plasmid pCR-PpCalp. To introduce the two single-point mutations, the pCR-PpCalp plasmid was subjected to mutagenesis using the GeneTailor Site-Directed Mutagenesis System (Invitrogen) and the mutagenic primer pair SP-PpCysMut0 and ASP-PpCysMut0. The mutagenized SmaI/BsiWI restriction fragment of the pCR-PpCalp fragment was finally subcloned into SmaI/BsiWI-digested pTDek1-3′, resulting in construct pArrow-DEK1° (Supplemental Fig. S7A). The construct was digested with SmaI and PacI prior to transformation into P. patens.
Complementation constructs were assembled to reinsert the Linker and LG3 sequences into the deleted loci. Constructs pCR-PpLinker and pCR-PpLG3 were used to reinsert the P. patens DEK1 Linker and DEK1 LG3 sequences into their native loci, respectively. In short, a DNA fragment (5,263 bp) spanning the Linker in addition to 5′ and 3′ flanking regions (nucleotides 7,368–12,630) was PCR amplified from genomic P. patens DNA using primers Linker5′-Fw and In25-R. Similarly, a DNA fragment (3,357 bp) spanning LG3 and 5′ and 3′ flanking regions (nucleotides 9,274–12,630) was PCR amplified using primers ArmSeq3 and In25-R. The resulting fragments were each cloned into the Zero Blunt PCR cloning vector, giving plasmid pCR-PpLinker (Supplemental Fig. S3) and pCR-PpLG3 (Supplemental Fig. S5A), respectively. We also built two vectors aimed to test heterologous LG3 sequences from Arabidopsis and Marchantia polymorpha. In these constructs, chimeric LG3 sequence from Arabidopsis (nucleotides 6,741–7,450; relative to the ATG start site; AT1G55350) and M. polymorpha (nucleotides 1,258,573–1,259,287; scaffold 26, Joint Genome Institute scaffold genomic data version 3.1) flanked by P. patens DEK1 5′ and 3′ targeting sequence were constructed as follows. To generate the Arabidopsis and M. polymorpha Linker In-Fusion inserts, forward and reverse gene-specific primers containing 5′ and 3′ 15-bp extensions complementary to the flanking regions of the P. patens LG3 sequence in the pCR-PpLG3 plasmid were used to PCR amplify Arabidopsis (primers SP-AtLG3-Inf and ASP-AtLG3-Inf) and M. polymorpha (primers SP-MpLG3-Inf and ASP-MpLG3-Inf) from genomic DNA. These inserts were each mixed with linearized vector pCR-PpLG3, produced by PCR amplification using primers SP_Inf2 and ASP_Inf2x to exclude the P. patens LG3 sequence. The inserts and linearized vector were ligated using In-Fusion cloning reactions, resulting in constructs pCR-AtLG3 and pCR-MpLG3 (Supplemental Fig. S5A). The complementation constructs were digested with EcoRI prior to P. patens protoplast transformation. An additional vector, pBHRF-AtLG3, was constructed to target the Arabidopsis LG3 sequence together with HRC into the dek1Δlg3 locus. First, we PCR amplified an In-Fusion fragment containing PpDEK1 nucleotides 8,347 to 11,544 with primers SP_Inf_6N and ASP_Inf_6N and genomic P. patens DNA as template. This insert was mixed with linearized vector pBHRF-ΔlinkerG1, produced by PCR using primers SP_Inf_5N and ASP_Inf_5N. The insert and linearized vector were ligated in an In-Fusion reaction resulting in plasmid pBHRF-Linker, containing the Pp DEK1 nucleotides 7,249 to 11,544 as the 5′ TGS. The Arabidopsis LG3 sequence fragment (nucleotides 6,741–7,450; relative to the ATG start site; AT1G55350) was amplified from genomic Arabidopsis DNA using primers SP-AtLG3_Inf and ASP-LG3_Inf. Vector pBHRF-Linker was PCR amplified to delete the P. patens LG3 sequence using primers SP_Inf2 and ASP_Inf2X. Finally, the linearized vector and the AtLG3 amplicon were ligated in an In-Fusion reaction creating vector pBHRF-AtLG3 (Supplemental Fig. S5B).
P. patens Transformation Procedure
P. patens protoplast production and transformation was performed according to Schaefer and Zrÿd (1997) as modified by Cove et al. (2009) with 15 µg of linearized plasmid DNA used per transformation. Briefly, transformed protoplast regeneration and selection was performed by transferring the tissue to different media according to the following sequence: 7 d of protoplast regeneration on Protoplast regeneration medium for the bottom layer (PRMB), 7 d of selection on BCDA medium supplemented with the appropriate antibiotic, 14 d of growth on BCDA medium, and 7 d on BCDA medium supplemented with the appropriate antibiotic. Resistant plants were individually transferred to fresh BCDA medium and used for genotyping after sufficient growth.
The Cre recombinase procedure to remove the resistant marker from the primary transformant was performed as described previously (Trouiller et al., 2006) with minor modifications. Transformed tissue was grown as the wild type, and protoplast production and transformation was performed using 20 µg of the plasmid pAct-Cre (Trouiller et al., 2006). Diluted protoplast suspensions were plated on cellophane-coated medium (approximately 25,000 counted protoplasts per 9-cm petri dish) to avoid mixing of regenerated plants. Protoplast regeneration and the test procedure were performed as follows: (1) 4 d of protoplast regeneration on PRMB medium; (2) 4 d of protoplast growth on BCDA medium; (3) individual plants transferred to fresh BCDA medium and growth for 8 d; and (4) replica plating of each individual plant onto BCDA medium and BCDA medium supplemented with the appropriate antibiotic. Strains showing loss of antibiotic resistance were selected and grown until sufficient tissue was available for PCR genotyping.
PCR Genotyping of Transformants
Each mutant was subjected to three rounds of PCR genotyping, except as stated otherwise, using the Phire Plant Direct PCR kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The primer sequences used for genotyping each set of transformants are provided in Supplemental Table S3. In the first round of genotyping, deletion mutants were genotyped for the loss of genomic sequence, while in the case of knock-in mutants, we genotyped for the gain of sequence. In the second PCR, we genotyped for proper 5′ and 3′ targeting at the locus using a primer annealing within the DNA fragment used for transformation and a primer annealing 5′ or 3′ to the 5′ or 3′ TGS, respectively. Finally, in the third PCR, we genotyped for single-copy insertions at the locus using primers annealing 5′ and 3′ to the 5′ and 3′ TGSs, respectively.
For dek1ΔlinkerG1 (Supplemental Fig. S1A), first genotyping was performed using primers Δlinker_1F and Δlinker_1R to assess the absence/presence of the Linker in hygromycin-resistant lines. Lines without a detectable PCR product, presumed to have lost the Linker, were tested in the second round of PCR genotyping to check for on-locus targeting using primers Δlinker_2F and 35S_rev1 for 5′ targeting and Term_F and Δlinker_2R for 3′ targeting. Targeted single-copy insertion was assessed in the third PCR genotyping using primers Δlinker_2F and Δlinker_2R. The same rationale was used to genotype dek1Δlinker3G transformants (Supplemental Fig. S2A) using the following primer sets: first genotyping, Δlinker_1F and Δlinker_1R; second genotyping, SP_LOOP_inverse and 35S_rev1 for 5′ targeting and Term_F and gDek1_rev for 3′ targeting; and third genotyping, SP_LOOP_inverse and gDek1_rev. In the case of dek1Δc-linker (Supplemental Fig. S6A), first genotyping was performed using primers ARMseq7 and ASP_PpCALP, showing a distinction between wild-type and transformed plants by a 478-bp reduction in the PCR fragment size with respect to the wild type, corresponding to the size of the deleted fragment. For second genotyping, primers TM2seq1 and 35S_rev1 for 5′ targeting and Term_F and ASP_PpCALP for 3′ targeting were used, while TM2seq1 and ASP_PpCALP were used for third genotyping, assessing single-copy on-locus insertions. For dek1Δlg3, where screening of transformed plants was based on phenotype, two rounds of PCR genotyping were performed (Supplemental Fig. S4A): first PCR using primers ARMseq6 and ASP_PpARM_inf1 that anneal within the targeting DNA, and second PCR using primers TM2seq1 and ASP_PpCALP that anneal outside the targeting DNA. Finally, for dek1° (Supplemental Fig. S7A), targeting was assessed using primers Null_1F and Null_1R with the PCR extension condition set to amplify only nontransformed plants (due to the presence of the resistance cassette in the transformed lines). G418R plants without a detectable wild-type band were assessed for on-locus targeting using primers Null_2F and 35S-rev1 for 5′ targeting and Term-F and Null_2R for 3′ targeting. Next, single-copy integration at the locus was assessed using primers Null_2F and Null_2R.
Molecular Characterization of Transformants
Genomic DNA for Southern-blot analysis was extracted using the Nucleon PhytoPure Genomic DNA Extraction Kit (GE Healthcare). Southern-blot analysis was performed as described by Perroud and Quatrano (2006) using 1 µg of DNA per digestion. Probes were digoxigenin labeled using the DIG Probe PCR synthesis kit (Roche) according to the manufacturer’s instructions. Templates for PCR amplification of probes were the corresponding DNA constructs used for transformation. The sequences of the primers used to generate the probes are provided in Supplemental Table S3, and the hybridization sites of each probe are schematically illustrated in Supplemental Figures S1A to S7A.
RT-PCR and DNA sequencing were used to analyze the mutant PpDEK1 cDNA to verify proper deletion/insertion and splicing of the transcript. Total RNA was isolated from protonema tissue using the Plant RNeasy Kit (Qiagen). DNaseI-treated total RNA (500 ng) was reverse transcribed using 100-unit SuperScript III Reverse Transcriptase (Invitrogen) primed with 200 ng of random hexamers at 55°C for 60 min. Phusion High-Fidelity DNA polymerase was used to amplify the DEK1 cDNA target using primers Ex7-F and Ex30-R, annealing in exons 7 and 30, respectively. After Exo SAP treatment to remove unused primers, the PCR product was sequenced with appropriate primers using BigDye version 3.1 chemistry (ABI) according to the SteP method (Platt et al., 2007). Extension products were precipitated using sodium acetate/ethanol, dissolved in Hi-Di formamide (ABI), and finally sequenced by Capillary Electrophoresis using the 3130xL Genetic Analyzer. DNA sequences were analyzed using Genomic Workbench version 7.
Real-Time Quantitative RT-PCR
Real-time quantitative RT-PCR was performed to determine the transcript level of DEK1 in the P. patens mutants relative to the wild-type control. RNA was extracted in two independent biological replicates using the Plant RNeasy Kit (Qiagen), and cDNA was prepared as described previously. Transcript level analysis was performed in duplicates using 0.2 µm forward and reverse primers and 1:4 diluted cDNA with the HOT FIREPol EvaGreen qPCR Mix Plus (ROX; Solis Biodyne) on the 7500 Fast Real-Time PCR Systems (ABI). Real-time quantitative RT-PCR cycling was performed as follows: 95°C for 15 min followed by 40 cycles of 95°C for 30 s, 58°C for 20 s, and 72°C for 32 s for amplification of all targets. The real-time quantitative RT-PCR data were analyzed using the LinRegPCR software version 11.0 (Ruijter et al., 2009) to obtain the threshold cycle number and the mean efficiency values for each sample and target, respectively. Relative mRNA levels of the targets were normalized to the transcript level of RSB (Pp3c18_1350V3.1; Xiao et al., 2011). Fold expression in mutant samples was calculated relative to the wild type according to the method of Pfaffl (2001). The following gene-specific primer pairs were used for target amplification: for DEK1, TMq-F and TMq-R; and for RSB, RSBq-F and RSBq-R. Each primer pair generated a single, expected PCR product based on post-PCR dissociation analyses.
Accession Numbers
Supplemental Table S1 provides the accession numbers for all sequences used in this study.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Vector construction, targeted DEK1 Linker deletion, and molecular characterization of the P. patens dek1ΔlinkerG1/cre mutant line.
Supplemental Figure S2. Vector construction, targeted insertion, and molecular characterization of the P. patens dek1ΔlinkerG3/cre#123 mutant line.
Supplemental Figure S3. Vector construction, targeted knock in of the P. patens DEK1 Linker, PCR genotyping, and gametophore morphology of the complemented line.
Supplemental Figure S4. Vector construction, targeted insertion of LinkerΔLG3, and molecular characterization of the P. patens dek1Δlg3 mutant line.
Supplemental Figure S5. Vector construction, targeted knock in of P. patens, M. polymorpha, and Arabidopsis DEK1 LG3, and PCR genotyping.
Supplemental Figure S6. Vector construction, targeted insertion, and molecular characterization of the P. patens dek1Δc-linker mutant line.
Supplemental Figure S7. Vector construction, targeted insertion, and molecular characterization of the P. patens dek1° mutant line.
Supplemental Table S1. Accession numbers for the orthologous DEK1 Linker protein sequences used in this study.
Supplemental Table S2. Amino acid sequence identity between each pair of DEK1 Linker sequences.
Supplemental Table S3. Oligonucleotide primers and their sequences used in this study.
Supplemental Data S1. Charophyte algae DEK1 Linker protein sequences used in this study.
Supplemental Data S2. Alignment of DEK1 Linker protein sequences in fasta format.
Supplementary Material
Acknowledgments
We thank the Imaging Centre, NMBU, for providing access to microscopes and technical assistance with microscopic analysis; Dr. Stefanie Reißmann and Dr. Regine Kahmann's group at the MPI for Terrestrial Microbiology for the use of confocal microscopy equipment; Dr. Rafi Ahmad at Hedmark University of Applied Sciences for valuable help with modeling of the DEK1 LG3 domain; and Charles Delwiche at the University of Maryland, College Park, for sharing the charophyte DEK1 sequences with us.
Glossary
- 2D
two-dimensional
- 3D
three-dimensional
- TM
transmembrane
- TGS
targeting sequence
- RT
reverse transcription
Footnotes
This work was supported by the Norwegian Research Council (grant no. FRIMEDBIO 240343 to O.-A.O. and support to A.E.A.), the Norwegian University for Life Sciences (to P.-F.P. and V.D.), and the Hedmark University of Applied Sciences (to W.J. and A.E.A.).
Articles can be viewed without a subscription.
References
- Aoyama T, Hiwatashi Y, Shigyo M, Kofuji R, Kubo M, Ito M, Hasebe M (2012) AP2-type transcription factors determine stem cell identity in the moss Physcomitrella patens. Development 139: 3120–3129 [DOI] [PubMed] [Google Scholar]
- Arthur JSC, Gauthier S, Elce JS (1995) Active site residues in m-calpain: identification by site-directed mutagenesis. FEBS Lett 368: 397–400 [DOI] [PubMed] [Google Scholar]
- Becraft PW, Li K, Dey N, Asuncion-Crabb Y (2002) The maize dek1 gene functions in embryonic pattern formation and cell fate specification. Development 129: 5217–5225 [DOI] [PubMed] [Google Scholar]
- Chandra NR, Prabu MM, Suguna K, Vijayan M (2001) Structural similarity and functional diversity in proteins containing the legume lectin fold. Protein Eng 14: 857–866 [DOI] [PubMed] [Google Scholar]
- Cove DJ, Perroud PF, Charron AJ, McDaniel SF, Khandelwal A, Quatrano RS (2009) The moss Physcomitrella patens: a novel model system for plant development and genomic studies. Cold Spring Harb Protoc 2009: pdb.emo115. [DOI] [PubMed] [Google Scholar]
- Demko V, Perroud PF, Johansen W, Delwiche CF, Cooper ED, Remme P, Ako AE, Kugler KG, Mayer KFX, Quatrano R, et al. (2014) Genetic analysis of DEFECTIVE KERNEL1 loop function in three-dimensional body patterning in Physcomitrella patens. Plant Physiol 166: 903–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Clos TW. (2013) Pentraxins: structure, function, and role in inflammation. ISRN Inflamm 2013: 379040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festucci-Buselli RA, Otoni WC, Joshi CP (2007) Structure, organization, and functions of cellulose synthase complexes in higher plants. Braz J Plant Physiol 19: 1–13 [Google Scholar]
- Frank MH, Edwards MB, Schultz ER, McKain MR, Fei Z, Sørensen I, Rose JKC, Scanlon MJ (2015) Dissecting the molecular signatures of apical cell-type shoot meristems from two ancient land plant lineages. New Phytol 207: 893–904 [DOI] [PubMed] [Google Scholar]
- Goss CA, Brockmann DJ, Bushoven JT, Roberts AW (2012) A CELLULOSE SYNTHASE (CESA) gene essential for gametophore morphogenesis in the moss Physcomitrella patens. Planta 235: 1355–1367 [DOI] [PubMed] [Google Scholar]
- Harrison CJ. (2015) Shooting through time: new insights from transcriptomic data. Trends Plant Sci 20: 468–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob-Wilk D, Kurek I, Hogan P, Delmer DP (2006) The cotton fiber zinc-binding domain of cellulose synthase A1 from Gossypium hirsutum displays rapid turnover in vitro and in vivo. Proc Natl Acad Sci USA 103: 12191–12196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KL, Faulkner C, Jeffree CE, Ingram GC (2008) The phytocalpain defective kernel 1 is a novel Arabidopsis growth regulator whose activity is regulated by proteolytic processing. Plant Cell 20: 2619–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorchid A, Ikura M (2002) How calpain is activated by calcium. Nat Struct Biol 9: 239–241 [DOI] [PubMed] [Google Scholar]
- Kofuji R, Hasebe M (2014) Eight types of stem cells in the life cycle of the moss Physcomitrella patens. Curr Opin Plant Biol 17: 13–21 [DOI] [PubMed] [Google Scholar]
- Landberg K, Pederson ERA, Viaene T, Bozorg B, Friml J, Jönsson H, Thelander M, Sundberg E (2013) The MOSS Physcomitrella patens reproductive organ development is highly organized, affected by the two SHI/STY genes and by the level of active auxin in the SHI/STY expression domain. Plant Physiol 162: 1406–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Demko V, Wilson RC, Johnson KA, Ahmad R, Perroud PF, Quatrano R, Zhao S, Shalchian-Tabrizi K, Otegui MS, et al. (2013) The catalytic domain CysPc of the DEK1 calpain is functionally conserved in land plants. Plant J 75: 742–754 [DOI] [PubMed] [Google Scholar]
- Lid SE, Gruis D, Jung R, Lorentzen JA, Ananiev E, Chamberlin M, Niu X, Meeley R, Nichols S, Olsen OA (2002) The defective kernel 1 (dek1) gene required for aleurone cell development in the endosperm of maize grains encodes a membrane protein of the calpain gene superfamily. Proc Natl Acad Sci USA 99: 5460–5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lid SE, Olsen L, Nestestog R, Aukerman M, Brown RC, Lemmon B, Mucha M, Opsahl-Sorteberg HG, Olsen OA (2005) Mutation in the Arabidopsis thaliana DEK1 calpain gene perturbs endosperm and embryo development while over-expression affects organ development globally. Planta 221: 339–351 [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL (2002) A Ca2+ switch aligns the active site of calpain. Cell 108: 649–660 [DOI] [PubMed] [Google Scholar]
- Olsen OA, Perroud PF, Johansen W, Demko V (2015) DEK1: missing piece in puzzle of plant development. Trends Plant Sci 20: 70–71 [DOI] [PubMed] [Google Scholar]
- Perroud PF, Quatrano RS (2006) The role of ARPC4 in tip growth and alignment of the polar axis in filaments of Physcomitrella patens. Cell Motil Cytoskeleton 63: 162–171 [DOI] [PubMed] [Google Scholar]
- Perroud PF, Cove DJ, Quatrano RS, McDaniel SF (2011) An experimental method to facilitate the identification of hybrid sporophytes in the moss Physcomitrella patens using fluorescent tagged lines. New Phytol 191: 301–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroud PF, Demko V, Johansen W, Wilson RC, Olsen OA, Quatrano RS (2014) Defective Kernel 1 (DEK1) is required for three-dimensional growth in Physcomitrella patens. New Phytol 203: 794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt AR, Woodhall RW, George AR Jr (2007) Improved DNA sequencing quality and efficiency using an optimized fast cycle sequencing protocol. Biotechniques 43: 58–62 [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires ND, Dolan L (2012) Morphological evolution in land plants: new designs with old genes. Philos Trans R Soc Lond B Biol Sci 367: 508–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge MJ, Bezanilla M (2010) Evolutionary crossroads in developmental biology: Physcomitrella patens. Development 137: 3535–3543 [DOI] [PubMed] [Google Scholar]
- Rudenko G, Hohenester E, Muller YA (2001) LG/LNS domains: multiple functions one business end? Trends Biochem Sci 26: 363–368 [DOI] [PubMed] [Google Scholar]
- Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, van den Hoff MJB, Moorman AFM (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders MJ, Hepler PK (1982) Calcium ionophore a23187 stimulates cytokinin-like mitosis in funaria. Science 217: 943–945 [DOI] [PubMed] [Google Scholar]
- Schaefer DG, Delacote F, Charlot F, Vrielynck N, Guyon-Debast A, Le Guin S, Neuhaus JM, Doutriaux MP, Nogué F (2010) RAD51 loss of function abolishes gene targeting and de-represses illegitimate integration in the moss Physcomitrella patens. DNA Repair (Amst) 9: 526–533 [DOI] [PubMed] [Google Scholar]
- Schaefer DG, Zrÿd JP (1997) Efficient gene targeting in the moss Physcomitrella patens. Plant J 11: 1195–1206 [DOI] [PubMed] [Google Scholar]
- Shinkai-Ouchi F, Koyama S, Ono Y, Hata S, Ojima K, Shindo M, duVerle D, Ueno M, Kitamura F, Doi N, et al. (2016) Predictions of cleavability of calpain proteolysis by quantitative structure-activity relationship analysis using newly determined cleavage sites and catalytic efficiencies of an oligopeptide array. Mol Cell Proteomics 15: 1262–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorimachi H, Mamitsuka H, Ono Y (2012) Understanding the substrate specificity of conventional calpains. Biol Chem 393: 853–871 [DOI] [PubMed] [Google Scholar]
- Strobl S, Fernandez-Catalan C, Braun M, Huber R, Masumoto H, Nakagawa K, Irie A, Sorimachi H, Bourenkow G, Bartunik H, et al. (2000) The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc Natl Acad Sci USA 97: 588–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P, Buzder-Lantos P, Tantos A, Farkas A, Szilágyi A, Bánóczi Z, Hudecz F, Friedrich P (2004) On the sequential determinants of calpain cleavage. J Biol Chem 279: 20775–20785 [DOI] [PubMed] [Google Scholar]
- Trouiller B, Schaefer DG, Charlot F, Nogué F (2006) MSH2 is essential for the preservation of genome integrity and prevents homeologous recombination in the moss Physcomitrella patens. Nucleic Acids Res 34: 232–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Barry JK, Min Z, Tordsen G, Rao AG, Olsen OA (2003) The calpain domain of the maize DEK1 protein contains the conserved catalytic triad and functions as a cysteine proteinase. J Biol Chem 278: 34467–34474 [DOI] [PubMed] [Google Scholar]
- Wendt A, Thompson VF, Goll DE (2004) Interaction of calpastatin with calpain: a review. Biol Chem 385: 465–472 [DOI] [PubMed] [Google Scholar]
- Xiao L, Wang H, Wan P, Kuang T, He Y (2011) Genome-wide transcriptome analysis of gametophyte development in Physcomitrella patens. BMC Plant Biol 11: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N. (2013) Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem Sci 38: 151–159 [DOI] [PubMed] [Google Scholar]
- Zhao S, Liang Z, Demko V, Wilson R, Johansen W, Olsen OA, Shalchian-Tabrizi K (2012) Massive expansion of the calpain gene family in unicellular eukaryotes. BMC Evol Biol 12: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.