

ABSTRACT
Myeloid-derived suppressor cells (MDSC) are a heterogeneous population of early myeloid cells that accumulate in the blood and tumors of patients with cancer. MDSC play a critical role during tumor evasion and promote immune suppression through variety of mechanisms, such as the generation of reactive oxygen and nitrogen species (ROS and RNS) and cytokines. AMPactivated protein kinase (AMPK) is an evolutionarily conserved serine/threonine kinase that regulates energy homeostasis and metabolic stress. However, the role of AMPK in the regulation of MDSC function remains largely unexplored. This study was designed to investigate whether treatment of MDSC with OSU-53, a PPAR-inactive derivative that stimulates AMPK kinase, can modulate MDSC function. Our results demonstrate that OSU-53 treatment increases the phosphorylation of AMPK, significantly reduces nitric oxide production, inhibits MDSC migration, and reduces the levels of IL-6 in murine MDSC cell line (MSC2 cells). OSU53 treatment mitigated the immune suppressive functions of murine MDSC, promoting T-cell proliferation. Although OSU-53 had a modest effect on tumor growth in mice inoculated with EMT-6 cells, importantly, administration of OSU53 significantly (p < 0.05) reduced the levels of MDSC in the spleens and tumors. Furthermore, mouse MDSC from EMT-6 tumor-bearing mice and human MDSC isolated from melanoma patients treated with OSU-53 showed a significant reduction in the expression of immune suppressive genes iNOS and arginase. In summary, these results demonstrate a novel role of AMPK in the regulation of MDSC functions and provide a rationale of combining OSU-53 with immune checkpoint inhibitors to augment their response in cancer patients.
KEYWORDS: AMPK, Immunotherapy, iNOS, MDSC, OSU-53
Abbreviations
- AICAR
5-aminoimidazole-4-carboxamide ribose
- AMPK
AMP-activated protein kinase
- IL-6
interleukin-6
- MDSC
myeloid-derived suppressor cell
- NOS
nitric oxide synthase
- TME
tumor microenvironment
- TNF-α
tumor necrosis factor α.
Introduction
The tumor microenvironment (TME) is composed of an extracellular matrix containing tumor cells, endothelial cells, fibroblasts, and a diverse population of immune cells.1 During the course of tumor progression, the TME is altered to facilitate the growth of cancer cells. A number of immune suppressive cells have been identified within the TME including myeloid-derived suppressor cells (MDSC), tumor-associated macrophages, and T-regulatory cells that can suppress the functions of innate and adaptive immune cells.2,3
MDSC are a heterogeneous group of cells consisting of granulocytes, monocytes, and dendritic cells at early stages of development.4,5 In humans, MDSC are characterized by the phenotypic markers CD33+CD11b+HLADRlow/neg with granulocytic and monocytic subsets being identified by the presence of CD15+ and CD14+, respectively.3,4 In mice, MDSC are identified as CD11b+/Gr-1+ cells with granulocytic and monocytic subsets classified by the expression of Ly6G and Ly6C, respectively.6 MDSC reside in the peripheral blood, lymphoid organs, and tumors of mice and their numbers correlate with tumor burden.7 MDSC can suppress immune cell functions via a variety of mechanisms including the generation of reactive oxygen species (ROS), nitric oxide (NO), indoleamine 2,3-dioxygenase (IDO), arginase and immunosuppressive cytokines (IL-6 and TGF-β) attenuating NK- and T-cell functions.6,8,9 Studies in murine models indicate that disruption of MDSC function can reverse immune tolerance to tumor antigens, stimulate antitumor immune responses and inhibit tumor growth.10
Immune therapies target cancer cells for destruction by activating the host immune system. However, the presence of immune suppressive cells such as MDSC in the TME can be a major impediment to immune-based therapies. Various experimental approaches have been explored to target MDSC in mouse models of cancer and this includes (1) agents that promote the differentiation of MDSC into mature myeloid cells (e.g., all trans retinoic acid, ATRA); (2) compounds that can block MDSC development (e.g., STAT3 inhibitors); (3) drugs that deplete MDSC (e.g., 5FU); and (4) direct inhibitors of MDSC immune suppressive function (e.g., iNOS inhibitors). Strategies targeting MDSC may be clinically used in combination with immune-based therapies for the treatment of cancer patients.11
AMP-activated protein kinase (AMPK) is an evolutionarily conserved serine/threonine kinase that regulates energy homeostasis and metabolic stress.12,13 The role of AMPK in regulating energy homeostasis is well established.13 In the liver, AMPK inhibits fatty acid synthesis and cholesterol synthesis.14 AMPK has also been shown to inactivate the mammalian target of rapamycin (mTOR) pathway via phosphorylation and activate the tuberous sclerosis complex-2 (TSC2).15 A potential role of AMPK in suppression of inflammatory responses has been suggested in studies using the pharmacological activator of AMPK, 5-aminoimidazole-4-carboxamide ribose (AICAR).16 Given AMPK's role as a master sensor and regulator of cellular energy, it is not surprising that this signaling pathway is deregulated in a variety of disease states, including cancer. Metformin a pharmacologic activator of AMPK has been shown to reduce risk of certain types of cancers in patients with type II diabetes.17,18 Several early stage clinical studies are now under way to investigate the potential of metformin to prevent different cancers or augment the efficacy of standard chemotherapy regimen.19
OSU-53 is a novel orally bioavailable thiazolidinedione derivative that has been shown to stimulate AMPK kinase in triple negative breast cancer cells.20 Preclinical studies using other pharmacological activators of AMPK such as metformin and AICAR have shown that these agents can inhibit carcinogen-induced tumorigenesis in mouse models.16,21 Conventional AMPK activators, including metformin, cause AMPK activation by generating an intracellular energy deficit via increased AMP:ATP ratios.22 This leads to metabolic stress that can induce AMPK independent cellular responses. To circumvent this problem, an allosteric AMPK activator OSU-53 was developed by our group.23 OSU-53 directly activates the kinase activity of recombinant AMPK α1β1γ2 by binding to the auto-inhibitory domain resulting in its activation. AMPK activation has also been shown to suppress the expression of the inflammatory cytokine IL-6, which promotes tumor growth by activating the Jak2/Stat3 signaling pathway.20,24 Treatment of a panel of breast and prostate cancer cells with OSU-53 reduced tumor growth by blocking oncogenic signaling and energy metabolism.25 Recently our group has demonstrated that OSU-53 reduced the growth of thyroid cancer cells and those cells harboring the activating RAS or BRAF mutations were more sensitive to OSU-53-mediated inhibition.26 In addition, OSU-53 can modulate signaling pathways downstream of the AMPK involved in the survival, mitochondrial biogenesis, and cytokine production.20
The present study was designed to investigate the effect of OSU-53 an AMPK activator, in the regulation of MDSC function. Our results demonstrate that OSU-53 treatment leads to the activation of phospho-AMPK, inhibits NO production, and reduces MDSC migration and cytokine production in vitro. Moreover, OSU-53 treatment reduced the immune suppressive function of murine and human MDSC and significantly reduced the frequency of MDSC in tumor-bearing mice. These results suggest that AMPK plays an important role in the regulation of MDSC function.
Results
OSU-53 treatment affects apoptosis of a murine MDSC cell line (MSC2)
The mouse MDSC cell line (MSC2) was used to examine the effects of the AMPK activator OSU-53, a PPAR-inactive derivative, on murine MDSC cell line in vitro (Fig. S1). MSC2 cells were treated overnight with various doses of OSU-53 (0–10 μM) or DMSO (vehicle) to determine whether OSU-53 was cytotoxic to MSC2 cells. Cells were stained with Annexin/PI and analyzed by flow cytometry to determine the percentage of apoptotic cells. OSU-53 treatment did not induce significant apoptosis up to a dose of 5.0 μM (Fig. 1A). However, there was an increase in the number of apoptotic cells at a dose of 10 μM. Similarly, OSU-53 was not cytotoxic to monocytes isolated from normal donors and did not induce any apoptosis at doses ranging between 0.5 μM and 10 μM as determined by annexin/PtdIns staining (Fig. 1B). Moreover, OSU-53 treatment did not induce apoptosis in EMT-6 tumor cells as determined by FACS analysis, described above (Fig. S2). Based on results of these experiments a dose range of 0–5.0 μM was selected for the subsequent experiments.
Figure 1.
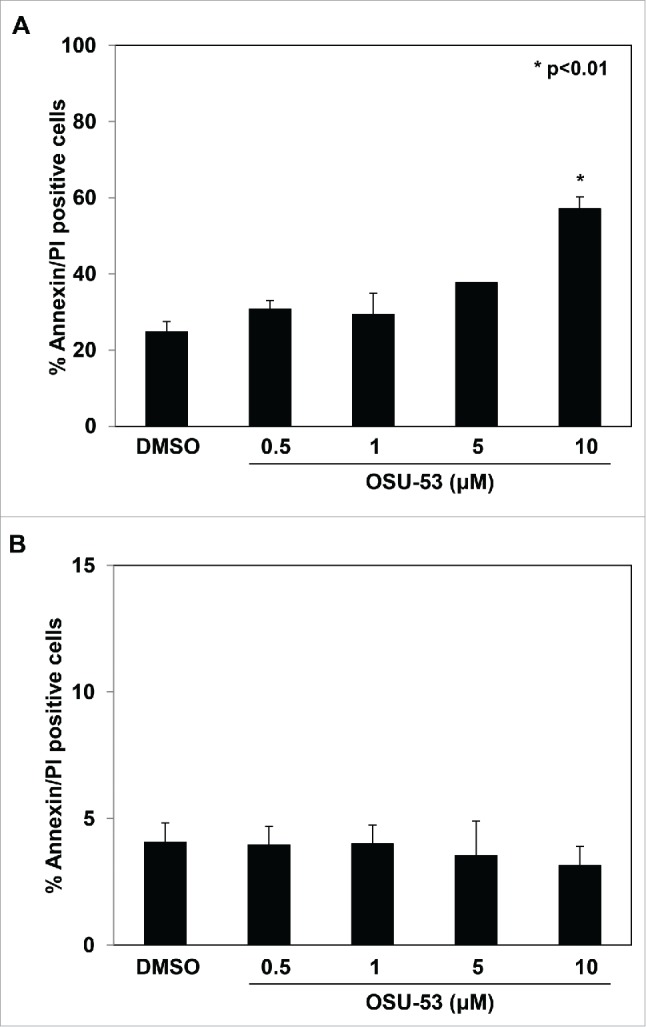
Effect of OSU-53 on the apoptosis of MSC2 cells. (A) MSC2 cells were treated with the indicated doses of OSU-53 or DMSO overnight and analyzed by FACS. (B) Monocytes isolated from healthy donors (n = 2) were treated overnight with the indicated doses of OSU-53 and DMSO (control). The cells were stained with AnnexinV/PI and analyzed via FACS to determine the percentage of apoptotic cells. Values represent mean ± SD.
OSU-53 treatment leads to the activation of AMPK in MDSC
OSU-53 has been shown to stimulate AMPK kinase in triple negative breast cancer cells.20 To investigate whether OSU-53 could activate AMPK activity in MDSC, MSC2 cells were treated with OSU-53 or DMSO for 12 h after which protein lysates were prepared. As shown by the immunoblot analysis, there was an increase in the levels of phosphorylated AMPK following the treatment of MSC2 with 5.0 μM of OSU-53 (Fig. 2 and Fig. S3). The overall levels of AMPK in MSC2 cells treated with OSU-53 remained unchanged.
Figure 2.
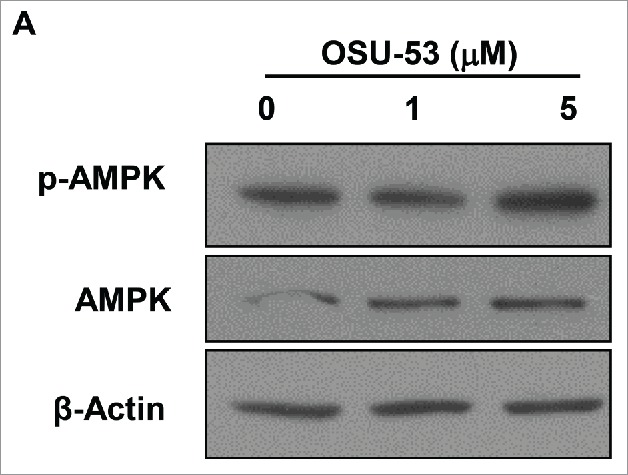
Increased expression of phosphorylated AMPK in MDSC treated with OSU53. (A) Immunoblot showing the expression of p-AMPK and total AMPK in MSC2 cells treated with the indicated doses of OSU-53 or DMSO for 12 h. Protein lysates from MSC2 cells were probed with p-AMPK, total AMPK, and ß-Actin antibodies.
OSU-53 treatment leads to an attenuation of NO production
NO is an important inhibitory molecule produced by MDSC that is involved in the suppression of NK-cell and T-cell function and is produced by MSC2 cells after LPS treatment.7 To examine the affect OSU-53 on the levels of NO, MSC2 cells were treated with the indicated doses of OSU-53 and stimulated with LPS for 24 h. Following the incubation, supernatants were collected and analyzed for levels of nitrite using the Griess reagent. Activation of MSC2 cells with LPS in the presence of DMSO resulted in an increase in the levels of nitrite from 2 μM to 25 μM. However, there was a significant reduction (p < 0.05) in the level of nitrite in the LPS stimulated MSC2 cells treated with 5 μM of OSU-53 (Fig. 3A). Furthermore, MSC2 cells treated with 5.0 μM OSU-53 showed a significant reduction in expression of iNOS (NOS2), a gene involved in the generation of NO (Fig. 3B). Similarly, MSC2 cells treated with another AMPK activator, metformin at concentrations of 2.5 mM–10 mM, showed a reduction in levels of NO production. The concentration of metformin required for NO inhibition was nearly 1,000-fold higher than OSU-53, demonstrating that OSU-53 and metformin may function via different mechanisms at inhibiting NO generation (Fig. S4).
Figure 3.
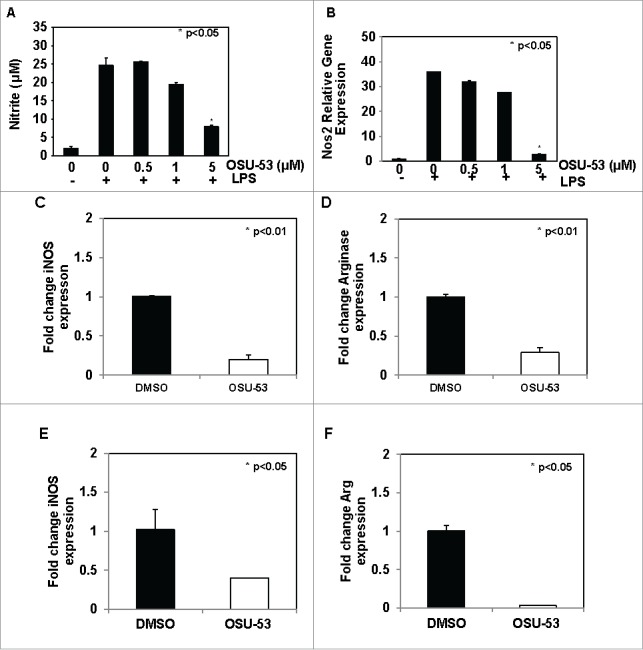
Reduction iNOS and arginase expression in murine and human MDSC treated with OSU-53. (A) MSC2 cells were treated with indicated doses of OSU-53 or DMSO (control) overnight and then stimulated with LPS. The levels of nitrite were measured in the supernatant using Griess reagent. (B) Fold change in NOS2 (iNOS) gene expression in MSC2 cells was treated with indicated doses of OSU-53 or DMSO. (C-D) iNOS and arginase gene expression in MDSC from EMT-6 tumor-bearing mice treated OSU-53 (5 µM) or DMSO. MDSC isolated from melanoma patients were treated overnight with DMSO or OSU-53 (5 µM). The expression of (E) inducible nitric oxide synthase and (F) arginase was determined using real time PCR. The bars show fold change in gene expression compared to DMSO (controls). Values represent mean ± SD. (p < 0.05).
NO and arginase are two key molecules that are involved in mediating the immune suppressive function of MDSC.27 MDSC isolated from EMT-6 tumor-bearing mice were treated with OSU-53 (5 μM) or DMSO showed a significant reduction (p < 0.01) in the expression of iNOS and arginase genes. Cox2 and PGE2 represent two other signaling pathways that are involved in the regulation of MDSC development and function.28,29 To determine whether AMPK activation affects the expression of Ptgs2, Ptger2, and Ptger4 genes, we performed RT-PCR on murine MDSC treated with OSU-53 or DMSO. There was a 30–40% reduction in the expression of these genes compared to DMSO-treated controls (Fig. S5).
MDSC accumulate in the blood and tumors of patients with cancer and are implicated in the failure of immune-based therapies, indicating that strategies aimed at depleting or blocking their immune suppressive function could be a successful strategy to improve the efficacy.3,30 Human MDSC isolated from melanoma patients were incubated overnight with OSU-53 (5.0 μM) or DMSO. The expression of iNOS and arginase genes was determined using RT-PCR. OSU-53 treatment resulted in a significant reduction in the expression of both iNOS (2.5-fold) and arginase (30-fold) genes in human MDSC compared to the DMSO-treated controls (Fig. 3E and F). These results demonstrate that OSU-53 can modulate immune suppressive functions of MDSC by inhibiting iNOS and arginase expression.
OSU-53 treatment reduces MDSC migration
Tumor cells secrete cytokines and chemokines that promote the recruitment of MDSC into the tumor stroma. To assess whether treatment of MDSC with OSU-53 affected their ability to migrate in response to factors produced by EMT6-HER2+ tumor cells. Migration assays were performed using MSC2 cells and conditioned media collected from EMT6-HER2+ cancer cells (Fig. 4A). MSC2 cells were treated with the indicated doses of OSU-53. Cell migration was allowed to proceed for 18 h, following which the inserts were stained, imaged, and analyzed. The numbers of MDSC were quantified using image J software. These assays showed a significant reduction (p < 0.001) in migration of MSC2 cells treated with OSU-53 compared to the DMSO-treated controls (Fig. 4B and C). These results suggest increased levels of p-AMPK in MDSC can impair their ability to migrate.
Figure 4.
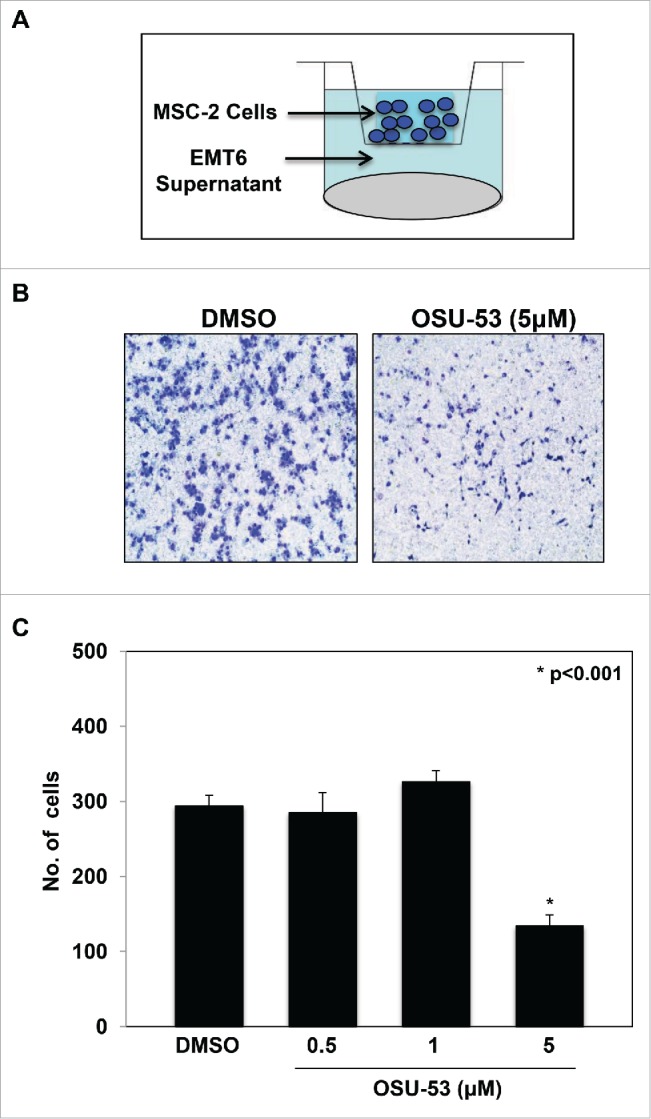
Reduced MSC2 migration in response to cytokines produced by tumor cells. (A) Schematic showing the experimental setup. MSC2 cells cultured in serum free media were treated with the indicated doses of OSU-53 for 12 h. The MSC2 cells were added into the transwell insert and migration was allowed to precede overnight in response to conditioned media from mouse EMT6-HER-2+ tumor cells. (B) Representative image of inserts showing MSC2 cells after migration. (C) Quantification of the number of migrated MSC2 cells. Values represent mean ± SD.
Reduced levels of IL-6 and TNF-α in OSU-53 treated cells
Various signaling pathways are involved in the generation, expansion, and regulation of MDSC functions. The ability of OSU-53 to affect the secretion of proinflammatory cytokines such as IL6 and TNFα was evaluated next. MSC2 cells were treated with the indicated doses of OSU-53 and were stimulated with LPS for 24 h. Following the incubation, supernatants were collected and analyzed for levels of various cytokines using a bead-based flow cytometry assay. The levels of IL-17, IL-10, IL-2, IL-4, IFNγ, IL-6, and TNF-α were analyzed. MSC2 cells activated with LPS did not produce any detectable levels of IL-17, IL-10, IL-2, IL-4, and INFγ (data not shown). Notably, there was a decrease in the levels of IL-6 and TNF-α produced by MSC2 following treatment with OSU-53. The treatment of MSC2 cells with OSU-53 (5 μM) resulted in a reduction in the levels of IL-6 (2.9-fold) and TNF-α (1.5-fold) (Fig. 5A and B). Both of these cytokines (IL-6 and TNFα) have been shown to have an important role in the regulation of MDSC function.5 These results are consistent with the previous studies that showed OSU-53 can decrease the production of IL-6 by triple negative breast cancer cells.20 However, to our knowledge, this is the first study that shows OSU- 53 can modulate IL6 and TNFα production by MDSC.
Figure 5.
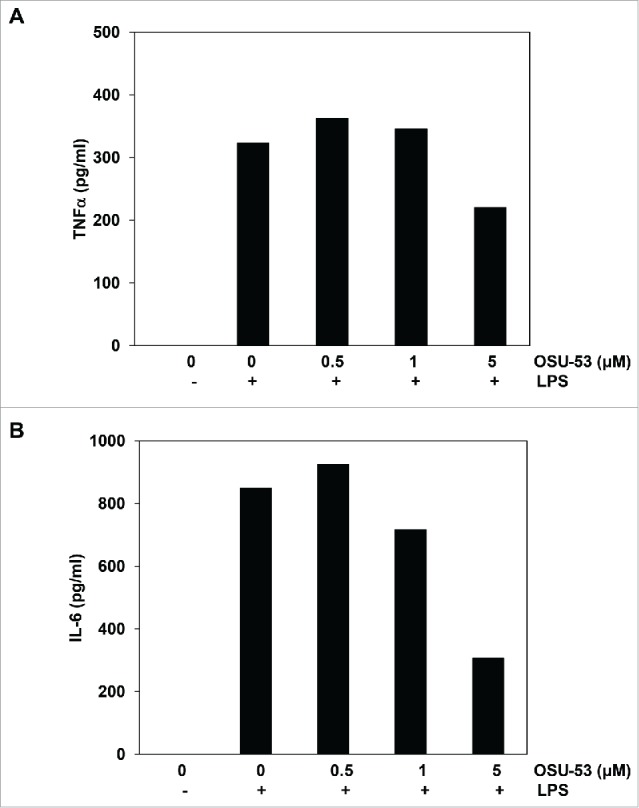
Levels of IL-6 and TNF-α in MSC2 cells treated with OSU-53. MSC2 cells were treated with the indicated doses of OSU-53 for 12 h, following which the cell free supernatant were collected and analyzed for levels of TNF-α and IL-6 by ELISA. (A) TNFα (pg/mL). (B) IL-6 (pg/mL).
OSU-53 reduces MDSC mediated T-cell proliferation
Our in vitro experiments using MSC-2 cells demonstrate that OSU-53 can attenuate MDSC functions by inhibiting the production of nitric oxide. Next, to examine whether OSU-53 can reduce the ability of MDSC to suppress T-cell proliferation, MDSC isolated from the spleens of EMT6 tumor-bearing mice were treated overnight with OSU-53 or DMSO (control), following which they were co-cultured with CFSE-labeled T cells. After a 3-day incubation period, T-cell proliferation was assessed by flow cytometry. MDSC treated with OSU-53 (5.0 μM) were less suppressive and had a reduced ability to inhibit T-cell proliferation compared to DMSO-treated controls. There was 35% decrease in the ability of OSU-53-treated MDSC to inhibit the proliferation of CD4+ T cells compared to the control (DMSO) (Fig. 6A). Similarly, there was a trend toward decreased CD8+ T proliferation following co-culture with OSU-53-treated MDSC (Fig. 6B). Together, these results demonstrate that OSU-53 mitigates the immune suppressive functions of murine MDSC.
Figure 6.
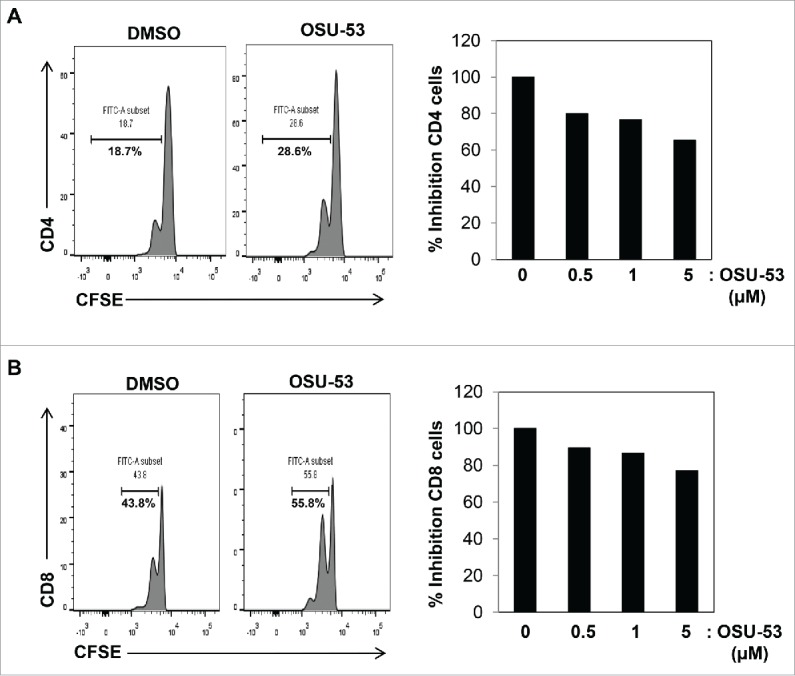
OSU-53 reduces MDSC mediated suppression of T-cell proliferation. MDSC isolated from tumor-bearing mice were treated with indicated doses DMSO or OSU-53 and co-cultured (2:1) with CFSE-labeled T cells activated with anti-CD3/CD28 beads for 3-days and proliferation was assessed by flow cytometry for CFSE staining. Cells were stained with anti-CD4+ or anti-CD8+ antibodies. Shown are representative histogram (left panels) and bar graphs (right panels). Results are from one representative experiment.
OSU-53 reduces the levels of MDSC in tumor-bearing mice
MDSC promote tumor growth by attenuating the antitumor function of T cells and NK cells in tumor-bearing mice and in cancer patients.5 To examine the effect of the AMPK activator OSU53 on the tumor growth and the frequency of MDSC, mice bearing EMT6 murine mammary carcinoma received OSU-53 (100 mg/kg) or vehicle via oral gavage daily for a period of 2 weeks as previously reported.20 The mice treated with OSU-53 showed a small but non-significant reduction in tumor volume (Fig. 7A). At the end of the study, spleens and tumors were analyzed for the presence of MDSC by FACS analysis. OSU-53 treatment resulted in a significant (p < 0.05) reduction of Cd11b+Gr-1+ MDSC in the spleen and tumor (Fig. 7C and D). This decrease in the frequency of MDSC was accompanied by a significant (p < 0.02) reduction in spleen weight (Fig. 7B). Furthermore, analysis of the spleens and tumors revealed that the levels of T cells and NK cells were also increased in mice treated with OSU-53 compared to vehicle-treated mice (Fig. S6A–H). Examination of tumors from OSU-53 or vehicle treated mice showed no major histopathological changes between the 2 groups of mice (Fig. S7). The lack of significant differences in tumor volume between vehicle- and OSU-53-treated mice suggests that the reduction of MDSC frequency is a direct effect of OSU-53 treatment on MDSC.
Figure 7.
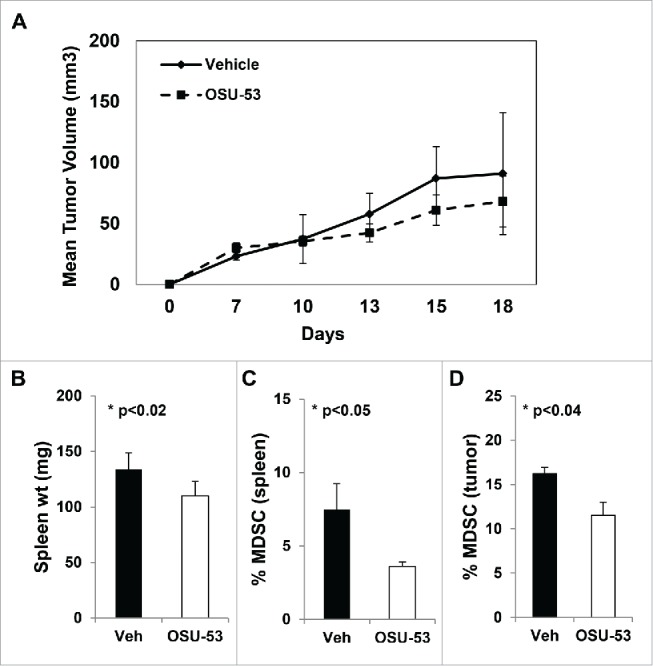
OSU-53 reduces MDSC frequency in tumor-bearing mice. Female Balb/c mice were inoculated with EMT-6-HER2 breast cancer cells in the mammary fat pad. The mice received OSU-53 (100 mg/kg) or vehicle via oral gavage for a period of 2 weeks. (A) Tumor growth in mice treated with OSU-53 or vehicle. (B) Weight of spleens from mice (C–D) FACS analysis of splenocytes and single cell suspensions prepared from tumors stained with CD11b and Gr1 and antibodies. Each group consisted of five mice. Values are the mean ± SE of tumor volumes at each time point, (p < 0.05).
MDSC generation is not affected by OSU-53
Cytokines like IL-6 and GMCSF are involved in generation and the expansion of MDSC during tumorigenesis.8 Therefore, the effect of OSU-53 on the ability of peripheral blood mononuclear cells (PBMC) to differentiate into MDSC was investigated. To this end, PBMCs isolated from healthy donors were cultured in media containing IL-6 and GM-CSF (10 ng/mL each) for a period of 6 d. PBMC were treated concurrently with OSU-53 (5 μM) or DMSO. This experiment showed that OSU-53 treatment led to a slight reduction in the ability PBMC to differentiate into MDSC in vitro (Fig. S8).
Discussion
AMPK is a conserved serine/threonine kinase that is activated in response to cellular stress.12 AMPK is an important signaling molecule involved in the regulation of multiple metabolic processes and is downregulated in cancer cells.31,32,33 However, the role of AMPK in the regulation of MDSC function remains largely unexplored. The results from the present study demonstrate that treatment with a novel allosteric AMPK activator OSU-53 leads to the activation of phospho-AMPK, inhibited NO production, reduced MDSC migration, and decreased production of immunosuppressive cytokines IL-6 and TNF-α in vitro. OSU-53 was also able to mitigate the immune suppressive functions of murine MDSC by reducing the expression of iNOS and arginase genes. Furthermore, tumor-bearing mice treated with OSU-53 showed a significant reduction in the levels of MDSC. Together, these results demonstrate a novel role of AMPK in the regulation of MDSC function and provide a rationale of combining AMPK activators with immune-based therapies, including immune checkpoint inhibitors.
OSU-53 is an orally bioavailable thiazolidinedione derivative that has been shown to stimulate AMPK kinase.20 Preclinical studies using other pharmacological activators of AMPK such as metformin and AICAR have shown that these agents can inhibit carcinogen-induced tumorigenesis in mouse models.16,21 Conventional AMPK activators, such as metformin, have low in vitro efficacy with IC50 in the millimolar range and are associated with gastrointestinal side effects.22 To circumvent these problems, an allosteric AMPK activator, OSU-53, was developed by our group.23 In contrast to metformin, OSU-53 is more potent with an IC50 in the low micromolar range compared to millimolar for metformin. More importantly, OSU-53 directly activates AMPK independent of its upstream kinase LKB1.20 This is in contrast to metformin that activates AMPK possibly via the inhibition of AMP deaminase.34 The results from the current study show the concentration of metformin required for NO inhibition was nearly 1,000-fold higher than OSU-53, demonstrating that OSU-53 and metformin may function via different mechanisms.
MDSC play a critical role during tumor evasion and promote immune suppression by the production of NO, ROS, arginase, indolamine 2,3-dioxygenase (IDO), and suppressive cytokines (e.g. IL10 and TGFβ).30 The frequency of circulating MDSC correlates with tumor burden and has prognostic value in a variety of solid tumors.9 Studies in murine models indicate that disruption of MDSC function can reverse immune tolerance to tumor antigens, stimulate antitumor immune responses, and markedly inhibit tumor growth.6 Given the ability of MDSC to suppress antitumor immune responses, this cell type has received significant interest as a potential biomarker and therapeutic target.35 Early clinical trials also suggest that reduction of circulating MDSC leads to improved disease specific survival.11
Considerable progress has been made in elucidating the mechanisms that are involved in the generation, expansion, and the signaling pathways involved in the regulation of MDSC function. However, MDSC function at the metabolic level still remains unclear. Immune cells including lymphocytes and myeloid cells modulate metabolism to fulfill their energy requirements. Thus, energy metabolism of immune effector cells is considered as potential target for immunotherapy.
Cell migration plays a crucial role during tumor invasion and metastasis.1 AMPK has been shown to modulate the expression of genes involved in migration.36 Our group has previously demonstrated that OSU-53 inhibits hypoxia-induced EMT and diminishes the capacity of breast cancer cells to migrate by reducing the expression of genes involved in migration, such as cadherin and vimentin.20,25 Consistent with those studies, the results from the present study demonstrate that OSU-53 can significantly impair the ability of MSC2 cells to migrate in response to cytokines produced by tumors cell in vitro. In addition, there was a significant reduction in the number of MDSC in tumor-bearing mice treated with OSU-53. AMPK activation can reduce the expression of genes that are involved in migration thereby impeding the recruitment of MDSC to the tumors. A recent study by Yan et al. provides further experimental evidence in which they showed augmented AMPK activity can inhibit cell migration by suppressing the phosphorylation of Pdlim5, a key protein involved in lamellipodia formation.36
MDSC can suppress the antitumor functions of T cells and NK cells via the generation of a ROS, NO, and a variety of immune suppressive cytokines.5 Our group has previously shown that NO generated by murine MDSC can inhibit phosphorylation of STAT1 thereby attenuating the NK- and T-cell functions.9 The JAK/STAT pathway is a key signaling pathway involved in the regulation of MDSC functions.37 Cytokines like IL-6 and GM-CSF are involved in the generation, expansion and in the regulation of MDSC functions. These cytokines can activate JAK/STAT signaling pathways leading to the activation of genes that are involved in mediating the suppressive function of MDSC.38,39 IL-6 has been shown to directly increase iNOS expression by activating the JAK2/STAT3 pathway in rat ventricular myocytes.40,41 MSC2 cells treated with OSU-53 showed a significant reduction in levels of NO production and levels of IL-6 in vitro, and this could be an additional mechanism via which OSU53 attenuates MDSC suppressive functions.
Studies by Pilon et al in myocytes have shown that one of the mechanisms via which AMPK activators like AICAR improves glucose homeostasis and insulin sensitivity is through the inhibition of iNOS, an enzyme whose levels are elevated during chronic inflammation.24 AMPK can also inhibit protein synthesis either through suppression of the mTOR-p70S6 kinase pathway or by inactivation of eukaryotic elongation factor 2.42 Additional mechanisms have been proposed by which AMPK activators can cause iNOS inhibition. AMPK may reduce iNOS protein content by promoting its ubiquitination, a process required for targeting iNOS through the proteasome proteolysis pathway. Inflammatory cytokines and LPS transcriptionally regulate iNOS expression through a complex network of intracellular pathways that involve NF-kB, Jak/Stat, and MAPK.43 PPAR ligands like rosiglitazone have been shown to reduce iNOS expression through transcriptional repression.44
The results from our in vitro experiments were further corroborated by the finding in murine MDSC that showed OSU-53 was able to attenuate their immune suppressive functions. OSU-53-treated murine MDSC were less immune suppressive and promoted T-cell proliferation. Moreover, tumor-bearing mice treated with OSU-53 showed a significant reduction in the levels of MDSC in the spleen and the tumors. This was accompanied by an increase in the frequency of T cells and NK cells in these mice. Similarly, human MDSC isolated from melanoma patients treated with OSU-53 showed a significant reduction in the expression of immune suppressive genes iNOS and arginase. Therefore, one mechanism via which OSU-53 may attenuate the immune suppressive function of MDSC involves the downregulation of iNOS and arginase expression. In addition, AMPK activation can impair the migration of MDSC to the spleen and into the tumors. AMPK-activating drugs are in clinical trials for prevention and treatment of a number of malignancies.45 Our results demonstrate that AMPK activation can inhibit the immune suppressive functions of MDSC and possibly combat tumor growth by enhancing the antitumor response of NK cell and T cells.
In summary, the results of this study demonstrate that AMPK plays an important role in the regulation of MDSC function. OSU-53 attenuates the immune suppressive functions of MDSC by inhibiting the expression of molecule(s) involved in immune suppression. MDSC contribute to cancer-induced immune suppression that can significantly impede the host response to immune-based therapies. Therefore, the combination of MDSC-modulating drugs like OSU-53 with mAbs (trastuzumab) or immune checkpoint inhibitors (PD-1/PDL1) represents a novel approach in immunotherapy.
Materials and methods
Cell lines and cell culture
A murine MDSC cell line, MSC-2 (gift from Gregoire Mignot), was cultured in RPMI medium (Life Technologies, Paisley, UK) containing 10% FBS and 1% sodium pyruvate. OSU-53 was synthesized according to a published procedure.23 DMSO was used as vehicle (control) for all the in vitro experiments since OSU-53 was reconstituted using DMSO
Annexin V/PI staining
Annexin/PtdIns staining was used to determine the cytotoxicity effect of OSU-53 on the MSC-2 cell line. MSC-2 cells were treated with various doses of DMSO (vehicle) or OSU-53 (0–10 μM). Monocytes were isolated from healthy donor PBMCs and were treated overnight with different doses of OSU-53 or DMSO. Cells were then harvested, re-suspended in annexin buffer, and stained with Annexin V and propidium iodine (PI). The cells were analyzed on an LSR-II flow cytometer (Becton-Dickinson, San José, CA) and the data were analyzed via FlowJo™ software (Treestar, Ashland, OR).
Immunoblot
MSC-2 cells were cultured with varying amounts of OSU-53 for 18 h. Cells were lysed with RIPA buffer containing protease and phosphatase (Sigma Aldrich, St. Louis, MO). The protein lysates were probed for AMPK, p-AMPK, and β–Actin antibodies (Cell Signaling).
Cell migration
MSC2 cells were treated with OSU-53 or DMSO. MSC2 cells (1 × 105) were plated in the top chamber of an 8 μm transwell assay. Media conditioned by the EMT6 cell line was used to stimulate MSC2 migration. Inserts were collected after 24 h and stained using the Dip Quick Stain Kit (Jorgensen Laboratories, Inc., Loveland, CO). Photographs were taken using a digital camera and cell numbers enumerated using image J software (2,048 × 1,536 pixels, Advanced Microscopy Group, Bothell, WA).
Nitric oxide (NO) estimation
MSC-2 cells treated with varying amounts of drug were cultured for 24 h. After this period, the supernatant was harvested and quantified for NO using Griess Reagent (Sigma Aldrich, St. Louis, MO). Equal parts Griess Reagent and sample were mixed together in a 96 well plate and absorbance values were recorded at 450 nm using a plate reader.
Cytokine analysis
MSC-2 cells treated with varying amounts of drug were cultured for 24 h. The supernatant were analyzed for presence of cytokines using the murine cytokine bead assay (BD Bioscience, San Jose, CA).
Real time PCR
Total RNA was extracted from MSC-2 cells, murine MDSC from EMT-6 tumor-bearing mice, and human MDSC from melanoma patients using the Trizol reagent (Life Technologies, Grand Island, NY, USA). Reverse transcription reactions were performed using 500 ng RNA in a 20 μL reaction with the high-capacity reverse transcription kit (Life Technologies). cDNA was used to measure the expression of murine or human Nos2 (iNOS) and arginase gene by quantitative-Real Time PCR using pre-designed primers (Integrated DNA Technologies, Coralville, IA). Pre-designed primers for murine or human βActin served as an internal control for each reaction (Life Technologies). Real Time PCR reactions were performed in triplicate using the ABI PRISM 7900HT fast Real Time PCR system with SYBR Green chemistry (Applied Biosystems).
Murine CFSE assay
MDSC isolated from the spleen of EMT6 tumor-bearing mice and T cells were isolated from a non-tumor-bearing mouse using the murine T-cell isolation kit (Stemcell Technologies, Inc.). MDSC were treated with DMSO or 5.0 μM OSU-53 overnight with 10 ng/mL IL-6 and GM-CSF. T cells were labeled with CFSE (Life Technologies) and incubated overnight with 10 ng/mL of IL-2 (Peprotech). T cells were non-specifically activated with anti-CD3/CD28 beads (Life Technologies) and co-cultured at 1:1, 2:1, and 4:1 ratios of T cells to MDSC. After 3 d cell proliferation was assessed by flow cytometry. APC anti-CD4+ and anti-CD8+ antibodies were used to identify T cell subsets (Biolegend, San Diego, CA).
Tumor study
Female 4–6 week old BALB/c mice (Jackson Laboratories, Bar Harbor, ME) were injected with 1 × 106 EMT6 cells in the mammary fat pad to generate tumors. OSU-53 (100 mg/kg) or vehicle was administered daily by oral gavage daily for a period of 2-weeks as previously described.25 These studies were conducted under a protocol approved by Ohio State University's Institutional Animal Care and Use Committee.
MDSC Isolation from tumor-bearing mice
Spleens and tumors were harvested aseptically from EMT-6 tumor-bearing mice. Single cell suspensions were prepared and stained with a panel of antibodies and analyzed by FACS. MDSC were isolated from spleens using the mouse MDSC isolation kit (Miltenyi Biotec, Auburn, CA).
MDSC isolation
Peripheral blood was obtained from melanoma patients receiving treatment at the Ohio State University Comprehensive Cancer Center under an Institutional Review Board (IRB) approved protocol (OSU-IRB-1999C0348). MDSC were isolated from fresh peripheral blood of metastatic melanoma patients by 30 min incubation with myeloid enrichment cocktail (Stemcell Technologies, Vancouver, BC) followed by gradient centrifugation using (Ficoll-Paque, GE Healthcare). The cells obtained from the buffy coat were suspended in MACS buffer incubated with anti-HLA-DR magnetic beads for 15 min at 4°C and isolated using MS-MACS column to obtain MDSC (CD33+CD11b+HLA-DR−/low).27 The MDSC consisted of both monocytic and the granulocytic subsets.
Generation of MDSC
PBMC were obtained from normal healthy adult blood donors (source leukocytes, American Red Cross, Columbus, OH). PBMC were separated from source leukocytes by density gradient centrifugation with Ficoll-Paque (GE Healthcare Bio-Sciences, Pittsburgh, PA) as previously described.46 PBMC were cultured in media containing IL-6 and GM-CSF (Peprotech, Rocky Hill, NJ) for a period of 6 d at 37°C in 5% CO2. During this process, the PBMC were treated with a dose of 5 μM OSU-53 or DMSO vehicle control. The media and drug were changed every 2–3 d.
Statistical analysis
Statistical differences between treatment groups were determined using an ANOVA model and Student's t-test. For tumor studies in mice, a linear mixed model was employed to model longitudinal tumor volume for mice under each treatment. Comparisons were done at each time point and averaged across all time points using statistics. The Holm–Bonferroni method was used for adjusting raw p-values for multiple comparisons across treatment groups.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health Grants P01CA95426, K24 CA93670 (W.E.C.), T32CA90338-27, P30 CA016058, Translational grant K12 CA133250 in experimental therapeutics National Cancer Institute, and Ohio State Translational Therapeutics Award TT-142. This work was supported by the Pelotonia Fellowship Program.
References
- 1.Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 2011; 17:320-9; PMID:21383745; http://dx.doi.org/ 10.1038/nm.2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013; 138:105-15; PMID:23216602; http://dx.doi.org/ 10.1111/imm.12036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci 2014; 1319:47-65; PMID:24965257; http://dx.doi.org/ 10.1111/nyas.12469 [DOI] [PubMed] [Google Scholar]
- 4.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/ 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trikha P, Carson WE. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta 2014. Aug; 1846(1):55-65; PMID:24727385; http://dx.doi.org/ 10.1016/j.bbcan.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 2008; 181:5791-802; PMID:18832739; http://dx.doi.org/ 10.4049/jimmunol.181.8.5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 2007; 13:828-35; PMID:17603493; http://dx.doi.org/ 10.1038/nm1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 2011; 32:19-25; PMID:21067974; http://dx.doi.org/ 10.1016/j.it.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mundy-Bosse BL, Young GS, Bauer T, Binkley E, Bloomston M, Bill MA, Bekaii-Saab T, Carson WE, Lesinski GB. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4+ T cells from patients with GI malignancy. Cancer Immunol Immunother 2011; 60:1269-79; PMID:21604071; http://dx.doi.org/ 10.1007/s00262-011-1029-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 2010; 70:3052-61; PMID:20388795; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3690 [DOI] [PubMed] [Google Scholar]
- 11.Wesolowski R, Markowitz J, Carson WE. Myeloid derived suppressor cells - a new therapeutic target in the treatment of cancer. J Immunother Cancer 2013; 1:10; PMID:24829747; http://dx.doi.org/ 10.1186/2051-1426-1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251-62; PMID:22436748; http://dx.doi.org/ 10.1038/nrm3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009; 458:1056-60; PMID:19262508; http://dx.doi.org/ 10.1038/nature07813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henin N, Vincent MF, Gruber HE, Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J 1995; 9:541-6; PMID:7737463 [DOI] [PubMed] [Google Scholar]
- 15.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577-90; PMID:14651849; http://dx.doi.org/ 10.1016/S0092-8674(03)00929-2 [DOI] [PubMed] [Google Scholar]
- 16.Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol 2005; 175:566-74; PMID:15972693; http://dx.doi.org/ 10.4049/jimmunol.175.1.566 [DOI] [PubMed] [Google Scholar]
- 17.Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: time for action. J Clin Oncol 2009; 27:3271-3; PMID:19487373; http://dx.doi.org/ 10.1200/JCO.2009.22.1630 [DOI] [PubMed] [Google Scholar]
- 18.Goodwin PJ, Stambolic V, Lemieux J, Chen BE, Parulekar WR, Gelmon KA, Hershman DL, Hobday TJ, Ligibel JA, Mayer IA et al.. Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat 2011; 126:215-20; PMID:20976543; http://dx.doi.org/ 10.1007/s10549-010-1224-1 [DOI] [PubMed] [Google Scholar]
- 19.Chae YK, Arya A, Malecek MK, Shin DS, Carneiro B, Chandra S, Kaplan J, Kalyan A, Altman JK, Platanias L et al.. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget 2016. Mar 19; PMID:27004404; http://dx.doi.org/ 10.18632/oncotarget.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee KH, Hsu EC, Guh JH, Yang HC, Wang D, Kulp SK, Shapiro CL, Chen CS. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. J Biol Chem 2011. Nov 11; 286(45):39247-58; PMID:21917926; http://dx.doi.org/ 10.1074/jbc.M111.264598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M et al.. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci U S A 2009; 106:12932-7; PMID:19625624; http://dx.doi.org/ 10.1073/pnas.0906606106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landgraf RR, Goswami D, Rajamohan F, Harris MS, Calabrese MF, Hoth LR, Magyar R, Pascal BD, Chalmers MJ, Busby SA et al.. Activation of AMP-activated protein kinase revealed by hydrogen/deuterium exchange mass spectrometry. Structure 2013; 21:1942-53; PMID:24076403; http://dx.doi.org/ 10.1016/j.str.2013.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guh JH, Chang WL, Yang J, Lee SL, Wei S, Wang D, Kulp SK, Chen CS. Development of novel adenosine monophosphate-activated protein kinase activators. J Med Chem 2010; 53:2552-61; PMID:20170185; http://dx.doi.org/ 10.1021/jm901773d [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Pilon G, Dallaire P, Marette A. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem 2004; 279:20767-74; PMID:14985344; http://dx.doi.org/ 10.1074/jbc.M401390200 [DOI] [PubMed] [Google Scholar]
- 25.Chou CC, Lee KH, Lai IL, Wang D, Mo X, Kulp SK, Shapiro CL, Chen CS. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res 2014; 74:4783-95; PMID:24994714; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plews RL, Mohd Yusof A, Wang C, Saji M, Zhang X, Chen CS, Ringel MD, Phay JE. A novel dual AMPK activator/mTOR inhibitor inhibits thyroid cancer cell growth. J Clin Endocrinol Metab 2015; 100:E748-56; PMID:25710562; http://dx.doi.org/ 10.1210/jc.2014-1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, McMichael EL, Duggan M, Campbell A, Keller K et al.. Myeloid-derived suppressor cells express Bruton's tyrosine kinase and can be depleted in tumor bearing hosts by ibrutinib treatment. Cancer Res 2016. Apr 15; 76(8):2125-36; PMID:26880800; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res 2011; 71:7463-70; PMID:22025564; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med 2005; 202:931-9; PMID:16186186; http://dx.doi.org/ 10.1084/jem.20050715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol 2006; 16:53-65; PMID:16168663; http://dx.doi.org/ 10.1016/j.semcancer.2005.07.005 [DOI] [PubMed] [Google Scholar]
- 31.Lee CW, Wong LL, Tse EY, Liu HF, Leong VY, Lee JM, Hardie DG, Ng IO, Ching YP. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res 2012; 72:4394-404; PMID:22728651; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, Suzuki K, Nakamoto M, Shimizu E, Minna JD et al.. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 2007; 26:5911-8; PMID:17384680; http://dx.doi.org/ 10.1038/sj.onc.1210418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, Potts PR. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 2015; 160:715-28; PMID:25679763; http://dx.doi.org/ 10.1016/j.cell.2015.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem 2011; 286:1-11; PMID:21059655; http://dx.doi.org/ 10.1074/jbc.M110.121806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider T, Sevko A, Heussel CP, Umansky L, Beckhove P, Dienemann H, Safi S, Utikal J, Hoffmann H, Umansky V. Serum inflammatory factors and circulating immunosuppressive cells are predictive markers for efficacy of radiofrequency ablation in non-small-cell lung cancer. Clin Exp Immunol 2015; 180:467-74; PMID:25644608; http://dx.doi.org/ 10.1111/cei.12596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan Y, Tsukamoto O, Nakano A, Kato H, Kioka H, Ito N, Higo S, Yamazaki S, Shintani Y, Matsuoka K et al.. Augmented AMPK activity inhibits cell migration by phosphorylating the novel substrate Pdlim5. Nat Commun 2015; 6:6137; PMID:25635515; http://dx.doi.org/ 10.1038/ncomms7137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity 2012; 36:503-14; PMID:22520844; http://dx.doi.org/ 10.1016/j.immuni.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol 2005; 174:636-45; PMID:15634881; http://dx.doi.org/ 10.4049/jimmunol.174.2.636 [DOI] [PubMed] [Google Scholar]
- 39.Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G et al.. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol 2003; 170:270-8; PMID:12496409; http://dx.doi.org/ 10.4049/jimmunol.170.1.270 [DOI] [PubMed] [Google Scholar]
- 40.Yu X, Kennedy RH, Liu SJ. JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J Biol Chem 2003; 278:16304-9; PMID:12595539; http://dx.doi.org/ 10.1074/jbc.M212321200 [DOI] [PubMed] [Google Scholar]
- 41.Xie QW, Whisnant R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med 1993; 177:1779-84; PMID:7684434; http://dx.doi.org/ 10.1084/jem.177.6.1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol 2002; 12:1419-23; PMID:12194824; http://dx.doi.org/ 10.1016/S0960-9822(02)01077-1 [DOI] [PubMed] [Google Scholar]
- 43.Blanchette J, Jaramillo M, Olivier M. Signalling events involved in interferon-gamma-inducible macrophage nitric oxide generation. Immunology 2003; 108:513-22; PMID:12667213; http://dx.doi.org/ 10.1046/j.1365-2567.2003.01620.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc Natl Acad Sci U S A 2003; 100:6712-7; PMID:12740443; http://dx.doi.org/ 10.1073/pnas.1031789100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab 2013; 24:469-80; PMID:23773243; http://dx.doi.org/ 10.1016/j.tem.2013.05.004 [DOI] [PubMed] [Google Scholar]
- 46.Varker KA, Kondadasula SV, Go MR, Lesinski GB, Ghosh-Berkebile R, Lehman A, Monk JP, Olencki T, Kendra K, Carson WE. Multiparametric flow cytometric analysis of signal transducer and activator of transcription 5 phosphorylation in immune cell subsets in vitro and following interleukin-2 immunotherapy. Clin Cancer Res 2006; 12:5850-8; PMID:17020993; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1159 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.