

ABSTRACT
Epigenome-wide DNA methylation association studies have identified highly replicable genomic loci sensitive to maternal smoking during gestation. The role of inter-individual genetic variation in influencing DNA methylation, leading to the possibility of confounding or bias of such associations, has not been assessed. We investigated whether the DNA methylation levels at the top 10 CpG sites previously associated with exposure to maternal smoking during gestation were associated with individual genetic variation at the genome-wide level. Genome-wide association tests between DNA methylation at the top 10 candidate CpG and genome-wide SNPs were performed in 736 case and control participants of the California Childhood Leukemia Study. Three of the strongest maternal-smoking sensitive CpG sites in newborns were significantly associated with SNPs located proximal to each gene: cg18146737 in the GFI1 gene with rs141819830 (P = 8.2×10−44), cg05575921 in the AHRR gene with rs148405299 (P = 5.3×10−10), and cg12803068 in the MYO1G gene with rs61087368 (P = 1.3×10−18). For the GFI1 CpG cg18146737, the underlying genetic variation at rs141819830 confounded the association between maternal smoking and DNA methylation in our data (the regression coefficient changed from −0.02 [P = 0.139] to −0.03 [P = 0.015] after including the genotype). Our results suggest that further studies using DNA methylation at cg18146737, cg05575921, or cg12803068 that aim to assess exposure to maternal smoking during gestation should include genotype at the corresponding SNP. New methods are required for adequate and routine inclusion of genotypic influence on DNA methylation in epigenome-wide association studies to control for potential confounding.
KEYWORDS: Biological markers, DNA methylation, genetics, prenatal exposure delayed effects, smoking
Introduction
Epigenome-wide DNA methylation association studies (EWAS) have been successful in identifying genomic loci sensitive to environmental exposures. For example, exposure to maternal smoking during gestation was significantly associated with DNA methylation status of more than 6,000 CpG sites in cord blood in a recent meta-analysis utilizing 6,685 participants across 13 birth cohorts from the Pregnancy And Childhood Epigenetics consortium.1 The results of such studies are of great public health importance as they provide opportunities to understand epigenetic mechanisms mediating the negative health effects of detrimental environmental exposures. They can also identify epigenetic biomarkers of a study subject's history of exposures, and potential epigenetic targets of future therapeutic compounds.
Results from EWAS, however, may be confounded by inter-individual heritable genetic variation that influences DNA methylation at particular genomic loci. Direct effects could arise from single nucleotide polymorphisms (SNPs) within specific CpG sites, though most variation is likely to be indirect due to neighboring polymorphic loci. Indeed, genetic sequences provide binding sites for transcription factors and, in turn, the binding of transcription factors influence the levels of DNA methylation.2 Heritable genetic variation that influences CpG methylation can therefore potentially confound associations between DNA methylation levels at a CpG site and an environmental exposure, if the distribution of SNP alleles differs between the exposed and the unexposed.3–6 This is an overlooked issue in most current EWAS.
Here, we inquired whether the DNA methylation levels at the top 10 CpG sites previously associated with exposure to maternal smoking during gestation1 were associated with individual genetic variation at the genome-wide level [i.e., to identify methyl-quantitative trait loci (methylQTLs)]. These 10 CpG sites are located in 4 unique gene regions at AHRR, GFI1, MYO1G, and CNTNAP2, and are hereafter referred to as the candidate CpG sites (Joubert, et al., 2016, see Supplementary Table 1).
Results
California Childhood Leukemia Study (CCLS) participants7 with complete data on demographic characteristics, smoking history, methylation, and genotyping data were included in the analyses (n = 736). About 54% of participants had childhood acute lymphoblastic leukemia, 59% were males, and approximately half were Hispanic (additional subject characteristics are reported in Table 1).
Table 1.
Principal characteristics of the 736 participants of the California Childhood Leukemia study and CpG sites associated with maternal smoking during gestation.
| n | % | |
|---|---|---|
| Childhood leukemia cases | 400 | 54.30 |
| Male participants | 439 | 59.60 |
| Non-Hispanic Whites | 216 | 30.5 |
| Hispanics | 400 | 54.4 |
| White/Caucasian | 132 | 18.6 |
| African American | 2 | 0.3 |
| Native American | 5 | 0.7 |
| Mixed or others | 245 | 34.6 |
| African American | 13 | 1.8 |
| Native American | 1 | 0.1 |
| Asian or Pacific Islander | 49 | 6.9 |
| Mixed or others | 46 | 6.5 |
| Exposed to maternal smoking during gestation (self-reported by mothers) |
65 |
8.80 |
| |
mean |
sd |
| Gestational age (years) | 39.3 | 2.2 |
| Birth weight (grams) | 3458.2 | 616.5 |
| Age at diagnosis (years) |
4.9 |
3.2 |
| Top ten CpG sites most associated with exposure to maternal smoking during gestation (from Joubert et al., 2016; values from the 450 arrays are presented) | β-value |
|
| mean | sd | |
| cg05575921 (AHRR) | 0.818 | 0.052 |
| cg12803068 (MYO1G) | 0.807 | 0.094 |
| cg04180046 (MYO1G) | 0.502 | 0.083 |
| cg25949550 (CNTNAP2) | 0.078 | 0.021 |
| cg09935388 (GFI1) | 0.749 | 0.109 |
| cg14179389 (GFI1) | 0.245 | 0.095 |
| cg22132788 (MYO1G) | 0.924 | 0.049 |
| cg12876356 (GFI1) | 0.786 | 0.123 |
| cg18146737 (GFI1) | 0.845 | 0.123 |
| cg19089201 (MYO1G) | 0.894 | 0.054 |
The top 10 SNP associations for each candidate CpG are reported in Supplementary Table 2, and they are annotated in the Supplementary Table 3. GWAS analyses identified SNPs that were strongly associated with methylation status at: cg18146737 (GFI1), cg05575921 (AHRR), and cg12803068 (MYO1G), suggesting that these SNPs may be methylQTLs (see Fig. 1). For each gene, the top SNPs were located within 720 kb of the candidate CpG and were, respectively, rs284180 on chromosome 1 [regression coefficient (β) = −0.04214, P = 3.9×10−9, lambda genomic inflation estimation = 1.028, Fig. 1, panel A], rs11745733 on chromosome 5 (β = 0.027, P = 6.7×10−10, lambda genomic inflation estimation = 1.00, Fig. 1, panel B), and rs6976664 on chromosome 7 (β = −0.056, P = 5.8×10−17, lambda genomic inflation estimation = 1.018, Fig. 1, panel C). GWA results for the other 7 candidate CpG sites did not suggest that any SNPs were strong methylQTLs (Supplementary Figs 1-7). Imputation to 1,000 Genomes Data was performed across a 600 kb region for each of the 3 methylQTLs. Fig. 2a–c shows gene locations and P-values resulting from the association tests within imputed data in regions that are centered on the 3 candidate CpG sites (±300 kb). Linkage disequilibrium (LD) plots are also displayed for all the tested SNPs. Stratification by ethnicity (Hispanic vs. non-Hispanic) did not produce notably different LD plots, and haplotype results are presented for both ethnicities together.
Figure 1.
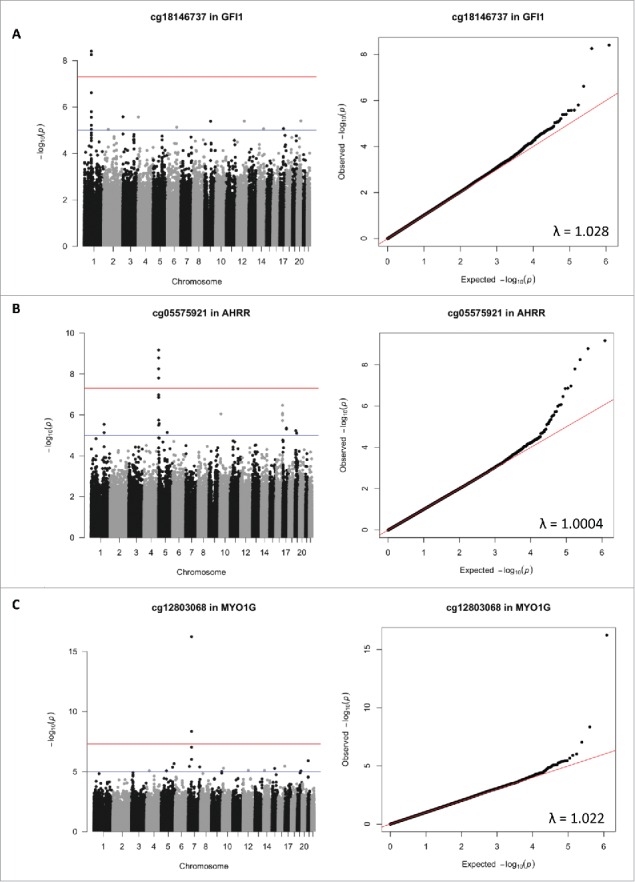
Manhattan and quantile-quantile plots (QQ-plots) of the results of genome-wide associations tests looking at genome-wide associations between 3 candidate CpG sites sensitive to smoking [cg18146737 in the GFI1 gene on the chromosome 1 (panel A), cg05575921 in the AHRR gene on the chromosome 5 (panel B), and cg12803068 in the MYO1G gene on the chromosome 7 (panel C)] and genotype at 606,588 SNPs throughout the genome in 736 participants of the California Childhood Leukemia Study. The genomic inflation estimation factors lambda are reported on the plots. On the Manhattan plots, the red line corresponds to the canonical threshold of genome-wide significance [-log10(5 × 10−08)], and the blue line represents suggestive significance [-log10(10−05)].
Figure 2.

Investigations of a ± 300 kb region around 3 CpG sites of interest (cg18146737, cg05575921, and cg12803068) for fine-mapping using imputed SNP data in 736 participants of the California Childhood Leukemia Study. The plots represent the P-values of the associations between DNA methylation at the CpG site of interest and genotype at the SNPs in the region. Canonical transcripts of the neighboring genes in the region are displayed based on the University of California, Santa Cruz Genome Browser data (hg19).26 Linkage disequilibrium plots are represented below (r2 values) for SNPs that were genotyped on array. The blue line is located at the location of the CpG site of interest.
Figure 2.

(Continued).
Figure 2.

(Continued).
cg18146737 in the GFI1 gene
Forty-five SNPs in the region were directly genotyped on-array and 1,222 were imputed. The most associated SNP with cg18146737 was rs115340020 (imputed SNP, β = −0.34, P = 1.7×10−46, surviving correction for multiple-testing), which was located 180,263 bp upstream of cg18146737. The second most associated SNP in the region, rs141819830, was in complete LD with rs115340020 (r2 = 1), and was located closer to cg18146737, positioned only 20,995 bp upstream; its regression coefficient was −0.34, and P-value 8.2×10−44 (see Fig. 2a). SNP rs141819830 corresponds to a deletion (ATTAGAG/A), with a minor allele frequency (MAF) of 0.01 overall and 0.02 in those of European ancestry, and it is located within a regulatory region for GFI1 in the promoter flanking region. Genotype variation at rs141819830 did confound the association between exposure to maternal smoking during gestation and DNA methylation at cg18146737 in our study population: the regression coefficient without rs141819830 genotype in the model was not significantly different from zero (β = −0.02, P = 0.139), and it became significantly different from zero (β = −0.03, P = 0.015) after including rs141819830 genotype in the regression model, see Table 2 and Fig. 3, panel A. The percentage of variation of the DNA methylation levels explained by exposure to smoking was 0.5%, and by rs141819830 genotype was 27.8%. Though we had adjusted for ancestry in the models, we also verified that the association between cg18146737 and rs141819830 was concordant in Hispanic and in non-Hispanic participants (in Hispanics: β = 0.38, P = 1.5 × 10−27; in non-Hispanics: β = 0.30, P = 6.8 × 10−19).
Table 2.
Tests of association between DNA methylation at the 3 CpG sites of interest and maternal smoking exposure during gestation while controlling (or not) for the genotype of the methylQTL (linear regression tests) in 736 participants of the California Childhood Leukemia Study.
| Regression coefficient | Standard Error | t-value | P | ||
|---|---|---|---|---|---|
| GFI1 (cg18146737) | |||||
| model without genotype | −0.02 | 0.02 | −1.48 | 0.139 | |
| model with genotype at rs141819830 | −0.03 | 0.01 | −2.43 | 0.015 | * |
| AHRR (cg05575921) | |||||
| model without genotype | −0.04 | 0.01 | −6.40 | 2.9E-10 | *** |
| model with genotype at rs148405299 | −0.04 | 0.01 | −6.38 | 3.2E-10 | *** |
| MYO1G (cg12803068) | |||||
| model without genotype | 0.04 | 0.01 | 3.28 | 0.001 | ** |
| model with genotype at rs61087358 | 0.03 | 0.01 | 2.86 | 0.004 | ** |
Models were adjusted for: leukemia case/control status, ancestry, cell-mixture, gender, gestational age and DNA methylation array batch number.
*P < 0.05
**P < 0.01
***P < 0.001
Figure 3.
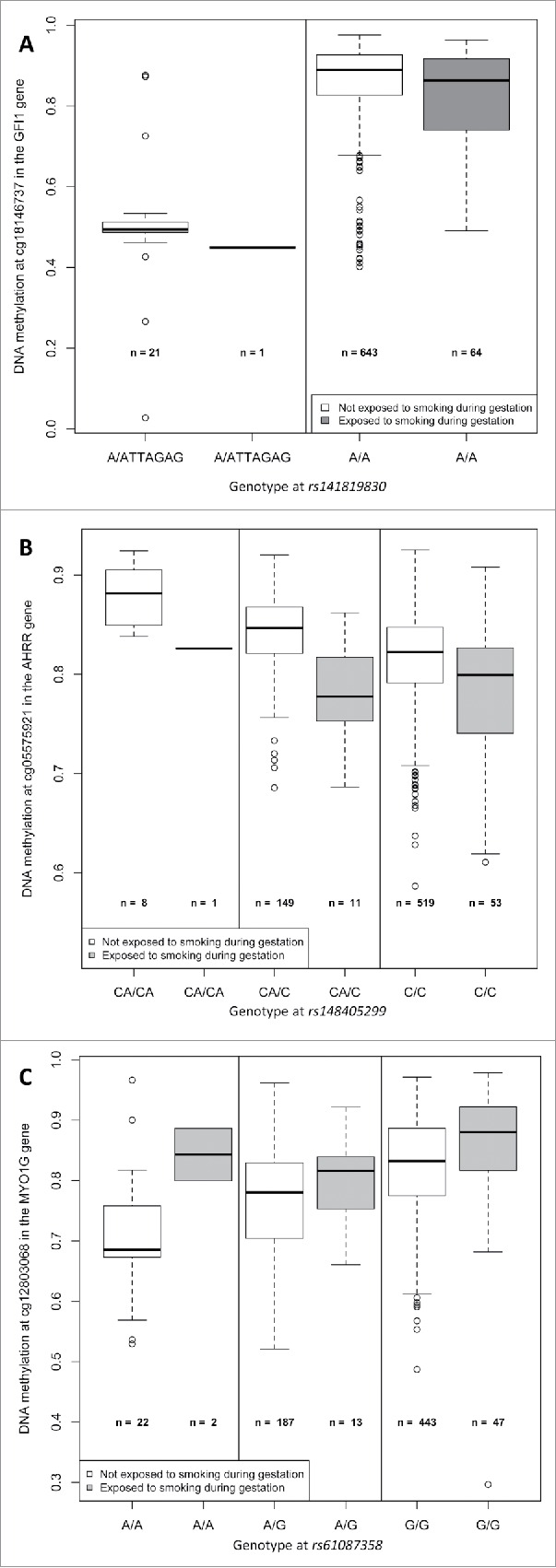
Boxplots representing the association between DNA methylation at the 3 CpG sites of interest (cg18146737, cg05575921, and cg12803068) and exposure to maternal smoking during gestation, by genotype at the corresponding strongest associated SNP (rs141819830, rs148405299, and rs61087368) in 736 participants of the California Childhood Leukemia Study.
cg05575921 in the AHRR gene
A hundred and thirty-five SNPs in the region (±300 kb of the CpG) were directly genotyped on-array and 2,722 were imputed. A broad peak of significantly associated SNPs encompassed cg05575921 (Fig. 2b). Two hundred and nineteen SNPs located within the peak with a P-value < 1 × 10−6 were in LD with each other (the mean r2 = 0.48, 25% of them had a r2 ≥ 0.72). The strongest associated SNP was rs148405299 (imputed SNP, β = 0.026, P = 5.3×10−10, surviving Bonferroni correction for multiple-testing), located in the AHRR gene, 6,392 bp upstream from cg05575921. There appeared to be a possible second association signal 169,238 bp upstream of the top hit SNP, centered around SNP rs34493940 (Fig. 2b). A conditional analysis including both the top SNP (rs148405299) and the SNP located at the left extremity of the peak (rs34493940) revealed that the latter was not independently associated with cg05575921, and that its observed association with cg05575921 was likely due to LD between the 2 SNPs (r2= 0.68). In 1,000 Genomes Data, rs148405299 corresponds to an insertion (C/CA). The allele frequency of the insertion is 0.07 overall and 0.14 in those of European ancestry, and it is located in an intron. In our study population, genotype variation at rs148405299 did not confound the association between exposure to maternal smoking during gestation and DNA methylation at cg05575921 [the regression coefficients was −0.04 (P = 3 × 10−10)] before and after including genotype in the model, see Table 2 and Fig. 3, panel B). The percentage of variation of the DNA methylation levels explained by exposure to smoking was 5.8%, and by rs148405299 was 5.3%.
cg12803068 in the MYO1G gene
One hundred and fifty-three SNPs in the region were directly genotyped on-array and 1,869 were imputed. A narrow association peak was observed within a single LD block (Fig. 2c). The top associated SNP was rs61087368 (imputed SNP, regression coefficient = 0.057, P = 1.3 × 10−18, surviving correction for multiple-testing), which was located 14,824 bp downstream from cg12803068. In 1,000 Genomes Data, the major allele was G and the minor allele was A, with a MAF of 0.13 overall and 0.21 in those of Europeans ancestry, and it is an intronic variant. Genotype variation at rs61087368 did not confound the association between exposure to maternal smoking during gestation and DNA methylation at cg12803068 in our data (the regression coefficients was 0.04 [P = 0.001] before and after genotype in the model, see Table 2 and Fig. 3, panel C). The percentage of variation of the DNA methylation levels explained by exposure to smoking was 1.4%, and by rs61087368 was 11.6%.
Discussion
In this study, we demonstrated for the first time that DNA methylation at 3 of the strongest maternal-smoking sensitive CpG sites in newborns1 was influenced by cis methyl-quantitative trait loci, including both SNPs and indels: cg18146737/rs141819830, cg05575921/rs148405299, and cg12803068/rs61087368. For cg18146737 in GFI1, the underlying genetic variant at rs141819830 was shown to confound the association between the environmental exposure (maternal smoking) and DNA methylation in our study population. In population-based studies it is likely that rare variants (like cg18146737 in GFI1 with a MAF of 0.01) may confound associations between an environmental exposure and DNA methylation levels more often than more common variants, as there are more chances that they are not evenly distributed between the exposed and the unexposed. Interestingly, loss of GFI1 causes neutropenia.8,9 It has been shown that smoking influences the incidence of neutropenia in the context of chemotherapy,10 which may result in part from a combination of genetic and epigenetic variation at the GFI1 locus. Further research is required to elucidate how variation at rs141819830 affects methylation at GFI1. None of the 3 SNPs/indels were previously associated with disease. Additionally, their DNA methylation levels were not associated with gene expression of the corresponding transcripts as far as we can determine (supplementary Table 4). However, our results support that further studies using DNA methylation at these candidate smoking exposure-sensitive CpG sites should take into account genetic variation at the corresponding SNPs when assessing the effect of an environmental exposure, as our study prove the concept that methylQTLs may confound the association between DNA methylation and exposure to smoking. Interestingly, our results demonstrated that genotype was a strong driver of DNA methylation at these candidate CpG sites, even stronger than exposure to maternal smoking during gestation.
Genetic variation influences the level of DNA methylation through the binding of transcription factors, which is influenced by both the genetic sequence of the binding sites and the abundance of the transcription factors.2 This concept has been quantified for the measurement of DNA methylation with the 450K array by Teh et al. (2014),4 who observed that, in general, the most variable DNA methylation probes used in the 450K array were associated with at least one SNP. In this study, other environmental variables were also associated with DNA methylation variation, independently from the genotype for about 75% of the tested differentially-methylated regions.4 In our study, both genetic variation and exposure to smoking were independently associated with DNA methylation levels as well. Genetic variation explained more of the variation in DNA methylation levels than exposure to smoking, which is concordant with the findings of Teh et al.
We observed that our top SNPs were located between approximately 6 and 20 kb away from the CpG of interest. Evidence from the literature on the typical distance between a CpG and a methylQTL is uncertain. Teh et al. previously observed an unlimited range of distances, with as many as 42% of the methylQTLs located on a different chromosome than the CpG (i.e., in trans). Other studies reported that most of the CpG/methylQTLs pairs were located within 3 to 5 kb of each other 5,11; however, one of these studies5 restricted the analysis to within a ±250 kb distance around the CpG of interest, excluding therefore the possibility to find far or trans associations.
Despite the growing interest in the scientific literature about exposures to smoking during gestation and their effects on DNA methylation at birth,1,12−17 this study is the first to combine genetic and epigenetic data in order to estimate the effects of exposure to maternal smoking during gestation on DNA methylation of candidate CpG sites while controlling for genotype of influencing methylQTLs. The identified methylQTLs were highly significantly associated with variation in DNA methylation at the 3 CpGs of interest, surviving a stringent Bonferroni P-value threshold of P < 8.2 × 10−9. Moreover, the relative proximity of the methylQTLs to the CpGs of interest further supports the biological plausibility of these associations. Future studies in different populations with both DNA methylation and SNP genotype data should, however, aim at replicating these findings to confirm these SNPs as methylQTLs for the corresponding CpGs. We carried out our analyses using a case/control study of childhood ALL due to the availability of DNA methylation, genome-wide SNP as well as smoking exposure data. However, this study design does present some limitations, such as the risk of recall bias for the self-reported maternal smoking during gestation for cases compared with controls. To address this issue, we performed separate analyses between smoking exposure and DNA methylation at the 3 CpG (cg18146737, cg05575921, and cg12803068) among cases and controls (supplementary Table 5); the direction and the size of the associations were concordant between previous findings by Joubert et al.1 and between cases and controls, reducing therefore the likelihood of a recall bias. Moreover, we cannot exclude that childhood ALL pathogenesis involves any of the methylQTLs or CpG sites studied here, although unlikely, as there were no previous reports on associations between any of the genes investigated here and childhood ALL. With regard to this potential issue, we nevertheless adjusted all of the models for case/control status.
Our results suggest that future studies using methylation at cg18146737, cg05575921, or cg12803068 that aim to assess exposure to maternal smoking during gestation should include genotype at the corresponding SNP/indel, in order to avoid potential confounding should allele distributions differ between the exposed and unexposed groups. Similarly, epigenome-wide DNA methylation association studies assessing environmental exposures or other outcomes should verify and control for heritable polymorphisms if the most associated CpG sites have corresponding methylQTLs. In addition, efforts should be made to address methods to routinely include genotype variation into epigenome-wide DNA methylation association studies as this is proven to be a major confounding factor.
Methods
Study population
This study was carried out using both childhood acute lymphoblastic leukemia (ALL) cases and healthy control participants from a case/control study—the California Childhood Leukemia Study, a California population-based case-control study that has recruited children with a diagnosis of acute lymphoblastic leukemia and matched healthy controls between 1996 and 2013. Case children were recruited in 9 major clinical centers in California at the time of the initial diagnosis. At the same time, a matched healthy control was randomly selected from the general population, using statewide birth certificate files. The controls were individually matched to the ALL cases on year and month of birth, sex, race, and Hispanic status—these data were documented in the birth records and computerized databases at the California Department of Public Health. Information on gestational age, gender, ethnic origin, and smoking history were collected through a questionnaire that was administered to the mothers at time of enrolment in the study, i.e., at time of their child's diagnosis (or at the equivalent age for controls). Neonatal dried blood spots (DBS) for the cases and controls were obtained from the California Department of Public Health. Participants' legal representatives provided informed consent. The California State and University of California IRBs have approved the study.
DNA isolation from DBS
The California Newborn Screening program has banked neonatal DBS leftover from testing for genetic disorders statewide since 1982. Five 14-mm diameter blood spot specimens were collected from infants on Guthrie cards by heel-stick between 12 hours and 6 days of age. DNA was extracted from neonatal DBS for 736 subjects (402 cases and 340 controls) according to the Qiagen QIAamp DNA Micro Kit protocol. Approximately 1/4 spot was used for SNP genotyping and 1/4 for DNA methylation testing.
Genome-wide SNP genotyping
Extracted DNA was genotyped using the Illumina Human OmniExpressV1 platform as previously described.18 A total of 666,932 SNPs were genotyped. Genotypes with call rates inferior to 98% were excluded. Any SNP with ≥10% missing values (n = 28,886), deviating from the Hardy-Weinberg equilibrium (HWE exact test's P < 0.001, n = 12,518), or with a minor allele frequency ≤1% were excluded of the analysis (n = 18,940). The total of SNPs remaining for genome-wide DNA methylation analyses was 606,588. There was no discordance between reported sex and that inferred from genotype data. The IMPUTE v2.3.1 software was used to perform imputation (with the standard Markov chain Monte Carlo algorithm and default settings for targeted imputation19) as previously described.20 The reference panel for the imputation contained all 1,000 Genomes Phase I integrated haplotypes.21 Information on population-based genetic variation was obtained from the 1,000 Genomes Database.21
Genome-wide DNA methylation arrays
Bisulfite treatment of DBS DNA was carried out using the EZ DNA Methylation-Direct™ Kits (Zymo). Genome-wide DNA methylation measurements were obtained for the 736 subjects using the Illumina HumanMethylation450 BeadChip® arrays (450K arrays), which include ∼450,000 CpG sites across the genome. Normalization of the data was performed in order to remove batch and plate-position effects according to the technique by Fortin et al.22
Statistical analyses
We performed genome-wide association tests between DNA methylation levels at each of the 10 candidate CpG sites and heterologous 606,588 polymorphic SNPs throughout the genome. For all SNP association analyses the predictor variable was DNA methylation status at the candidate CpG, and the outcome of interest was genotype. Additionally, potential confounding factors were included in the models, including, neonatal blood cards' cell-mixture estimated by the Refactor method, which uses an unsupervised feature selection step followed by a sparse principal component analysis, according to Rahmani et al.23; each individual's ancestry (estimated by the first 2 dimensions of a principal component analysis of a representative random sample of independent SNPs, based on the methods described by Walsh et al.24); sex; gestational age; childhood leukemia case/control status; and the methylation array batch numbers. Top associated SNPs (n=3) with a P-value < 5×10−8 (i.e., the canonical GWAS threshold of significance) were selected for fine-mapping, with investigation of ±300 kb regions around the CpG of interest using imputed SNP data. Associated SNPs from this fine mapping were considered significant if they survived Bonferroni correction for multiple testing, with a threshold of P < 8.2×10−9 calculated by dividing 0.05 by the total number of tests in the study [i.e., 606,588 SNPs × 10 GWASes + 2,722 + 1,869 + 1,222 (3 regional associations tests using imputed SNP data), total = 6,071,693 tests]. R-squared linkage disequilibrium (LD) plots were generated for the fine-mapping regions; any differences of LD blocks between ethnicities were visually assessed. The LD plots were mapped to the UCSC genome browser (version hg19), and canonical gene transcripts were represented only.
When significant associations between a candidate CpG site and a SNP were found, we tested whether exposure to maternal smoking during gestation was associated with differential methylation, while controlling for the genotype of the associated SNP. In other words, we tested whether the genotype confounded the association between smoking exposure and DNA methylation. However, due to the case/control design of the CCLS, we investigated whether the exposure to maternal smoking during pregnancy might have presented a recall bias in cases. We formally performed first tests of association between exposure to smoking and DNA methylation at the 3 CpG sites separately in cases and in controls, and results are provided in supplementary Table 5. Statistical significance was reached for most but not all of the tested associations—probably due to the small sample size of exposed and to some small effect of exposure on DNA methylation, in particular in cg18146737. However, because the direction of association and the magnitude were roughly similar between cases and in controls, and were concordant with previous findings from Joubert et al.1 (see Table 2), we excluded a recall bias, and we pooled cases and controls for the subsequent analyses. For each of the SNP-associated CpGs, we built 2 successive linear regression models, with smoking exposure as the predictor variable and DNA methylation as the outcome (including the covariates described above), with and without including the SNP genotype. Interactions between smoking exposure and genotype were investigated. We used likelihood ratios tests to select the best-fitted model (cutoff likelihood ratio test's P-value 0.05) and analysis of covariance to estimate the percentage of variance explained by the exposure to smoking variable and by the genotype among the best-fitted model.
Associations between DNA methylation and RNA expression at the top 10 candidate CpG sites associated with exposure to smoking during gestation were tested in peripheral blood mononucleated cells of 20 healthy males (young and old). The data are publicly available on the Gene Expression Omnibus data base (accession number GSE49065). DNA methylation was measured by the 450K arrays, and gene transcripts were analyzed by the Affymetrix Human Gene 1.1 ST Array. Simple unadjusted linear regression was performed for each CpG site.
GWASes were performed with PLINK v1.90b3.34. Other statistical analyses were performed with R 3.2.1,25 using additionally the ‘LDHeatmap’ and ‘qqman’ packages.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Hong Quach and Diana Quach for their help in the genotyping.
Funding
This work was supported by the National Institute of Environmental Health Sciences and the Environmental Protection Agency [grant number: P01ES018172, P50ES018172, RD83451101, and RD83615901 to C.M. and J.L.W.]; The National Institute of Environmental Health Sciences [grant number: R01ES09137 to C.M. and J.L.W.]; The National Cancer Institute [grant number: R01CA155461 to J.L.W.]; The Tobacco Related Disease Research Program [grant 18CA-0127 to J.L.W.]; The Swiss Science National Foundation [grants number: P2LAP3_158674 to S.G.]; And the Sutter-Stöttner Foundation to S.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Environmental Protection Agency.
References
- 1.Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, Reese SE, Markunas CA, Richmond RC, Xu CJ, et al.. DNA methylation in newborns and maternal smoking in pregnancy: Genome-wide Consortium Meta-analysis. Am J Hum Genet 2016; 98(4):680-96[Internet] [cited 2016 Apr 6]; PMID:27040690; http://dx.doi.org/ 10.1016/j.ajhg.2016.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, Buil A, Ongen H, Yurovsky A, Bryois J, Giger T, Romano L, Planchon A, et al.. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife 2013; 2:e00523; PMID:23755361; http://dx.doi.org/ 10.7554/eLife.00523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al.. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013; 31:142-7; PMID:23334450; http://dx.doi.org/ 10.1038/nbt.2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teh AL, Pan H, Chen L, Ong ML, Dogra S, Wong J, MacIsaac JL, Mah SM, McEwen LM, Saw SM, et al.. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res 2014; 24:1064-74; PMID:24709820; http://dx.doi.org/ 10.1101/gr.171439.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol 2014; 15:R37; PMID:24555846; http://dx.doi.org/ 10.1186/gb-2014-15-2-r37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y, Li X, Aryee MJ, Ekström TJ, Padyukov L, Klareskog L, Vandiver A, Moore AZ, Tanaka T, Ferrucci L, et al.. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am J Hum Genet 2014; 94:485-95; PMID:24656863; http://dx.doi.org/ 10.1016/j.ajhg.2014.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metayer C, Zhang L, Wiemels JL, Bartley K, Schiffman J, Ma X, Aldrich MC, Chang JS, Selvin S, Fu CH, et al.. Tobacco smoke exposure and the risk of childhood acute lymphoblastic and myeloid leukemias by cytogenetic subtype. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol 2013; 22:1600-11; PMID:23853208; http://dx.doi.org/ 10.1158/1055-9965.EPI-13-0350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Person RE, Li FQ, Duan Z, Benson KF, Wechsler J, Papadaki HA, Eliopoulos G, Kaufman C, Bertolone SJ, Nakamoto B, et al.. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet 2003; 34:308-12; PMID:12778173; http://dx.doi.org/ 10.1038/ng1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, Orkin SH. Gfi−1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 2004; 431:1002-7; PMID:15457180; http://dx.doi.org/ 10.1038/nature02994 [DOI] [PubMed] [Google Scholar]
- 10.Kanai M, Morita S, Matsumoto S, Nishimura T, Hatano E, Yazumi S, Sasaki T, Yasuda H, Kitano T, Misawa A, et al.. A history of smoking is inversely correlated with the incidence of gemcitabine-induced neutropenia. Ann Oncol Off J Eur Soc Med Oncol ESMO 2009; 20:1397-401; http://dx.doi.org/ 10.1093/annonc/mdp008 [DOI] [PubMed] [Google Scholar]
- 11.Banovich NE, Lan X, McVicker G, van de Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK, Gilad Y. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. Plos Genet 2014; 10:e1004663; PMID:25233095; http://dx.doi.org/ 10.1371/journal.pgen.1004663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joubert BR, Håberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun Ø, Cupul-Uicab LA, et al.. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2012; 120:1425-31; PMID:22851337; http://dx.doi.org/ 10.1289/ehp.1205412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joubert BR, Haberg SE, Bell DA, Nilsen RM, Vollset SE, Midttun O, Ueland PM, Wu MC, Nystad W, Peddada SD, et al.. Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer Epidemiol Biomarkers Prev 2014; 23:1007-17; PMID:24740201; http://dx.doi.org/ 10.1158/1055-9965.EPI-13-1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markunas CA, Xu Z, Harlid S, Wade PA, Lie RT, Taylor JA, Wilcox AJ. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2014; 122(10):1147-53[Internet] [cited 2015 Sep 9]; Available from: http://ehp.niehs.nih.gov/1307892; PMID:24906187; http://dx.doi.org/ 10.1289/ehp.1307892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KW, Richmond R, Hu P, French L, Shin J, Bourdon C, Reischl E, Waldenberger M, Zeilinger S, Gaunt T, et al.. Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ Health Perspect 2015; 123:193-9; PMID:25325234; http://dx.doi.org/ 10.1289/ehp.1408614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Küpers LK, Xu X, Jankipersadsing SA, Vaez A, la Bastide-van Gemert S, Scholtens S, Nolte IM, Richmond RC, Relton CL, Felix JF, et al.. DNA methylation mediates the effect of maternal smoking during pregnancy on birthweight of the offspring. Int J Epidemiol 2015; 44(4):1224-37:dyv048; PMID:25862628; http://dx.doi.org/ 10.1093/ije/dyv048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richmond RC, Simpkin AJ, Woodward G, Gaunt TR, Lyttleton O, McArdle WL, Ring SM, Smith AD, Timpson NJ, Tilling K, et al.. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon longitudinal study of parents and children (ALSPAC). Hum Mol Genet 2015; 24:2201-17; PMID:25552657; http://dx.doi.org/ 10.1093/hmg/ddu739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh KM, Chokkalingam AP, Hsu LI, Metayer C, de Smith AJ, Jacobs DI, Dahl GV, Loh ML, Smirnov IV, Bartley K, et al.. Associations between genome-wide native american ancestry, known risk alleles and B-cell ALL risk in Hispanic children. Leukemia 2013; 27:2416-9; PMID:23615557; http://dx.doi.org/ 10.1038/leu.2013.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5:e1000529; PMID:19543373; http://dx.doi.org/ 10.1371/journal.pgen.1000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh KM, de Smith AJ, Chokkalingam AP, Metayer C, Dahl GV, Hsu L, Barcellos LF, Wiemels JL, Buffler PA. Novel childhood ALL susceptibility locus BMI1-PIP4K2A is specifically associated with the hyperdiploid subtype. Blood 2013; 121:4808-9; PMID:23744494; http://dx.doi.org/ 10.1182/blood-2013-04-495390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium 1000 Genomes Project, others . A map of human genome variation from population-scale sequencing. Nature 2010; 467:1061-1073; PMID:20981092; http://dx.doi.org/ 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, Hansen KD. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol 2014; 15:503; PMID:25599564; http://dx.doi.org/ 10.1186/s13059-014-0503-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahmani E, Zaitlen N, Baran Y, Eng C, Hu D, Galanter J, Oh S, Burchard EG, Eskin E, Zou J, et al.. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods [Internet] 2016; 13(5):443-5.[cited 2016 Apr 4]; advance online publication. PMID:27018579; http://dx.doi.org/ 10.1038/nmeth.3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh KM, de Smith AJ, Welch TC, Smirnov I, Cunningham MJ, Ma X, Chokkalingam AP, Dahl GV, Roberts W, Barcellos LF, et al.. Genomic ancestry and somatic alterations correlate with age at diagnosis in Hispanic children with B-cell acute lymphoblastic leukemia. Am J Hematol 2014; 89:721-5; PMID:24753091; http://dx.doi.org/ 10.1002/ajh.23727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.R Core Team R: A Language and Environment for Statistical Computing [Internet]. 2013; Available from: http://www.R-project.org/ [Google Scholar]
- 26.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002 Jun; 12(6):996-1006; PMID:12045153; http://dx.doi.org/ 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.