

ABSTRACT
Carcinoembryonic antigen (CEA) is a cell surface antigen highly expressed in various cancer cell types and in healthy tissues. It has the potential to be a target for chimeric antigen receptor (CAR)-modified T-cell therapy; however, the safety of this approach in terms of on-target/off-tumor effects needs to be determined. To address this issue in a clinically relevant model, we used a mouse model in which the T cells expressing CEA-specific CAR were transferred into tumor-bearing CEA-transgenic (Tg) mice that physiologically expressed CEA as a self-antigen. The adoptive transfer in conjunction with lymphodepleting and myeloablative preconditioning mediated significant tumor regression but caused weight loss in CEA-Tg, but not in wild-type mice. The weight loss was not associated with overt inflammation in the CEA-expressing gastrointestinal tract but was associated with malnutrition, reflected in elevated systemic levels of cytokines linked to anorexia, which could be controlled by the administration of an anti-IL-6 receptor monoclonal antibody without compromising efficacy. The apparent relationship between lymphodepleting and myeloablative preconditioning, efficacy, and off-tumor toxicity of CAR-T cells would necessitate the development of CEA-specific CAR-T cells with improved signaling domains that require less stringent preconditioning for their efficacy. Taken together, these results suggest that CEA-specific CAR-based adoptive T-cell therapy may be effective for patients with CEA+ solid tumors. Distinguishing the fine line between therapeutic efficacy and off-tumor toxicity would involve further modifications of CAR-T cells and preconditioning regimens.
KEYWORDS: Adoptive T-cell therapy, carcinoembryonic antigen, chimeric antigen receptor, cytokine release syndrome, neurotoxicity, preconditioning
Abbreviations
- CAR
chimeric antigen receptor
- CEA
carcinoembryonic antigen
- CRS
cytokine release syndrome
- GPI
glycosylphosphatidylinositol
- mAb
monoclonal antibody
- MHC
major histocompatibility complex
- TCR
T-cell receptor
- Tg
transgenic
- WT
wild-type
Introduction
The adoptive transfer of T cells genetically modified to express an artificial receptor consisting of the variable fragment of an antibody specific for a cell surface molecule linked to T-cell signaling molecule, termed chimeric antigen receptor (CAR), is emerging as a promising approach for cancer immunotherapy. CAR consists of a single-chain variable fragment (scFv) as an ectodomain, a short hinge, a transmembrane domain, and an endodomain with one (first generation), two (second generation), or three (third generation) signaling domains derived from CD3ζ and costimulatory molecules. CAR-T cells have advantages over T-cell receptor (TCR)-modified T cells in that they recognize tumor cells without major histocompatibility complex (MHC) restrictions, allowing for improved patient targeting and overcoming tumor escape mechanisms of MHC loss and altered peptide processing, which is an issue commonly observed in human carcinomas.1 Recent clinical trials of adoptive therapy with CAR-T cells targeting CD19 have shown an impressive efficacy in patients with hematologic malignancies,2-6 which leads to the expectations that this approach could be extended to include treatment for solid tumors. Although the efficacy of CAR-T-cell therapy for the treatment of hematological malignancies cannot be doubted, substantial risks and toxicities in response to CD19 CAR therapy include B-cell aplasia, cytokine release syndrome (CRS), and tumor lysis syndrome.7 Although B-cell aplasia as a consequence of “on-target/off-tumor” toxicity 8 has been well tolerated and is treatable with immunoglobulin replacement therapy,4 this type of toxicity may have adverse consequences if the target antigen is expressed in non-replaceable organs. Therefore, when this approach is applied to solid tumors, on-target/off-tumor toxicity should be always considered in selecting target antigens and tested in preclinical studies using appropriate animal models, wherein the target antigen should be expressed as a self-antigen and spatiotemporally similar to expression in humans.
Carcinoembryonic antigen (CEA) is a glycosylphosphatidylinositol-anchored 180-kDa glycoprotein that is highly expressed on the cell surfaces of many human tumors of epithelial origin, including carcinomas of the colon, stomach, pancreas, ovaries, and lungs.9 Although CEA is also expressed on the normal epithelial cells of pulmonary and gastrointestinal tracts, its expression is restricted to the apical surface of the epithelial cell membranes facing the lumen in normal adult tissues so that it is invisible to immune cells.10 After neoplastic transformation, luminal epithelial cells lose the apical polarity of CEA expression,11 resulting in CEA gaining access to blood capillaries resulting in increased serum levels of soluble CEA, where it can be used as an excellent marker to identify CEA+ carcinomas.12 This clear contrast of the localization pattern makes CEA a plausible tumor-associated antigen target for immunotherapy of cancer.13,14 Although a study using T cells transduced with TCR against CEA was halted after all patients developed severe transient colitis caused by the destruction of normal epithelial cells,15 it seems a logical sequence given that the non-polarized expression of MHC Class I molecules bound to CEA peptides on CEA-expressing healthy epithelial cells16 and the cross presentation by stromal cells that do not express CEA. Thus, CEA still remains a plausible target in CAR-T-cell therapy, but the on-target/off-tumor toxicity and the efficacy of CAR-T-cell therapy in a clinically relevant animal model that mimics the human situation should be tested before being applied to humans. In the present study, we addressed these issues in a clinically relevant mouse model using CEA-transgenic (CEA-Tg) mice, which closely mimics the human situation regarding the physiological expression of CEA as a self-antigen on epithelial cells of various organs including the gastrointestinal tract. Our results support the feasibility of CAR-T-cell therapy targeting CEA but also underscore potential risks of developing liver toxicity in addition to CRS.
Results
Mouse T cells transduced with CEA-specific CAR target CEA+, but not CEA−, tumors
Mouse T cells were engineered to express a CAR composed of anti-CEA scFv F11-39 in the ectodomain and CD28 and CD3ζ signaling endodomains using lentiviral vectors (Fig. 1A). Both CD4+ and CD8+ T cells expressed CAR that could bind CEA on the cell surfaces (Fig. 1B). Phenotypic analysis revealed that CAR+ T cells did not express CD127 and consisted of CD62L positive and negative cells, indicating they were mixtures of early effectors that would become memory cells and mature effectors (Fig. 1B).17 CAR-T cells also demonstrated CEA-specific lysis upon co-incubation in vitro with CEA+ MC32a tumor cells, but not with the parental CEA− MC38 tumor cells (Fig. 1C). This CEA-specific lysis by CAR-T cells was accompanied by an increase in the levels of IL-2-, IFNγ-, TNFα-, and CD107a-expressing cells (Fig. 1D).
Figure 1.

Design and characterization of a CEA-specific CAR. (A) Schematic representation of the retroviral vector encoding CEA-specific CAR and its introduction into T cells. (B) Phenotypic analysis of CEA-specific CAR-T cells. CEA-specific CARs on mouse T cells transduced using retroviral vectors were analyzed after staining with biotinylated-CEA followed by staining with anti-CD4+, CD8+, CD62L, and CD127. (C) Cytotoxic activity of CEA-specific CAR-T cells against CEA− (MC38) and CEA+ (MC32a) gastric tumor cell lines in 6-h 51Cr-release assays. (D) Cytokine production profile of CEA-specific CAR-T cells cultured with MC38 and MC32a tumor cell lines in 6-h intracellular cytokine staining assays.
Adoptive therapy with CEA-specific CAR-T cells induced tumor regression in a CEA-dependent manner
Having confirmed the functionality of CAR-T cells in vitro, we proceeded to evaluate the therapeutic potential and safety of CAR-T cells in a clinically relevant mouse model using mice transgenic for human CEA (CEA-Tg mice) that physiologically express CEA.18 CEA-Tg mice transduced with the complete gene for CEA, including the flanking regulatory elements, were fully immunocompetent and showed cell-type-specific expression of CEA, reflecting the human situation (Fig. S1A and B), which is consistent with previous studies.18,19 CEA-Tg mice bearing 7-d-old CEA+ tumors (MC32a), some of which also received lymphodepleting or myeloablative preconditioning, were adoptively transferred with CAR-T cells expressing the congenic marker CD45.1 (Fig. 2A). As shown in Fig. 2B, CAR-T cells exerted potent antitumor effects in CEA-Tg mice, as well as in WT mice, especially in those that received preconditioning. This antitumor effect was CEA-specific, since the growth of the CEA− tumor MC38, a parental cell line of MC32a, was completely unhampered by the transfer. The marginal, if any, antitumor effect of CAR-T cells seen in mice that did not receive preconditioning was closely associated with rapid disappearance of transferred CAR-T cells in the peripheral blood (Fig. 2C).
Figure 2.
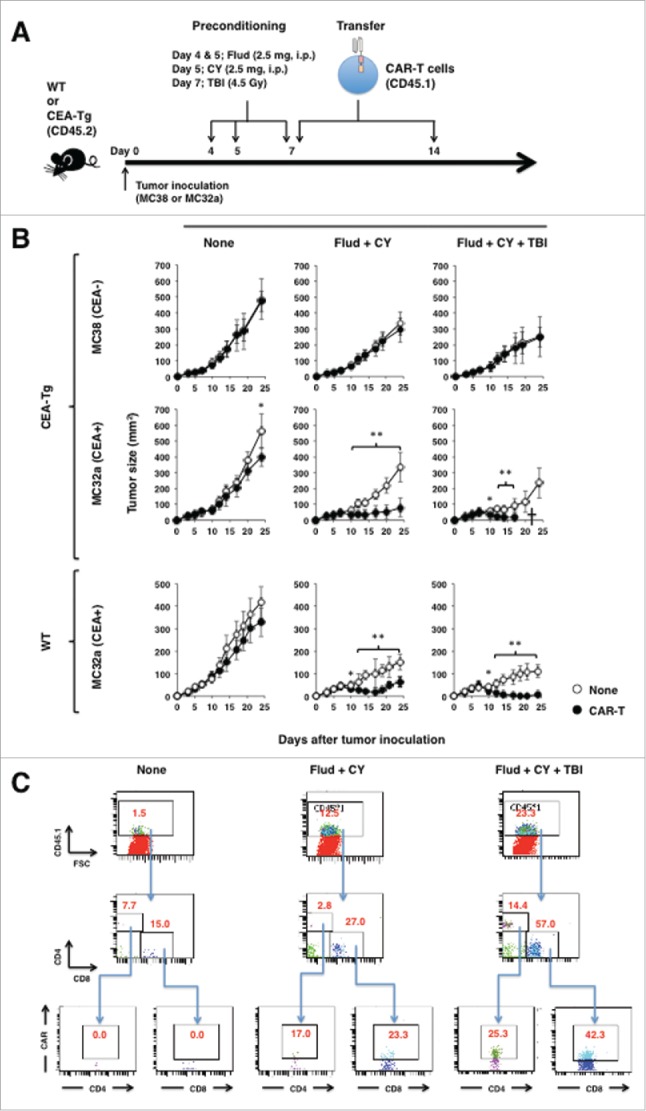
Adoptive transfer of CEA-specific CAR-T cells eradicated established tumors in an antigen-specific manner. (A) Schematic representation of the adoptive transfer experiment using CEA-specific CAR-T cells. (B) Tumor growth curves of mice transferred with CAR-T cells. WT and CEA-Tg mice bearing 7-d-old MC38 or MC32a tumors were transferred with CEA-specific CAR-T cells. Some mice also received lymphodepleting preconditioning as indicated. Tumor volumes were measured by a calliper using the formula (length × width) at the indicated time points (n = 5). *p < 0.05, **p < 0.01. (C) In vivo persistency of transferred CAR-T cell in tumor-bearing CEA-Tg mice. Peripheral blood of CEA-Tg mice as in (A) was collected at day 17 after tumor inoculation by retro-orbital bleeding, pooled (n = 5), and subjected to flow cytometry analysis.
Adoptive transfer with CEA-specific CAR-T cells induced severe weight loss without overt inflammation in the gastrointestinal tract in CEA-Tg mice, but not in WT mice
We noticed that the CEA-Tg mice, but not the WT mice, that were preconditioned and transferred with CAR-T cells showed debilitation and suffered from severe weight loss (Fig. 3A). It was possible that CAR-T cells prepared from T cells of WT mice that were not tolerant to CEA might react to CEA expressing tissues and caused weight loss. To rule out this possibility, CAR-T cells were prepared from T cells of CEA-Tg mice (Fig. S2A) and transferred into tumor-bearing CEA-Tg mice that also received fludarabine, cyclophosphamide, and total body irradiation. As shown in Fig. 3B, adoptively transferred CAR-T cells from CEA-Tg mice and WT mice induced weight loss with equal kinetics and to indistinguishable levels. The efficacy of tumor growth inhibition by these T-cell preparations was also similar (Fig. S2B). On the other hand, CEA-Tg mice transferred with mock-transduced T cells from WT mice did not show any signs of deliberation nor suffered from severe weight loss (Fig. 3B, open circle). Next we sought to determine whether the preconditioning might have allowed the transferred CAR-T cells to access normal cells expressing CEA and induced inflammation. Hematoxylin and eosin (H&E) staining revealed that there was inflammation in the lungs of CEA-Tg and WT mice to which CAR-T cells had been transferred, but the severities were comparable to each other (Fig. 4A). On the other hand, there was no overt inflammation in the tissues other than the lungs that expressed CEA. We went on to further determine whether the transferred CAR-T cells infiltrated into those tissues in a manner dependent on CEA expression. Because we could not detect CAR-T cells by using biotinylated-CEA possibly due to loss of binding ability of CAR to CEA upon tissue fixation, CD45.1+ cells, about 27% of which express CAR (Fig. 2B), were used as a surrogate for CAR-T cells infiltrating into the corresponding tissues. This was indeed the case in that the CAR-T cells infiltrated into the lungs, small intestines, and large intestines of CEA-Tg and, to a lesser extent, WT mice that had received fludarabine, cyclophosphamide, and total body irradiation (Fig. 4B). However, the degree of infiltration did not correlate with the severity of inflammation and there were no signs of tissue destruction.
Figure 3.
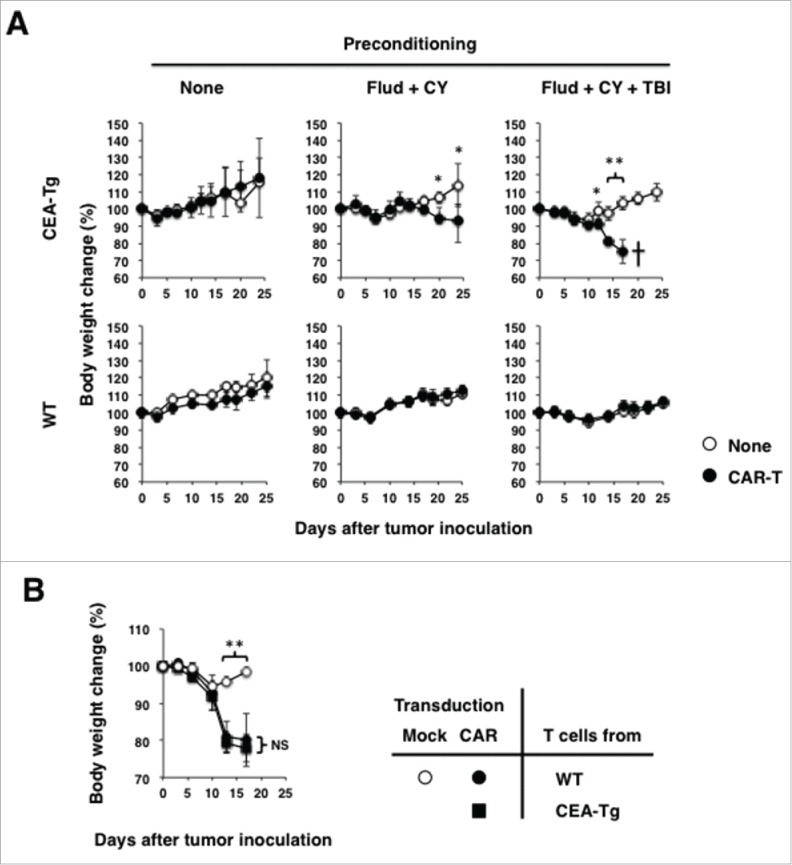
Increased efficacy of tumor growth suppression by CEA-specific CAR-T cells in preconditioned mice is associated with severe weight loss and eventual death of CEA-Tg mice, but not WT mice. (A) Body weight changes of CEA-Tg and WT mice from Fig. 2 were analyzed at the indicated time points (n = 5). (B) CAR-T cells prepared from CEA-Tg and WT mice and those mock-transduced T cells from WT mice were transferred into MC32a tumor-bearing CEA-Tg mice that also received fludarabine, cyclophosphamide, and total body irradiation. Body weight changes of CEA-Tg were analyzed at indicated time points. *p < 0.05, **p < 0.01.
Figure 4.
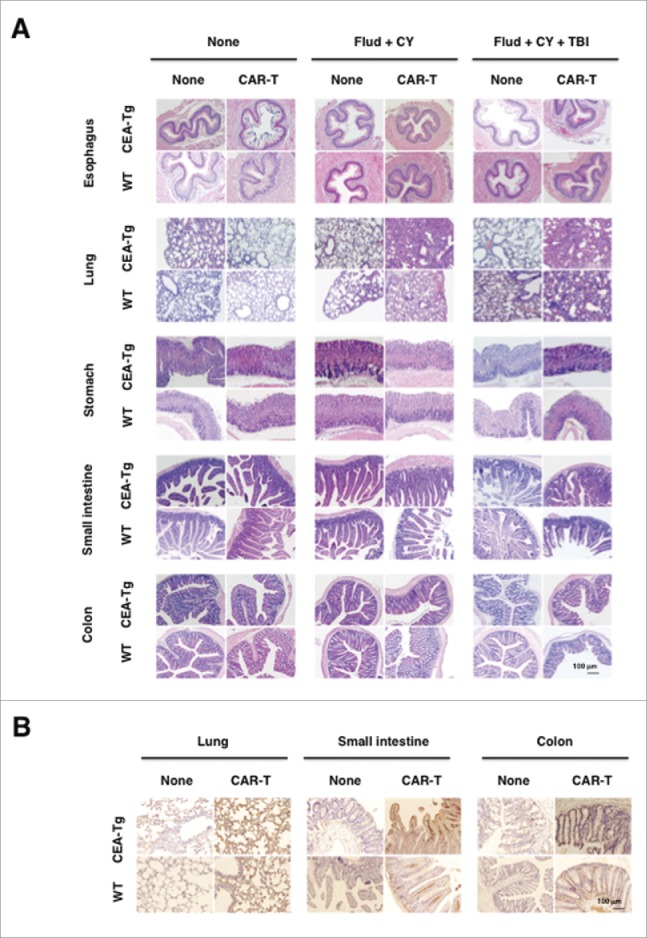
Severe weight loss and the eventual death of CEA-Tg mice transferred with CEA-specific CAR-T cells were not associated with inflammation in CEA-expressing tissues. Cryostat sections of organs collected from mice as in Fig. 2A at 17 day after tumor inoculation were subjected to H&E staining (A) and CD45.1 immunohistochemistry (B).
Anorexia induced by increased levels of pro-inflammatory cytokines caused weight loss in CEA-Tg mice, but not in WT mice, treated with CAR-T cells
Adoptive cell therapy using CAR-modified T cells is often associated with CRS,20 which is closely linked to sickness behaviors including anorexia that leads to weight loss.21 Because the CEA-Tg mice that received myeloablative preconditioning suffered from the most severe weight loss that led eventual death, the sera of this group of mice were subjected to cytokine bead arrays in order to explore the underlying mechanisms. As shown in Fig. 5A, serum levels of IL-1α, IL-6, and TNFα, most of which have been shown to cause anorexia in humans as well as in mice,22-24 significantly increased in CEA-Tg mice, but not in WT mice. Furthermore, this was associated with anorexia and malnutrition as judged by food consumption and serum levels of total protein, albumin, and glucose (Fig. 5B and C). Although both WT and CEA-Tg mice exhibited anorexia up until day 10, which was possibly caused by preconditioning, the WT mice recovered from anorexia thereafter, whereas the CEA-Tg mice continued to suffer from anorexia.
Figure 5.

Weight loss seen in preconditioned CEA-Tg mice transferred with CEA-specific CAR-T cells was caused by anorexia. (A) Increased serum levels of cytokines associated with anorexia. Pooled serum samples from mice (n = 5) were collected at day 17 after tumor inoculation and subjected to analysis by cytokine beads arrays. (B) Reduced diet consumption of preconditioned CEA-Tg mice transferred with CEA-specific CAR-T cells. Diet consumption of CEA-Tg and WT mice were measured at the indicated time points and the measured quantity was divided by the number of mice to determine the quantity of consumed food by each mouse. (C) CEA-Tg mice, but not WT mice, suffered from malnutrition and liver function of CEA-Tg and WT mice. Pooled serum samples (n = 5) were subjected to serum chemistry analysis.
CEA-Tg mice partially recovered from anorexia by administration of anti-IL-6 receptor antibody and escaped from eventual death without compromising the efficacy of CAR-T-cell therapy
Clinical trials of adoptive T-cell therapy with CAR-modified T cells targeting hematological malignancies revealed that the humanized anti-IL-6 receptor (anti-IL-6R) monoclonal antibody, tocilizumab, can successfully control CRS without compromising the efficacy of adoptive T-cell therapy.25 Therefore, we next sought to determine whether the administration of anti-IL-6R antibody is effective in controlling anorexia in CEA-Tg mice bearing solid tumors. We treated CEA-Tg mice with the anti-IL-6R monoclonal antibody starting on day 10, when the WT mice recovered from anorexia. This treatment resulted in an increase in food consumption (Fig. 6A) and the correction of malnutrition (Fig. 6B), which was associated with a partial recovery from weight loss (Fig. 6C) and completely escaped from eventual death. Moreover, the efficacy of CAR-T cells against CEA+ tumors was unaffected by the administration (Fig. 6D).
Figure 6.

Administration of anti-IL-6R antibody successfully controlled cytokine releasing syndrome-like symptoms caused by on-target/off-tumor toxicity of CEA-specific CAR-T cells without compromising the antitumor efficacy. CEA-Tg mice bearing 7-d-old MC32a tumors that received myeloablative preconditioning and CAR-T cells were treated with the anti-IL-6R antibody (MR16-1) weekly starting on day 10. (A) Recovery from anorexia of CEA-Tg mice treated with the anti-IL-6R antibody. Diet consumption of CEA-Tg mice treated with MR16-1 or control rat IgG was measured at the indicated time points and the quantity of consumed food by each mouse was obtained as in Fig. 5B. (B) Recovery from malnutrition, but not liver function, of CEA-Tg mice treated with the anti-IL-6R antibody. Pooled serum samples collected from mice (n = 5) at day 17 after tumor inoculation were subjected to serum chemistry analysis. (C) Recovery from body weight loss in CEA-Tg mice treated with the anti-IL-6R antibody. Body weight changes of CEA-Tg mice treated with MR16-1 or control rat IgG were analyzed at the indicated time points (n = 5). *p < 0.05, **p < 0.01. D: Treatment of CEA-Tg mice with the anti-IL-6R antibody did not compromise the antitumor efficacy of CAR-T cells. Tumor growth curves of CEA-Tg mice treated with CAR-T cells together with or without anti-IL-6R (n = 5) are shown. *p < 0.05, **p < 0.01.
Ectopic expression of CEA in the liver
We noticed that the CEA-Tg mice that received preconditioning and were transferred with CAR-T cells displayed abnormal liver functions (Fig. 5C). It has been shown that CEA was expressed in the apical cytoplasm and along the luminal surface of bile duct epithelial cells in normal human liver tissues, indicating that CEA accumulates in and is excreted by bile ducts.26 Accordingly, we observed that the livers of CEA-Tg mice expressed CEA not restricted to bile duct epithelial cells but interspersed with the stroma (Fig. S3A). This was associated with CAR-T cell infiltration (Fig. S3B) and focal inflammatory cell infiltration (Fig. S3C).
Discussion
Recent clinical trials of adoptive cell therapy using CAR-modified T cells targeting CD19 have shown remarkable efficacy in patients with hematologic malignancies. 2-6 In contrast, the effect of CAR-T cells on solid tumors has been demonstrated to be limited, 27-30 with one clinical trial using first-generation CAR-T cells specific for GD2 in patients with neuroblastoma and showing prolonged complete remission.31 In addition to CRS, which is often associated with the clinical efficacy of CAR-T cell therapy for hematologic malignancies,20 two clinical trials with CAR-T cells targeting solid tumors have revealed considerable toxicity toward normal tissues; 28,32 the so-called on-target/off-tumor toxicity may have dreadful consequences when the target antigen is expressed in non-replaceable organs. In this regard, studying the efficacy and safety of CAR-T cells in animal models is still imperative for the validation of CAR constructs under therapeutic settings in the treatment of solid tumors. Many studies performed in immunodeficient mice bearing human cancer xenografts treated with human CAR-T cells,33-35 or in immunocompetent mice bearing syngeneic tumor cells ectopically expressing human tumor associated antigens treated with mouse CAR-T cells,36-38 have demonstrated the potential benefits of the CAR-T-cell approach. However, the contribution of the xenogeneic incompatibility in the former and immunogenicity of human antigen to which the hosts are non-tolerant in the latter may unfaithfully reflect the ability of CAR-T cells to control tumor growth. Moreover, these models hardly addressed the safety issues. In the present study, we overcame these disadvantages and reproduced the human situation by using a CEA-Tg mouse that expressed CEA as a self-antigen in a manner similar to the human counterpart 18 and was tolerant to CEA,39 into which mouse T cells transduced with second-generation CAR consisting of CEA-specific scFv in the ectodomain, and CD28 and CD3ζ in the signaling endodomain were adoptively transferred.
Systematic comparison of the various trials is difficult due to differences in the constructs and vectors of the CAR and the dose of CAR-T cells. However, a consensus view has been reached that lymphodepletion or myeloablation prior to CAR-T cell infusion is mandatory to shape a desirable environment for the adoptively transferred T cells.40 In supporting this notion, our results also demonstrated that the adoptive transfer of CEA-specific CAR-T cells in conjunction with, but not without, these preconditioning manipulations mediated significant regression of CEA+, but not CEA−, tumors in CEA-Tg mice. As expected, this was associated with the persistence of the adoptively transferred CAR-T cells, which is a key factor for efficient antitumor activity.31,41,42
Although CEA-specific CAR-T cells exerted potent antitumor activities toward CEA+ tumors with equal efficiency in CEA-Tg and WT mice, only CEA-Tg mice suffered from severe weight loss that led to eventual death. Our results that adoptive transfer of CAR-T cells prepared from T cells from CEA-Tg mice as well as WT mice that were non-tolerant to CEA equally induced weight loss in CEA-Tg mice clearly indicate that the specificity of CAR was responsible for this adverse effect. Therefore, this weight loss may be due to targeting healthy tissues that express CEA by the adoptively transferred CAR-T cells, since the CEA-Tg mice used in this study expressed CEA in a manner that was parallel to the human situation, i.e., in normal epithelial cells of the lungs and gastrointestinal tract with polarized expression at the luminal side and prominent expression on highly differentiated epithelial cells in upper colonic crypts. Indeed, immunohistochemical analysis revealed that the adoptively transferred CAR-T cells infiltrated into the lungs, small intestines, and colons of CEA-Tg mice. However, H&E staining revealed that these tissues were of healthy morphology without signs of extensive inflammation. The infiltration of adoptively transferred CAR-T cells seemed to have been caused by preconditioning, which led to antigen-nonspecific infiltration of CAR-T cells into those tissues, since the WT mice also exhibited such infiltration to a slightly lesser extent. It was possible that underlying mechanisms by which CAR-T cells infiltrate into tissues include specificity of CAR to CEA, reactivity of intrinsic TCR to CEA, and non-specificity to CEA. Nevertheless, those CAR-T cells that infiltrated into CEA+ tissues in CEA-Tg mice and CEA− tissues in WT mice showed no indication of extensive activation. We showed that the weight loss was due to, at least partly, malnutrition caused by anorexia as judged by food consumption and serum chemistry analysis. Serum levels of cytokines linked to anorexia were increased in CEA-Tg mice transferred with CAR-T cells, reminiscent of CRS seen in patients treated with CD19-CAR.20 Since the transferred CAR-T cells did not induce CRS-like symptoms in WT mice bearing CEA+ tumors undergoing tumor destruction, a causal relationship between CAR-T cells and tumors seemed unlikely. It is possible that preconditioning may have allowed CAR-T cells to infiltrate into various tissues and, in the case of CEA-Tg mice, react with CEA on healthy tissues to express CD40L and activate local macrophages to produce inflammatory cytokines including IL-6. In line with this, the treatment of anti-IL-6R antibody resulted in an increase in food consumption and the correction of malnutrition, which was associated with recovery from weight loss and complete escape from eventual death without compromising the efficacy of CAR-T cells in killing CEA+ tumors.
Our results are partially inconsistent with those of previous studies using the same CEA-Tg mice, which did not suffer from any weight loss upon adoptive transfer with CAR-T cells after non-myeloablative lymphodepletion preconditioning,43,44 whereas we repeatedly observed weight loss in the CEA-Tg mice receiving lymphodepletion and CAR-T cells, albeit of a less severe nature than was observed in the CEA-Tg mice receiving myeloablation and CAR-T cells. In the former study, the authors used SCA431 CAR consisting of CD3ζ and CD28 signaling domains similar to our F39-11 CAR but with a moderate affinity to CEA (SCA431: 37 nM vs. F39-11: 0.55 nM). It is possible that our high affinity CAR reduced the activation threshold of T cells, manifesting adverse impacts. However, there seemed to be no causal relationship between weight loss and colitis, and our high affinity CAR-T cells did not induce overt inflammation in the colonic tissues of CEA-Tg mice receiving myeloablative preconditioning. On the other hand, CD4+ T cells expressing SCA431 CAR induced severe colitis upon adoptive transfer into different CEA-Tg mice with CEA-expression levels equivalent to those of humans (i.e., lower than our CEA-Tg mice)45 after 5-Gy total-body irradiation.46 Although the reasons for these discrepancies remain unknown, they could involve the differences in populations of commensal bacteria found in mice housed in different institutions. It appears that increasing the strength of preconditioning manifested on-target/off-tumor toxicity irrespective of affinities of CAR to CEA.
We found that CEA-Tg mice expressed CEA in the livers, albeit at a very low level as compared to in the colons. This ectopic expression of CEA seen in livers of CEA-Tg mice did not seem to be an artifact of transgene insertion, since low but non-negligible levels of CEA mRNA were detected in human livers, and bile duct epithelial cells have been shown to express CEA.26 The expression of CEA in the liver raises a concern about the risks of liver toxicity. Accordingly, in association with the infiltration of CAR-T cells into the liver, CEA-Tg mice transferred with CAR-T cells exhibited increased levels of aspartate aminotransferase, alanine transaminase, and lactate dehydrogenase, and decreased levels of choline esterase in the serum, which was never reported in preclinical mouse models.43,44 It is noteworthy that treatment with the anti-IL-6R antibody failed to correct the liver dysfunction induced by CAR-T cells. It remains possible that these toxicities seen in CEA-Tg mice may be an overestimation in that the expression levels of CEA in CEA-Tg mice used in the present study are 3- to 10-fold higher than in healthy humans; 18 however, those adverse events shown in the present study need to be watched when applying CEA-specific CAR-T-cell therapy in clinic.
The CAR-T cells used in our and other studies favor lymphodepleting/myeloablative preconditioning for prolonged in vivo persistency and clinical efficacy.31,41,42 The apparent relationship between the preconditioning, efficacy, and off-tumor toxicity of CAR-T cells would necessitate the development of CEA-specific CAR-T cells with improved signaling domains that do not require preconditioning for their efficacy, given the type of signals delivered by CAR greatly affects the in vivo persistency and resistance of CAR-T cells to immunosuppression by tumors. Taken as a whole, these results suggest that CEA-specific CAR-based adoptive T-cell therapy may be effective for patients with CEA+ solid tumors. Distinguishing the fine line between therapeutic efficacy and off-tumor toxicity would include further modifications of CAR-T cells and preconditioning regimens.
Materials and methods
Animals
C57BL/6 mice obtained from Japan SLC, Inc., C57BL/6 (CD45.1) congenic mice (in house), and CEA transgenic mice (C57BL/6 background; kindly provided by Dr. W. Zimmermann18) were fed with a standard diet, housed under specific pathogen free conditions, and used at 5–8 weeks of age. All animal experiments were conducted under protocols approved by the Animal Care and Use Committee of Mie University Life Science Center.
Antibodies and reagents
The following antibodies were used for cell surface and intracellular stainings: APC-conjugated anti-CD8+ (53–6.7), PerCP-Cy5.5-conjugated anti-CD4+ (RM4-5), and APC-Cy7-conjugated anti-TNFα (MP6-XT22) were purchased from BD Biosciences; PE-conjugated-anti-IL-2 (JES6-5H4), PE-CD107a-conjugated (1D4B), and PE-Cy7-conjugated anti-CD45.1 (A20) were purchased from BioLegend; FITC-conjugated-anti-IFNγ (XMG1.2) was purchased from eBioscience. A rat anti-mouse IL-6 receptor mAb (MR16-1) was kindly provided by Chugai Pharmaceutical Co.
Cell lines
Murine colon carcinoma MC38 cells and MC38 cells expressing human CEA (designated MC32a) by retroviral transduction with CEA cDNA were kindly provided by Dr. Greiner.47 These cell lines were cultured in RPMI-1640 supplemented with 10% FCS, 5 × 10−5 M 2-mercaptoethanol, 100-μg/mL streptomycin, and 100-U/mL penicillin.
Vector construction and preparation of virus solutions
A scFv of monoclonal antibody (mAb) F39-11 specific to CEA in the VL-VH orientation,48 along with a CD8α hinge, CD28 transmembrane domain, plus CD3ζ and CD28 signaling domains were cloned into a pMS3 retroviral vector.49 The murine stem cell virus LTR was used to drive CAR expression. The vesicular stomatitis virus G-pseudotyped (VSV-G) retroviruses were transiently obtained by conventional methods using G3T-hi cells (Takara Bio, Shiga, Japan). The GP+E86 cells (ATCC® CRL 9642TM) were transduced with transiently produced VSV-G retroviruses to produce ecotropic retroviruses.
Generation of CAR-T cells
Whole spleen cells (1.5 × 107 cells/5 mL) from C57BL/6 (CD45.1) congeneic mice were stimulated with immobilized anti-CD3 (1 µg/mL; 145-2C11) and soluble anti-CD28 (1 µg/mL; 37.51) in one well of a six-well plate. One day after the stimulation (on day 1), 5 × 105 cells were transduced with the viral vector by using the RetroNectin-bound virus infection method, wherein virus solutions were preloaded onto RetroNectin (Takara Bio)-coated wells of a 24-well plate containing 1-mL culture medium as described.50 On day 3, the cells were transferred to a 50-mL flask containing 10-mL culture medium for expansion. On day 5, the cells were harvested and used for experiments. Recombinant human IL-2 (Novartis) at 60 IU/mL was added during culturing. In some experiments, CAR-T cells were prepared using T cells from CEA-Tg mice. Recombinant CEA (Abnova) was biotinylated using the Biotin Labeling Kit (Dojindo) according to the manufacturer protocol and, together with anti-CD4+, anti-CD8+, and anti-CD45.1, used for the detection of CAR on CD4+/CD8+ T cells.
In vitro functional assays
Cytotoxicity was analyzed by standard chromium release assays as previously described.51 Briefly, CAR-T cells (1 × 105–3 × 106 cells/0.2 mL/well) were co-cultured with tumor cells (1 × 105 cells/0.2 mL/well) for 6 h. Cytokine production was analyzed using intracellular cytokine flow cytometry as previously described.52 Briefly, CAR-T cells (1 × 105 cells/0.2 mL/well) were co-cultured with tumor cells (5 × 104 cells/0.2 mL/well) for 5 h. In both cases, antigen-specific activation of CAR-T cells was induced using MC32a tumor cells, with MC38 tumor cells serving as the negative control.
Immunohistochemical staining
Paraffin-embedded sections of mouse tissues fixed with 10% buffered formalin were subjected to H&E staining, and immunohistochemical staining was performed according to the standard avidin–biotin immunoperoxidase complex technique (VECTASTAIN Elite ABC kit PK-6102, Vectastain Laboratories, Inc. Burlingame, CA, USA). Mouse monoclonal anti-CEA/CD66e (CB30; Cell Signaling Technology) or CD45.1 (A20, Abcam ab25078) was used as the primary antibodies and developed with DAB.
Tumor challenge and treatment
CEA-Tg mice and WT mice were injected subcutaneously with 2.5 × 106 MC32a or MC38 tumor cells in 200-µL PBS. Tumor-bearing mice were randomly assigned to three groups that received CAR-T cells only, lymphodepleting preconditioning (intraperitoneal injection of 2.5-mg fludarabine on days 4 and 5, and 2.5-mg cyclophosphamide on day 5), or myeloablative preconditioning (lymphodepleting preconditioning plus 4.5-Gy total body irradiation under anesthesia with a lead shield around the tumor on day 7 before adaptive transfer with CAR-T cells). On days 7 and 14, those mice were injected with 5 × 106 CAR-T cells in 200-µL PBS or PBS alone intravenously. In some experiments, tumor-bearing mice that received CAR-T cells were also injected with 0.5-mg anti-IL-6R antibody (MR16-1) or control rat IgG intravenously once a week starting on day 10. Tumor growth was monitored twice a week, and tumor size was determined as the mean length of two right-angled diameters measured using microcallipers. Mice were euthanized for humane reasons when tumors grew to 40 mm in the longest diameter.
Serum measurements
Mice were anaesthetized with isoflurane and peripheral blood samples were harvested from the orbital plexus. Serum was obtained by centrifugation, separated into two aliquots, and each of them stored at −80°C until use. One aliquot was shipped to the WAKO Pure Chemical Industries (Tokyo, Japan) for serum chemistry analysis. The other aliquot was subjected to cytokine bead arrays (Bio-Plex, Multiplex Immunoassay, Bio-Rad Laboratories).
Statistics
Data are presented as means ± SEM where error bars are shown. Statistical analysis was performed by unpaired two-tailed Student's t-tests using Microsoft Excel. p values of less than 0.05 were considered statistically significant. All experiments were conducted at least three times and one of the representative results is shown.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by JSPS KAKENHI Grant Numbers 25670553 to TK and 24591899 to LW.
References
- 1.Vitale M, Pelusi G, Taroni B, Gobbi G, Micheloni C, Rezzani R, Donato F, Wang X, Ferrone S. HLA class I antigen down-regulation in primary ovary carcinoma lesions: association with disease stage. Clin Cancer Res 2005; 11:67-72; PMID:15671529 [PubMed] [Google Scholar]
- 2.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M et al.. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra25; PMID:24553386; http://dx.doi.org/ 10.1126/scitranslmed.3008226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM et al.. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015; 33:540-9; PMID:25154820; http://dx.doi.org/ 10.1200/JCO.2014.56.2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al.. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Eng J Med 2014; 371:1507-17; PMID:25317870; http://dx.doi.org/21832238 10.1056/NEJMoa1407222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3:95ra73; PMID:21832238; http://dx.doi.org/ 10.1126/scitranslmed.3002842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN et al.. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385:517-28; PMID:25319501; http://dx.doi.org/ 10.1016/S0140-6736(14)61403-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF et al.. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Eng J Med 2013; 368:1509-18; PMID:23527958; http://dx.doi.org/25488419 10.1056/NEJMoa1215134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casucci M, Hawkins RE, Dotti G, Bondanza A. Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother 2015; 64:123-30; PMID:25488419; http://dx.doi.org/ 10.1007/s00262-014-1641-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fichera A, Michelassi F, Arenas RB. Selective expression of carcinoembryonic antigen promoter in cancer cell lines: targeting strategy for gene therapy in colorectal cancer. Dis Colon Rectum 1998; 41:747-54; PMID:9645743; http://dx.doi.org/ 10.1007/BF02236263 [DOI] [PubMed] [Google Scholar]
- 10.Nap M, Mollgard K, Burtin P, Fleuren GJ. Immunohistochemistry of carcino-embryonic antigen in the embryo, fetus and adult. Tumour Biol 1988; 9:145-53; PMID:3399813; http://dx.doi.org/ 10.1159/000217555 [DOI] [PubMed] [Google Scholar]
- 11.Yan Z, Deng X, Chen M, Xu Y, Ahram M, Sloane BF, Friedman E. Oncogenic c-Ki-ras but not oncogenic c-Ha-ras up-regulates CEA expression and disrupts basolateral polarity in colon epithelial cells. J Biol Chem 1997; 272:27902-7; PMID:9346938; http://dx.doi.org/ 10.1074/jbc.272.44.27902 [DOI] [PubMed] [Google Scholar]
- 12.Beauchemin N, Arabzadeh A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Met Rev 2013; 32:643-71; PMID:23903773; http://dx.doi.org/10632322 10.1007/s10555-013-9444-6 [DOI] [PubMed] [Google Scholar]
- 13.Nolan KF, Yun CO, Akamatsu Y, Murphy JC, Leung SO, Beecham EJ, Junghans RP. Bypassing immunization: optimized design of “designer T cells” against carcinoembryonic antigen (CEA)-expressing tumors, and lack of suppression by soluble CEA. Clin Cancer Res 1999; 5:3928-41; PMID:10632322 [PubMed] [Google Scholar]
- 14.Emtage PC, Lo AS, Gomes EM, Liu DL, Gonzalo-Daganzo RM, Junghans RP. Second-generation anti-carcinoembryonic antigen designer T cells resist activation-induced cell death, proliferate on tumor contact, secrete cytokines, and exhibit superior antitumor activity in vivo: a preclinical evaluation. Clin Cancer Res 2008; 14:8112-22; PMID:19088026; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-4910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM et al.. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011; 19:620-6; PMID:21157437; http://dx.doi.org/ 10.1038/mt.2010.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bos R, van Duikeren S, Morreau H, Franken K, Schumacher TN, Haanen JB, van der Burg SH, Melief CJ, Offringa R. Balancing between antitumor efficacy and autoimmune pathology in T-cell-mediated targeting of carcinoembryonic antigen. Cancer Res 2008; 68:8446-55; PMID:18922918; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1864 [DOI] [PubMed] [Google Scholar]
- 17.Opata MM, Stephens R. Early decision: effector and effector memory T cell differentiation in chronic infection. Curr Immunol Rev 2013; 9:190-206; PMID:24790593; http://dx.doi.org/ 10.2174/1573395509666131126231209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eades-Perner AM, van der Putten H, Hirth A, Thompson J, Neumaier M, von Kleist S, Zimmermann W. Mice transgenic for the human carcinoembryonic antigen gene maintain its spatiotemporal expression pattern. Cancer Res 1994; 54:4169-76; PMID:8033149 [PubMed] [Google Scholar]
- 19.Thompson JA, Eades-Perner AM, Ditter M, Muller WJ, Zimmermann W. Expression of transgenic carcinoembryonic antigen (CEA) in tumor-prone mice: an animal model for CEA-directed tumor immunotherapy. Int J Cancer J Int Du Cancer 1997; 72:197-202; PMID:9212243; http://dx.doi.org/25999455 10.1002/(SICI)1097-0215(19970703)72:1%3c197::AID-IJC28%3e3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
- 20.Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015; 125:4017-23; PMID:25999455; http://dx.doi.org/ 10.1182/blood-2014-12-580068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 2002; 25:154-9; PMID:11852148; http://dx.doi.org/ 10.1016/S0166-2236(00)02088-9 [DOI] [PubMed] [Google Scholar]
- 22.Solmi M, Veronese N, Favaro A, Santonastaso P, Manzato E, Sergi G, Correll CU. Inflammatory cytokines and anorexia nervosa: A meta-analysis of cross-sectional and longitudinal studies. Psychoneuroendocrinol 2015; 51:237-52; PMID:25462897; http://dx.doi.org/24963216 10.1016/j.psyneuen.2014.09.031 [DOI] [PubMed] [Google Scholar]
- 23.Stella J, Croney C, Buffington T. Effects of stressors on the behavior and physiology of domestic cats. Appl Anim Behav Sci 2013; 143:157-63; PMID:25210211; http://dx.doi.org/24963216 10.1016/j.applanim.2012.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi YK, Jung KY, Woo S-M, Yun YJ, Jun C-Y, Park JH, Shin YC, Cho SG, Ko SG. Effect of Sipjeondaebo-tang on cancer-induced anorexia and cachexia in CT-26 tumor-bearing mice. Mediators Inflamm 2014; 2014:736563; PMID:24963216; http://dx.doi.org/ 10.1155/2014/736563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J 2014; 20:119-22; PMID:24667956; http://dx.doi.org/ 10.1097/PPO.0000000000000035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerber MA, Thung SN. Carcinoembryonic antigen in normal and diseased liver tissue. Am J Pathol 1978; 92:671-9; PMID:356625 [PMC free article] [PubMed] [Google Scholar]
- 27.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR et al.. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 2007; 15:825-33; PMID:17299405; http://dx.doi.org/ 10.1038/sj.mt.6300104 [DOI] [PubMed] [Google Scholar]
- 28.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R et al.. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 2013; 21:904-12; PMID:23423337; http://dx.doi.org/ 10.1038/mt.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006; 24:e20-2; PMID:16648493; http://dx.doi.org/ 10.1200/JCO.2006.05.9964 [DOI] [PubMed] [Google Scholar]
- 30.Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, Khare PD, Thorn M, Ma Q, Stainken BF et al.. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res 2015; 21:3149-59; PMID:25850950; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Khare PD, Thorn M, Ma Q, Stainken BF et al.. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011; 118:6050-6; PMID:21984804; http://dx.doi.org/ 10.1182/blood-2011-05-354449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2009; 18:843-51; PMID:20179677; http://dx.doi.org/25941351 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, Newick K, Sun J, Lo A, Baradet T. et al.. Generation of Potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol Res 2015; 3:815-26; PMID:25941351; http://dx.doi.org/ 10.1158/2326-6066.CIR-15-0054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 2015; 21:524-9; PMID:25849134; http://dx.doi.org/ 10.1038/nm.3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hillerdal V, Ramachandran M, Leja J, Essand M. Systemic treatment with CAR-engineered T cells against PSCA delays subcutaneous tumor growth and prolongs survival of mice. BMC Cancer 2014; 14:30; PMID:24438073; http://dx.doi.org/ 10.1186/1471-2407-14-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, Rosenberg SA. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res 2012; 18:1672-83; PMID:22291136; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kofler DM, Chmielewski M, Rappl G, Hombach A, Riet T, Schmidt A, Hombach AA, Wendtner CM, Abken H. CD28 costimulation impairs the efficacy of a redirected T-cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing lck activatio. Mol Ther 2011; 19:760-7; PMID:21326215; http://dx.doi.org/ 10.1038/mt.2011.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, Feldman SA, Restifo NP, Rosenberg SA. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest 2010; 120:3953-68; PMID:20978347; http://dx.doi.org/ 10.1172/JCI43490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saha A, Chatterjee SK, Foon KA, Primus FJ, Sreedharan S, Mohanty K, Bhattacharya-Chatterjee M. Dendritic cells pulsed with an anti-idiotype antibody mimicking carcinoembryonic antigen (CEA) can reverse immunological tolerance to CEA and induce antitumor immunity in CEA transgenic mice. Cancer Res 2004; 64:4995-5003; PMID:15256474; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-0626 [DOI] [PubMed] [Google Scholar]
- 40.Barrett DM, Grupp SA, June CH. Chimeric antigen receptor- and TCR-modified T Cells enter main street and wall street. J Immunol 2015; 195:755-61; PMID:26188068; http://dx.doi.org/ 10.4049/jimmunol.1500751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G et al.. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17:1453-64; PMID:19384291; http://dx.doi.org/ 10.1038/mt.2009.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berry LJ, Moeller M, Darcy PK. Adoptive immunotherapy for cancer: the next generation of gene-engineered immune cells. Tissue Antigens 2009; 74:277-89; PMID:19775368; http://dx.doi.org/ 10.1111/j.1399-0039.2009.01336.x [DOI] [PubMed] [Google Scholar]
- 43.Chmielewski M, Hahn O, Rappl G, Nowak M, Schmidt-Wolf IH, Hombach AA, Abken H. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterol 2012; 143:1095-107.e2; PMID:22750462; http://dx.doi.org/22378346 10.1053/j.gastro.2012.06.037 [DOI] [PubMed] [Google Scholar]
- 44.Chmielewski M, Rappl G, Hombach AA, Abken H. T cells redirected by a CD3ζ chimeric antigen receptor can establish self-antigen-specific tumour protection in the long term. Gene Ther 2013; 20:177-86; PMID:22378346; http://dx.doi.org/ 10.1038/gt.2012.21 [DOI] [PubMed] [Google Scholar]
- 45.Chan CH, Stanners CP. Novel mouse model for carcinoembryonic antigen-based therapy. Mol Ther 2004; 9:775-85; PMID:15194045; http://dx.doi.org/ 10.1016/j.ymthe.2004.03.009 [DOI] [PubMed] [Google Scholar]
- 46.Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther 2014; 22:1018-28; PMID:24686242; http://dx.doi.org/ 10.1038/mt.2014.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaharoff DA, Hance KW, Rogers CJ, Schlom J, Greiner JW. Intratumoral immunotherapy of established solid tumors with chitosan/IL-12. J Immunother 2010; 33:697-705; PMID:20664357; http://dx.doi.org/ 10.1097/CJI.0b013e3181eb826d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuroki M, Hachimine K, Huang J, Shibaguchi H, Kinugasa T, Maekawa S, Kuroki M. Re-targeting of cytotoxic T lymphocytes and/or natural killer cells to CEA-expressing tumor cells with anti-CEA antibody activity. Anticancer Res 2005; 25:3725-32; PMID:16302732 [PubMed] [Google Scholar]
- 49.Okamoto S, Amaishi Y, Goto Y, Ikeda H, Fujiwara H, Kuzushima K, Yasukawa M, Shiku H, Mineno J. A promising vector for TCR gene therapy: differential effect of siRNA, 2A Peptide, and Disulfide Bond on the introduced TCR expression. Mol Ther Nucleic Acids 2012; 1:e63; PMID:23250361; http://dx.doi.org/ 10.1038/mtna.2012.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwamura K, Kato T, Miyahara Y, Naota H, Mineno J, Ikeda H, Shiku H. siRNA-mediated silencing of PD-1 ligands enhances tumor-specific human T-cell effector functions. Gene Ther 2012; 19:959-66; PMID:22113316; http://dx.doi.org/ 10.1038/gt.2011.185 [DOI] [PubMed] [Google Scholar]
- 51.Toda M, Wang L, Ogura S, Torii M, Kurachi M, Kakimi K, Nishikawa H, Matsushima K, Shiku H, Kuribayashi K et al.. UV irradiation of immunized mice induces type 1 regulatory T cells that suppress tumor antigen specific cytotoxic T lymphocyte responses. Int J Cancer 2011; 129:1126-36; PMID:21710495; http://dx.doi.org/ 10.1002/ijc.25775 [DOI] [PubMed] [Google Scholar]
- 52.Torii M, Wang L, Ma N, Saito K, Hori T, Sato-Ueshima M, Koyama Y, Nishikawa H, Katayama N, Mizoguchi A et al.. Thioredoxin suppresses airway inflammation independently of systemic Th1/Th2 immune modulation. Eur J Immunol 2010; 40:787-96; PMID:20017193; http://dx.doi.org/ 10.1002/eji.200939724 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.