

Abstract
Cellular transitions require dramatic changes in gene expression that are supported by regulated mRNA decay and new transcription. The maternal‐to‐zygotic transition is a conserved developmental progression during which thousands of maternal mRNAs are cleared by post‐transcriptional mechanisms. Although some maternal mRNAs are targeted for degradation by microRNAs, this pathway does not fully explain mRNA clearance. We investigated how codon identity and translation affect mRNA stability during development and homeostasis. We show that the codon triplet contains translation‐dependent regulatory information that influences transcript decay. Codon composition shapes maternal mRNA clearance during the maternal‐to‐zygotic transition in zebrafish, Xenopus, mouse, and Drosophila, and gene expression during homeostasis across human tissues. Some synonymous codons show consistent stabilizing or destabilizing effects, suggesting that amino acid composition influences mRNA stability. Codon composition affects both polyadenylation status and translation efficiency. Thus, the ribosome interprets two codes within the mRNA: the genetic code which specifies the amino acid sequence and a conserved “codon optimality code” that shapes mRNA stability and translation efficiency across vertebrates.
Keywords: codon optimality, decay, maternal‐to‐zygotic transition, translation, zebrafish
Subject Categories: Development & Differentiation, Protein Biosynthesis & Quality Control, RNA Biology
Introduction
During biological transitions, cells install a new transcriptional program while removing the previous program. This process depends on the post‐transcriptional control of persistent transcripts as new mRNAs are transcribed. In early embryonic development, maternally deposited mRNAs and proteins govern the embryonic development of most animals until the zygotic genome is activated (Tadros & Lipshitz, 2009; Yartseva & Giraldez, 2015). Following egg activation and fertilization, the embryo initiates the maternal‐to‐zygotic transition (MZT). During this universal transition, the transcriptome undergoes a drastic remodeling resulting in the clearance of thousands of maternal mRNAs and activation of the zygotic genome. 3′UTRs regulate mRNA stability across different biological processes (Giraldez et al, 2006; Bushati et al, 2008; Bartel, 2009; Lund et al, 2009; Rice, 2015; Rusek et al, 2015). For example, conserved microRNA families (miR‐430/‐427 and miR‐309) are responsible for the degradation of a subset of maternal mRNAs in zebrafish (Giraldez et al, 2006), Xenopus (Lund et al, 2009), and Drosophila (Bushati et al, 2008), respectively. Similarly, in Drosophila, the RNA binding protein SMAUG functions to destabilize maternal mRNAs (Tadros et al, 2007). Despite the identification of these factors, thousands of maternal mRNAs are cleared by unknown mechanisms (Tadros & Lipshitz, 2009; Yartseva & Giraldez, 2015). While most of the factors identified are conserved and regulate mRNA stability through the 3′UTR, the role of the coding sequence (CDS) in regulating transcript stability during the MZT transition and tissue homeostasis across animals is largely unexplored (Richter & Coller, 2015; Yartseva & Giraldez, 2015; Mishima & Tomari, 2016).
The ribosome represents the most abundant RNA binding complex in the cell (Doudna & Rath, 2002), translating mRNA sequences into amino acids. Translation is also important for quality control mechanisms (Shoemaker & Green, 2012; Pechmann et al, 2013), such as nonsense‐mediated decay (NMD), no‐go decay (NGD), and nonstop decay (NSD), which surveil translation to regulate defective transcripts. Recently, specific codons have been associated with transcript stability (optimal codons) or instability (non‐optimal codons) in bacteria and yeast (Presnyak et al, 2015; Boel et al, 2016). The degeneracy of the genetic code allows multiple codons to encode for the same amino acid (Gamow, 1954), and silent codon substitutions can influence protein expression (Ikemura, 1982; Kudla et al, 2009). While codon usage bias exists across all domains of life (Novoa & Ribas de Pouplana, 2012), the evolutionary pressures exerted by the codon usage on gene expression and growth are distinct between unicellular and multicellular systems. Thus, it is unclear whether codon identity regulates mRNA stability across vertebrates, and if so, what is the identity of the codons conferring regulation, the conservation of the regulatory code, and the biological implications of this mechanism across animals. Interestingly, during the review process of this manuscript, it has been shown that differential use of synonymous codons shapes maternal mRNA clearance during MZT in zebrafish (Mishima & Tomari, 2016). This analysis looks at the codon adaptation index, which compares the effect of different codons encoding the same amino acid. However, it is still unknown what is the relative effect of the 61 coding codons independent of the amino acid encoded (non‐synonymous codons), whether all synonymous codons for a given amino acid have similar effects on mRNA stability and whether the translation‐dependent regulatory mechanism of mRNA stability during MZT is conserved across animals. Here, we investigate how translation of specific codons shapes mRNA expression during the MZT and homeostasis, and define a regulatory layer within the genetic code across vertebrates, that we term the “codon optimality code”.
Results
To investigate whether translation of specific codons influences mRNA stability in vertebrates, we developed two complementary approaches. First, we tested whether blocking translation alters the codon composition of an mRNA population due to differential mRNA decay (Fig 1A). To this end, we constructed a library of mRNA sequences by PCR assembly of random fragments of the transcriptome (~300–500 nt in length) into a reporter DNA that shares (i) identical 5′ and 3′UTR sequences, (ii) a GFP translation start sequence (26 nt) which we can target with a specific morpholino oligo (MO) to block translation (Giraldez et al, 2006) (Fig EV1A), (iii) stop codons in all three frames within the 3′UTR, and (iv) Illumina adapter sequences to facilitate generation of a sequencing library (Fig 1A). We generated mRNAs from this reporter library in vitro and injected these highly diverse mRNA sequences (Fig EV1B) into zebrafish and Xenopus embryos at the one‐cell stage, with the goal to define conserved regulatory sequences between these species. Using high‐throughput sequencing, we quantified the occurrence of each codon in the reporter library 8–9 h after injection with (−MO) or without (+MO) translation. We calculated the reporter codon stabilizing index (rCSI) as the statistical significance of the enrichment (+) or depletion (−) of each codon between these conditions (−MO versus +MO) in triplicates (F‐value, ANOVA). We found differential accumulation of individual codons in the reporter libraries to be dependent on translation, suggesting that translation of specific codons might influence mRNA stability in vertebrates (Fig 1B and C) (8 h versus 8 +MO zebrafish, 9 h versus 9 +MO Xenopus, P < 1e‐100, one‐way ANOVA). For example, the GGU codon was significantly enriched when translation was allowed (−MO), suggesting that GGU stabilizes mRNA in a translation‐dependent manner (Figs 1B and EV1C) (P < 1e‐300, one‐way ANOVA). In contrast, the codon CAC was significantly depleted in the reporter mRNA library when translation was allowed (−MO), suggesting that it lowers mRNA stability in a translational‐dependent manner (Figs 1B and EV1C) (P < 1e‐300, one‐way ANOVA). We observed a strong correlation of the rCSI in zebrafish and Xenopus, suggesting that codon‐mediated transcript stability in the context of translation is conserved across vertebrates (Fig 1C). Comparing the codon composition of the reporter library between late (8 hpi zebrafish, 9 hpi Xenopus) and early stages (2 hpi zebrafish, 1 hpi Xenopus) yielded similar results, suggesting that the effect of different codons on mRNA stability is not due to an off‐target effect of the GFP MO (Fig EV1E and F). Together, these results suggest that translation of specific codons affects mRNA stability in vertebrates.
Figure 1. Translation modulates stability of mRNA in zebrafish and Xenopus .
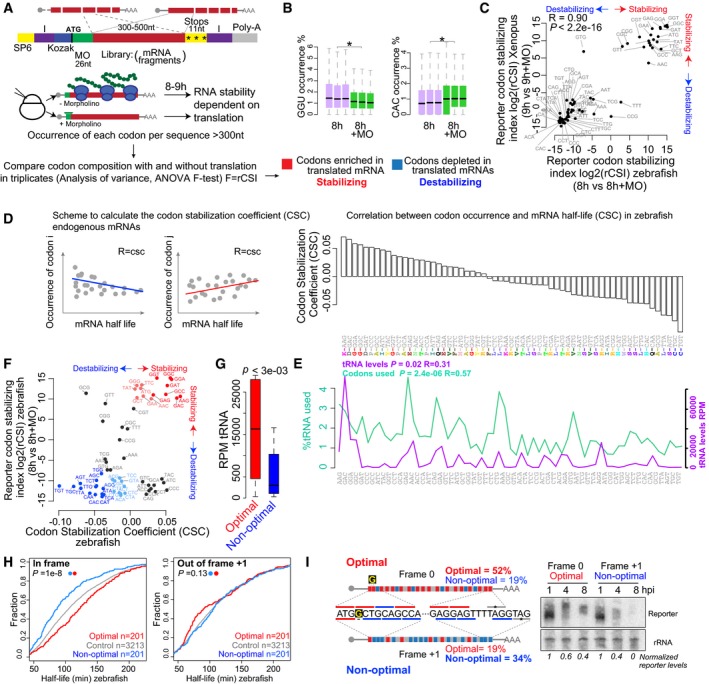
- Scheme of the reporter library which includes random fragments of the transcriptome in the CDS. Transcripts share the same 5′ and 3′UTR and 26 nt within the translation start site, which are recognized by a specific translation blocking morpholino (MO) (green). I, Illumina adapters, purple. SP6, SP6 promoter. mRNAs are injected at the one‐cell stage in zebrafish and Xenopus, with or without a translation blocking morpholino. Reporter mRNA library is analyzed 8–9 h post‐injection using high‐throughput sequencing. The analysis of variance (ANOVA) for the occurrence of each codon in the presence (−MO) or the absence (+MO) of translation is performed between triplicates in zebrafish and Xenopus to calculate the reporter codon stabilizing index (rCSI). If codons cause differential mRNA stability in a translation‐dependent manner, potential stabilizing codons should be enriched in translated mRNAs (−MO versus +MO) (+rCSI), and destabilizing codons depleted (−MO versus +MO) (−rCSI).
- Box plots showing the occurrence of the GGU and the CAC codons in the presence (+MO) and absence of MO in the reporter mRNA library 8 h post‐injection (hpi) (triplicates). The box defines the first and third quartiles, with the median indicated with a thick black line and vertical lines indicate the variability outside the upper and lower quartiles. *P < 1e‐100, one‐way ANOVA.
- Biplots of the reporter codon stabilizing index (rCSI) in the presence and the absence of translation (+MO) in zebrafish and in Xenopus embryos, P < 2.2e‐16, R = 0.9, Spearman rank correlation. The rCSI is the ANOVA F‐test value for the comparison of codon occurrences (e.g. panel B) between treatments in triplicates. This value is multiplied by −1 for depleted/destabilizing codons.
- Scheme of the codon stabilization coefficient (CSC) calculated as the Pearson correlation coefficient between the occurrence of each codon and the half‐life of each mRNA. Bar plots ranking the 61 coding codons based on their codon stability coefficient (CSC). The encoded amino acids are indicated.
- Line plots of the relative demand of each codon in the zebrafish transcriptome at 6 hpf (% used), and the RNA levels for each tRNA (RPM). The P‐value indicates the significant correlation with the CSC.
- Biplots of the rCSI comparing 8 h versus 8 h + MO and CSC in zebrafish embryos, P < 1.2e‐4, R = 0.4, Spearman rank correlation. Based on the consistent identification by both methods, we defined optimal codons highlighted in red and non‐optimal codons in blue. The intensity of the color represents the strength (light, weak; dark, strong) based on different CSC values.
- Box plot of tRNA levels (RPM) for optimal and non‐optimal codons (P = 3e‐03, Wilcoxon rank‐sum test). The box defines the first and third quartiles, with the median indicated with a thick black line and vertical lines indicate the variability outside the upper and lower quartiles.
- Cumulative distributions of the half‐life of the maternal mRNAs in the absence of zygotic transcription in zebrafish. Shown are all mRNAs (gray), mRNAs with the highest fraction (top quartile) of optimal codons and the lowest fraction of non‐optimal codons (red), mRNAs with the highest fraction of non‐optimal codons and the lowest fraction of optimal codons (blue), in frame (left), or 1 nucleotide out of frame +1 (right) (P = 1e‐8, in frame; P = 0.13, 1 nt out of frame, Wilcoxon rank‐sum test).
- Diagram of two transcripts that only differ in a single nucleotide (G in yellow) which creates a frameshift, making one transcript enriched in optimal codons and the other in non‐optimal codons. Time‐course Northern blot shows that non‐optimal transcripts decay faster than their optimal counterparts. The radioactive intensity is shown in italics and normalized to the 1 hpi sample for each injection.
Figure EV1. Translation modulates stability of mRNA in zebrafish and Xenopus .
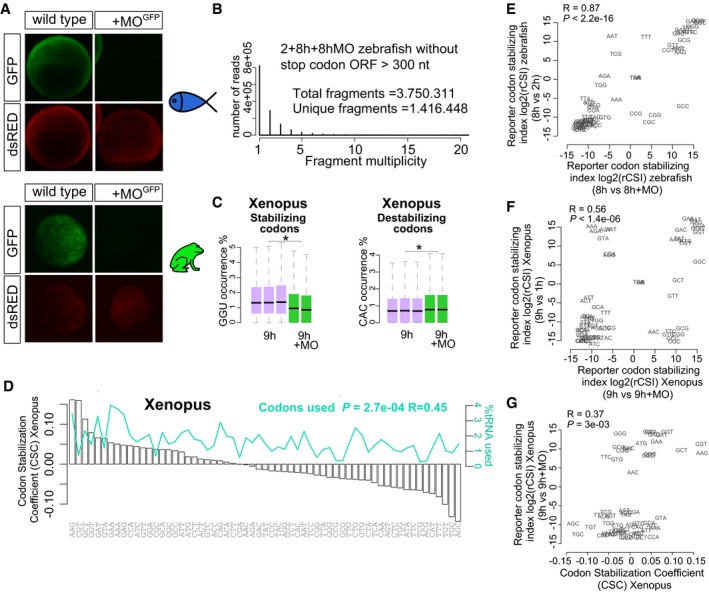
-
AGFP reporter expression (green) and control dsRed expression (red) at 8 hpf and 9.5 hpf in zebrafish and Xenopus, respectively. The GFP reporter is repressed by co‐injection with the morpholino which recognizes the GFP translation initiation sequence (MO). dsRed is not recognized by the MO. This morpholino is complementary to the translation initiation site in the reporter mRNA library used in Fig 1A.
-
BBar plot shows distribution of fragment multiplicity for fragments longer that 300 nt without stop codons at 2, 8 h, and 8 h + MO in zebrafish embryos, showing a high degree of complexity of the reporter library mRNA.
-
CBox plots show the occurrence of the optimal codon GGU and non‐optimal codon CAC in injected mRNA at 1, 9 h, and 9 h + MO in Xenopus embryos (*P < 1e‐300 and *P < 1e‐300, respectively, one‐way ANOVA). The box defines the first and third quartiles, with the median indicated with a thick black line and vertical lines indicate the variability outside the upper and lower quartiles. The analysis of variance (ANOVA) for the occurrence of each codon in the presence (−MO) or the absence (+MO) of translation is used to calculate the reporter codon stabilizing index (rCSI). If codons cause differential mRNA stability in a translation‐dependent manner, potential stabilizing codons should be enriched in translated mRNAs (−MO versus +MO) (+rCSI), and destabilizing codons depleted (−MO versus +MO) (−rCSI).
-
DBar plots ranking the 61 coding codons from highest to lowest correlation with mRNA half‐life (CSC). The relative abundance of each codon present in the expressed transcriptome at 9 hpf in Xenopus is shown (green).
-
E, FBiplots of the rCSI derived by comparing codon enrichment or depletion in two different conditions: x‐axis, in the presence and the absence of translation (±MO); y‐axis, in a late versus earlier time points (E) in zebrafish or (F) in Xenopus. Both comparisons yield similar results, ruling out nonspecific effects of the MO. One hour or 2 h is too early to observe the full effects of translational regulation in mRNA stability.
-
GBiplots of the rCSI (9 h/9 h + MO) from Xenopus embryos and the codon stabilization coefficient (CSC) in Xenopus for each codon, P = 3e‐3, R = 0.37, Spearman rank correlation.
Analysis of the reporter library allowed us to investigate the effects derived from the coding sequence and isolate them from potential regulatory effects of the 5′ and 3′UTRs, since every mRNA shares the same UTR sequence. To ensure the exogenous nature of the reporter library did not influence our results, we used a second approach to analyze the effect of individual codons on the stability of endogenous maternal mRNAs in vivo in zebrafish and Xenopus. To avoid confounding effects due to newly transcribed mRNAs, we calculated the half‐life for each mRNA after blocking zygotic transcription with the RNA pol II inhibitor α‐amanitin (Hamatani et al, 2004) (Table EV1). We calculated the Pearson correlation coefficient between mRNA half‐life and codon occurrence, a metric previously defined in yeast as codon stability coefficient (CSC) (Presnyak et al, 2015) (Figs 1D and EV1D). We observed that some codons were correlated with mRNA half‐life while others were anticorrelated, consistent with different effects on mRNA stability. Similar to previous observations in yeast, each individual codon only elicits a small effect on the overall stability of the mRNA (Presnyak et al, 2015). We observed a significant positive correlation for the CSC between zebrafish and Xenopus (P < 2.2e‐03, R = 0.65, Spearman), but not with yeast (P = 0.02, R = −0.27 with Fish, P < 5e‐03, R = −0.35, Spearman) (Presnyak et al, 2015), suggesting that the regulatory activity of multiple codons is shared between animals but different from yeast.
Comparison of the two methods revealed consistent effects for most codons on mRNA stability observed in the reporter library (rCSI) and the endogenous mRNAs (CSC) (Figs 1F and EV1G) (zebrafish, P = 1e‐3, R = 0.4, Spearman; Xenopus, P = 3e‐3, R = 0.4, Spearman). Because each method has distinct biases aforementioned (i.e. exogenous expression, UTR regulation), we adopted a conservative approach and selected those codons displaying consistent effects in both methods. Based on the consistent effects of each codon on the reporter library and endogenous mRNAs, we defined: optimal codons, with potential stabilizing effects (rCSI > 250 and CSC > 0) and non‐optimal codons with potential destabilizing effects (rCSI < −250 and CSC < 0) (Fig 1F). We next performed sequence analysis of tRNA levels in the zebrafish embryos using highly processive thermostable group II intron reverse transcriptase that allows unwinding of the tRNA secondary structure (TGIRT) (Zheng et al, 2015). This revealed that optimal codons are decoded by more abundant tRNAs than non‐optimal ones (P = 3e‐03, Wilcoxon rank‐sum test) (Fig 1G and E). Analysis of the codon usage in the transcriptome indicated that optimal codons are more commonly used in the transcriptome (P = 2.4e‐06, R = 0.57, Spearman) (Fig 1G), suggesting that codon optimality may be in part related to tRNA availability and codon usage (dos Reis et al, 2004; Pechmann & Frydman, 2013; Presnyak et al, 2015; Richter & Coller, 2015).
Two lines of evidence indicate that translation of specific codons indeed regulates mRNA half‐life and is dependent on translation. We observed a longer half‐life for mRNAs enriched in optimal codons (defined in Fig 1F) than those enriched non‐optimal codons in both zebrafish and Xenopus (Figs 1H and EV2C) (P = 1e‐8, Wilcoxon rank‐sum test). Based on different CSC confidence thresholds, we defined two populations within the optimal (red) and non‐optimal (blue) codons (light = weak, 0 < CSC < 0.03; dark = strong, CSC > 0.03, Fig 1F), which indeed showed different effects on mRNA half‐life (Fig EV2A). This effect depends on the codons in frame 0 and is not observed in transcripts that are enriched in optimal or non‐optimal codons in reading frames +1 or +2, consistent with translation‐dependent regulation rather than regulation by a sequence motif that is independent of the reading frame (Figs 1H and EV2B and C) (P = 0.11, Wilcoxon rank‐sum test). Second, we analyzed whether different optimality determines the rate of decay of two sets of reporter mRNAs. Each pair of reporters differs in a single nucleotide insertion after the translation start codon, causing a frameshift that results in opposite codon optimality. Non‐optimal reporters were less stable over time compared to their optimal counterparts (Figs 1I and EV2D and E). When translation was inhibited with cycloheximide, the differential stability is lost (Fig EV2D). Together, these results define a codon optimality code by which translation of specific codons modulates mRNA stability in vertebrates.
Figure EV2. Translation of specific codons modulates mRNA stability in vertebrates.
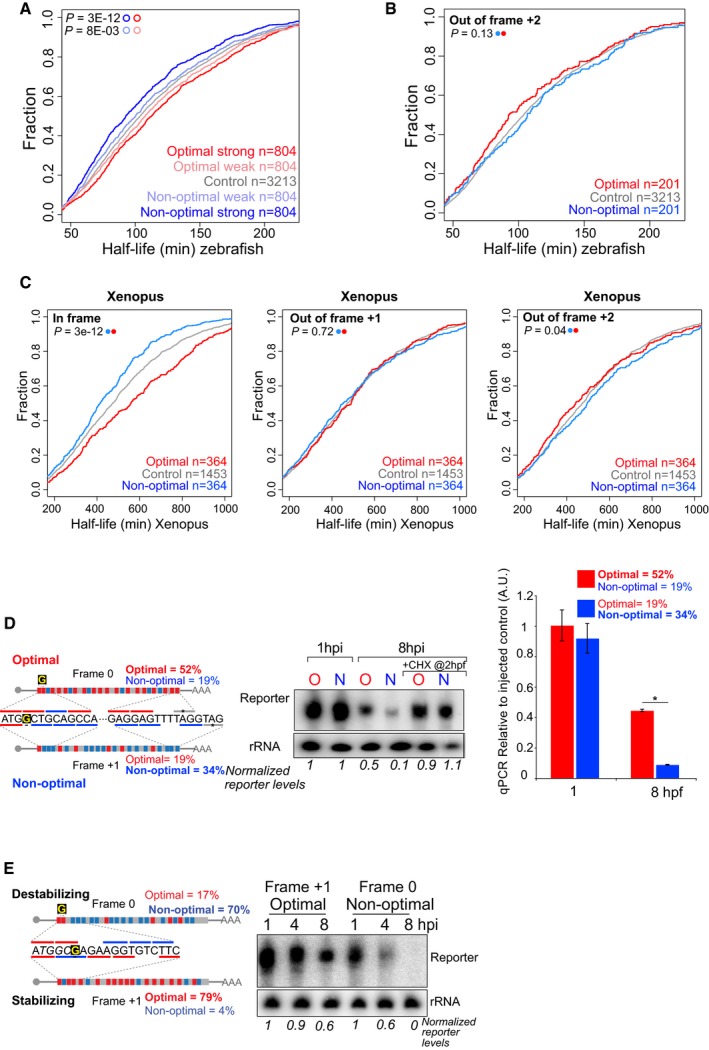
-
ACumulative distributions of the half‐life of the maternal mRNAs in the absence of zygotic transcription in zebrafish. Represented are mRNAs with the highest fraction of strong optimal codons (red, CSC value > 0.03), weak optimal codons (light red, 0.03 > CSC > 0), weak non‐optimal (light blue −0.03 < CSC < 0), or strong non‐optimal (blue CSC < −0.03). These four groups are non‐overlapping. The control group contains all the maternal transcripts (gray) for which we could calculate the half‐life. This result shows that the effects of the codon optimality (half‐life depending in the codon composition) are gradual and depend on the strength of the codons.
-
BCumulative distributions of the half‐life of the maternal mRNAs in the absence of zygotic transcription in zebrafish. The red line represents genes enriched in optimal codons quantified 2 nucleotides out frame. The blue one is enriched non‐optimal codons also counting in 2 nucleotides out frame. The gray lines represented all mRNAs. This result shows that the effect of codon optimality depends on the reading frame those codons are analyzed and as such depends on translation rather than a sequence motif independent of the reading frame. The P‐values are shown, and the color code determines the conditions that were compared using a Wilcoxon rank‐sum test.
-
CCumulative plots shown as described in (B), for Xenopus embryos, counting the codons in frame (left panel), 1 nucleotide out frame (middle) and 2 nucleotides out frame (right panel). These results indicate codon optimality shapes mRNA half‐life in a codon‐dependent manner in Xenopus embryos. The group of transcripts enriched in either optimal or non‐optimal in each of the three frames is different, meaning that the 364 optimal transcripts in frame are different from the 364 enriched in optimal codons when the codons composition was calculated in 1 or 2 nucleotides out of frame.
-
D, EDiagram of two transcripts that only differ in a single nucleotide (G in yellow) which creates a frameshift. One is enriched in optimal codons and the other in non‐optimal codons due to a change in the reading frame. A time‐course Northern blot shows that non‐optimal (N) transcripts decay faster than their optimal (O) counterparts. These differences are lost when translation is blocked by cycloheximide (CHX) (D). The radioactive intensity is shown in italics and normalized to the 1 hpi sample for each injection. Right, qRT–PCR for the two exogenous transcripts with opposite optimality from Fig 1I. Non‐optimal transcripts decay faster than their optimal counterparts (GFP was used as internal control). Results are shown as the averages ± standard error of the mean for three replicates. *P < 0.0001.
Codon identity regulates the maternal program of mRNA decay
Programs of maternal mRNA clearance can be distinguished based on their independence (maternal program) or dependence on zygotic transcription (zygotic program) (Tadros & Lipshitz, 2009; Yartseva & Giraldez, 2015). While miR‐430 plays a critical role in the zygotic program (Giraldez et al, 2006; Bazzini et al, 2012), the effectors and regulatory sequences in the maternal program are poorly understood (Yartseva & Giraldez, 2015). To determine whether codon optimality regulates mRNA stability/clearance during the MZT, we analyzed the fold change in mRNA levels before (2 hpf) and after zygotic genome activation (6 hpf). We compared the rate of maternal mRNA clearance for transcripts containing putative miR‐430 target sites (6, 7 or 8 mer complementary to miR‐430) (Bazzini et al, 2012), optimal‐ and non‐optimal transcripts in (i) wild‐type embryos, (ii) in embryos where we blocked the zygotic program by inhibiting transcription with α‐amanitin, and (iii) in embryos where we blocked de novo translation with cycloheximide (CHX) at 2 hpf, allowing the initial translation of the maternal mRNAs (0–2 h), but preventing translation thereafter (Fig 2A). Three lines of evidence indicate that codon identity shapes maternal mRNA decay. First, mRNAs enriched in non‐optimal codons had a higher rate of decay than those enriched in optimal codons in wild‐type embryos (Fig 2B) (P = 5e‐76, Wilcoxon rank‐sum test). Interestingly, the degree of regulation by non‐optimal codons is similar to that observed in transcripts containing putative miR‐430 target sites (Fig 2B), suggesting that optimality has a strong regulatory effect on mRNA stability (P = 0.61, Wilcoxon rank‐sum test). Second, in contrast to miR‐430, the effect of codon identity on mRNA stability is independent of transcription, indicating that codon‐mediated regulation of mRNA stability is part of the maternal program of RNA decay (Fig 2B) (P = 2e‐48, Wilcoxon rank‐sum test) consistent with a recently published study (Mishima & Tomari, 2016). Third, the effect of codon optimality‐mediated stability/decay was abolished after blocking translation with cycloheximide (Fig 2B) (P = 0.83, Wilcoxon rank‐sum test), consistent with a translation‐dependent regulation of mRNA stability. These results were recapitulated in individual endogenous transcripts using cycloheximide or a targeted translation blocking morpholino to inhibit their translation, reducing the translation‐dependent stabilization/decay of the endogenous mRNA (Fig 2C). These results indicate that the effect of codon identity is part of the maternal program of mRNA clearance and requires translation to regulate mRNA decay during the MZT (Mishima & Tomari, 2016).
Figure 2. Codon identity regulates the maternal program of mRNA decay.
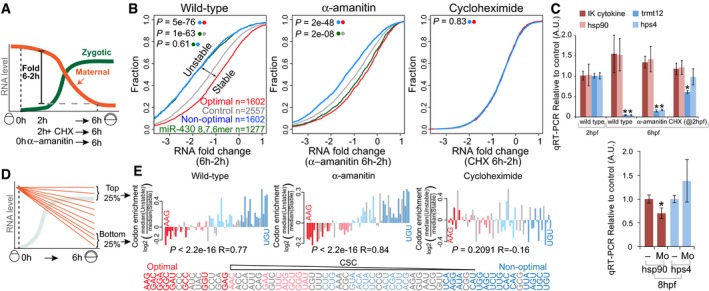
- Diagram illustrating the maternal‐to‐zygotic transition in zebrafish, depicting the analysis of mRNA fold changes between 2 and 6 hpf. Embryos were bathed in cycloheximide (CHX) at 2 hpf or injected with the RNA Pol II inhibitor α‐amanitin at 0 hpf.
- Cumulative distributions of mRNA fold change (6–2 h) in wild‐type embryos, embryos injected with α‐amanitin, and embryos treated with the translation elongation inhibitor cycloheximide. The different curves represent transcripts: enriched in non‐optimal codons (top quartile, blue), containing at least one miR‐430 complementary site (6, 7 or 8 mer) (green), enriched in optimal codons (top quartile, red) and a control set of maternal genes that do not follow any of the described criteria (gray). P‐values are indicated for the comparisons shown with the colored dots (Wilcoxon rank‐sum test).
- qRT–PCR analysis for predicted optimal (red) and non‐optimal (blue) transcripts relative to the controls (cdk2ap2 and taf15). IK cytokine, constitutive heat shock protein 90 (hsp90ab), tRNA methyltransferase 12 homolog (trmt12), and Hermansky–Pudlak syndrome 4 (hps4). Results are shown as the average expression ± standard deviation from three biological replicates (top panel) and six biological replicates (bottom panel) (Mo, embryos were injected with a morpholino complementary to the translation start site of the endogenous mRNA). *P < 0.0001 compared to 2 h (top panel). *P < 0.005 compared to uninjected embryos (bottom panel).
- Schematic representing the maternal mRNA decay dynamics and the transcript groups analyzed.
- Bar plot representing relative codon distributions between the most unstable (quartile) and the most stable (quartile, n = 1,458) maternal mRNAs in wild‐type, α‐amanitin‐, and cycloheximide (CHX)‐treated embryos. Codons were sorted by their CSC value (Fig 1D). P‐values are indicated for the comparisons between enrichment/depletion in each condition and CSC (Spearman rank test). Optimal codons were highlighted in red and non‐optimal in blue (light, weak; dark, strong, as Fig 1F). Note the depletion of optimal (red) and enrichment of non‐optimal (blue) codons in unstable mRNAs in wild‐type and α‐amanitin‐treated embryos but not in CHX‐treated embryos, consistent with a translation‐mediated mechanism of maternal mRNA decay through codon optimality.
The results presented above indicate that a high density of non‐optimal codons destabilizes mRNAs during the MZT; however, the extent to which codon identity regulates maternal mRNA degradation is unclear. We reasoned that if codon identity plays a major role in maternal mRNA turnover, those mRNAs that are most strongly degraded should be enriched for non‐optimal codons and depleted of optimal codons independently of potential regulatory elements in their 5′ and 3′UTRs. Alternatively, if codon identity represents a minor mechanism of maternal mRNA decay, then we expect that codon optimality effects would be overshadowed by transcripts under other regulatory mechanisms with neutral codon bias. We analyzed mRNA decay in untreated embryos, in embryos where the zygotic program of mRNA decay is inhibited by α‐amanitin treatment, and in embryos where translation is inhibited by CHX treatment (Fig 2A). We first compared the codon composition of transcripts in the top and bottom quartiles of mRNA fold change during the MZT (between 2 and 6 hpf) (Fig 2D) (2,916 genes). In wild‐type and α‐amanitin‐treated embryos, the most unstable mRNAs were enriched in non‐optimal codons and depleted of optimal codons (Fig 2E). In α‐amanitin‐treated embryos, where the zygotic program of mRNA decay is not active, 22 of 24 non‐optimal codons were enriched in the most unstable mRNAs compared to the most stable ones (Fig 2E). Conversely, 14 of 15 optimal codons were depleted within the most unstable mRNAs compared to the most stable ones (Fig 2E). As a consequence, we observed a significant correlation between codon stability coefficient (CSC) and codons enriched or depleted in unstable transcripts (P = 2.2e‐16, R = 0.77 wild type, and P = 2.2e‐16, R = 0.84, α‐amanitin, Spearman) (Fig 2E). In contrast, no significant correlation was observed in CHX‐treated embryos (P = 0.2 R = −0.16, Spearman). These results indicate that codon identity represents a major, translation‐dependent mechanism regulating maternal mRNA stability during the MZT.
Codon identity is a conserved mechanism shaping mRNA stability during the MZT
Zebrafish and Xenopus share a significant degree of conservation in the codon optimality code (Fig 1C). To determine whether differential mRNA stability across the MZT is regulated by codon optimality across animal species, we analyzed mRNA decay across Xenopus, mice, and Drosophila (Table EV2). First, we applied the codon optimality values defined in zebrafish to predict the transcripts enriched in optimal and non‐optimal codons in these species, then we compared mRNA levels before and after the MZT (Fig 3A). We observed that mouse, Xenopus, and Drosophila transcripts enriched for zebrafish optimal codons were significantly more stable than transcripts enriched for non‐optimal codons (P = 6e‐50 Xenopus, 2e‐15 mouse, 2e‐19 Drosophila, Wilcoxon rank‐sum test) (Fig 3C–F). Conversely, genes enriched in non‐optimal codons were less stable. Genes enriched in optimal codons were enriched in GO terms related with housekeeping processes such as translation, protein folding, and regulation of actin dynamics (Fig EV3B and C). Second, to determine whether codon optimality is a major mechanism of regulation for a significant fraction of the maternal transcriptome, we analyzed the codon composition of top (most stable) and bottom (least stable) quartiles of mRNAs, and compared these to the codon optimality in zebrafish (Fig 3B). We observed that optimal codons were depleted and non‐optimal codons were enriched in unstable mRNAs, consistent with a conserved role of codon optimality in the regulation of mRNA stability during the MZT across animals (Fig 3G–J) (P = 6.8e‐12 zebrafish, 5e‐11 Xenopus, 1e‐03 mouse, 3e‐04 Drosophila, Spearman). While the effect of non‐optimal codons appears to be weaker in mouse that in other species, future experiments will be needed to determine whether these differences are due to the predicted code used to infer optimality in mice or the differences in timing and cell division that might make other mechanisms more prevalent during this transition (Svoboda et al, 2015). Together, these results indicate that codon optimality‐mediated control of mRNA stability is a conserved mechanism to regulate differential mRNA stability during the MZT across animals.
Figure 3. Codon identity is a conserved mechanism shaping mRNA stability during the MZT.
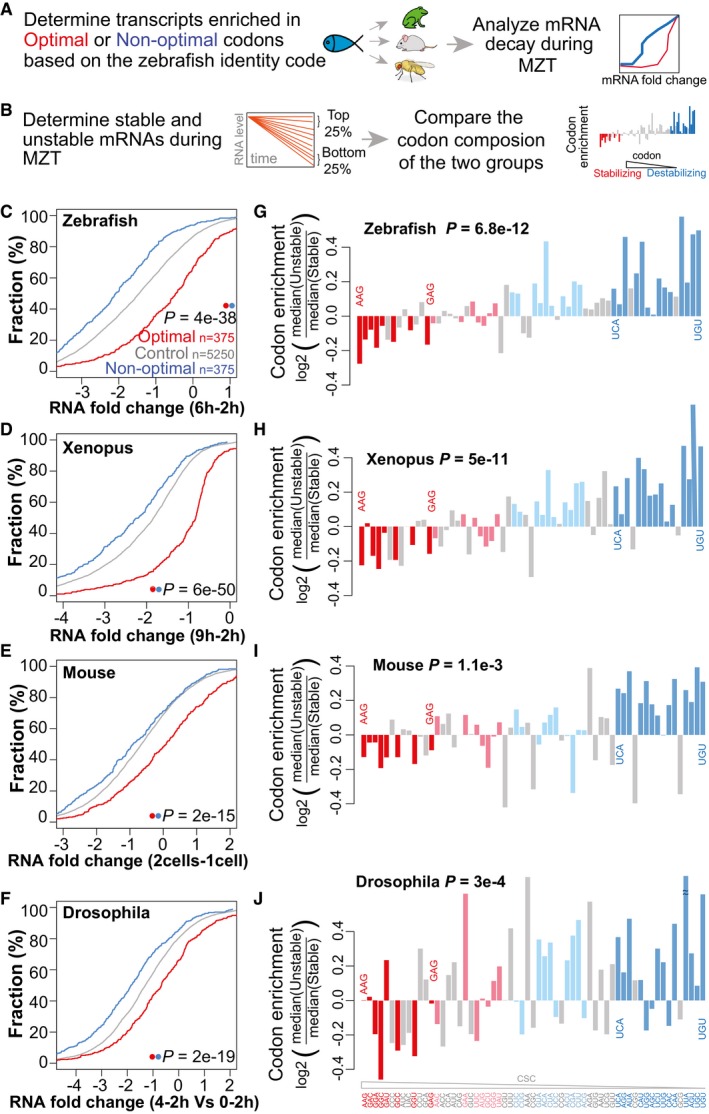
-
ASchematic representation of the analysis workflow for the panels (C–F). Optimal and non‐optimal transcripts are predicted based on the zebrafish optimality code (Fig 1F) and mRNA decay during the MZT in each species was analyzed.
-
BSchematic representation of the analysis workflow for the panels (G–J). The top and bottom quartiles of mRNAs, ranked by stability during the MZT were determined for each species. The codon composition of each group was compared and correlated with the codon optimality defined in zebrafish (CSC).
-
C–FCumulative distributions of mRNA fold change for optimal (red), non‐optimal (blue), and control transcripts across zebrafish (C), Xenopus (D), mouse (E), and Drosophila (F).
-
G–JCodon enrichment/depletion between the most unstable (quartile) and the most stable (quartile, n = 1,500) maternal mRNAs during the MZT in zebrafish (G), Xenopus (H), mouse (I), and Drosophila (J). Most optimal codons (red: light, weak; dark, strong) are depleted, and non‐optimal (blue: light, weak; dark, strong) codons are enriched in unstable mRNAs compared to stable ones. P‐values are indicated for the comparisons between enrichment/depletion in each species and codon optimality defined in zebrafish (CSC) (Spearman rank test).
Figure EV3. Characterization of genes enriched in optimal and non‐optimal codons.
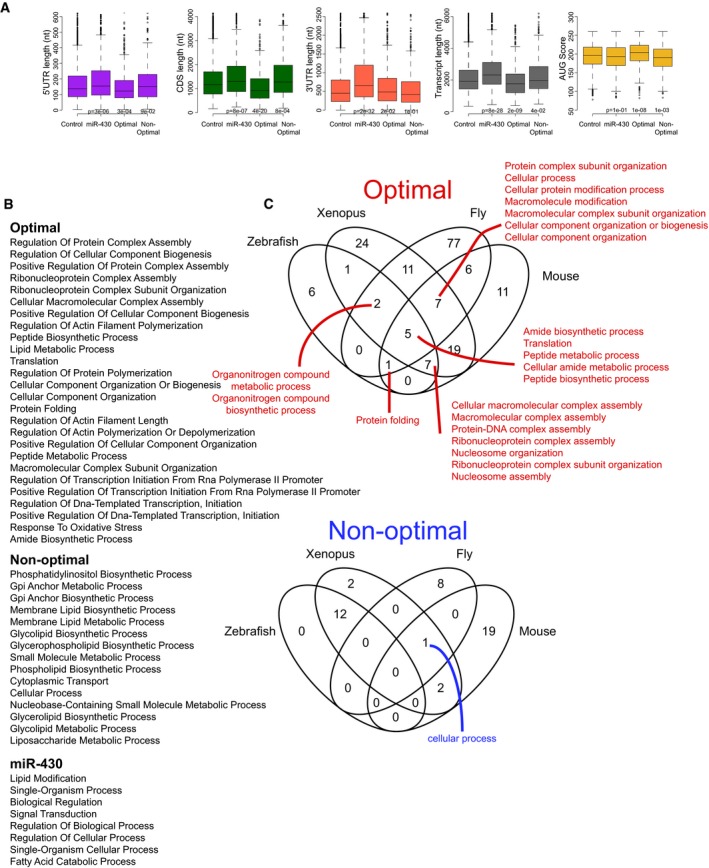
- Box plot showing different transcript features of genes enriched in optimal codons, in non‐optimal codons and genes with predicted miR‐430 target sites. For example, miR‐430 targets have significantly longer 3′UTRs. The box defines the first and third quartiles, with the median indicated with a thick black line and vertical lines indicate the variability outside the upper and lower quartiles.
- Enrichment for Gene ontology functions among optimal, non‐optimal, and miR‐430 target sites genes.
- Gene ontology functions enriched in orthologous genes in zebrafish, Xenopus, mouse, and Drosophila that share the same level of optimality.
Codon composition shapes mRNA levels during homeostasis across vertebrates
Codon bias correlates with gene expression across multiple species (Ikemura, 1982; Akashi, 1994; Akashi & Gojobori, 2002; Drummond & Wilke, 2008; Kudla et al, 2009; Novoa & Ribas de Pouplana, 2012). Because codon optimality showed a strong effect on mRNA half‐life during the MZT, we investigated whether it can also influence steady‐state mRNA levels across vertebrates. To this end, we calculated the correlation between codon occurrence and mRNA levels in zebrafish, Xenopus, and 32 human tissues (Uhlen et al, 2015) (Fig 4A). We observed a significant correlation between codon stability score and mRNA expression levels across zebrafish embryos (Fig 4B), organs (Fig 4B), Xenopus (Fig 4B), and human tissues (Fig 4C). Comparison with the optimality values defined during embryogenesis in zebrafish and Xenopus revealed that highly expressed genes are enriched in optimal codons and depleted in non‐optimal ones in zebrafish and Xenopus (Fig 4B and C). These results indicate that preferential use of specific codons in highly expressed genes is conserved across vertebrates, and remarkably, it corresponds with the CSC defined during embryogenesis (Figs 1D and 4B and C). Based on the differential half‐life of mRNAs with different codon optimality for exogenous (Figs 1C and I, and EV2D and E) and endogenous mRNAs (Figs 1D and H, 2, 3, and EV2C), these results suggest that codon optimality shapes steady‐state mRNA levels across tissues in vertebrates.
Figure 4. Codon and amino acid composition shape mRNA levels across vertebrates.
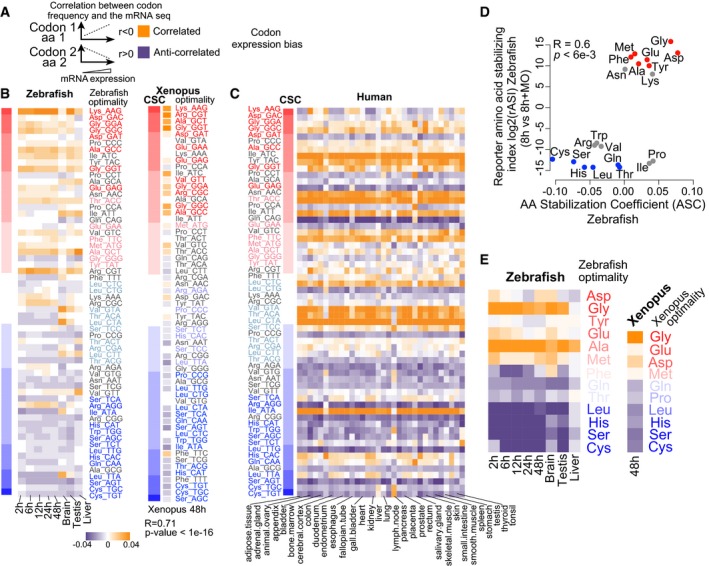
- Diagram explaining how codon/amino acid expression bias was computed: Correlation was calculated between the frequency of each codon/amino acid and mRNA levels per gene. Orange means correlation and purple anticorrelation.
- Heatmap showing the codon expression bias across the transcriptome for different developmental time points (2, 6, 12, 24 and 48 hpf) and adult organs in zebrafish and Xenopus at 48 h (right). The codon optimality and CSC calculated during embryogenesis are shown to the right (red, optimal to blue, non‐optimal). Heatmap shows the correlation of codon occurrence and Xenopus mRNA expression at 48 h. On the left, a heat map (red‐blue) shows the CSC values for zebrafish and Xenopus. The codons are sorted by their CSC values. For most tissues, optimal codons are correlated with highly expressed genes (orange), while non‐optimal codons are anticorrelated with high levels of expression (purple). We also observe a correlation between mRNA expression and the CSC across different samples (2 h: P = 1e‐07 R = 0.6, 6 h: P = 2e‐16 R = 0.7, 12 h: P = 3e‐09 R = 0.6, 24 h: P = 2e‐07 R = 0.6, 48 h: P = 1e‐07 R = 0.6, brain: P = 1e‐04 R = 0.4, testis: P = 1e‐07 R = 0.6, liver: P = 1e‐03 R = 0.4, Spearman rank test).
- Heatmap of the codon composition bias showing the correlation between the CSC in zebrafish (left) with gene expression across 32 human tissues. The codon optimality is labeled as in (B).
- Biplots of the reporter amino acid stabilizing index (rASI) comparing 8 h versus 8 h + MO and amino acid stabilization coefficient (ASC) in zebrafish embryos, P < 6e‐3, R = 0.6, Spearman rank correlation. Based on the consistent identification by both methods, we defined optimal amino acids highlighted in red and non‐optimal amino acids in blue.
- Heatmap showing the amino bias across the transcriptome for different developmental time points (2, 6, 12, 24 and 48 hpf) and adult organs in zebrafish, and Xenopus at 48 h (right). The optimal and non‐optimal amino acids are shown to the right (red, optimal to blue, non‐optimal).
Synonymous codons can share the same effects on mRNA stability
The analysis presented above indicates that specific codons influence mRNA stability during embryogenesis (Figs 1, 2, 3), and are correlated with mRNA expression levels and optimality during tissue homeostasis (Fig 4). Previous studies have shown a strong amino acid bias in the transcriptome. This bias has been explained in terms of tRNA expression, tRNA demand, translational accuracy, and the metabolic “cost” of different amino acids (Akashi, 1994; Craig & Weber, 1998; Baudouin‐Cornu et al, 2001; Akashi & Gojobori, 2002; Pechmann & Frydman, 2013). Our experimental assays of codon identity on mRNA stability raised the possibility that synonymous codons might share similar effects on mRNA levels, resulting in amino acid optimality. This could explain the amino acids bias in the transcriptome due to the consistent effect of synonymous codons on mRNA stability. To test this hypothesis, we first analyzed whether the encoded amino acid influences mRNA stability in zebrafish and Xenopus using two approaches parallel to the codon analysis undertaken in Fig 1. First, we analyzed the reporter amino acid stabilizing index (rASI) as the statistical significance of the enrichment (+) or depletion (−) of synonymous codons for each amino acid between these conditions (−MO versus +MO) (F‐value, ANOVA). Second, we analyzed the amino acid stabilization coefficient (ASC, the Pearson correlation coefficient between mRNA half‐life and the amino acids encoded in that transcript). Interestingly, both methods revealed that several encoded amino acids are correlated with mRNA stability. Similar to the analysis preformed at the levels of individual codons, we defined optimal amino acids that are enriched in the library and correlated with longer mRNA half‐life, indicating a potential stabilizing effect (rASI > 1,000 and ASC > 0) and non‐optimal codons with potential destabilizing effects (rASI < −1,000 and ASC < 0) (Figs 4D and EV4A). While this analysis depends in the relative use and optimality for each synonymous codon, there are clear examples of amino acids that are encoded by either optimal (asparagine) or non‐optimal (histidine) codons. These results indicate that amino acids can also be defined as optimal and non‐optimal, influencing mRNA stability when they are decoded, yet future experiments will be needed to uncouple codon and amino acid optimality. This observation adds a novel layer of complexity into the codon optimality, where the encoded amino acid might affect the stability of the translated mRNA.
Figure EV4. Codon optimality regulates poly(A) tail length.
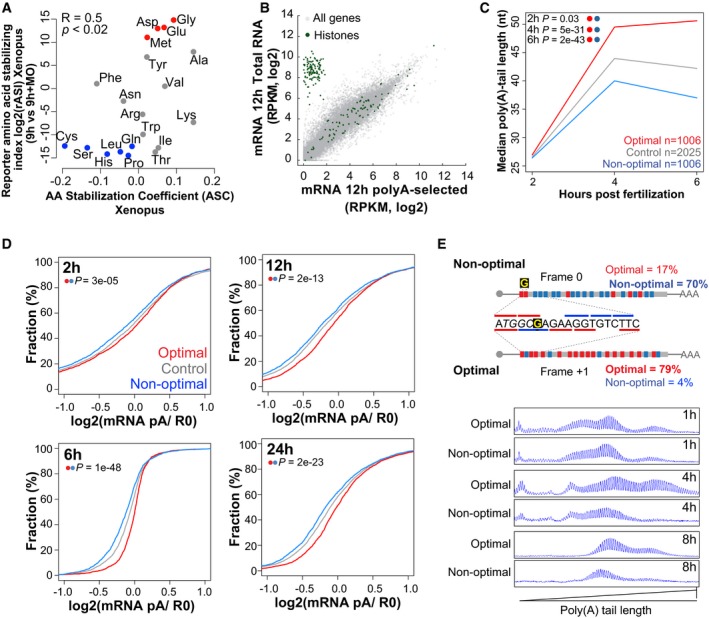
- Biplots of the reporter amino acid stabilizing index (rASI) comparing 9 h versus 9 h + MO and amino acid stabilization coefficient (ASC) in Xenopus embryos, P < 0.02, R = 0.5, Spearman rank correlation. Based on the consistent identification by both methods, we defined optimal amino acids highlighted in red and non‐optimal amino acid in blue.
- Scatter plot showing expression levels (log2 RPKM) of purified poly(A) mRNA and total RNA at 12 hpf. Histone genes (that lack a poly(A) tail) are highlighted in green.
- Line plot showing the poly(A) tail length of control mRNAs (gray) based on PAL‐seq data, mRNAs enriched in optimal codons (red), or in non‐optimal (blue) at 2, 4, and 6 hpf (P = 0.03, P = 5e‐31, P = 2e‐43, Wilcoxon rank‐sum test, respectively).
- Cumulative distributions of the ratio of purified poly(A)+/total RNA for genes enriched in optimal codons (red) (n = 1,943 at 2 h, 1,791 at 6 h, 2,207 at 12 h, and 2,469 at 24 h) or non‐optimal codons (blue) (n = 1,941 at 2 h, 1,790 at 6 h, 2,208 at 12 h, and 2,462 at 24 h) or genes that were not included in any of the other two groups (gray) at 2, 6, 12, and 24 hpf (P = 3e‐05, P = 1e‐48, P = 2e‐13, P = 2e‐23, Wilcoxon rank‐sum test, respectively).
- Single nucleotide resolution electrophoresis for poly(A) length for two sets of transcripts that only differ in a single nucleotide insertion G, but contains opposite codon optimality due to a change in the reading frame (Fig EV2E). This result shows that at 1 h the poly(A) tail length of both constructs is similar; however, by 4 and 8 h the length of the poly(A) tail is shorter in the transcript enriched for non‐optimal codons.
To evaluate whether amino acid optimality can partially explain the amino acid bias in gene expression in homeostasis, we calculated the correlation between amino acid occurrence and mRNA levels in zebrafish and Xenopus (Fig 4A). We found that optimal amino acids are preferentially used in highly expressed genes and non‐optimal amino acids are preferentially used in lowly expressed genes in both zebrafish and Xenopus (Fig 4E). These results suggest that part of the amino acid bias in the transcriptome can be explained by amino acid optimality and the concordant effect of synonymous codons on mRNA stability.
Codon composition affects poly(A) tail length and translation efficiency
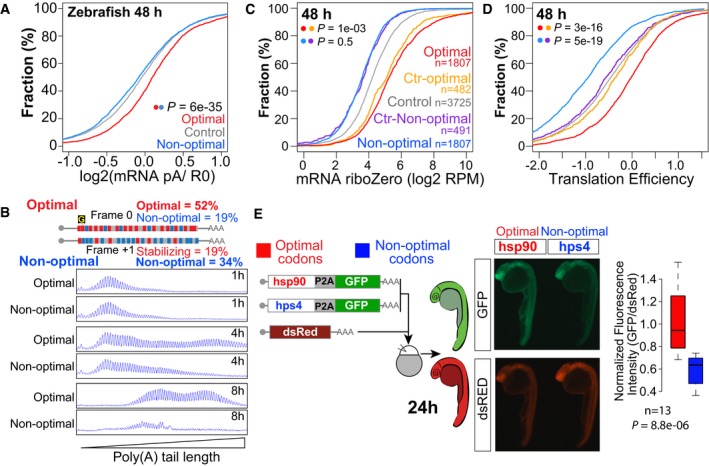
The length of the poly(A) tail influences translation and stability of mRNAs (Eckmann et al, 2011; Weill et al, 2012; Subtelny et al, 2014). Three lines of evidence indicate that codon identity modulates the polyadenylation status of the mRNA. First, we analyzed the polyadenylation status of mRNAs with different codon optimality content across different embryonic stages (2, 6, 12, 24, and 48 hpf). To this end, we calculated the fraction of deadenylated mRNAs by comparing the levels of poly(A+)‐containing mRNA (oligo(dT) purified) with total RNA depleted of ribosomal RNA. This provides an indirect measurement of the polyadenylation status and represents the proportion of deadenylated and polyadenylated mRNAs rather than a direct measurement of their length (Bazzini et al, 2012; Park et al, 2016). As a control, we evaluated histone mRNAs, which are typically deadenylated, and found that they displayed a low poly(A)/total RNA ratio (Fig EV4B) supporting the use of the poly(A+)/total RNA ratio as a proxy of the polyadenylation status. We found that mRNAs enriched in non‐optimal codons display a lower polyadenylation status (poly(A)/total RNA ratio) compared to those enriched in optimal codons across developmental stages (Figs 5A and EV4D) (P = 6e‐35, Wilcoxon rank‐sum test). Second, we analyzed the poly(A) tail length of mRNAs throughout early zebrafish embryogenesis using previously generated PAL‐seq data (Subtelny et al, 2014). We found that mRNAs enriched in non‐optimal codons had shorter poly(A) tails at 4 and 6 hpf, whereas mRNAs enriched in optimal codons had longer poly(A) tails (Fig EV4C) (P = 5e‐31, P = 2e‐43, Wilcoxon rank‐sum test). Their initial poly(A) tail lengths were comparable (P = 0.03, Wilcoxon rank‐sum test) (Fig EV4C), consistent with similar polyadenylation status for both mRNA populations by 2 hpf (Fig EV4D) (P = 3e‐05, Wilcoxon rank‐sum test). Third, we analyzed the poly(A) tail length using high‐resolution electrophoresis (Bazzini et al, 2012) for two sets of reporter mRNAs that had opposite codon optimality depending on the frame in which they were translated (Figs 1I and EV2E). Non‐optimal reporters displayed shorter poly(A) tails (Figs 5B and EV4E) and faster decay over time than their optimal counterparts, although their initial poly(A) status was comparable at 1 hpi (Figs 1I, 5B, EV2E, and EV4E). Together, consistent with recent reports (Mishima & Tomari, 2016), these results indicate that a higher ratio of non‐optimal/optimal codons is associated with a shorter poly(A) tail, mRNA destabilization and decay (Fig 6A).
Figure 5. Codon optimality regulates poly(A) tail length and translation efficiency.
-
ACumulative distributions of the ratio of poly(A) selected and a total RNA at 48 hpf.
-
BSingle nucleotide resolution electrophoresis for poly(A) tail length for two sets of transcripts that only differ in a single nucleotide insertion (“G”), but contain opposite codon optimality due to a change in the reading frame (Fig 1I).
-
C, DTotal mRNA (C) and translation efficiency (D) for genes enriched in non‐optimal codons (n = 2,611) and in optimal codons at 48 hpf (n = 2,612). Control sets of genes that have similar RNA level distributions as each group are shown (purple and yellow, f.g). P‐values are indicated for the comparisons shown with the colored dots (Wilcoxon rank‐sum test). Note how optimal mRNAs have a higher polyadenylation status (A) and translation efficiency than non‐optimal mRNAs (D).
-
EFluorescence microscopy images of representative embryos at 24 hpf expressing a bicistronic reporter encoding an optimal or non‐optimal coding sequence followed by a cis‐acting hydrolase elements (P2A; CHYSEL) and GFP. P2A causes ribosome skipping, what allows us to use GFP as a readout of the translation of the upstream polypeptide. dsRed mRNA is co‐injected as a control, and it is shown below. The level of GFP is higher in the optimal reporter (hsp90) than the non‐optimal one (hps4). Box plot displays fluorescence quantification of 24‐h embryos injected with each reporter. GFP fluorescence intensity was normalized to dsRed intensity in each embryo, n = 13 pairs, P = 8.8e‐06, Wilcoxon rank‐sum test. The box defines the first and third quartiles, with the median indicated with a thick black line and vertical lines indicate the variability outside the upper and lower quartiles.
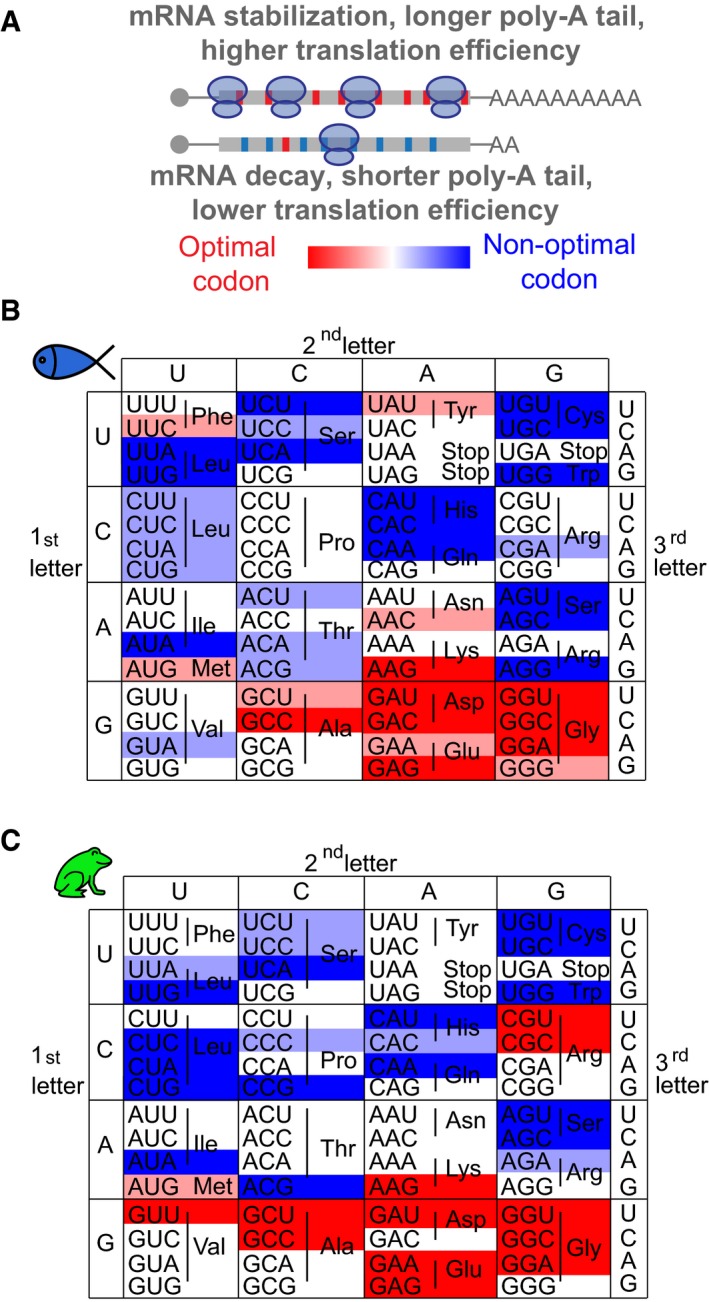
Figure 6. “Codon optimality code” in vertebrates.
-
AmRNAs enriched in optimal (red) codons have longer poly(A) tails, higher translation efficiency (TE), and higher stability compared to genes enriched in non‐optimal codons (blue).
-
B, CHeatmap showing the codon optimality code underlying the genetic code in zebrafish (B) and Xenopus (C). Based on the consistent identification by two methods (rCSI and CSC), we defined optimal codons in red and non‐optimal codons in blue. The intensity of the color represents the strength (light, weak; dark, strong) based on different CSC values, which corresponds to different cumulative effects in mRNA half‐life (Fig 1F).
Translation efficiency across mRNAs in vivo can vary over 250‐fold between different genes. Ribosome footprinting provides a quantitative measurement of translation and, upon normalization to total RNA, allows us to calculate the translation efficiency for each gene (Ingolia et al, 2009). While the codon adaptation index (Sharp & Li, 1987) only explains a small portion of the variance in protein per mRNA in yeast (Ingolia et al, 2009) and bacteria (Plotkin & Kudla, 2011), differences in codon optimality in yeast for individual reporter genes result in different protein output and inferred translation efficiency (Presnyak et al, 2015). Furthermore, it has been shown that codon composition can influence translation efficiency (Hussmann et al, 2015). To investigate the effect of codon optimality in translation in vivo in vertebrates, we measured translation efficiency using ribosome footprinting across different developmental time points in zebrafish embryos (2, 6, 12, 24 and 48 hpf) (Bazzini et al, 2014). Because codon optimality influences mRNA levels, we used a control population with a similar mRNA level distribution (48 h P = 1e‐3 for optimal, P = 0.5 for non‐optimal, Wilcoxon rank‐sum test) (Figs 5C and EV5A). mRNAs enriched in optimal codons had significantly higher translation efficiency than the control set across time points (48 h P = 3e‐16, Wilcoxon rank‐sum test) (Figs 5D and EV5A). In contrast, mRNAs enriched in non‐optimal codons displayed lower translation efficiency than their control set (48 h P = 5e‐19, Wilcoxon rank‐sum test) (Figs 5D and EV5A). Consistent with these observations, ribosome profiling of individual genes with different optimality displays dramatically different ribosome occupancy, with higher translation of optimal genes (Fig EV5B). To determine whether protein production is indeed affected by the codon optimality of individual genes, we compared GFP expression from two reporter mRNAs consisting of an optimal or non‐optimal coding sequence followed by a ribosome skipping sequence (P2A) (Donnelly et al, 2001; de Felipe et al, 2006) and GFP. This allowed the analysis of protein production in vivo independently of the half‐life of the protein analyzed. We observed higher translation of the optimal reporter compared to the non‐optimal one (Fig 5E), while no difference was observed in the expression levels of the co‐injected dsRed control mRNA (Fig 5E). Taken together, these results indicate that codon optimality shapes translation efficiency and protein output in vivo.
Figure EV5. Codon optimality regulates translation efficiency.
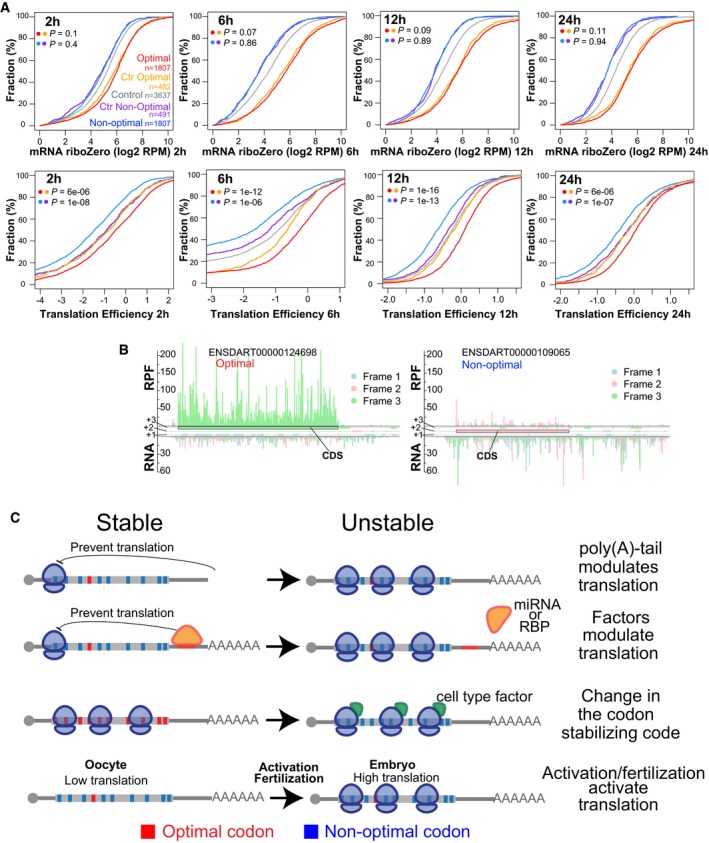
- Cumulative distributions of mRNA levels and translation efficiency at 2, 6, 12, and 24 hpf for genes enriched in optimal (red) or non‐optimal codons (blue). Also plotted are two control sets of genes that have similar RNA level distribution to those enriched in optimal or non‐optimal codons (yellow for optimal and purple for non‐optimal), and the rest of the analyzed genes (gray). Number of analyzed genes and P‐values obtained after comparing the cumulative distribution between of the experimental set and the control set as indicated by the colors, Wilcoxon rank‐sum test.
- Subcodon profile plot showing ribosome protected fragments (RPF) and input reads (total RNA) aligned to each of the transcripts shown. Reads are colored based on the frame (1, 2, or 3) position relative to the transcript. All putative ORFs (distal AUG‐Stop) were also colored for each respective frame (blue, pink, and green boxes). The annotated ORF is marked. Note that the scale for both transcripts is the same. The gene enriched in optimal codons presents a higher translation efficiency because contain a higher ration of RPF/RNA than the gene enriched in non‐optimal codons.
- Speculative model representing how different factors could modulate mRNA expression due to codon optimality. The length of the poly(A) tail as well as factors such microRNA or RNA binding protein can modulate translation and consequently modulate the effects of codon optimality on mRNA stability. Alternatively, specific cellular factors may affect the codon optimality code and so change the gene expression across tissues by modifying mRNA stability. For example, in the oocyte the level of translation is low. After egg activation and/or fertilization translation is activated, consequently a gene enriched in non‐optimal codons may become unstable after translation is activated.
Discussion
Our study identifies mRNA translation and codon optimality as a conserved mechanism shaping maternal mRNA clearance during early embryogenesis. mRNAs with a high proportion of non‐optimal codons are less stable, whereas the most stable mRNAs show an enrichment of optimal codons (Figs 1 and 2). Based on our current analysis, we propose that this mechanism is a key part of the maternal program of mRNA regulation and modulates the stability and decay of several thousand maternal mRNAs in zebrafish (Figs 2 and 3). A recent analysis of codons on mRNA stability in zebrafish by Mishima and Tomari relay on the codon adaptation index, which only allows to measure the relative effects of synonymous codons, but does not allow comparison of codons encoding for different amino acids (Mishima & Tomari, 2016). In contrast, our study directly measures the relative effects of all codons on mRNA stability in a translation‐dependent manner, allowing direct comparison of synonymous and non‐synonymous codons on mRNA stability. The strength of the regulation and number of genes modulated by codon optimality are comparable to that observed by microRNAs during this process (Fig 2). It is likely that these mechanisms function in a fraction of genes with additional regulatory motifs in the 3′UTR and RNA binding proteins to modulate mRNA stability during the MZT (Tadros & Lipshitz, 2009; Yartseva & Giraldez, 2015). Indeed, it has been shown that the effect of the codon adaptation index on mRNA stability is stronger in transcripts with shorter 3′UTRs (Mishima & Tomari, 2016), suggesting that longer UTRs might harbor regulatory elements that antagonize the effect of codon optimality. Interestingly, maternal mRNAs deposited in the egg are stable until egg activation, fertilization, or zygotic genome activation, depending on the specific mRNA (Mendez & Richter, 2001; Clark et al, 2007; Yartseva & Giraldez, 2015). Codon optimality could provide a mechanism to trigger mRNA decay upon activation of translation following fertilization and might explain the tight control between the simultaneous activation of translation and mRNA deadenylation/destabilization upon fertilization (Fig EV5C) (Richter & Coller, 2015). Conversely, factors that modulate translation could influence mRNA stability via codon optimality, thus providing a valuable mechanism for feedback regulation in the cell (Fig EV5C). For example, this mechanism could modulate degradation/stability once mRNAs are localized to distinct subcellular regions and initiate translation, such as during synaptic regulation (Kiebler & DesGroseillers, 2000; Martin & Ephrussi, 2009), axis specification during development (Johnstone & Lasko, 2001), or germ cell specification (Slaidina & Lehmann, 2014).
By analyzing the effect of translation on mRNA stability using a reporter library and endogenous mRNA half‐life (Fig 1), we defined a codon optimality code during early embryogenesis (Fig 6). Our results provide evidence for the conservation of codon optimality (Presnyak et al, 2015) across vertebrates (Figs 1, 3 and 4), identify physiological contexts where this mechanism regulates gene expression during the MZT (Figs 2 and 3), and suggest that specific amino acids are associated with differential mRNA stabilization (Fig 4). The bias between codons or amino acids, and mRNA expression levels has been previously recognized across species and is thought to result from selection for efficient, accurate translation, and folding of highly expressed genes (Ikemura, 1982; Akashi, 1994; Akashi & Gojobori, 2002; Drummond & Wilke, 2008; Kudla et al, 2009; Novoa & Ribas de Pouplana, 2012). The amino acid optimality code (Fig 6) provides an alternative perspective on sequence changes between paralogs in evolution and human disease. Individual gene sequence might be determined not only by the function of the protein but also by its optimality with respect to mRNA stability and translation efficiency potentially driving sequence evolution. Based on the strong effect of codon optimality on mRNA stability and the relationship between codon bias and optimality, codon optimality is a strong post‐transcriptional regulatory mechanism in vertebrates that shapes gene expression across tissues. This mechanism can explain the previously recognized relationship between codons used or amino acids encoded in the transcriptome and mRNA expression levels across metazoans in both prokaryotes and eukaryotic cells (Ikemura, 1982; Akashi & Gojobori, 2002; Akashi, 2003; Hussmann et al, 2015). However, metazoans have different evolutionary constraints regarding adaptation, metabolic and nutrients availability than yeast or prokaryotes. We observe that there is a correlation between tRNA levels and optimality, with non‐optimal codons associated with lower tRNA levels (Fig 1). Indeed, previous studies of tRNAs in tumor cells have found that different gene networks driving proliferation and cell differentiation have different codon usage, and the activation of these programs is influenced by different tRNA levels (Gingold et al, 2014), suggesting that they are under the regulation of codon optimality. The codon optimality code defined during early embryogenesis (Fig 1) correlates with codon bias in homeostasis (Fig 4), suggesting that the code is conserved between early embryogenesis (MZT) and somatic tissues. However, further experiments, defining codon optimality code in specific cell types or under different physiological conditions, that is, stress, will define how the code changes with cell identity and cellular state. In addition to tRNA levels, tRNA modification might also play a key role in optimality (Gustilo et al, 2008; Novoa et al, 2012). Previous studies in yeast have linked the effect of codon optimality to the translocation speed of the ribosome, which also influences protein folding (Presnyak et al, 2015; Yu et al, 2015). Reduced codon optimality coincides with shorter poly(A) tail length (Fig 5). In addition, we found the genes enriched in non‐optimal codon present lower translation efficiency, suggesting that codon optimality also influences the level of translation (Fig 5). However, we cannot distinguish whether the poly(A) length changes are the cause or consequence of the translation differences caused by codon optimality. In addition, we cannot exclude that additional features that influence translation initiation are co‐selected with optimal codons (Fig EV3A). Our results are consistent with the role of codon optimality in translation efficiency and the speed of ribosome translocation in yeast (Novoa & Ribas de Pouplana, 2012; Hussmann et al, 2015; Presnyak et al, 2015). Based on the slower translocation rates in yeast over non‐optimal codons (Presnyak et al, 2015), we speculate that slower ribosome translocation might allow the recruitment of accessory proteins that engage the deadenylation machinery. While we observe a correlation between tRNA levels and the extremes of optimality, it is likely that this code integrates multiple inputs of translation from tRNA availability and accuracy (Akashi, 1994; Ishimura et al, 2014; Hussmann et al, 2015), tRNA modification (Gustilo et al, 2008; Novoa et al, 2012), to peptide bond formation, amino acid identity, and other factors influencing the translocation rate and ultimately elongation (Baragana et al, 2015; Faller et al, 2015). Future studies will be needed to determine how the ribosome or accessory proteins decode codon optimality (Richter & Coller, 2015), and how this code is regulated across cellular transitions, tissues, and pathological states.
Materials and Methods
Reporter library preparation
Zebrafish embryos were collected at 2, 8, and 10 h post‐fertilization (hpf), and 120 ng of total RNA was isolated using TRIzol following manufacturer instructions. Poly(A) mRNAs were purified using the Dynabeads mRNA Purification kit (Invitrogen, cat 610.06). Poly(A) purified mRNAs were reverse transcribed using SuperScript™ III Reverse Transcriptase (Invitrogen) with RT‐Oligo (GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCCTAgCTAcCTANNNNNNNNS) according to the manufacturer (Invitrogen). After adding the reverse transcriptase, the reaction was incubated at 25°C for 10 min, followed by 30 min at 42°C and 20 min at 50°C. The reaction was inactivated at 75°C for 15 min and treated with RNase H for 20 min at 37°C.
The cDNA molecules were purified using AmPureXp beads. The total 20 μl of the cDNA reaction was incubated with 36 μl AmPureXp beads (Beckman Coulter) and 15 μl of water at room temperature for 30 min with gentle mixing. The tube was exposed to a magnetic wall and the liquid discarded. The magnetic beads were washed two times with EtOH 70% and dried for 5 min at room temperature. The cDNA was then eluted from the beads using 10 μl of preheated water at 37°C and incubated for 10 min at 40°C.
The 5′ adaptor was ligated to the cDNA using 3 μl of the cDNA, 1 μl CircLigase buffer, 0.5 μl ATP 1 mM, 0.5 μl MnCl2 50 mM, 4 μl 50% PEG6000, 0.5 μl Adaptor 100 μM (/5Phos/gACAGCTCCTCGCCCTTGCTCACCATTTCGCCGTTAGA/3InvdT/), and 0.5 μl Circ Ligase™ ssDNA Ligase (Epicentre). The reaction was incubated at 60°C for 2 h, followed by 1 h at 68°C and 10 min at 80°C, then purified using AmPureXp (Beckman Coulter) as described above. The purified molecules were eluted in 20 μl of water.
Final sequences were added by PCR. Amplification was done using Phusion High‐Fidelity DNA Polymerase (Thermo Scientific) and oligos: ATTTAGGTGACACTATAGAAGtGCTACACGACGCTCTTCCGATCTTCTAACGGCGAA ATGGTGAG and (T)60GTGACTGGAGTTCAG. PCR was carried out for initial 3 min at 98°C, followed by three cycles of: 80 s at 98°C, 15 s at 52°C, and 45 s at 72°C. This was followed by 20 cycles of 10 s at 98°C, and 45 s at 72°C, with a final incubation at 72°C for 5 min. The reaction was separated on agarose gel and DNA fragments between 500 and 900 nt were extracted and purified using MinElute columns (Qiagen) and eluted in 10 μl.
Reporter library injection and sample preparation
A total of 1,000 pl of a 10 ng/μl reporter library was injected into wild‐type zebrafish embryos at the one‐cell stage. To block translation of the reporter library in a subset of the embryos, GFP morpholino (Giraldez et al, 2006) was co‐injected with the reporter library at 250 μM. One‐cell stage Xenopus embryos were injected with 2 nl of the 10 ng/μl reporter library, with a subset also co‐injected with 250 μM GFP morpholino. A total of 25 zebrafish and Xenopus embryos injected only with the reporter library were collected at the 16‐cell or 4‐cell stage, respectively. A total of 25 zebrafish embryos injected with the reporter library alone and co‐injected with the MO were collected at 8 h post‐injection (hpi) (50% epiboly) by triplicated. A total of 25 Xenopus embryos were collected at 9.5 hpi (Stage 11). Total RNA was isolated using TRIzol (Invitrogen). The library mRNAs were reverse transcribed using SuperScript™ III Reverse Transcriptase (Invitrogen) with RT‐Oligo (GTGACTGGAGTTCAGACGTGTG) following the manufacturer's instructions (Invitrogen). The cDNA molecules were purified using AmPureXp beads as described above. Amplification was performed using Phusion High‐Fidelity DNA Polymerase (Thermo Scientific) using oligo: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT in combination with any of the following barcoded oligos.
Index_14 = CAAGCAGAAGACGGCATACGAGATggaactGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTctagctac,
Index_15 = CAAGCAGAAGACGGCATACGAGATtgacatGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTctagctac,
Index_22 = CAAGCAGAAGACGGCATACGAGATcgtacgGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTctagctac,
Index_25 = CAAGCAGAAGACGGCATACGAGATatcagtGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTctagctac.
PCR was carried out for initial 3 min at 98°C, followed by different number of cycles of 10 s denaturation at 98°C, 10 s at 50°C, and 45 s at 72°C. A final incubation at 72°C for 5 min was done. For all zebrafish samples, 12 cycles were done, for the Xenopus 20 cycles were done, except for 4‐cell stage where 14 were used.
The PCR fragments were purified with AmPureXp beads (Beckman Coulter) as described above. Libraries were sequenced (together with unrelated libraries) using Illumina HiSeq2000 (2 × 75 nt) using less that ~5% of a lane per sample and never more that four samples per lane due to the low‐complexity regions at the 5′ and 3′ fragment ends.
Reporter library analysis
Sequences that did not contain the exact first 26‐nt expected translated sequence (ATGGTGAGCAAGGGCGAGGAGCTGTC) and the exact added stop codons (CTAGCTACCTA) were discarded. The remaining sequences were mapped with Bowtie2 to zebrafish Ensembl release 80 cDNA (longest transcript per gene) using the following parameters: –local, –no‐mixed, –no‐discordant, –no‐overlap, –norc, –no‐unal, ‐I 200, ‐X 600 (Langmead & Salzberg, 2012; Cunningham et al, 2014). The fragment sequences spanning the full locus delineated by each pair of reads were extracted using SAMtools and BEDTools (Li et al, 2009; Quinlan & Hall, 2010). After mapping, the 5′ 26‐nt translated sequence was added back to all fragments. All sequences with stop codons in frame were discarded. Each of the coding codons (61) was counted, and codon frequencies were computed using the length of the coding sequence. Frequencies of each codons were compared between conditions (e.g. 8 h with and 8 h without MO, or 8 h versus 2 h) using the three replicates using a analysis of the variance (ANOVA). The F‐value was multiplied by −1 if the average of medians was higher in the presence of the MO. This final positive or negative F‐value was termed as reporter codon stabilizing index (rCSI). Therefore, optimal codon displayed a higher average of the medians in the absence of MO than in the presence of MO and therefore present a positive rCSI. Non‐optimal displayed a higher average of the medians in the presence of MO than in the absence of MO and so presented a negative rCSI. In the case of Xenopus 9 h with MO, one sample was discarded for low‐quality reasons. The codons used (Fig 1E) were calculated by multiplying the number of each codon by the level of mRNA at 6 h. The same rCSI calculations were done at the level of amino acid, calculating the reporter amino acid stabilizing index (rASI).
RNA‐seq during early zebrafish and Xenopus embryogenesis
Embryos were injected with 0.2 ng of α‐amanitin (Sigma Aldrich) at one‐cell stage. The Xenopus embryos were also co‐injected with a U1‐U2 morpholinos as means to block splicing as a control for an unrelated experiment. Twenty‐five embryos were collected and total RNA isolated with TRIzol. The zebrafish samples were spiked in with Yeast RNA for normalization purposes. Depletion of ribosomal RNA was performed with Ribo‐Zero Gold kits (Epicentre) according to the published protocol. Strand‐specific TruSeq Illumina RNA sequencing libraries were constructed by the Yale Center for Genome Analysis and sequenced in Illumina HiSeq 2000/2500 machines to yield single‐end 76‐nt reads. Raw reads were initially filtered by aligning permissively to a ribosomal DNA index using Bowtie v0.12.953 with switches ‐seedlen 25 ‐n 3 ‐k 1 ‐y ‐e 10,000. Unaligned reads were then aligned to the zebrafish Zv9 (UCSC danRer7) genome sequence using Tophat v2.0.754 with default parameters. Sequencing data are available at the Sequence Read Archive (SRA) under the accession number SRP072296.
mRNA half‐lives of maternal mRNA were calculated based on the linear regression of mRNA decay from 2.5, 3, 4, 6, and 8 hpf using log2(RPKM) expression data (Lee et al, 2013). Maternal genes were selected with half‐life longer than 10 min and shorter than 250 min, and Pearson correlation between log2(RPKM) and decay time < −0.8. In Xenopus, 2, 5, 7, and 9 hpf samples were used with the same correlation cutoff.
To calculate the codon stability coefficient (CSC) (Presnyak et al, 2015) in zebrafish and Xenopus, the occurrence of each of the 61 coding codon per gene was calculated and the Pearson correlation coefficient with the respect the mRNA half‐life calculated. Optimal codons were defined as the codons with rCSI higher than 250 and a positive CSC. Non‐optimal codons presented rCSI lower than −250 and negative CSC. The optimal and non‐optimal were also splitted into weak and strong: the strong ones were defined as the codon with a CSC higher than 0.03 (optimal) or lower than −0.03 (non‐optimal) and the weak ones had 0 < CSC < 0.03 optimal and 0 > CSC > −0.03 non‐optimal codons. Similar to the CSC, the amino acid stability coefficient (ASC) in zebrafish and Xenopus were calculated.
Maternal‐to‐zygotic transition analysis
For the maternal‐to‐zygotic transition analysis, RPKM were calculated from zebrafish, Xenopus, mouse, and Drosophila melanogaster (Daines et al, 2011; Lee et al, 2013; Abe et al, 2015). The genes enriched in optimal or non‐optimal codons were defined as the top quartile transcripts based on the proportion of optimal codons. The miR‐430 target group was defined as transcripts with miR430 target sites in the 3′UTR (8, 7, or 6 mer). The lengths of the 5′UTR, 3′UTR, ORF, and transcripts were calculated for each group as well as the enrichment of GO terms.
For the maternal‐to‐zygotic transition analysis in multiple species, the top 6,000 most highly expressed genes before genome activation in each species were analyzed. The top quartile of optimal or non‐optimal based on the zebrafish codon optimality code was defined and the fold change before and after the zygotic genome activation calculated.
Quantitative RT–PCR
cDNAs were generated from 900 ng of total RNA using SuperScript® III First‐Strand Synthesis System (ThermoFisher Scientific) and following the manufacturer's instructions. A total of 5 μl from a 1/50 dilution of the cDNA reaction was used to determine the expression of ik cytokine, hsp90, trmt12, hps4, taf15 and cdk2ap2 (Fig 2C), GFP and optimal and non‐optimal transcripts by using the Power SYBR Green PCR Master Mix Kit (Applied Biosystems) and the ViiA 7 instrument (Applied Biosystems). The PCR cycling profile consisted of incubation at 50°C for 2 min, followed by a denaturing step at 95°C for 10 min and 40 cycles at 95°C for 15 s and 60°C for 1 min. The following primers were used: ik cytokine: forward, 5′‐GATGGCCATGAGGTGGAG‐3′ and reverse, 5′‐ATGGTGGCGTGTTTTTGAG‐3′; hsp90: forward, 5′‐CCTGACGACATCTCCAATGA‐3′ and reverse, 5′‐CGACGAGGGATGAAGAGAAG‐3′; trmt12: forward, 5′‐TCCCAGGATGGTTATCGAAC‐3′ and reverse, 5′‐AGAACATGCACTTGGTGACG‐3′; hps4: forward, 5′‐TGTGTCGCTGTGTGTCTGAA‐3′ and reverse: 5′‐ATCAGCTGGTCGAGGACATC‐3′; taf15: forward, 5′‐GCTGCAGCATTGAAAACCTTCAGTC‐3′ and reverse, 5′‐TCCATCACACACACACACTAAACGC‐3′; cdk2ap2: forward, 5′‐AGGATCTTGTGGCTCTTCTCCATCAC‐3′ and reverse, 5′‐TTTCACGGCTCATCTCCTCAATGAC‐3′; gfp: forward, 5′‐GAAGTTCGAGGGCGACAC‐3′ and reverse, 5′‐CCGTCCTCCTTGAAGTCG‐3′; optimal and non‐optimal: forward, 5′‐CCACCTCCACTTCCATGTTT‐3′ and reverse, 5′‐TGGACACTTCTGCATCCTCA‐3′.
Morpholino and reporter experiment
0.5 mM of each morpholino (hsp90ab1 5′‐TTCTTGGCGCATTTCTTCAGGCATC; hps4 5′‐CGGCCAAAACAGCACTTTCAGCCAT, Gene tools) were injected in one‐cell stage zebrafish embryos and collected (n = 10) at 8 hpf. Quantitative RT–PCR was done as described before.
Hsp90 and hps4 ORFs (927 nt and 930 nt starting by the codon after ATG) were PCR amplified using cDNA from 2 hpf zebrafish embryos:
Fw‐hsp90 5′‐GCAAGGGCGAGGAGCTGTTCCCTGAAGAAATGCGCCAAGA, Rv‐hsp90 5′‐ACGTCTCCAGCCTGCTTCAGCAGGCTGAAGTTAGTAGCTCCGCTTCCGTCGTTGGTCAGGCTCTTGTAA; Fw‐hps4 5′‐GCAAGGGCGAGGAGCTGTTCGCTGAAAGTGCTGTTTTGG, Rv‐hps4 5′‐ACGTCTCCAGCCTGCTTCAGCAGGCTGAAGTTAGTAGCTCCGCTTCCCGGCATTGTTTGATAGGACA.
GFP was amplified from a transgenic C. elegans line expressing a version of GFP codon optimized for C. elegans (Fw‐Ce‐GFP 5′‐TCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACCTAGTAAAGGAGAAGAACTTTTC, Rv‐Ce‐GFP 5′‐GTGCTCTTCCGATCCTACCTACTATTTGTATAGTTCATCCAT). These PCRs were then purified and combined to be used as template for a second PCR (Fw 5′‐ATCGAATTCTCTAACGGCGAAATGGTGAGCAAGGGCGAGGAGCTGTTC, Rv 5′‐ATTGGATCCTCTAACGGCGAAATGGTGAGCAAGGGCGAGGAGCTGTTC). These PCR products were cloned in pCS2+ (EcoRI/BamHI). The final clones were sequenced and confirmed for hsp90/hps4‐P2A‐GFP fusions.
Codons and amino acid bias in homeostasis
The occurrence of each 61 coding codons and 20 amino acids was quantified from the annotated ORF and normalized by its length. The Pearson correlation between the occurrence of each codon or amino acid per gene and its RNA level was calculated.
Zebrafish tissues were isolated under the dissecting scope and immediately frozen. RNA was isolated, library constructed and sequence as described before.
The human tissue data were extracted from RNA‐seq of coding RNA from tissue samples of 122 human individuals representing 32 different tissues (Uhlen et al, 2015).
Poly(A) analysis
The length of the poly(A) tail was extracted from published tables with no further analysis(Subtelny et al, 2014). To calculate the ratio of deadenylated mRNA, the RPKM calculated from enriched in poly(A) RNA was divided by the RPKM of total RNA coming from the same embryos (Bazzini et al, 2014).
The poly(A) tail length of injected mRNA was visualized with a modified version of the high‐resolution poly(A) tail assay (Hire‐PAT) (Bazzini et al, 2012). RNA was extracted, a 3′ adaptor ligated (5rApp‐AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC‐3ddC). cDNA was produced with following oligo: CTATAGCGAGACCCCCCCCCCTTCAGACGTGTGCTCTTCCGATCT. And PCR with specific forward oligo and reverser oligo FAM‐GGTAATACGACTCACTATAGCGAGACCCCCCCCCCTT were done. PCR fragments were analyzed in capillary electrophoresis.
Injected mRNA
Three‐hundred‐nt‐long mRNA fragments were search through the zebrafish transcriptome to have high proportion of optimal codons in one frame and high proportion of non‐optimal codons in one nucleotide out of frame. In parallel, artificial sequences were designed to follow the same criteria (optimal codon in one frame and non‐optimal in another one).
Designed sequence, the G in bold was removed to change the frame: ATGgcGAGAAGGTGTCTTCGTTTATGGCAACGGAGGCGTAGGAGCATATGCATATGGAGGAGTAGGCGGTGCTGGTTATGTTGCCGACATGGTCTATGTATATGTCTTCGGAGGCGCCTTCGCTGCCGCCAAGGCTTATGCTGCGGGAGGGGGAGGGGCCAACGACAAGGTCTATGCTGCGGAAGGGGTAGGTGGTGCTGCTGCTGTCTATGGAGGGGATGCCGGAGGTGCTGCGGTCTTCGACATGGGTGCCGTCATGGAAGGCGACATGGCCAAGGAAGGAGGCAAGGGCAACGTTGCGGCCAACGCtaaAtag
Two nested PCRs were performed, first to amplify the specific fragment and second to add the SP6 promoter and poly(A) tail using the following oligos:
Fish_Frame:gatcttctaacggcgaaatgGCTGCAGCCACCAAGTCCG
Fish_Out‐Frame: gatcttctaacggcgaaatgCTGCAGCCACCAAGTCCGA
Fish_reverse: gtgactggagttcagacgtgtgctcttccgatcctacctaAAACTCCTCAAGATCATCAAAC
Designed_Frame gatcttctaacggcgaaatggcAGAAGGTGTCTTCG
Designed_OUT‐Frame: gatcttctaacggcgaaatggcGAGAAGGTGTCTTCG
Designed_ reverse: gtgactggagttcagacgtgtgctcttccgatcctacctaGCGTTGGCCGCAACGTTG
Sp6‐ ATTTAGGTGACACTATAGAAGtGCTACACGACGCTCTTCCGATCTTCTAACGGCG
Poly(A) (T)60GTGACTGGAGTTCAG
Northern blot: RNA were separated on 8% polyacrylamide/URE gel, transfer to membrane and hybridize with 32P label probe: CATTTCGCCGTTAGAAGATCGGAAGAGCGTCGTGTAG.
Gene ontology
Enrichment for functions among optimal and non‐optimal gene sets was performed by first mapping each gene to its Gene ontology (GO) (Ashburner et al, 2000) using Biomart (Smedley et al, 2015), restricting the onlotogies to “biological processes”. Functional enrichments were tested with Fisher's exact tests using all genes as background as implemented in the GOATOOLS. False discovery rate was applied to correct for multiple testing. For zebrafish, miR‐430 target genes were analyzed as a third set along with optimal and non‐optimal gene sets.
Construction of tRNA sequencing libraries
The tRNA sequencing libraries were constructed by using TGIRT‐seq, which gives full‐length reads of tRNAs (Qin et al, 2016). Briefly, the template‐switching reverse transcription reactions were done by using an initial template–primer substrate consisting of a 34‐nt RNA oligonucleotide, which contains an Illumina Read 2 primer‐binding site and a 3′‐blocking group (C3 Spacer, 3SpC3; IDT), annealed to a complementary 35‐nt DNA primer that leaves an equimolar mixture of A, C, G, or T single nucleotide 3′ overhangs. Reactions were done in 20 μl of reaction medium containing 30–100 ng tRNA, 100 nM template–primer substrate, 1 μM TGIRT enzyme (TGIRT‐III; InGex), and 1 mM dNTPs (an equimolar mix of dATP, dCTP, dGTP, and dTTP) in 450 mM NaCl, 5 mM MgCl2, 20 mM Tris–HCl, pH 7.5, and 5 mM dithiothreitol (DTT). DTT was prepared from a frozen 1 M stock solution. Reactions were assembled by adding all components, except dNTPs, to a sterile PCR tube containing tRNAs with the TGIRT enzyme added last. After pre‐incubating at room temperature for 30 min, reactions were initiated by adding dNTPs and incubated for 30 min at 60°C. cDNA synthesis was terminated by adding 5 M NaOH to a final concentration of 0.25 M, incubating at 95°C for 3 min, and then neutralizing with 5 M HCl. The resulting cDNAs were purified with a MinElute Reaction Cleanup Kit (QIAGEN) and ligated at their 3′‐end to a 5′‐adenlyated/3′‐blocked (C3 spacer, 3SpC3; IDT) adapter by using Thermostable 5′ AppDNA/RNA Ligase (New England Biolabs) according to the manufacturer's recommendations. The ligated cDNA products were re‐purified with a MinElute column and amplified by PCR by using Phusion High‐Fidelity DNA polymerase (Thermo Fisher Scientific) with 200 nM of Illumina multiplex and 200 nM of barcode primers. PCR was done with initial denaturation at 98°C for 5 s followed by eight cycles of 98°C for 5 s, 60°C for 10 s and 72°C for 10 s. The PCR products were purified by using the Agencourt AMPure XP (Beckman Coulter) and sequenced on a NextSeq 500 instrument (Illumina) to obtain 75‐nt paired‐end reads.
Bioinformatic analysis for tRNA
Illumina TruSeq DNA adapter and primer sequences were trimmed from the reads, and reads < 18 nt after trimming were discarded. Reads were then mapped by using TopHat v2.0.10 (default settings) to the zebrafish genome reference sequence (Ensembl Zv9) (Langmead & Salzberg, 2012). Unmapped reads from this first pass (Pass 1) were remapped to the Zv9 reference genome by Bowtie2 v2.1.0 with local alignment (default settings) to improve the mapping rate for those reads that contain post‐transcriptionally added nucleotides [e.g. CCA and poly(U)], untrimmed adapter sequences, and non‐templated nucleotides added to the 3′‐end of the cDNAs by TGIRT enzymes (Pass 2) (Kim et al, 2013). To improve the mapping rate for tRNAs, mapped reads from Passes 1 and 2 were intersected with tRNA annotations from the Genomic tRNA Database (Lowe & Eddy, 1997) to collect both uniquely and multiply mapped tRNAs reads. These were then combined with unmapped reads after Pass 2, and the combined reads were mapped to zebrafish tRNA reference sequences (UCSC genome browser website) using Bowtie2 local alignment with default settings. Because similar or identical tRNAs with the same anticodon can be multiply mapped to different tRNA loci by Bowtie2, mapped tRNA reads with MAPQ ≥ 1 were combined according to their tRNA anticodon prior to calculating the tRNA read counts. Only those features with 10 or more mapped reads were counted.
Ribosome profiling
Zebrafish ribosome profiling used were previously published by our laboratory (Bazzini et al, 2014).
Author contributions
AAB and AJG designed and conceived the project and interpreted the data. AAB performed the experiments and data analysis. FdV, AAB and MKK designed and performed the experiments in Xenopus. MAM‐M performed the qRT–PCR and reporter experiments. YQ and JY cloned and quantified the tRNA level. CEV and TGJ provided data processing and custom scripts. AAB and AJG wrote the manuscript with input from the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
We thank Alan Lambowitz for his generous support on the tRNA analysis. We thank D. Dimaio, J. Galan, and K. Bilguver for intellectual and sequencing support. A. Enright for computational analysis. We thank Angela Andersen for feedback on the manuscript and copyediting. Y, Mishima, J. Beaudoin, D. Cifuentes, H. Codore, E. Fleming, M. Lee, C. Takacs, T. Van Den Elzen, V. Yartseva, G.Urbonaite, and members of the Giraldez lab for reagents and discussions. Supported by the Pew Fellows Program in Biomedical Sciences (AAB), the James Hudson Brown‐Alexander Brown Coxe Postdoctoral Fellowship (FdV), P2GEP3_148600 the Swiss National Science Foundation (CEV), T32GM007499 (TGJ), the US National Institutes of Health (grants R21 HD073768, R01 HD081379 GM103789, R01 GM102251, R01 GM101108, and GM081602 to AJG and grant R01 HD081379 to MKK; R01 GM37949 and GM37951 to Alan Lambowitz). MKK is supported by the Edward Mallinckrodt Jr. Foundation. AJG is supported by the Pew Scholars Program in the Biomedical Sciences, the March of Dimes, the Yale Scholars Program and the Whitman fellowship funds provided by E.E. Just, L.B. Lemann, E. Evelyn and M. Spiegel, the H. Keffer Hartline and E.F. MacNichol Jr at the Marine Biological Laboratory in Woods Hole, MA.
The EMBO Journal (2016) 35: 2087–2103
See also: J Martinez & B Zagrovic (October 2016)
Contributor Information
Ariel A Bazzini, Email: arb@stowers.org.
Antonio J Giraldez, Email: antonio.giraldez@yale.edu.
References
- Abe K, Yamamoto R, Franke V, Cao M, Suzuki Y, Suzuki MG, Vlahovicek K, Svoboda P, Schultz RM, Aoki F (2015) The first murine zygotic transcription is promiscuous and uncoupled from splicing and 3′ processing. EMBO J 34: 1523–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H (1994) Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics 136: 927–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H, Gojobori T (2002) Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis . Proc Natl Acad Sci USA 99: 3695–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H (2003) Translational selection and yeast proteome evolution. Genetics 164: 1291–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel‐Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, Proto WR, Blagborough AM, Meister S, Wirjanata G, Ruecker A, Upton LM, Abraham TS, Almeida MJ, Pradhan A, Porzelle A, Martínez MS, Bolscher JM, Woodland A, Luksch T et al (2015) A novel multiple‐stage antimalarial agent that inhibits protein synthesis. Nature 522: 315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudouin‐Cornu P, Surdin‐Kerjan Y, Marliere P, Thomas D (2001) Molecular evolution of protein atomic composition. Science 293: 297–300 [DOI] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, Giraldez AJ (2012) Ribosome profiling shows that miR‐430 reduces translation before causing mRNA decay in zebrafish. Science 336: 233–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, Obermayer B, Fleming ES, Vejnar CE, Lee MT, Rajewsky N, Walther TC, Giraldez AJ (2014) Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J 33: 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boel G, Letso R, Neely H, Price WN, Wong KH, Su M, Luff JD, Valecha M, Everett JK, Acton TB, Xiao R, Montelione GT, Aalberts DP, Hunt JF (2016) Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 529: 358–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Stark A, Brennecke J, Cohen SM (2008) Temporal reciprocity of miRNAs and their targets during the maternal‐to‐zygotic transition in Drosophila . Curr Biol 18: 501–506 [DOI] [PubMed] [Google Scholar]
- Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, Pollard DA, Sackton TB, Larracuente AM, Singh ND, Abad JP, Abt DN, Adryan B, Aguade M, Akashi H, Anderson WW et al (2007) Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218 [DOI] [PubMed] [Google Scholar]
- Craig CL, Weber RS (1998) Selection costs of amino acid substitutions in ColE1 and ColIa gene clusters harbored by Escherichia coli . Mol Biol Evol 15: 774–776 [DOI] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho‐Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Girón CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Kähäri AK, Keenan S et al (2014) Ensembl 2015. Nucleic Acids Res 43: D662–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daines B, Wang H, Wang L, Li Y, Han Y, Emmert D, Gelbart W, Wang X, Li W, Gibbs R, Chen R (2011) The Drosophila melanogaster transcriptome by paired‐end RNA sequencing. Genome Res 21: 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD (2001) The “cleavage” activities of foot‐and‐mouth disease virus 2A site‐directed mutants and naturally occurring “2A‐like” sequences. J Gen Virol 82: 1027–1041 [DOI] [PubMed] [Google Scholar]
- Doudna JA, Rath VL (2002) Structure and function of the eukaryotic ribosome: the next frontier. Cell 109: 153–156 [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO (2008) Mistranslation‐induced protein misfolding as a dominant constraint on coding‐sequence evolution. Cell 134: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann CR, Rammelt C, Wahle E (2011) Control of poly(A) tail length. Wires RNA 2: 348–361 [DOI] [PubMed] [Google Scholar]
- Faller WJ, Jackson TJ, Knight JR, Ridgway RA, Jamieson T, Karim SA, Jones C, Radulescu S, Huels DJ, Myant KB, Dudek KM, Casey HA, Scopelliti A, Cordero JB, Vidal M, Pende M, Ryazanov AG, Sonenberg N, Meyuhas O, Hall MN et al (2015) mTORC1‐mediated translational elongation limits intestinal tumour initiation and growth. Nature 517: 497–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe P, Luke GA, Hughes LE, Gani D, Halpin C, Ryan MD (2006) E unum pluribus: multiple proteins from a self‐processing polyprotein. Trends Biotechnol 24: 68–75 [DOI] [PubMed] [Google Scholar]
- Gamow G (1954) Possible relation between deoxyribonucleic acid and protein structures. Nature 173: 318 [Google Scholar]
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sorensen KD, Andersen LD, Andersen CL, Hulleman E, Wurdinger T, Ralfkiær E, Helin K, Grønbæk K, Orntoft T, Waszak SM, Dahan O et al (2014) A dual program for translation regulation in cellular proliferation and differentiation. Cell 158: 1281–1292 [DOI] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, Schier AF (2006) Zebrafish MiR‐430 promotes deadenylation and clearance of maternal mRNAs. Science 312: 75–79 [DOI] [PubMed] [Google Scholar]
- Gustilo EM, Vendeix FA, Agris PF (2008) tRNA's modifications bring order to gene expression. Curr Opin Microbiol 11: 134–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamatani T, Carter MG, Sharov AA, Ko MS (2004) Dynamics of global gene expression changes during mouse preimplantation development. Dev Cell 6: 117–131 [DOI] [PubMed] [Google Scholar]
- Hussmann JA, Patchett S, Johnson A, Sawyer S, Press WH (2015) Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet 11: e1005732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T (1982) Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J Mol Biol 158: 573–597 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS (2009) Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL (2014) RNA function. Ribosome stalling induced by mutation of a CNS‐specific tRNA causes neurodegeneration. Science 345: 455–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone O, Lasko P (2001) Translational regulation and RNA localization in Drosophila oocytes and embryos. Annu Rev Genet 35: 365–406 [DOI] [PubMed] [Google Scholar]
- Kiebler MA, DesGroseillers L (2000) Molecular insights into mRNA transport and local translation in the mammalian nervous system. Neuron 25: 19–28 [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB (2009) Coding‐sequence determinants of gene expression in Escherichia coli . Science 324: 255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Bonneau AR, Takacs CM, Bazzini AA, DiVito KR, Fleming ES, Giraldez AJ (2013) Nanog, Pou5f1 and SoxB1 activate zygotic gene expression during the maternal‐to‐zygotic transition. Nature 503: 360–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR (1997) tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Liu M, Hartley RS, Sheets MD, Dahlberg JE (2009) Deadenylation of maternal mRNAs mediated by miR‐427 in Xenopus laevis embryos. RNA 15: 2351–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Ephrussi A (2009) mRNA localization: gene expression in the spatial dimension. Cell 136: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Richter JD (2001) Translational control by CPEB: a means to the end. Nat Rev Mol Cell Biol 2: 521–529 [DOI] [PubMed] [Google Scholar]
- Mishima Y, Tomari Y (2016) Codon usage and 3′ UTR length determine maternal mRNA stability in zebrafish. Mol Cell 61: 874–885 [DOI] [PubMed] [Google Scholar]
- Novoa EM, Ribas de Pouplana L (2012) Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet 28: 574–581 [DOI] [PubMed] [Google Scholar]
- Novoa EM, Pavon‐Eternod M, Pan T, Ribas de Pouplana L (2012) A role for tRNA modifications in genome structure and codon usage. Cell 149: 202–213 [DOI] [PubMed] [Google Scholar]
- Park JE, Yi H, Kim Y, Chang H, Kim VN (2016) Regulation of poly(A) tail and translation during the somatic cell cycle. Mol Cell 62: 462–471 [DOI] [PubMed] [Google Scholar]
- Pechmann S, Frydman J (2013) Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol 20: 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Willmund F, Frydman J (2013) The ribosome as a hub for protein quality control. Mol Cell 49: 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12: 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, Coller J (2015) Codon optimality is a major determinant of mRNA stability. Cell 160: 1111–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Yao J, Wu DC, Nottingham RM, Mohr S, Hunicke‐Smith S, Lambowitz AM (2016) High‐throughput sequencing of human plasma RNA by using thermostable group II intron reverse transcriptases. RNA 22: 111–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, Savva R, Wernisch L (2004) Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32: 5036–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AP (2015) Roles of microRNAs and long‐noncoding RNAs in human immunodeficiency virus replication. Wiley Interdiscip Rev RNA 6: 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Coller J (2015) Pausing on polyribosomes: make way for elongation in translational control. Cell 163: 292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusek AM, Abba M, Eljaszewicz A, Moniuszko M, Niklinski J, Allgayer H (2015) MicroRNA modulators of epigenetic regulation, the tumor microenvironment and the immune system in lung cancer. Mol Cancer 14: 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH (1987) The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Green R (2012) Translation drives mRNA quality control. Nat Struct Mol Biol 19: 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaidina M, Lehmann R (2014) Translational control in germline stem cell development. J Cell Biol 207: 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G, Bardou P, Beck T, Blake A, Bonierbale M, Brookes AJ, Bucci G, Buetti I, Burge S, Cabau C, Carlson JW et al (2015) The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res 43: W589–W598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, Bartel DP (2014) Poly(A)‐tail profiling reveals an embryonic switch in translational control. Nature 508: 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda P, Franke V, Schultz RM (2015) Sculpting the transcriptome during the oocyte‐to‐embryo transition in mouse. Curr Top Dev Biol 113: 305–349 [DOI] [PubMed] [Google Scholar]
- Tadros W, Goldman AL, Babak T, Menzies F, Vardy L, Orr‐Weaver T, Hughes TR, Westwood JT, Smibert CA, Lipshitz HD (2007) SMAUG is a major regulator of maternal mRNA destabilization in Drosophila and its translation is activated by the PAN GU kinase. Dev Cell 12: 143–155 [DOI] [PubMed] [Google Scholar]
- Tadros W, Lipshitz HD (2009) The maternal‐to‐zygotic transition: a play in two acts. Development 136: 3033–3042 [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- Weill L, Belloc E, Bava FA, Mendez R (2012) Translational control by changes in poly(A) tail length: recycling mRNAs. Nat Struct Mol Biol 19: 577–585 [DOI] [PubMed] [Google Scholar]
- Yartseva V, Giraldez AJ (2015) The maternal‐to‐zygotic transition during vertebrate development: a model for reprogramming. Curr Top Dev Biol 113: 191–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CH, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y (2015) Codon usage influences the local rate of translation elongation to regulate co‐translational protein folding. Mol Cell 59: 744–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM, Pan T (2015) Efficient and quantitative high‐throughput tRNA sequencing. Nat Methods 12: 835–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File