

Abstract
Intermediate neural progenitors (INPs) need to avoid both dedifferentiation and differentiation during neurogenesis, but the underlying mechanisms are not well understood. In Drosophila, the Ets protein Pointed P1 (PntP1) is required to generate INPs from type II neuroblasts. Here, we investigated how PntP1 promotes INP generation. By generating pntP1-specific mutants and using RNAi knockdown, we show that the loss of PntP1 leads to both an increase in type II neuroblast number and the elimination of INPs. The elimination of INPs results from the premature differentiation of INPs due to ectopic Prospero expression in newly generated immature INPs (imINPs), whereas the increase in type II neuroblasts results from the dedifferentiation of imINPs due to loss of Earmuff at later stages of imINP development. Furthermore, reducing Buttonhead enhances the loss of INPs in pntP1 mutants, suggesting that PntP1 and Buttonhead act cooperatively to prevent premature INP differentiation. Our results demonstrate that PntP1 prevents both the premature differentiation and the dedifferentiation of INPs by regulating the expression of distinct target genes at different stages of imINP development.
KEY WORDS: Drosophila, Neuroblast, Intermediate neural progenitor, Earmuff, Pointed, Buttonhead
Summary: The Ets transcription factor Pointed maintains the balance between differentiation and dedifferentiation of intermediate neural progenitors by acting through two distinct pathways.
INTRODUCTION
The generation of brain complexity in higher order animals involves the production of intermediate neural progenitors (INPs) from neural stem cells (NSCs) (Lui et al., 2011; Pontious et al., 2008). INPs transiently proliferate to amplify the NSC output. However, INPs have very limited developmental potential and can generate only fate-restricted progeny. Defects in maintaining INP proliferation owing to precocious differentiation and cell cycle exit could lead to reduced brain complexity and brain malformation (Colasante et al., 2015; Quinn et al., 2007; Reillo et al., 2011), whereas the aberrant dedifferentiation of INPs may result in tumorigenic overgrowth (Liu et al., 2011; Walton et al., 2009). Therefore, INPs need to avoid not only differentiation but also dedifferentiation in order to produce a precise number of progeny with specific cell fates. It is crucial to decipher the mechanisms that prevent INP differentiation and dedifferentiation in order to understand the generation of brain complexity and the formation of brain tumors.
In Drosophila larval brains, INPs have similar roles to mammalian INPs in amplifying neuronal output from type II neuroblasts (NBs). Unlike ganglion mother cells (GMCs) generated from type I NBs, which divide only once and produce two neurons (Hartenstein et al., 2008), each INP generated from type II NBs produces ∼10 neurons by dividing in multiple rounds, thereby generating several GMCs (Bello et al., 2008; Boone and Doe, 2008; Bowman et al., 2008; Wang et al., 2014). INPs must maintain their self-renewal and avoid differentiation while dividing to produce neurons. However, newly generated INPs are immature and are prone to dedifferentiate into NBs if they fail to differentiate into mature INPs (Bowman et al., 2008; Eroglu et al., 2014; Koe et al., 2014). Thus, Drosophila INPs provide an excellent model for studying how INPs avoid dedifferentiation and premature differentiation.
In recent years, studies have begun to identify key genetic programs that prevent the dedifferentiation or premature differentiation of INPs. For example, preventing the dedifferentiation of immature INPs (imINPs) requires the cell fate determinants Brain tumor (Brat) and Numb, the Fez family transcription factor Earmuff (Erm), as well as the SWI/SNF and Histone deacetylase 3 (HDAC3) chromatin remodeling complexes (Bowman et al., 2008; Eroglu et al., 2014; Koe et al., 2014; Weng et al., 2010). The loss of any of these factors could result in imINP dedifferentiation and supernumerary type II NBs. By contrast, preventing the premature differentiation of INPs requires the suppression of the homeodomain protein Prospero (Pros) in INPs. Our recent studies show that the Sp8 family transcription factor Buttonhead (Btd) is required to suppress Pros in imINPs (Xie et al., 2014). The loss of Btd results in ectopic Pros expression in imINPs and the premature differentiation of INPs into GMCs. However, our understanding of the mechanisms that prevent the dedifferentiation and premature differentiation of INPs is still incomplete.
Our previous studies revealed that the Ets family transcription factor Pointed P1 (PntP1) is specifically expressed in type II NBs and imINPs (Zhu et al., 2011). Functional analyses show that inhibiting PntP1 activity by its antagonist Yan (Anterior open – FlyBase) transforms type II NBs into type I NBs and eliminates INPs, whereas PntP1 misexpression transforms type I NBs into type II NBs and promotes INP generation, suggesting that PntP1 is required for type II NB specification and INP generation (Zhu et al., 2011). However, it is still unknown exactly how PntP1 promotes of INP generation
In this study, we investigated PntP1 function in imINPs by taking advantage of our novel pntP1-specific mutant alleles and using RNAi knockdown. We show that PntP1 prevents both the dedifferentiation and premature differentiation of INPs by regulating the expression of distinct target genes at different stages of imINP development. Our work reveals the mechanistic details of the PntP1-mediated generation of INPs in type II NB lineages and provides novel insights into the mechanisms that maintain the balance between INP dedifferentiation and differentiation.
RESULTS
Partial loss of PntP1 results in the elimination of INPs and an increase in the number of type II NBs
The only currently available pntP1-specific allele is a null allele, pntΔ33, in which the pntP1-specific exon 1, including part of the coding region, is deleted (O'Neill et al., 1994). However, pntΔ33 is not ideal for loss-of-function phenotypic analyses in Drosophila larval type II NB lineages for two reasons. First, PntP1 proteins perdure in pntΔ33 mutant type II NB clones even at late third instar larval stages (Zhu et al., 2011). Second, the embryonic lethality of pntΔ33 makes it impossible to examine type II NB lineage development in pntΔ33 homozygous mutant larvae. Therefore, we decided to generate novel pntP1-specific mutant alleles using CRISPR/Cas9 technology (Jinek et al., 2012; Port et al., 2014; Sebo et al., 2014) to investigate how PntP1 regulates INP generation. We chose two 20 nt sequences that are 249 and 536 bp downstream of the translation start codon as gRNA targets (Fig. 1Aa,b). We generated two mutant lines, named pntP182 and pntP190, which carry small indels. These indels cause frame shifts and premature stop codons, resulting in deletions of 535 and 445 amino acids at the C-terminus, including the Ets DNA-binding domain, in the pntP182 and pntP190 mutants, respectively (Fig. 1Ab,c). Both pntP190 and pntP182 homozygotes are late third instar larval lethal, indicating that they are likely to be hypomorphic alleles.
Fig. 1.

Partial loss of PntP1 increases the number of type II NBs and eliminates INPs. (A) The generation of Drosophila pntP1-specific mutants. (a) The gene structure of pntP1 and gRNA target sites (arrowheads). Gray boxes, untranslated regions; orange boxes, coding regions. (b) Sequences of gRNA target sites (red) and indels in pntP182 and pntP190 mutants. (c) The protein structure of PntP1 and truncated PntP1 proteins generated from pntP182 and pntP190 mutants. Gray areas, non-specific sequences caused by frame shifts. (B,B′) A wild-type (WT) brain lobe. Type II NB lineages are labeled with mCD8-GFP driven by pntP1-GAL4. Arrows, Ase− type II NBs; arrowheads, Dpn+ Ase+ INPs. (C-F′) pntP1 mutant brains of the indicated genotypes. Brains are stained with phalloidin to outline NBs. The number of Ase− type II NBs (arrows) is significantly increased in pntP190 (C,C′), pntP190/pntΔ88 (E,E′) and pntP182/pntΔ88 (F,F′) mutants but not in pntP182 (D,D′). Dashed circles outline type II NB lineages that generated GMCs (open arrowheads) but not INPs. Insets are enlarged views of lineages without INPs. (G-H′) Pnt knockdown leads to ectopic Ase expression in type II NBs (green arrows) and elimination of INPs in most lineages (dashed lines). Yellow arrows, Ase− NBs; open arrowheads, GMCs. (I-K) Quantification of the number of Ase− type II NBs (I), percentage of Ase− NBs with INPs (J) and number of INPs (K) in pnt mutants. (L-O) Quantification of the total number of type II NBs (L), percentage of lineages with Ase+ NBs (M), percentage of lineages with INPs (N) and number of INPs (O) in Pnt knockdown brains. Sample sizes are indicated above each bar. *P<0.05, **P<0.01. (P) Schematic diagrams of wild-type type II NB lineages and phenotypes in pnt mutant or Pnt knockdown type II NB lineages. Scale bars: 20 μm.
We then examined type II NB lineages in pntP190 and pntP182 homozygous mutant larvae in order to investigate how the loss of PntP1 would affect type II NB lineage development. In wild-type larval brains, there are only eight type II NBs per lobe, which can be distinguished from type I NBs by the absence of the proneural protein Asense (Ase). Each type II NB lineage contains ∼20-25 mature INPs, which express both Ase and the bHLH protein Deadpan (Dpn) (Fig. 1B,B′,I-K,P) (Bello et al., 2008; Boone and Doe, 2008; Bowman et al., 2008). We found that in pntP190 mutant brains, the number of Ase− type II NBs is doubled. However, the number of INPs was reduced by 50% and ∼25% of Ase− type II NBs did not produce mature INPs. Instead, only a few Ase+ Dpn− cells that resembled GMCs were generated (Fig. 1C,C′,I-K,P). In pntP182 mutants, the loss of INPs was even more severe. There were only two or three INPs per Ase− NB on average and ∼75% of the Ase− type II NBs did not produce INPs (Fig. 1D,D′,I-K,P). However, in contrast to the increase in the number of Ase− type II NBs in pntP190 mutants, the number of Ase− type II NBs was reduced to about six per lobe in the pntP182 mutants. Because PntP1 is required to suppress Ase expression in type II NBs (Zhu et al., 2011), it is possible that some type II NBs might have become Ase+ in the pntP182 mutants (and possibly in pntP190 mutants as well), as observed in Pnt knockdown type II NBs (see below).
To verify whether the phenotypes observed in the pntP190 and pntP182 mutants are indeed caused by mutations of pntP1, we examined phenotypes in pntP190/pntΔ88 or pntP182/pntΔ88 transheterozygotes and compared them with phenotypes in Pnt knockdown type II NB lineages. pntΔ88 carries a deletion that covers the entire pntP1 transcript, including the 3′ exons shared by other pnt transcripts (Brunner et al., 1994). We found that pntP190/pntΔ88 mutants had similar, but more severe, phenotypes compared with pntP190 mutants (Fig. 1E,E′,I-K,P). In pntP190/pntΔ88 mutants, the number of Ase− type II NBs was further increased to ∼25 per lobe and the number of INPs was reduced by nearly 70%. In the pntP182/pntΔ88 mutants, we observed a similar loss of INPs as in the pntP182 mutants, but a slight increase in the number of Ase− type II NBs (Fig. 1F,F′,I-K,P). Consistently, we observed a 50-100% increase in the total number of type II NBs and the complete elimination of INPs in over 90% of type II NB lineages when Pnt was knocked down by two independent UAS-pnt RNAi lines driven by type II NB lineage-specific pntP1-GAL4. Furthermore, Ase was ectopically expressed in 60-80% of Pnt knockdown type II NBs (Fig. 1G-H′,L-P), which is in accord with our previous results showing that the Yan-mediated inhibition of Pnt activity leads to ectopic ase activation in type II NBs. These data show that the loss of PntP1 leads to not only the transformation of type II NBs into type I NBs as we previously reported (Zhu et al., 2011) but also the elimination of INPs and the generation of extra type II NBs (Fig. 1P).
PntP1 suppresses the expression of Pros in Ase− imINPs and prevents the premature differentiation of INPs into GMCs
We then examined the expression of Pros in imINPs and the identity of the Ase+ progeny in pnt mutant or Pnt knockdown type II NB lineages without INPs to investigate why the loss of PntP1 led to the elimination of mature INPs. During INP maturation, newly generated imINPs first differentiate from an Ase− state to an Ase+ state before they fully mature. Pros is not normally expressed in type II NBs or imINPs (Fig. 2A-A″) (Bowman et al., 2008). Our recent studies show that in the absence of Btd, Ase− imINPs ectopically express nuclear Pros, which promotes the premature differentiation of INPs into GMCs and eliminates mature INPs (Xie et al., 2014). Therefore, we wondered whether the elimination of INPs resulting from the loss of PntP1 was also due to the Pros-mediated premature differentiation of INPs into GMCs, which express both Ase and nuclear Pros. We examined Pros expression in the Pnt knockdown and pntP182 mutant type II NB lineages that showed a more consistent loss of INPs. Indeed, unlike in normal type II NB lineages, nuclear Pros was consistently expressed in newly generated Ase− imINPs, which can be identified as Ase− cells adjacent to the NBs, in the Pnt knockdown and pntP182 mutant type II NB lineages. Furthermore, the Ase+ progeny that were generated in lineages without mature INPs also expressed nuclear Pros, indicating that they were GMCs (Fig. 2B-D″). These results suggest that PntP1 is required to suppress Pros expression in newly generated imINPs and to prevent imINPs from prematurely differentiating into GMCs.
Fig. 2.
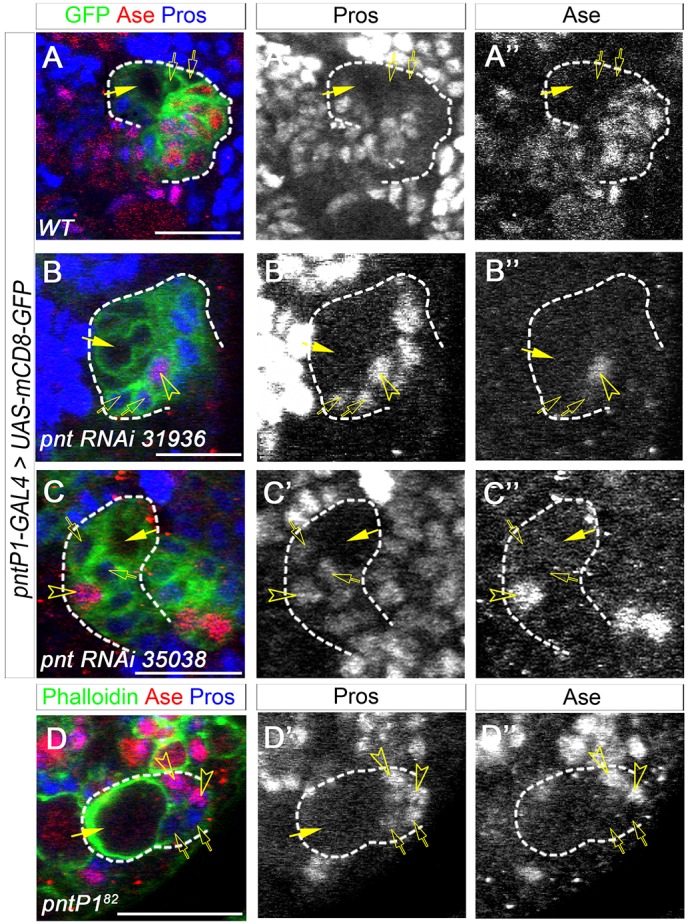
Loss of PntP1 results in ectopic nuclear Pros expression in imINPs. (A-A″) Pros is not expressed in Ase− imINPs (open arrows) in a wild-type GFP-labeled type II NB lineage. Arrows, type II NBs. (B-D″) Nuclear Pros is detected in Ase− imINPs (open arrows) and Ase+ progeny (open arrowheads) in Pnt knockdown (B-C″) and pntP182 mutant (D-D″) type II NB lineages. Scale bars: 20 μm.
Ectopic pros expression in imINPs is responsible for the loss of INPs
Given that the main function of Pros is to promote cell cycle exit and differentiation (Choksi et al., 2006; Maurange et al., 2008), it is very likely that the loss of INPs resulting from the loss of PntP1 is due to the ectopic expression of nuclear Pros in imINPs. To test this, we examined whether reducing Pros expression could rescue the loss of INPs in Pnt knockdown type II NB lineages. Therefore, we knocked down Pnt in a pros17 heterozygous background. In the pros17 heterozygous mutants, type II NB lineages produced similar numbers of INPs as in the wild type (Fig. 3A-B″,E,F). However, unlike the Pnt knockdown in the wild-type background, which eliminated INPs in nearly all type II NB lineages, Pnt knockdown in pros17 heterozygotes only eliminated INPs in ∼50% of lineages and there were still an average of about six INPs per lineage (Fig. 3C-F), suggesting that reducing Pros expression partially rescued the loss of INPs in the Pnt knockdown type II NB lineages. These results indicate that the ectopic nuclear Pros expression in imINPs is responsible for the loss of INPs that results from PntP1 loss.
Fig. 3.

Reducing Pros rescues the loss of INPs resulting from Pnt knockdown. (A-D″) Type II NB lineages are labeled with mCD8-GFP driven by pntP1-GAL4. Arrows, type II NBs; arrowheads, INPs. (A-B″) Wild-type (A-A″) or pros17 heterozygous (B-B″) brains. Each type II NB lineage has multiple mature INPs. (C-C″) INPs are largely eliminated in Pnt knockdown type II NB lineages. Note that most type II NBs become Ase+ (green arrows). (D-D″) INPs are produced in a subset of type II NB lineages and Ase remains suppressed in most type II NBs (arrows) when Pnt is knocked down in pros17/+ heterozygotes. (E-G) Quantification of the percentage of lineages with INPs (E), the number of INPs (F) and the percentage of lineages with Ase− NBs (G). **P<0.01. Scale bars: 20 μm.
In addition to the rescue of INPs, we found that reducing Pros expression partially restored the suppression of Ase in Pnt knockdown type II NBs. When Pnt was knocked down in the wild-type background, only 25% of type II NBs remained Ase−. However, when Pnt was knocked down in the pros17 heterozygous background, which still increased the total number of type II NBs to 15.8±3.1 per lobe (mean±s.d., n=13), ∼70% of type II NBs were Ase− (Fig. 3C-D″,G). Because nuclear Pros is not detected in the Pnt knockdown type II NBs, the restoration of Ase suppression in Pnt knockdown type II NBs is likely to be an indirect effect. We previously reported that maintaining PntP1 expression in type II NBs may require a feedback signal from INPs, and the loss INPs could lead to a loss/reduction of PntP1 expression in type II NBs (Xie et al., 2014). Therefore, one possibility is that the rescue of INPs by the reduction of Pros might help restore the feedback signal, which might in turn partially restore the expression of PntP1 and the suppression of Ase in the NBs, as occurs in btd mutant clones (Xie et al., 2014). However, we could not detect PntP1 proteins after knocking down PntP1 in either the wild-type or pros17 heterozygous background, suggesting that the partially restored PntP1 expression could still be below the detection limit (data not shown)
PntP1 and Btd genetically interact to inhibit premature INP differentiation and suppress Ase expression in type II NBs
Because the loss of PntP1 or Btd leads to similar Pros-mediated premature differentiation of INPs, we next investigated whether PntP1 and Btd function in the same pathway by performing genetic interaction tests. We examined whether reducing the expression of Btd would enhance the loss of INPs in pntP190 homozygous mutants, which show a less severe loss of INPs and might be more sensitive to the reduction in Btd expression. We used btdXG81 or btd-GAL4 heterozygous mutants to reduce Btd expression. btdXG81 is a missense loss-of-function allele, and btd-GAL4 is a lethal p{GAL4} insertion allele, in which GAL4 is integrated into the btd promoter (Estella and Mann, 2010; Wimmer et al., 1993). We labeled type II NB lineages with mCD8-GFP driven by btd-GAL4, which is only expressed in type II NB lineages on the dorsal side of larval brains (Xie et al., 2014), to quantify the number of INPs in btd-GAL4/+; pntP190 mutants. For the pntP190 or btdXG81/+; pntP190 mutants, we focused on Ase− type II NBs for the quantification because we could not identify type II NBs if they became Ase+. In the pntP190 homozygous mutants the average number of INPs per Ase− NB was reduced by ∼50%, and ∼25% of the Ase− NBs did not have associated INPs (Fig. 4A,A′,C,C′,G,H), whereas btdXG81 or btd-GAL4 heterozygous mutants did not exhibit an obvious loss of INPs (Fig. 4B,B′,E,E′,G,H). However, the loss of INPs in the pntP190 mutant type II NB lineages was dramatically enhanced in the btdXG81 or btd-GAL4 heterozygous mutant background. In the btd-GAL4/+; pntP190 or btdXG81/+; pntP190 mutants, the number of INPs was reduced by more than 80%, and ∼90% of NBs did not have associated INPs (Fig. 4D,D′,F,F′,G,H). The enhancement of INP loss in pntP190 mutants by the reduction of Btd suggests that Btd and PntP1 genetically interact to prevent the premature differentiation of INPs and that Btd and PntP1 are likely to function in the same pathway.
Fig. 4.

PntP1 and Btd genetically interact to suppress premature differentiation of INPs and Ase expression in type II NBs. (A-F′) Type II NB lineages are labeled with either mCD8-GFP driven by pntP1-GAL4 (A) or btd-GAL4 (E,F) or by phalloidin staining (C,B,D). Arrows, type II NBs; arrowheads, INPs. (A-B′,E,E′) Wild-type (A,A′), btdXG81/+ (B,B′) and btd-GAL4/+ (E,E′) larval brains. Each type II NBs is associated with multiple INPs. (C,C′) pntP190 mutant brain showing an increased number of Ase− type II NBs and loss of INPs in a small subset of type II NB lineages (dashed circles indicate examples). (D,D′,F,F′) btdXG81/+ pntP190 (D,D′) or btd-GAL4/+ pntP190 (F,F′) mutants have decreased numbers of Ase− type II NBs and no INPs in most type II NB lineages (dashed lines). However, the total number of GFP-labeled NBs is increased in btd-GAL4/+ pntP190 mutants but the majority of them become Ase+ (green arrows) (F,F′). (G-J) Quantification of the number of INPs per lineage (for wild type, btdXG81/+ and btd-GAL4/+, and btd-GAL4/+ pntP190) or per Ase− NB (for pntP190 or btdXG81/+ pntP190) (G), percentage of lineages (or Ase− NBs) with INPs (H), and the total number of type II NBs (I) or the number of Ase− type II NBs (J). **P<0.01. Scale bars: 20 μm.
Interestingly, in addition to enhancing the loss of INPs, reducing Btd expression also promoted ectopic activation of ase in pntP190 mutant type II NBs. The pntP190 mutants usually had ∼13 Ase− NBs per lobe. However, the number of Ase− type II NBs was reduced to four to six per lobe in the btdXG81/+; pntP190 or btd-GAL4/+; pntP190 mutants, although the btdXG81 and btd-GAL4 heterozygotes had the same number of Ase− type II NBs as the wild type (Fig. 4C-F′,I,J). By labeling type II NB lineages with mCD8-GFP driven by btd-GAL4, we found that the total number of type II NBs in the btd-GAL4/+; pntP190 mutants was still significantly increased compared with the wild type, similar to the pntP190 mutants, but over 60% of the type II NBs were Ase+ (Fig. 4F,F′,I,J), indicating that the reduction of Ase− type II NBs in the btd-GAL4/+; pntP190 mutants (and probably in the btdXG81/+; pntP190 mutants as well) was due to the ectopic activation of ase in the NBs. This ectopic ase activation is likely to have occurred throughout type II NB lineage development because the ectopic Ase expression could already be observed at 1 day after larval hatching, and the number of Ase+ type II NBs continued to increase as the total number of type II NBs increased from early to late larval stages (Fig. S1). The ectopic ase activation in pntP190 mutant type II NBs resulting from the reduction of Btd expression suggests that PntP1 and Btd function cooperatively to suppress Ase expression in type II NBs.
Pnt knockdown in Ase− imINPs leads to the generation of extra type II NBs
In addition to the elimination of INPs, the loss of PntP1 also increased the number of type II NBs. We next investigated the cellular origin of the extra type II NBs. Several previous studies have shown that imINPs are not fully committed to their cell fate and can revert back to the NB fate in the absence of tumor suppressors such as Brat, Numb or Erm (Bowman et al., 2008; Weng et al., 2010). Because PntP1 is highly expressed in imINPs (Zhu et al., 2011), we wondered whether the generation of ectopic type II NBs resulting from the loss of PntP1 was also due to imINP dedifferentiation. To test this, we tried to knockdown Pnt in imINPs using erm-GAL4(III) or erm-GAL4(II) (Xiao et al., 2012). erm-GAL4(III) is mainly expressed in Ase+ imINPs, whereas erm-GAL4(II) is expressed in both Ase− and Ase+ imINPs, except for the newly generated Ase− imINPs. Our results showed that Pnt knockdown by erm-GAL4(III) did not produce any extra type II NBs in third instar larval brains (Fig. 5A-B″,E), whereas Pnt knockdown by erm-GAL4(II) significantly increased the number of type II NBs to ∼16 per brain lobe (Fig. 5C-E). These results suggest that the generation of extra type II NBs resulting from the loss of PntP1 is likely to be due to dedifferentiation of Ase− imINPs into type II NBs. The dedifferentiation of imINPs is likely to occur at a low frequency because the total number of type II NBs was only increased by 100-200% at the third instar larval stages when pntP1 was mutated or knocked down, and individual ectopic type II NB lineages were often well separated (e.g. Fig. 1G-H′, Fig. 4F). However, the dedifferentiation could occur at any stage during development, as indicated by the existence of lineages with multiple NBs at early and late larval stages (e.g. the second GFP-labeled lineage from the left in Fig. 4F and the second GFP-labeled lineage from the right in Fig. S1A).
Fig. 5.
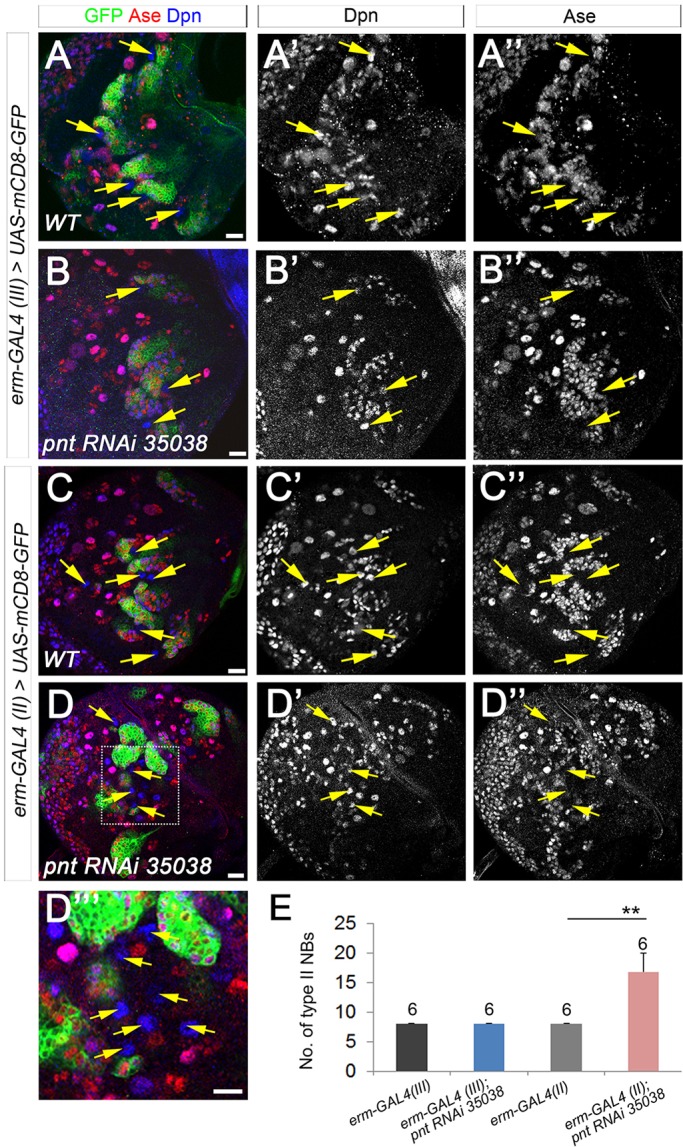
Knockdown of Pnt in Ase− imINPs increases the number of type II NBs. (A-A″,C-C″) Wild-type type II NB lineages labeled with mCD8-GFP driven by erm-GAL4(III) (A-A″) or erm-GAL4(II) (C-C″). Arrows, type II NBs. (B-B″) Knockdown of Pnt by erm-GAL4(III) does not lead to the generation of extra type II NBs (arrows). (D-D″′) Knockdown of Pnt by erm-GAL4(II) results in an increased number of type II NBs (arrows). (D″′) An enlarged view of the boxed area in D. (E) Quantification of total numbers of type II NBs. **P<0.01. Scale bars: 20 μm.
Erm expression is lost/reduced in Pnt knockdown type II NB lineages
Why does the loss of PntP1 lead to the dedifferentiation of imINPs into type II NBs? Our previous studies show that the misexpression of PntP1 in type I NB lineages is sufficient to induce the expression of Erm in INP-like cells, but that inhibiting PntP1 activity with Yan abolishes the expression of Erm in type II NB lineages (Zhu et al., 2011). Furthermore, Erm and PntP1 are co-expressed in imINPs (Janssens et al., 2014; Zhu et al., 2011), and loss of Erm similarly leads to an increase in the number of type II NBs. Therefore, Erm could be a PntP1 target, and the dedifferentiation of imINPs resulting from the loss of PntP1 could be due to the loss of Erm in imINPs. To test this, we examined Erm expression in Pnt knockdown type II NB lineages by immunostaining. In normal type II NB lineages, Erm is expressed in Ase− and Ase+ imINPs (Fig. 6A-A″) (Janssens et al., 2014). However, we did not observe any obvious Erm staining in Pnt knockdown type II NB lineages, even when Ase was not ectopically expressed in the NBs (Fig. 6B-D), indicating that the expression of Erm was largely abolished by Pnt knockdown and the loss of Erm expression was not due to the transformation of type II NBs into type I-like NBs. The loss of Erm expression in Pnt knockdown type II NB lineages provides evidence to support the hypothesis that Erm is a PntP1 target.
Fig. 6.
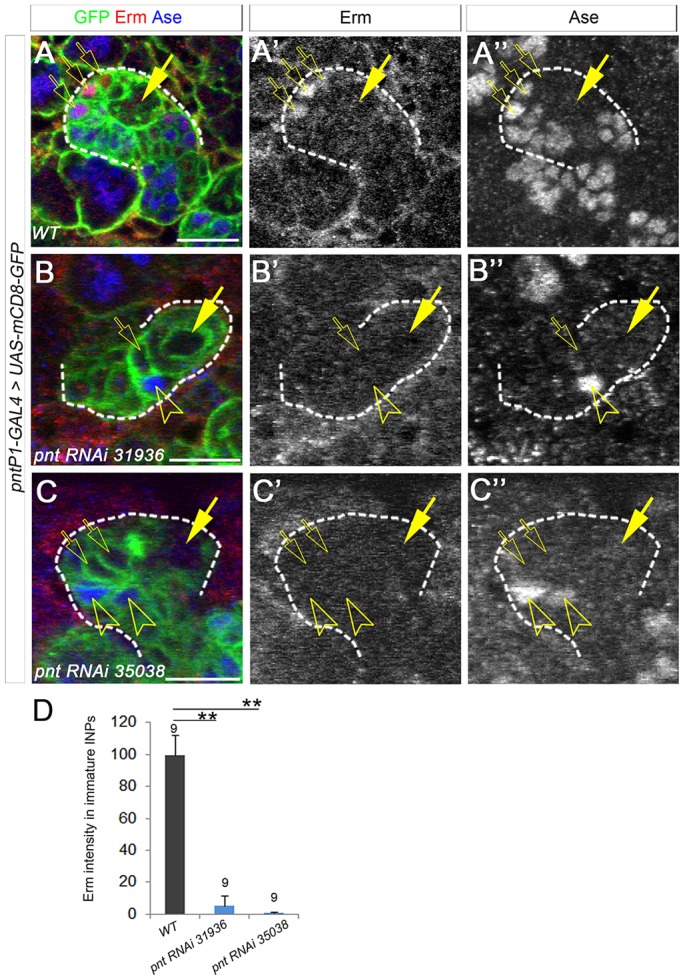
Erm is largely abolished in Pnt knockdown type II NB lineages. (A-A″) Erm is expressed in Ase− and Ase+ imINPs (open arrows) in a wild-type type II NB lineage labeled by GFP. Arrows, type II NBs. (B-C″) Erm is not detected in Ase− imINPs (open arrows) or Ase+ progeny (open arrowheads) when Pnt is knocked down. (D) Quantification of Erm staining intensity. **P<0.01. Scale bars: 20 μm.
The loss of Erm accounts for the generation of extra type II NBs that result from the loss of PntP1
To determine whether the loss of Erm expression was indeed responsible for the dedifferentiation of imINPs resulting from the loss of PntP1, we tested genetic interactions between erm and pntP1 by examining whether reducing Erm expression would further increase the generation of extra type II NBs resulting from the partial loss of PntP1 and if restoring Erm expression would suppress the generation of extra type II NBs. If the dedifferentiation of imINPs resulting from the loss of PntP1 is indeed due to the loss or reduction of Erm expression, then removing one wild-type copy of the erm gene would further increase the number of type II NBs. By contrast, restoring Erm expression in imINPs should suppress the dedifferentiation of imINPs and thus reduce the number of type II NBs.
Indeed, we found that although erm2 heterozygous mutants did not have any extra type II NBs (Fig. 7A,B,K), knockdown of Pnt in erm2 heterozygous mutants led to a significant increase in the total number of type II NBs (including both Ase− and Ase+ type II NBs) compared with Pnt knockdown in the wild-type background. Similar increases were observed when two independent UAS-pnt RNAi lines were used (Fig. 7C-F,K). Consistently, the removal of one wild-type copy of erm doubled the number of Ase− type II NBs in pntP190 mutant larvae (Fig. 7G,H,L). By contrast, when erm-GAL4(II) drove the expression of UAS-erm in imINPs, the generation of extra type II NBs in the pntP190 mutants was significantly suppressed (Fig. 7I,J,L). The total number of Ase− type II NBs in the pntP190 mutants was reduced to ten per lobe when UAS-erm was expressed in imINPs, suggesting that restoring Erm expression largely prevents the dedifferentiation of imINPs.
Fig. 7.

Loss of Erm is responsible for the generation of ectopic type II NBs that result from the loss of PntP1. Type II NB lineages are labeled with mCD8-GFP driven by either pntP1-GAL4 (A-F) or erm-GAL4 (I) or by phalloidin staining (G,H,J). Arrows, type II NBs. (A,B) Wild-type (A) and erm2 heterozygous mutant (B) brains have eight type II NBs (only seven and four are shown, respectively). (C-F) Knockdown of Pnt in erm2 heterozygotes (E,F) leads to the generation of more type II NBs than in the wild type (C,D). (G,H) The generation of ectopic Ase− type II NBs in pntP190 homozygous brains is enhanced in the erm2 heterozygous background. (I,J) Expression of Erm driven by erm-GAL4(II) reduces the number of Ase− type II NBs in pntP190 mutant brains (J) but not in the wild type (I). (K,L) Quantification of the total number of type II NBs (K) and the number of Ase− type II NBs (L). **P<0.01. (M) Working model of the role of PntP1 in preventing the premature differentiation and dedifferentiation of imINPs. PntP1 functions together with Btd to suppress Ase and Pros in the NB and in newly generated imINPs, respectively, to specify type II NBs and prevent premature differentiation of INPs. At later stages of imINP development, PntP1 activates erm to promote INP maturation. N, neuron.
Taken together, the enhancement and the suppression of the generation of ectopic type II NBs by reducing or maintaining Erm expression, respectively, indicate that the loss of Erm expression accounts for the dedifferentiation of imINPs into type II NBs that results from the loss of PntP1.
DISCUSSION
We have previously shown that PntP1 is required for the generation of INPs in type II NB lineages (Zhu et al., 2011), but the underlying mechanisms have not been elucidated. We show in this study that PntP1 has at least two distinct roles in INPs: preventing the premature differentiation of INPs into GMCs by acting together with Btd to suppress Pros expression in newly generated imINPs; and preventing the dedifferentiation of INPs into type II NBs by activating Erm expression late in imINP development. Therefore, PntP1 is able to maintain the balance between INP differentiation and dedifferentiation by functioning through two distinct pathways (Fig. 7M).
Our results show that a reduction in PntP1 function leads to both a loss of INPs and an increase in the number of type II NBs without completely transforming all type II NBs into type I-like NBs. The loss of INPs is particularly obvious in the pntP182 mutant and Pnt knockdown type II NB lineages, possibly because there is a more severe reduction of PntP1 function in the pntP182 mutants or after Pnt knockdown. We provide several lines of evidence to demonstrate that the loss of INPs is due to the Pros-mediated premature differentiation of INPs. First, the loss of INPs is not due to the transformation of type II NBs into type I NBs because the loss of INPs occurs independently of the ectopic Ase expression in the NBs. Second, when INPs are lost, the type II NBs generate GMCs instead. Third, nuclear Pros is ectopically activated in the newly generated imINPs when Pnt is lost. Fourth, the loss of INPs can be almost fully rescued by reducing Pros expression. Therefore, PntP1 normally inhibits Pros expression in the newly generated imINPs so that the imINPs can differentiate into mature INPs and undergo self-renewing divisions instead of becoming GMCs and exiting the cell cycle.
Although it remains to be investigated exactly how PntP1 suppresses Pros expression, our work suggests that it does so by functioning together with Btd, which is also required to suppress Pros in imINPs (Xie et al., 2014). We show that reducing Btd expression enhances the loss of INPs in pntP190 mutants. Although reducing Btd expression also enhances the ectopic activation of Ase in type II NBs, enhancement of INP loss also occurs in lineages without ectopic Ase expression. The genetic interaction between Btd and PntP1 suggests that they function in the same pathway to suppress Pros expression. However, it is unlikely that there is a direct regulatory relationship between Btd and PntP1 for several reasons. First, although Btd and PntP1 show similar expression patterns in type II NB lineages, Btd, but not PntP1, is also expressed in a subset of type I NB lineages. Second, we did not observe obvious changes in the expression of mCD8-GFP driven by btd-GAL4 in the pntP190 mutant or Pnt knockdown type II NB lineages (data not shown). Third, PntP1 expression is maintained in the majority of btd mutant type II NB clones (Xie et al., 2014). Fourth, PntP1 promotes the generation of INPs from type I NBs only when Btd is coexpressed, but not when PntP1 is expressed alone (Xie et al., 2014). It is well documented that Ets family proteins are able to bind to other transcription factors and that this partnership can enhance binding to the promoters of target genes and contribute to the functional specificity of Ets proteins (Hollenhorst et al., 2011). Therefore, one interesting possibility is that PntP1 and Btd physically interact and bind cooperatively to the promoters of their target genes.
In addition to preventing premature differentiation of INPs, our phenotypic analyses in the pntP1 mutants and Pnt knockdown type II NB lineages suggest that PntP1 also prevents imINP dedifferentiation. We show that there is a significant increase in the number of type II NBs in pntP190 homozygous or pntP190/pntΔ88 transheterozygous mutants or after knocking down Pnt. The extra type II NBs are likely to be derived from the dedifferentiation of Ase− imINPs, as knocking down Pnt by erm-GAL4(II) but not erm-GAL4(III) leads to an increase in the number of type II NBs. We also provide several lines of evidence to show that the dedifferentiation of imINPs is at least in part due to the loss of Erm in imINPs. First, Erm expression in imINPs is lost after Pnt knockdown and the loss of Erm occurs independently of the transformation of type II NBs into type I NBs. Second, reducing the expression of Erm significantly enhances the generation of extra type II NBs resulting from the loss of PntP1, whereas maintaining Erm expression in Ase− imINPs significantly suppresses the generation of extra type II NBs in the pntP190 mutants. Our results are consistent with a recent report showing that knockdown of Pnt by the type II NB driver wor-GAL4 ase-GAL80 increases the number of type II NBs, and reducing Pnt expression enhances the generation of ectopic type II NBs in erm mutants (Komori et al., 2014). However, our work identifies additional cellular and molecular mechanisms by which extra type II NBs are generated following the loss of PntP1.
One of the defining features of type II NBs is the lack of Ase expression. We previously reported that PntP1 was responsible for the suppression of Ase in type II NBs (Zhu et al., 2011). However, a recent study argues that PntP1 is only required for INP specification, but not type II NB specification, by showing that knockdown of Pnt by wor-GAL4 ase-GAL80 only increases the number of type II NBs but does not ectopically activate Ase in type II NBs (Komori et al., 2014). Here, we show that Pnt knockdown by pntP1-GAL4 is sufficient to ectopically activate Ase in ∼80% of type II NBs, confirming that PntP1 indeed specifies type II NB identity. The discrepancy could be due to differences in the efficiency of Pnt knockdown by RNAi involving different GAL4 drivers. However, our results show that reducing Pros expression not only rescues the loss of INPs but also restores the suppression of Ase expression in Pnt knockdown type II NBs, raising the question of whether PntP1 specifies type II NBs by directly acting in the NBs or by indirectly promoting INP generation. Our previous results show that PntP1 misexpression is able to suppress Ase expression in type I NBs, even if INPs are not generated, suggesting that PntP1 is likely to act in the NBs to suppress Ase expression. However, INPs are also likely to be involved in maintaining type II NB identity by providing a feedback signal to maintain PntP1 expression in the NB, as we proposed in a previous study (Xie et al., 2014). One candidate feedback signal could be the Notch ligand. Our recent studies suggest that Notch signaling is required to maintain PntP1 expression and type II NB identity (Li et al., 2016; Zhu et al., 2012). INPs could provide the ligand to activate Notch in the NBs, as suggested in a recent study (Song and Lu, 2011). This Notch-mediated feedback mechanism for maintaining neural progenitor cells is also conserved in mammals (Campos et al., 2001; Lui et al., 2011; Yoon et al., 2008).
However, it is surprising that Ase remains suppressed at least in a subset of pntP190 or pntP182 mutant type II NBs. Given that PntP1 is a transcription factor, one might expect that the deletion of the Ets DNA-binding domain in the pntP190 and pntP182 mutants would lead to a complete loss of PntP1 function and to ectopic activation of Ase in all type II NBs. However, our genetic data suggest they are likely to be hypomorphic alleles because the phenotypes in the pntP190 and pntP182 mutants are weaker than in the pntP190/pntΔ88 transheterozygotes. Furthermore, the pntP190 and pntP182 mutants show different degrees of severity in phenotype, which also indicates that deletion of the Ets DNA-binding domain does not completely abolish PntP1 function. Therefore, the truncated PntP1 protein generated in the pntP190 or pntP182 mutants could still be partially functional and the DNA-binding domain of PntP1 might be dispensable for function. Our results show that reducing Btd expression significantly enhances the ectopic activation of Ase in pntP190 mutant type II NBs. Although we do not rule out the possibility that the enhanced ectopic Ase activation could be secondary to the enhanced loss of INPs and their feedback signal(s), Btd and PntP1 might actually function together to suppress Ase. In support of this notion, we previously showed that Btd overexpression was able to partially suppress Ase expression in a subset of type I NBs in larval brains (Xie et al., 2014). The truncated PntP1 proteins generated from the pntP190 or pntP182 alleles might still be able to interact with Btd to regulate target gene expression.
In summary, our studies shed new light on the mechanistic details of the PntP1-mediated generation of INPs as well as type II NB specification. However, it remains to be investigated how PntP1 and Btd act together to specify type II NBs and inhibit Pros expression in INPs. Understanding this might rely on identification of the direct targets of PntP1 and Btd.
MATERIALS AND METHODS
Fly stocks
UAS-pnt RNAi lines (#31936 and #35038, Bloomington Drosophila Stock Center) were used for Pnt knockdown. Type II NB lineage-specific pntP1-GAL4 (named GAL414-94 previously) (Zhu et al., 2011) and erm-GAL4(II) or (III) (Pfeiffer et al., 2008; Xiao et al., 2012) were used to drive the expression of UAS-transgenes in type II NB lineages or in imINPs, respectively. Other fly lines include: pntΔ88/Tm6Tb (Brunner et al., 1994), which was used for pntP1 loss-of-function phenotypic analyses; pros17/TM6Tb (Doe et al., 1991) for reducing Pros expression; btdXG81, FRT19A/FM7c, Kr-GFP and btd-GAL4, FRT19A/FM7c, Kr-GFP (Estella and Mann, 2010; Wimmer et al., 1993) for reducing Btd expression; and erm2/Cyo, act-GFP (Weng et al., 2010) for reducing Erm expression.
Generation of pntP1 mutants using the CRISPR/Cas9 system
gRNA targets were selected using an online design tool (http://tools.flycrispr.molbio.wisc.edu/targetFinder/). The gRNA-expressing vectors were generated by a series of PCR reactions (see the supplementary Materials and Methods) (Sebo et al., 2014) and injected into y1, P[vas-Cas9.S]ZH-2A, w1118 (BDSC #52669) at 1 µg/ml (Rainbow Transgenic Flies, Camarillo, CA, USA) for generating mutant lines. Mutations were detected by PCR amplification of genomic DNAs followed by sequencing using pntP1-specific primers.
UAS-transgene expression
For RNAi knockdown or misexpression of transgenes, larvae were raised at 30°C after hatching. UAS-dcr2 was co-expressed with UAS-RNAi transgenes to enhance the efficiency of RNAi knockdown. Phenotypes were examined at third instar larval stages. To examine whether reducing Pros expression could rescue the loss of INPs resulting from Pnt RNAi knockdown, UAS-pnt RNAi was first recombined with pntP1-GAL4. Then, the UAS-pnt RNAi pntP1-GAL4 recombinant flies were crossed with either wild type or pros17/Tm6,Tb and their progenies raised at 25°C.
Immunostaining, confocal microscopy and statistical analyses
Dissection, fixation and immunostaining of larval brains were performed as described (Lee and Luo, 1999). Primary antibodies used for immunostaining include: guinea pig anti-Ase (1:5000; Brand et al., 1993) and rabbit anti-Dpn (1:500; Bier et al., 1992) (both gifts from Y. N. Jan), rat anti-mCD8 (Life Technologies, clone #5H10; 1:100), mouse anti-Pros (Developmental Studies Hybridoma Bank; 1:20) and rabbit anti-Erm (1:50; Janssens et al., 2014; a gift from H.Y. Wang). Secondary antibodies conjugated to Cy2, Cy3, Cy5 or DyLight 647 (Jackson ImmunoResearch) were used at 1:100 (Cy2) or 1:500 (Cy3, Cy5 or DyLight 647). A Zeiss LSM510 confocal microscope was used to acquire images, which were processed with Adobe Photoshop. Two-tailed Student's t-tests were used for statistical analyses.
Acknowledgements
We thank Drs Y. N. Jan, H. Y. Wang, C. Y. Lee, the Bloomington Drosophila Stock Center, and the Developmental Studies Hybridoma Bank for antibodies and fly stocks; M. Pan and X. Jackson for technical support; Dr F. Pignoni and S.Z. lab members for thoughtful discussion and comments.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
S.Z. and Y.X. designed the project and approaches, interpreted data and wrote the paper. Y.X., X.L., Y.H. and X.D. conducted experiments and analyzed the data. K.O., A.U. and L.C. provided technical support. Y.P. designed the strategy for generating pntP1 mutants. Y.H., X.D., K.O., Y.P. and L.C. contributed to manuscript editing.
Funding
This work was supported by a March of Dimes Foundation Basil O'Connor Starter Scholar Research Award [#5-FY14-59 to S.Z.]; and the National Institute of Neurological Disorders and Stroke of the National Institutes of Health [R01NS085232 to S.Z.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.137281.supplemental
References
- Bello B. C., Izergina N., Caussinus E. and Reichert H. (2008). Amplification of neural stem cell proliferation by intermediate progenitor cells in Drosophila brain development. Neural Dev. 3, 5 10.1186/1749-8104-3-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier E., Vaessin H., Younger-Shepherd S., Jan L. Y. and Jan Y. N. (1992). deadpan, an essential pan-neural gene in Drosophila, encodes a helix-loop-helix protein similar to the hairy gene product. Genes Dev. 6, 2137-2151. 10.1101/gad.6.11.2137 [DOI] [PubMed] [Google Scholar]
- Boone J. Q. and Doe C. Q. (2008). Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cells. Dev. Neurobiol. 68, 1185-1195. 10.1002/dneu.20648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman S. K., Rolland V., Betschinger J., Kinsey K. A., Emery G. and Knoblich J. A. (2008). The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev. Cell 14, 535-546. 10.1016/j.devcel.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M., Jarman A. P., Jan L. Y. and Jan Y. N. (1993). asense is a Drosophila neural precursor gene and is capable of initiating sense organ formation. Development 119, 1-17. [DOI] [PubMed] [Google Scholar]
- Brunner D., Ducker K., Oellers N., Hafen E., Scholz H. and Klambt C. (1994). The ETS domain protein pointed-P2 is a target of MAP kinase in the sevenless signal transduction pathway. Nature 370, 386-389. 10.1038/370386a0 [DOI] [PubMed] [Google Scholar]
- Campos L. S., Duarte A. J., Branco T. and Henrique D. (2001). mDll1 and mDll3 expression in the developing mouse brain: role in the establishment of the early cortex. J. Neurosci. Res. 64, 590-598. 10.1002/jnr.1111 [DOI] [PubMed] [Google Scholar]
- Choksi S. P., Southall T. D., Bossing T., Edoff K., de Wit E., Fischer B. E., van Steensel B., Micklem G. and Brand A. H. (2006). Prospero acts as a binary switch between self-renewal and differentiation in Drosophila neural stem cells. Dev. Cell 11, 775-789. 10.1016/j.devcel.2006.09.015 [DOI] [PubMed] [Google Scholar]
- Colasante G., Simonet J. C., Calogero R., Crispi S., Sessa A., Cho G., Golden J. A. and Broccoli V. (2015). ARX regulates cortical intermediate progenitor cell expansion and upper layer neuron formation through repression of Cdkn1c. Cereb. Cortex 25, 322-335. 10.1093/cercor/bht222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe C. Q., Chu-LaGraff Q., Wright D. M. and Scott M. P. (1991). The prospero gene specifies cell fates in the Drosophila central nervous system. Cell 65, 451-464. 10.1016/0092-8674(91)90463-9 [DOI] [PubMed] [Google Scholar]
- Eroglu E., Burkard T. R., Jiang Y., Saini N., Homem C. C. F., Reichert H. and Knoblich J. A. (2014). SWI/SNF complex prevents lineage reversion and induces temporal patterning in neural stem cells. Cell 156, 1259-1273. 10.1016/j.cell.2014.01.053 [DOI] [PubMed] [Google Scholar]
- Estella C. and Mann R. S. (2010). Non-redundant selector and growth-promoting functions of two sister genes, buttonhead and Sp1, in Drosophila leg development. PLoS Genet. 6, e1001001 10.1371/journal.pgen.1001001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenstein V., Spindler S., Pereanu W. and Fung S. (2008). The development of the Drosophila larval brain. Adv. Exp. Med. Biol. 628, 1-31. 10.1007/978-0-387-78261-4_1 [DOI] [PubMed] [Google Scholar]
- Hollenhorst P. C., McIntosh L. P. and Graves B. J. (2011). Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 80, 437-471. 10.1146/annurev.biochem.79.081507.103945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens D. H., Komori H., Grbac D., Chen K., Koe C. T., Wang H. and Lee C.-Y. (2014). Earmuff restricts progenitor cell potential by attenuating the competence to respond to self-renewal factors. Development 141, 1036-1046. 10.1242/dev.106534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A. and Charpentier E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816-821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koe C. T., Li S., Rossi F., Wong J. J., Wang Y., Zhang Z., Chen K., Aw S. S., Richardson H. E., Robson P. et al. (2014). The Brm-HDAC3-Erm repressor complex suppresses dedifferentiation in Drosophila type II neuroblast lineages. ELife 3, e01906 10.7554/eLife.01906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori H., Xiao Q., Janssens D. H., Dou Y. and Lee C.-Y. (2014). Trithorax maintains the functional heterogeneity of neural stem cells through the transcription factor buttonhead. ELife 3, e03502 10.7554/eLife.03502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. and Luo L. (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451-461. 10.1016/S0896-6273(00)80701-1 [DOI] [PubMed] [Google Scholar]
- Li X., Xie Y. and Zhu S. (2016). Notch maintains Drosophila type II neuroblasts by suppressing the expression of the Fez transcription factor Earmuff. Development 143, 2511-2521. 10.1242/dev.136184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Sage J. C., Miller M. R., Verhaak R. G., Hippenmeyer S., Vogel H., Foreman O., Bronson R. T., Nishiyama A., Luo L. et al. (2011). Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209-221. 10.1016/j.cell.2011.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui J. H., Hansen D. V. and Kriegstein A. R. (2011). Development and evolution of the human neocortex. Cell 146, 18-36. 10.1016/j.cell.2011.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurange C., Cheng L. and Gould A. P. (2008). Temporal transcription factors and their targets schedule the end of neural proliferation in Drosophila. Cell 133, 891-902. 10.1016/j.cell.2008.03.034 [DOI] [PubMed] [Google Scholar]
- O'Neill E. M., Rebay I., Tjian R. and Rubin G. M. (1994). The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell 78, 137-147. 10.1016/0092-8674(94)90580-0 [DOI] [PubMed] [Google Scholar]
- Pfeiffer B. D., Jenett A., Hammonds A. S., Ngo T. T., Misra S., Murphy C., Scully A., Carlson J. W., Wan K. H., Laverty T. R. et al. (2008). Tools for neuroanatomy and neurogenetics in Drosophila. Proc. Natl. Acad. Sci. USA 105, 9715-9720. 10.1073/pnas.0803697105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontious A., Kowalczyk T., Englund C. and Hevner R. F. (2008). Role of intermediate progenitor cells in cerebral cortex development. Dev. Neurosci. 30, 24-32. 10.1159/000109848 [DOI] [PubMed] [Google Scholar]
- Port F., Chen H.-M., Lee T. and Bullock S. L. (2014). Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 111, E2967-E2976. 10.1073/pnas.1405500111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn J. C., Molinek M., Martynoga B. S., Zaki P. A., Faedo A., Bulfone A., Hevner R. F., West J. D. and Price D. J. (2007). Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev. Biol. 302, 50-65. 10.1016/j.ydbio.2006.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reillo I., de Juan Romero C., Garcia-Cabezas M. A. and Borrell V. (2011). A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb. Cortex 21, 1674-1694. 10.1093/cercor/bhq238 [DOI] [PubMed] [Google Scholar]
- Sebo Z. L., Lee H. B., Peng Y. and Guo Y. (2014). A simplified and efficient germline-specific CRISPR/Cas9 system for Drosophila genomic engineering. Fly (Austin) 8, 52-57. 10.4161/fly.26828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y. and Lu B. (2011). Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor-forming stem cells in Drosophila. Genes Dev. 25, 2644-2658. 10.1101/gad.171959.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton N. M., Snyder G. E., Park D., Kobeissy F., Scheffler B. and Steindler D. A. (2009). Gliotypic neural stem cells transiently adopt tumorigenic properties during normal differentiation. Stem Cells 27, 280-289. 10.1634/stemcells.2008-0842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-C., Yang J. S., Johnston R., Ren Q., Lee Y.-J., Luan H., Brody T., Odenwald W. F. and Lee T. (2014). Drosophila intermediate neural progenitors produce lineage-dependent related series of diverse neurons. Development 141, 253-258. 10.1242/dev.103069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M., Golden K. L. and Lee C.-Y. (2010). dFezf/Earmuff maintains the restricted developmental potential of intermediate neural progenitors in Drosophila. Dev. Cell 18, 126-135. 10.1016/j.devcel.2009.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer E. A., Jäckle H., Pfeifle C. and Cohen S. M. (1993). A Drosophila homologue of human Sp1 is a head-specific segmentation gene. Nature 366, 690-694. 10.1038/366690a0 [DOI] [PubMed] [Google Scholar]
- Xiao Q., Komori H. and Lee C.-Y. (2012). klumpfuss distinguishes stem cells from progenitor cells during asymmetric neuroblast division. Development 139, 2670-2680. 10.1242/dev.081687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Li X., Zhang X., Mei S., Li H., Urso A. and Zhu S. (2014). The Drosophila Sp8 transcription factor Buttonhead prevents premature differentiation of intermediate neural progenitors. ELife 3, e03596 10.7554/elife.03596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K.-J., Koo B.-K., Im S.-K., Jeong H.-W., Ghim J., Kwon M.-C., Moon J.-S., Miyata T. and Kong Y.-Y. (2008). Mind bomb 1-expressing intermediate progenitors generate notch signaling to maintain radial glial cells. Neuron 58, 519-531. 10.1016/j.neuron.2008.03.018 [DOI] [PubMed] [Google Scholar]
- Zhu S., Barshow S., Wildonger J., Jan L. Y. and Jan Y.-N. (2011). Ets transcription factor Pointed promotes the generation of intermediate neural progenitors in Drosophila larval brains. Proc. Natl. Acad. Sci. USA 108, 20615-20620. 10.1073/pnas.1118595109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Wildonger J., Barshow S., Younger S., Huang Y. and Lee T. (2012). The bHLH repressor Deadpan regulates the self-renewal and specification of Drosophila larval neural stem cells independently of Notch. PLoS ONE 7, e46724 10.1371/journal.pone.0046724 [DOI] [PMC free article] [PubMed] [Google Scholar]