

ABSTRACT
Adoptive immunotherapy for cancer treatment is an emerging field of study. Till now, several tumor-derived, peptide-specific T cell responses have been harnessed for treating cancers. However, the contribution of lipid-specific T cells in tumor immunity has been understudied. CD1 molecules, which present self- and foreign lipid antigens to T cells, are divided into group 1 (CD1a, CD1b, and CD1c) and group 2 (CD1d). Although the role of CD1d-restricted natural killer T cells (NKT) in several tumor models has been well established, the contribution of group 1 CD1-restricted T cells in tumor immunity remains obscure due to the lack of group 1 CD1 expression in mice. In this study, we used a double transgenic mouse model expressing human group 1 CD1 molecules (hCD1Tg) and a CD1b-restricted, self-lipid reactive T cell receptor (HJ1Tg) to study the potential role of group 1 CD1-restricted autoreactive T cells in antitumor response. We found that HJ1 T cells recognized phospholipids and responded more potently to lipid extracted from tumor cells than the equivalent amount of lipids extracted from normal cells. Additionally, the autoreactivity of HJ1 T cells was enhanced upon treatment with various intracellular toll-like receptor (TLR) agonists, including CpG oligodeoxynucleotides (ODN), R848, and poly (I:C). Interestingly, the adoptive transfer of HJ1 T cells conferred protection against the CD1b-transfected murine T cell lymphoma (RMA-S/CD1b) and CpG ODN enhanced the antitumor effect. Thus, this study, for the first time, demonstrates the antitumor potential of CD1b-autoreactive T cells and their potential use in adoptive immunotherapy.
KEYWORDS: Autoreactive T cells, CD1, CpG, self-lipids, tumor immunity
Abbreviations
- CTL
Cytotoxic T Lymphocytes
- DC
Dendritic Cell
- iNKT
Invariant Natural Killer T
- MDSC
Myeloid-Derived Suppressor Cell
- ODN
oligodeoxynucleotides
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PI
phosphatidylinositol
- PS
phosphatidylserine
- pLPE
lysophosphatidylethanolamine
- TLR
Toll-Like Receptor
- Treg
Regulatory T cells
Introduction
The use of immunotherapy to treat cancer is emerging as a viable treatment option. Several studies have demonstrated that the presence of tumor antigen-specific cytotoxic CD8+ T lymphocytes (CTL) is associated with better disease prognosis.1,2 The secretion of IFNγ and direct cytotoxicity largely mediate their antitumor effects.1 Over the years, unique/mutated proteins in tumor cells have been identified that serve as neo-peptide antigens for T cells.3,4 In addition, lipid biosynthesis is altered in cancer cells, thus affecting the lipid species present.5-7 In fact, it has been shown that tumors not only harbor structurally different lipids compared to normal cells, but also, in several cases, accumulate fatty acids, cholesterol, and phospholipid species.5-7 Thus, it is plausible that self-lipid-reactive T cells could contribute to antitumor immunity through either recognizing novel tumor lipids or responding to accumulated self-lipids.
CD1 molecules present both self- and foreign lipid antigens to T cells and are divided into group 1 and group 2 based on sequence homology.8-10 Group 1 CD1 molecules consist of CD1a, CD1b, and CD1c, whereas CD1d belongs to group 2.8,9 Although humans express all CD1 isoforms, mice only express CD1d.11 Thus, it is not surprising that most of the knowledge about the role of CD1-restricted T cells in tumor immunity comes from the study of CD1d-restricted natural killer T (NKT) cells. Type I NKT cells (also called invariant NKT cells) contribute mostly to antitumor immunity by exhibiting direct cytotoxicity toward CD1d+ tumors and/or by the production of IFNγ, which can subsequently activate NK cells, CTLs, and dendritic cells (DCs).12-14 For example, in a xenograft tumor model, the adoptive transfer of iNKT cells into mice harboring a CD1d-expressing B cell lymphoblast (C1R) resulted in smaller tumor size compared to iNKT cells transferred into mice with CD1d negative C1R tumors.15 However, a few studies also demonstrated the inhibition of antitumor activity by iNKT cells in T and B cell lymphoma models.16,17 Type II NKT cells (also called variant NKT cells), on the other hand, are traditionally known to play an immunosuppressive role as a result of IL-13 secretion, which promotes activation of TGF-β-secreting myeloid-derived suppressor cells (MDSCs) and at the same time inhibits cytotoxic capacity of CD8+ T cells and iNKT cells.18-21 Interestingly, a previous study showed that the immunosuppressive effect of type II NKT cells is reversed in the presence of CpG oligodeoxynucleotides (ODN), a TLR 9 agonist, leading to an increased ratio of IFNγ to IL-13 production.22 These data suggested that CD1d-restricted NKT cells could potentially be used in adoptive immunotherapy to treat tumors.
A recent study showed that CD1c-restricted T cells recognized tumor-derived methylated lysophosphatidic acid and lysed CD1c+ leukemia cells, highlighting the role of group 1 CD1-restricted T cells in tumor immunity.23 However, the contribution of group 1 CD1-restricted T cells to antitumor immunity in vivo remains largely unknown. In general, group 1 CD1-restricted T cells have more diverse TCR usage compared to iNKT cells and a large proportion is reactive to self-lipids.24-29 In fact, about 1/10–1/300 of all circulating T cells in humans are autoreactive group 1 CD1-restricted T cells.30 An early study of group 1 CD1-autoreactive T cells was reported in multiple sclerosis patients, who had a higher frequency of sulfatide and GM1-specific CD1b-restricted T cells compared to normal controls.25 Subsequently, it was demonstrated that sulfatide could be presented by all group 1 CD1 isoforms to prime T cells responses.29 In addition, recent studies have shown that CD1a-restricted human T cell clones recognize skin-derived apolar oils and phospholipids; autoreactive CD1b-restricted human T cell clones recognize a range of phospholipids, whereas CD1c-reactive clones can bind cholesterol and its esters.31-33 Considering that these lipids accumulate in most tumors, it is imperative to explore the role of autoreactive group 1 CD1-restricted T cells in tumor immunity. This would shed light on the potential use of these T cells in cancer immunotherapy.
In this study, we used double transgenic mice (hCD1Tg/HJ1Tg) expressing human group 1 CD1 molecules34 and CD1b-autoreactive HJ1 TCR35 to study the function of group 1 CD1-restricted autoreactive T cells in the context of tumor immunity. We found that HJ1 T cells recognized various phospholipids, especially phosphatidylethanolamine (PE) and ether-linked phophatidylethanolamine but not sphingolipids. Additionally, the autoreactivity of HJ1 T cells was enhanced upon treatment with various TLR agonists. HJ1 T cells also showed cytotoxicity toward a T cell lymphoma cell line (RMA-S) transfected with CD1b and responded more strongly to lipids extracted from tumor cells compared to those from normal cells. Consequently, adoptive transfer of HJ1 T cells exhibited protection against RMA-S/CD1b tumors in vivo but not to the parental RMA-S cells. This antitumor effect was enhanced in the presence of CpG ODN. Thus, this is the first study to demonstrate the antitumor potential of CD1b-autoreactive T cells.
Results
CD1b-autoreactive HJ1 T cell hybridoma selectively responds to phospholipids
Our previous studies showed that HJ1 T cells share several characteristics with NKT cells, in that they exhibit preactivated phenotypes, are polyfunctional and recognize lipid antigens conserved between humans and mice.35 It has been shown that NKT cells respond to two major lipid families: phospholipids and sphingolipids.10 Thus, in this study, we wanted to determine the antigens that CD1b-autoreactive HJ1 T cells responded to. We generated HJ1 T cell hybridoma and tested their reactivity to a panel of phospholipids and sphingolipids, known to stimulate NKT cells, in a CD1b plate bound assay. A value of 1 was assigned to IL-2 secreted when HJ1 T cell hybridoma were incubated with CD1b, without the addition of exogenous lipids. HJ1 T cells selectively responded to glycerophospholipids, including PE, phosphatidylglycerol (PG), phosphatidylcholine (PC), phosphatidylinositol (PI) but not phosphatidylserine (PS). They were also activated by ether-linked lysophosphatidylethanolamine (pLPE) (Fig. 1A), which has been implicated in the thymic selection of iNKT cells.36 However, HJ1 T cells did not respond to β-D-glucosylceramides and sulfatides (Fig. 1A). In addition, HJ1 T cells demonstrated a dose-dependent response to PE. The response was CD1b-specific, as PE presented by the other CD1 isoform (CD1d), did not activate HJ1 T cells. As a negative control, PE-loaded CD1b did not stimulate a CD1d-restricted iNKT cell hybridoma, 2C12 (Fig. 1B). Collectively, these data suggested that HJ1 T cells could be activated by various phospholipid species in their native forms upon being directly presented by CD1b.
Figure 1.
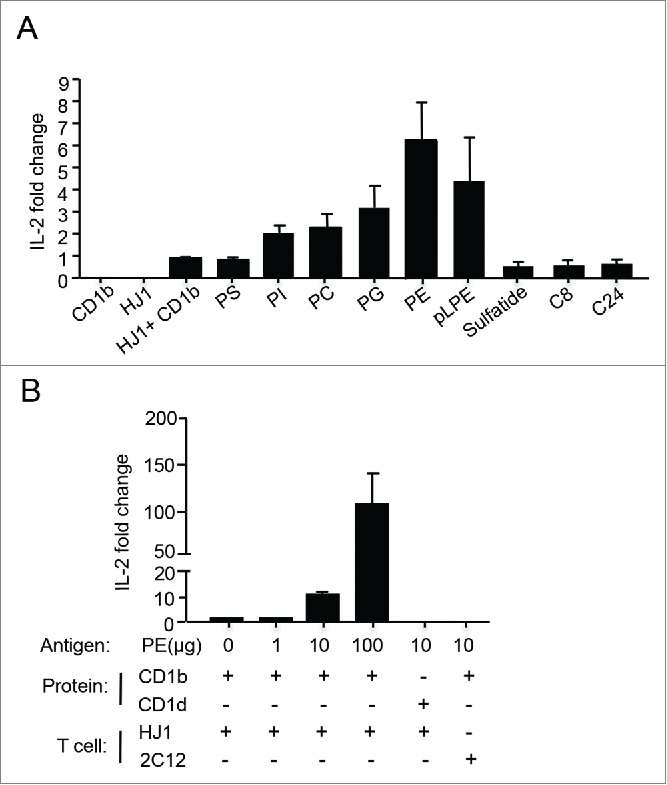
HJ1 T cell hybridoma responds to self-lipids in a CD1b-dependent manner. Plate-bound complexes of lipid/CD1b were incubated with 5 × 104 HJ1 T cell hybridoma for 24 h. IL-2 in the supernatant was measured by ELISA. HJ1 incubated with CD1b without added antigens had a value of 1 and fold change was calculated using this value as reference. (A) The responses of HJ1 T cell hybridoma to a panel of self-lipid antigens. Data shown are representative of four independent experiments, and are the mean ± SEM. (B) PE activates HJ1 T cell hybridoma in a dose-dependent and CD1b-specific manner. The incubation of plate-bound PE/CD1b with iNKT hybridoma 2C12, and plate-bound PE/CD1d with HJ1 T cell hybridoma served as controls. Data shown are representative of three independent experiments and error bars represent the mean ± SEM.
HJ1 T cells recognize tumor-derived lipids and lyse CD1b-expressing tumors
Since HJ1 T cells recognize phospholipids and several phospholipid species like PE and PC are abundant in lymphoma cells,37 we next determined whether HJ1 T cells could recognize lipids isolated from both human (Molt-4) and murine (RMA-S) lymphoma cells. As the murine-derived RMA-S T cell tumor line does not inherently express CD1b, we stably transfected RMA-S cells with CD1b encoding cDNA. The expression of CD1b on both transfected and untransfected RMA-S cells is depicted in Fig. 2B. A Folch extraction protocol was used to extract lipids from tumor cells (Molt-4 and RMA-S/CD1b) and normal cells (B6 thymocytes and human T cells). Plate-bound CD1b preincubated with serially diluted amounts of the cellular lipids was used to stimulate HJ1 T cell hybridoma. We found that not only did HJ1 T cells secrete IL-2 upon incubation with CD1b loaded with lipids extracted from Molt-4 (Fig. 2A) and RMA-S/CD1b (Fig. 2C) lymphoma cells, but they also exhibited a stronger response to tumor-derived lipids compared to normal T cell-derived lipids (Fig. 2A and C). In addition, HJ1 T cells could lyse CD1b-expressing RMA-S cells but not the parental RMA-S cell line (Fig. 2D). These data suggest that lipid antigens derived from tumor cells can be presented by CD1b, leading to the activation of HJ1 T cells. As such, HJ1 T cells effectively kill CD1b-expressing tumor cells.
Figure 2.
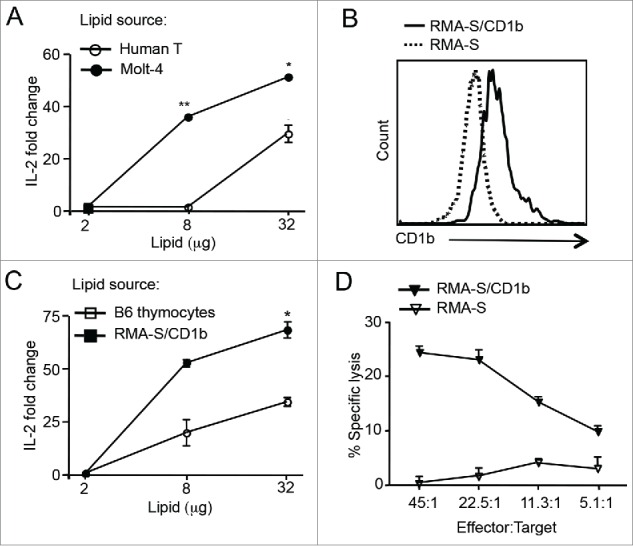
HJ1 T cells can recognize tumor-derived lipids and kill CD1b-expressing RMA-S T cell lymphoma. (A) Extracted lipids from PBMC-derived human T cells (open circle) and Molt-4 cells (black circle) were loaded on to purified CD1b protein and subsequently incubated with 5 × 104 HJ1 T cell hybridoma cells. Twenty-four hours later, IL-2 in the supernatant was measured by ELISA. HJ1 incubated with CD1b without added antigens had a value of 1 and fold change was calculated using this value as reference. (B) Surface expression of CD1b on RMA-S cells and RMA-S/CD1b transfectants was determined by flow cytometry. RMA-S/CD1b transfectants stained with anti-CD1b antibody are depicted by the black line. RMA-S cell stained with anti-CD1b antibody are represented by the dotted line. (C) Extracted lipids from B6 thymocytes (open square) and RMA-S/CD1b cells (black square) were used to stimulate HJ1 T cell hybridoma as in (A). (D) HJ1 CTL effectors were incubated at different ratios with 51Cr labeled RMA-S (open triangle) or RMA-S/CD1b (black triangle) targets for 4 h. Supernatant was collected and assayed for the presence of 51Cr. Data are representative of at least two independent experiments. Error bars represent the mean ± SEM. ***p < 0.005; **p < 0.01; *p < 0.05.
CpG, an intracellular TLR ligand and a potent antitumor agent, enhances autoreactivity of HJ1 T cells
Since HJ1 T cells recognized lipids from tumors and killed CD1b+ RMA-S cells, we hypothesized that HJ1 T cells may play a role in antitumor immunity. Additionally, we also proposed that enhancing HJ1 T cell autoreactivity would lead to a stronger protective response against tumors. Our previous study showed that extracellular TLR ligands enhanced HJ1 T cell autoreactivity.35 Thus, we examined whether CpG ODN, an intracellular TLR 9 agonist, known to have antitumor effects,38 could enhance HJ1 T cell autoreactivity. We also compared the effect of other TLR agonists on the reactivity of HJ1 T cells. We detected the highest production of IFNγ by HJ1 T cells in response to CpG ODN-treated hCD1Tg BMDCs compared to other TLR ligand treatments, such as poly (I:C) (TLR3) and R848 (TLR7/8) (Fig. 3A). In addition, treatment with R848 but not poly (I:C) or CpG ODN led to IL-13 secretion by HJ1 T cells in the presence of hCD1Tg BMDCs (Fig. 3B). Pam3Cys, the TLR 2 ligand, served as a positive control since we had previously showed that HJ1 T cell autoreactivity was enhanced in its presence.35
Figure 3.
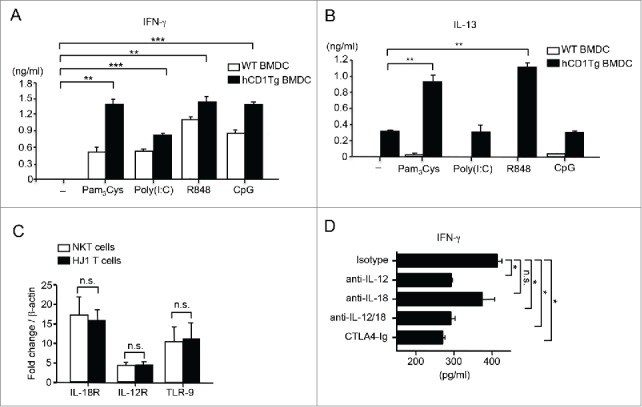
CpG ODN can enhance cytokine secretion by HJ1 T cells in vitro. WT (open bars) and hCD1Tg BMDC (black bars) were either left untreated or pretreated with TLR agonists- Pam3Cys, poly (I:C), R848 and CpG ODN prior to co-culture with enriched hepatic HJ1 T cells from hCD1Tg/HJ1Tg/Rag2−/− mice. ELISA was performed to detect IFNγ (A) and IL-13 (B) secretion in the supernatant 24 h later. Data are representative of two independent experiments. (C) The expression of IL-12R, IL-18R, and TLR 9 on purified iNKT cells (open bars) and HJ1 T cells (black bars) was determined by detecting mRNA levels relative to β-actin using quantitative RT-PCR. (D) Hepatic HJ1 T cells were co-cultured with CpG ODN-treated hCD1Tg BMDC for 48 h in the presence of anti-IL-12 and/or anti-IL-18 or in the presence of CTLA-4 Ig. IFNγ ELISA was performed upon collection of supernatant. Data shown are representative of two independent experiments and error bars represent the mean ± SEM. ***p < 0.005; **p < 0.01; *p < 0.05.
Because CpG ODN induced the highest CD1b-dependent IFNγ to IL-13 production from HJ1 T cells and because of its known antitumor effects, we subsequently investigated the mechanism by which CpG ODN enhances HJ1 T cell autoreactivity. It has been shown that the concomitant secretion of IL-12 and IL-18 from TLR-stimulated DCs is sufficient to activate iNKT cells.39 To determine whether similar mechanisms were involved in CpG ODN-mediated enhancement of HJ1 T cell autoreactivity, we first determined whether HJ1 T cells expressed receptors for IL-12 and IL-18 by RT-PCR. We found that HJ1 T cells expressed both IL-12 receptor and IL-18 receptor at a similar level to that of iNKT cells (Fig. 3C). Consequently, when CpG ODN-treated hCD1Tg BMDCs were co-cultured with HJ1 T cells in the presence of blocking antibodies against IL-12 and/ or IL-18, we observed that neutralization of IL-12 but not IL-18 lead to decreased IFNγ secretion. Also, addition of CTLA-4 Ig resulted in decreased IFNγ secretion by HJ1 T cells (Fig. 3D). These data suggested that DC-derived IL-12 and the interaction of CD28 and co-stimulatory molecules on DCs play an important role in HJ1 T cell activation by CpG ODN. Interestingly, purified HJ1 T cells with expressed TLR 9 at similar levels to iNKT cells (Fig. 3C). This suggested that CpG ODN could perhaps directly activate HJ1 T cells in the absence of CD1b-expressing DCs. This could explain the production of IFNγ by HJ1 T cells when stimulated with CpG-ODN-treated WT BMDCs, which do not express CD1b (Fig. 3A).
To examine whether CpG ODN could activate HJ1 T cells in vivo, we administered (i.v.) CpG ODN or PBS to hCD1Tg/HJ1Tg/Rag2−/− mice and compared the CD69 expression and IFNγ production in these two treatment groups. Hepatic HJ1 T cells from CpG ODN-injected mice had higher CD69 expression as well as IFNγ secretion compared to PBS-treated controls (Fig. 4A and B). Taken together, our results showed that CpG ODN could activate HJ1 T cells both in vitro and in vivo, supporting the potential use of CpG ODN to boost antitumor effects of HJ1 T cells in adoptive immunotherapy.
Figure 4.
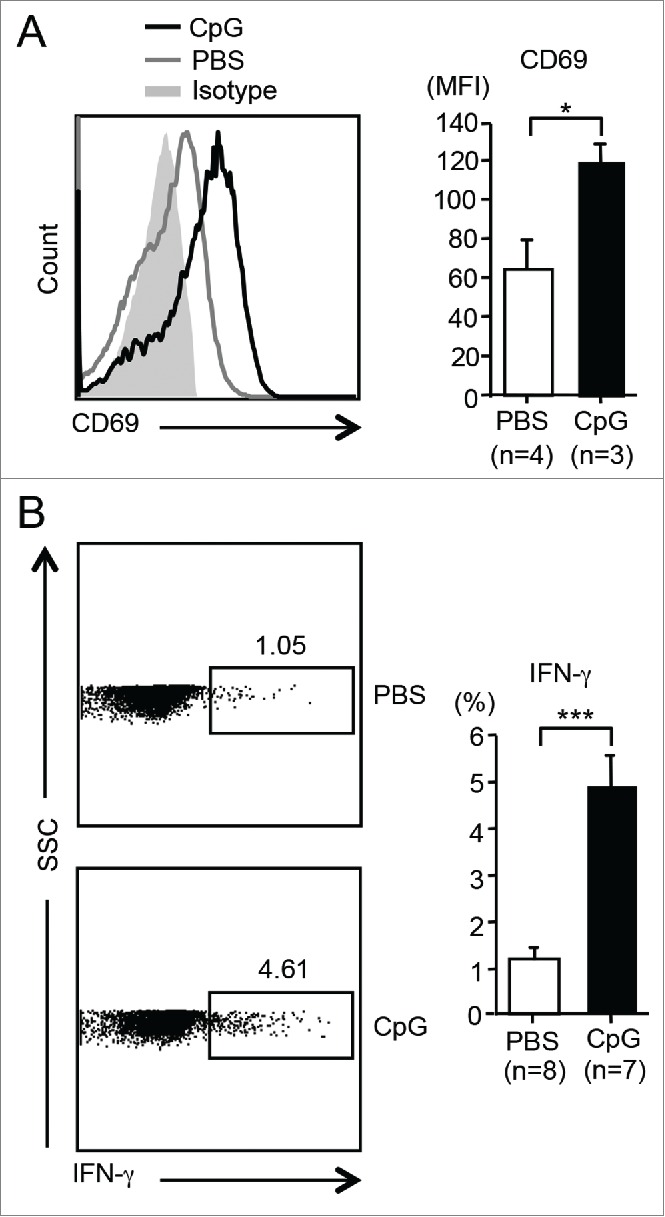
CpG ODN can activate HJ1 T cells in vivo. HJ1Tg/hCD1Tg/Rag−/− mice were injected (i.v.) with either 40 μg (in 100 μL PBS) of CpG or PBS. Twelve hours later, the activation and cytokine production of hepatic HJ1 T cells was determined by flow cytometry. (A) Representative histograms show the expression of CD69 on HJ1 T cells from CpG ODN-injected (black line) or PBS-injected mice (gray line). Gray solid area indicates isotype control. To the right, CD69 expression, on HJ1 T cells in PBS (open bar) or CpG ODN (black bar) injected mice, is depicted in bar graphs. Error bars represent mean ± SEM of the mean fluorescence intensity of CD69. (B) Representative dot-plots of IFNγ-producing HJ1 T cells in the PBS or CpG ODN-injected HJ1Tg/hCD1Tg/Rag−/− mice (left). Bar graphs (right) depict IFNγ secretion by HJ1 T cells upon PBS (open bar) and CpG ODN (black bar) injection. Mean ± SEM is shown for the bar graphs. ***p < 0.005; **p < 0.01; *p < 0.05.
HJ1 T cells exhibit in vivo antitumor immunity, which is enhanced in the presence of CpG ODN
To explore the antitumor potential of HJ1 T cells in vivo and to test whether CpG ODN treatment can augment the antitumor effect of HJ1 T cells, hCD1Tg (CD45.1+) mice were subcutaneously inoculated with either 5 × 106 RMA-S or RMA-S/CD1b tumor cells. Four days later, 2–4 × 106 in vitro expanded HJ1 T cells (CD45.2+) with or without CpG ODN were injected in to the tumors. This process was repeated 6 d later and tumor size was measured on indicated days. Mice were sacrificed on day 14 after tumor inoculation. We found that mice inoculated with CD1b-expressing tumors (RMA-S/CD1b) that received intra-tumor injection of HJ1 T cells and CpG ODN had the smallest tumors. As expected, administration of CpG ODN reduced the tumor size, albeit to a lesser extent than the combination treatment. Injection of HJ1 T cells alone into mice bearing CD1b-expressing tumors also slowed tumor growth to a considerable amount (Fig. 5A). In contrast, HJ1 T cells did not exert a detectable antitumor effect in mice inoculated with RMA-S (CD1b−) tumor cells (Fig. 5A). Interestingly, in RMA-S tumor-bearing mice, combination treatment of CpG ODN and HJ1 T cells did not enhance protection compared to RMA-S/CD1b tumor-bearing mice treated with just CpG ODN, suggesting the observed antitumor effect of HJ1 T cells is mainly CD1b dependent (Fig. 5A). Additionally, we observed no difference in tumor growth in mice that received RMA-S and RMA-S/CD1b cells without treatment.
Figure 5.

HJ1 T cells exhibit in vivo antitumor immunity, which is enhanced in the presence of CpG. 5 × 106 RMA-S or RMA-S/CD1b cells were injected (s.c.) in to hCD1Tg (CD45.1+) mice. Tumor size was measured on day 4 after tumor injection and every other day thereafter. HJ1 T cells were transferred with or without CpG ODN (40 μg in 100 μL of PBS) on days 4 and 10. Arrows represent days when HJ1 and or CpG ODN were injected in to tumors. (A) Tumor volumes are shown as mean ± SEM. Tumor size of mice injected with RMA-S cells is depicted by open square, mice injected with RMA-S/CD1b cells with open circle, mice injected with RMA-S cells + HJ1 T cells with black square, mice injected with RMA-S + HJ1 T cells + CpG ODN with black diamond, mice injected with RMA-S/CD1b + HJ1 T cells with black circle, mice injected with RMA-S/CD1b cells + CpG ODN with black triangle and mice injected with RMA-S/CD1b + CpG ODN + HJ1 T cells with black upside down triangle. Mice were killed at day 14 after tumor injection and tumor-infiltrating leukocytes were prepared and stained with mAbs to CD45.1, CD8α, TCRβ, NK1.1, and IFNγ to quantify the (B) percentage and number of CD8+ IFNγ+ T cells. (C) The percent of IFNγ+ NK cells was also quantified. Also, tumor infiltrating leukocytes were stained with mAbs to CD45.1, TCRβ, CD4+, Foxp3, CD11b, Ly6C, CD11c, and CD86 to quantify the percent of Treg (D), the percent of monocytic (E) and granulocytic (F) MDSCs, and CD86 expression on DCs (G). Bar graphs depict the mean ± SEM of the percentages and numbers. Open bars represent mice that received RMA-S tumor cells + HJ1 T cells, light gray bars represent mice with RMA-S/CD1b tumors + HJ1 T cells, dark gray bars depict mice with RMA-S/CD1b tumors + CpG ODN and black bars represent mice with RMA-S/CD1b tumors + CpG ODN + HJ1 T cells. ***p < 0.001; **p < 0.01; *p < 0.05.
Next, we attempted to delineate the mechanism(s) by which HJ1 T cells contributed to antitumor immunity. Upon examination of tumor infiltrating leukocytes, we could not detect HJ1 T cells (CD45.2+TCRβ+). This phenomenon has also been observed with iNKT cells in tumor models40; in spite of the presence of a phenotype, the adoptively transferred iNKT cells were untraceable. However, the percentage and total number of endogenous CD8+ IFNγ producing T cells (recipient mice were CD45.1+ while HJ1 T cells were CD45.2+) were highest in mice that received HJ1 T cells along with CpG ODN compared to all other groups. Additionally, adoptive transfer of HJ1 T cells alone into CD1b+ tumors resulted in higher IFNγ secretion by endogenous CD8+ T cells compared to HJ1 T cells transferred into CD1b− tumors. Furthermore, CD107a, a surrogate marker for cytolytic activity, was increased on endogenous CD8+ T cells in RMA-S/CD1b tumor-bearing mice injected with HJ1 T cells, regardless of the presence of CpG ODN, compared to CD1b+ tumor-bearing mice that did not receive HJ1 T cells (Fig. S1, left panel). This suggested that transactivation of CD8+ T cells by HJ1 T cells might play a role in protection against tumor growth (Fig. 5B). Also, there was no significant difference in IFNγ secretion by NK cells among the treatment groups (Fig. 5C). Additionally, HJ1 T cells with or without CpG ODN administration did not alter the expression of CD107a on NK cells compared to untreated CD1b+ tumor-bearing mice. (Fig. S1, right panel). This could be attributed to the use of type B CpG ODN in our assays, since it is known that type A CpG ODN activates NK cells most potently. Furthermore, there were no significant differences in the percentage of CD4+ regulatory T cells (Fig. 5D) and monocytic and granulocytic MDSCs (Fig. 5E and F) in the tumors of different groups. Even though the expression of CD86 co-stimulatory molecule increased on DCs from CpG ODN-treated mice, there was no difference between CpG ODN and HJ1 + CpG ODN groups (Fig. 5G). These suggested that HJ1 T cells, and thus CD1b-autoreactive T cells might play a protective role in tumor immunity potentially by directly lysing tumor cells in the initial stages and later by activating other CD8+ T cells.
Discussion
In this study, we demonstrated that CD1b-autoreactive HJ1 T cells recognized a panel of phospholipids including ether-linked phospholipids. Unlike both types of CD1d-restricted NKT cells, which are activated by β-linked glycosphingolipids, HJ1 T cells were not activated by sphingolipids.22,41–43 A recent study also suggests that mammalian α-linked glycosphingolipids can activate type I NKT cells.44 Although ether-linked forms of pLPE and lysophaphatidic have been implicated in the selection of type I NKT cells, nothing is known about the selecting self-lipids for type II NKT cells and group 1 CD1-reactive T cells.36 This suggests that there are both overlapping and unique antigens recognized by CD1b-restricted T cells and NKT cells. Interestingly, RMA-S tumor-derived lipids are known to activate a type II NKT cell clone.45 We also found that HJ1 T cells were activated to a greater extent in the presence of both human (Molt-4) and murine (RMA/S-CD1b) T cell lymphoma-derived lipids. Even though the nature of the antigen remains unknown, it can be speculated that phospholipids species present in tumor cells might be activating the HJ1 T cells. In fact, studies have reported that phospholipids accumulate in several breast and brain cancers and lymphomas.6,37 Since group 1 CD1 molecules are expressed on both acute myelogenous leukemia and acute lymphoblastic leukemia cells, group 1 CD1-restricted T cell responses are promising targets for adoptive leukemia immunotherapy.23
HJ1 T cells were not only directly activated by TCR-CD1b interactions in the presence of self-lipids, but their autoreactivity was enhanced by various TLR agonists that target either cell surface TLRs (e.g., Pam3Cys for TLR2)35 or intracellular TLRs (e.g., CpG ODN for TLR9). Our previous study showed that TLR2-mediated signaling enhanced the autoreactivity of HJ1 T cells by stimulating DCs to produce IL-12 and upregulate CD86/CD80 expression.35 Similarly, the enhanced HJ1 T cell response by CpG ODN was partially a result of DC-derived IL-12 and engagement of co-stimulatory molecules on DCs and CD28 on HJ1 T cells. However, blocking IL-12 did not completely abrogate cytokine secretion by these T cells. This could be a result of direct HJ1 activation by CpG ODN. This could also explain the substantial IFNγ production by HJ1 T cells when stimulated with CpG ODN-pulsed wildtype DC, which does not express CD1b. Taken together, the data suggested that not only could HJ1 T cells be protective against tumors by directly recognizing tumor lipids, but also that the response could be enhanced in the presence of CpG ODN. Interestingly, both CpG and IL-12 have been used in pre-clinical studies as anticancer therapy.38,46 Both these immunostimulatory agents work to increase the production of IFNγ by innate and adaptive immune cells. However, clinical trials have yielded mixed results, mostly due to over stimulation of the immune system and related adverse side effects or the lack of apparent benefits over existing treatment options.38,46 Nevertheless, recent efforts have been directed toward delivering these agents to the local tumor microenvironment and in conjunction with other immunotherapies like T cell adoptive transfer.
In vivo adoptive transfer of HJ1 T cells resulted in protection against CD1b-expressing RMA-S tumors. Additionally, the co-administration of HJ1 T cells and CpG ODN resulted in the greatest reduction of tumors. Even though no HJ1 T cells could be detected in the tumors when mice were sacrificed, the adoptive transfer of these T cells did show protection. The inability to track adoptively transferred T cells, in spite of a phenotype change observed, is a phenomenon that has been previously observed with NKT cells in tumor models.40 Although the reason for the disappearance of these cells is not well understood, it can be speculated that the unique properties of these innate-like T cells or absence of sufficient antigenic stimulation or metabolic stress-induced cell death could be possible explanations for this phenomenon. Since we observed that HJ1 T cells offered more protection against tumors that expressed CD1b compared to CD1b− tumors, we propose that this is a result of direct cytotoxicity toward tumor cells in the initial phases, and later through the enhanced activation of endogenous CD8+ T cells. Co-administration of HJ1 T cells and CpG ODN led to increased IFNγ secretion by CD8+ T cells compared to administration of HJ1 T cells alone, suggesting CpG ODN treatment further enhances HJ1 T cell mediated activation of CD8+ T cells. Our results further suggested that CpG ODN-mediated enhanced protection was largely CD1b dependent since administration of HJ1 T cells along with CpG in to RMA-S tumors did not enhance protection over the administration of CpG alone. Even though in in vitro experiments, CpG treatment led to some cytokine secretion by HJ1 T cells in the absence of CD1b, a strong CD1b dependency was observed in vivo. This could result from accumulation of DC-derived cytokines in the immediate vicinity of HJ1 T cells under in vitro conditions. However, in vivo, the level of cytokines present within close proximity of HJ1 T cells might not be sufficient to trigger a CD1b independent activation of HJ1 T cells. Since HJ1 T cell presence did not activate NK cells, it is possible the cytokines from HJ1 T cells enhanced DC maturation and function, which subsequently activated CD8+ T cells. Although RMA-S/CD1b tumor cells have low surface expression of MHC Ia molecules,47 our data showed that the response mounted by endogenous CD8+ T cells was able to contribute to antitumor immunity.
The physiological function of group 1 CD1 autoreactive T cells still remains largely unknown. Like some CD1b-autoreactive T cells in humans,32 HJ1 T cells used in this study recognized several types of phospholipids in a CD1b-specific manner. We found that the cytokine-producing capacity of HJ1 T cells was enhanced by various TLR agonists, suggesting a means to modulate the function of CD1b autoreactive T cells in a variety of settings. In addition, we found that HJ1 T cells exhibited an antitumor effect, which was potentiated by the treatment of CpG ODN. Furthermore, the adoptive transfer of HJT 1 T cells into immunocompetent recipients revealed the transactivation capacity of autoreactive group 1 CD1-restricted T cells. Altogether, our data not only shed light on the potential self-antigens that can activate CD1b-autoreactive T cells, but also revealed their antitumor potential. The findings from this study are significant since a large proportion of group 1 CD1-restricted T cells in humans are autoreactive. Thus, harnessing the therapeutic potential of these T cells could represent a promising target to develop novel therapies for the clinic.
Materials and methods
Mice used and ethics statement
C57BL/6 (B6) and CD45.1 congenic B6 mice were purchased from the Jackson Laboratory. hCD1Tg (in B6 background) and HJ1Tg/hCD1Tg/Rag2−/− mice were generated as previously described.34,35 Vα14 transgenic mice were provided by Dr. Albert Bendelac (University of Chicago) and crossed onto B6 background. All animals were housed in a specific pathogen-free facility at Northwestern University. All animal work was approved by the Institutional Animal Care and Use Committee.
Human cell isolation and processing
The study protocol was approved by the IRB at Northwestern University and informed consent was obtained from the participating donor prior to their inclusion in this study. Blood was drawn from a healthy donor with no known clinical abnormalities. 30 mL of whole blood was obtained by venipuncture using heparinized blood collection tubes (BD, 368661) and PBMC were immediately isolated using Histopaque-1077 (Sigma, 10771-500ML). A total of about 2 × 106 PBMC/mL of blood was isolated as determined by counting using a hemocytometer. T cells from total PBMC were enriched using biotinylated anti-CD3 antibody (eBioscience, 13-0037-82) and subsequently by positive selection with streptavidin microbeads following manufacturer's protocol (Miltenyi, 130-048-101). On the same day, lipids from T cells were extracted using the Folch method.48
Reagents and antibodies
CpG ODN 1826 (5′-tccatgacgttcctgacgtt-3′) was synthesized at Integrated Device Technology. Purified CD1b and CD1d proteins were provided by the National Institutes of Health tetramer facility. Lipids antigens phosphatidylethanolamine (PE, 840021), phosphatidylinositol (PI, 840042), phosphatidylserine (PS, 870336), phosphatidylcholine (PC, 840051), phosphatidylglycerol (PG, 841138), synthetic 1-O-1′-(Z)-octadecenyl-2-hydroxy-sn-glycero-3-lysophosphoethanolamine (pLPE), synthetic D-glucosyl-ß-1,1′-N-octanoyl-D-erythro-sphingosine (C8, 860540), and synthetic D-glucosyl-ß-1,1′-N-nervonoyl-D-erythro-sphingosine (C24, 860549) were purchased from Avanti Polar Lipids. Bovine ceramide-galactoside-3-sulfate (sulfatides) was purchased from Matreya (1049). Fluorochrome-labeled and purified monoclonal antibodies against mouse TCR-β (109229), CD8-α (100752), CD69 (104508), CD45.2 (109806), CD45.1 (110726), CD1b (329108) CD11b (101224), CD11c (117310), CD86 (105014), NK1.1 (108708), and IFNγ (505806) were purchased from BioLegend. Anti-CD107a (553793) was purchased from BD Biosciences.
Generation and stimulation of HJ1 T cell hybridoma
The mouse HJ1 T cell hybridoma line was generated by cell fusion between BW5147 α−β− thymoma cell line and HJ1 T cell blasts as described.49 Lipids were either purchased commercially or extracted from tumor cell lines and normal cells using chloroform–methanol extraction (Folch method). To immobilize lipid/CD1b complexes, 1.5–2 μg/mL of CD1b protein (in PBS) were incubated with 10 μg/mL or at the indicated concentration of lipid antigen for 4 h. 96-well Immulon plates (Thermo Fisher Scientific, 3455) were then coated for 2 h at 37°C with 100 μL of CD1b protein loaded with or without lipid. Subsequently, wells were washed three times with RPMI-10 media (RPMI 1640 supplemented with 10% FBS, 1% penicillin-streptomycin, 20 mM L-glutamine, and 50 μM 2-mercaptoethanol). HJ1 T cell hybridoma (5 × 104 cells/well) were stimulated with plate-bound CD1b/lipid complexes for 24 h and IL-2 levels in culture supernatants were measured by ELISA (BD, 554424 and 554426).
Cell preparations
Hepatic leukocyte cell suspensions were prepared from HJ1Tg/hCD1Tg/Rag2−/− mice. For in vitro BMDC co-culture experiments, HJ1 T cells were enriched through depletion of CD11b+ (BioLegend, 101206) and MHC II+ (BioLegend, 114406) cells using mAb and anti-FITC magnetic beads following the manufacturer's instructions (Miltenyi, 130-048-701). Greater than 90% of the enriched populations were TCRβ+. BMDCs were prepared using 10 ng/mL GM-CSF (PeproTech, 315–03) and 2 ng/mL IL-4 (PeproTech, 214–14). Tumor-infiltrating cells were isolated by physical disruption of tissue followed by digestion with 1 mg/mL collagenase IV (Sigma, C5138-1G) and 100 μg/mL DNase (Sigma, 11284932001) at 37°C for 30 min.
Flow cytometry
For cell surface staining, cells were pre-incubated with purified 2.4G2 mAb and then stained with the appropriate combinations of mAbs. For intracellular cytokine staining, cells were stimulated with 50 ng/mL phorbol 12-myristate 13-acetate (Enzo Life Sciences, BML-PE160-0005) and 0.5 μg/mL ionomycin (Sigma, I9657-5MG) for 4 h followed by addition of brefeldin A (BioLegend, 420601) for 2 h. For CD107a staining, antibody was added along with PMA/ionomycin. For other antibodies, cells were surface stained after stimulation and fixed with 4% paraformaldehyde (Sigma, 158127-3KG), permeabilized with 0.1% saponin (Sigma, S7900), followed by anti-cytokine mAb staining. Flow cytometry was performed with a FACSCanto II (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Detection of cytokine secretion
BMDC were either untreated or pretreated with a variety of toll-like receptor (TLR) ligands: Pam3Cys (100 ng/mL), poly (I:C) (10 μg/mL), R848 (10 μg/mL), CpG (10 μg/mL) overnight. Enriched 1–2 × 105 HJ1 T cells were then cultured with either untreated or pretreated 1 × 105 BMDCs for 48 h in RPMI-10 media. For blocking experiments, 20 μg/mL of CTLA4-Ig (eBioscience, 16-1521-82) or 10 μg/mL anti-IL-12 (eBioscience, 16-7123-85), anti-IL-18 (R&D Systems, AF122) or isotype matched control antibodies were added along with HJ1 T cells and BMDCs. IFNγ and IL-13 levels in culture supernatants were measured by ELISA (eBioscience, anti-IFNγ: 16-7312-38 and 16-7311-85) and BD CBA (558349).
Generation of RMA-S/CD1b transfectants
Human CD1B cDNA clone in pCMV/hygro vector was purchased from SinoBiological Inc. (HG11831-G-N). DH5α E. coli cells were transformed with the CD1b-expression plasmid and the plasmid DNA was extracted using Qiagen Midi Prep kit (12145). Plasmid DNA (5 μg) was then electroporated into RMA-S cells and stable transfectants was selected for hygromycin B-resistance (240 μg/mL, Invitrogen, 10687010). Subsequently, CD1b expression on transfected RMA-S cells was determined by flow cytometry.
Cytotoxicity assay
HJ1 CTL effectors were established by stimulating splenocytes from HJ1Tg/hCD1Tg/Rag2−/− mice with 1 μg/mL Concanavalin A (Sigma, C5275-5MG) for 3 d in RPMI-10, followed by culture in supplemented Mischell Dutton media with 20 U/mL IL-2 (Peprotech, 212–12) for an additional 4 d. RMA-S and RMA-S/CD1b transfectants (1 × 106 cells) were labeled with 50 μCi [51Cr] sodium chromate (MP Biomedicals, 016201502) for 1 h at 37°C. 51Cr-labeled target cells (1×104 cells/well) were incubated with HJ1 CTL effectors for 4 h at 37°C. Supernatant was collected to assay for 51Cr release. The percentage of specific lysis was calculated as [(experimental release – spontaneous release)/(maximum release-spontaneous release)] ×100.
mRNA detection
HJ1 T cells were isolated from the livers of HJ1Tg/hCD1Tg/Rag2−/− mice and NKT cells were isolated from the livers of Vα14 transgenic mice. HJ1 T cells were purified using Miltenyi positive selection with APC conjugated α-CD3 antibody, whereas NKT cells were purified using APC conjugated CD1d/α-GalCer tetramer. Subsequently, anti-APC microbeads (Miltenyi, 130-090-855) were used to derive a >95% pure population of both T cells. mRNA was then extracted from the cells with the Qiagen RNeasy kit (74104) followed by reverse transcription to cDNA using Superscript® III (Invitrogen, 18080085). Primers against targets of interest were mixed with cDNA and iQ™ SYBR® Green Supermix (Bio-Rad, 1708880). Quantitative PCR was performed using a Bio-Rad iQ5 cycler. The following primer sets were used: β-actin forward 5′ CTTCTTTGCAGCTCCTTCGTT, β-actin reverse 5′ AGGAGTCCTTCTGACCCATTC; IL18Rα forward 5′ TCACCGATCACAAATTCATGTGG, IL-18Rα reverse 5′ TGGTGGCTGTTTCA TTCCTGT; IL-12Rβ1 forward 5′ TGCCGCTACTTCTCCTCAG, IL-12Rβ1 reverse 5′ ACTTCATGGTTCGGTTCCCAA; TLR9 forward 5′ ATGGTTCTCCGTCGAAGGACT, TLR9 reverse 5′ GAGGCTTCAGCTCACAGGG.
In vitro expansion of HJ1 T cells
Lymphocytes isolated from the spleen and lymph nodes of HJ1Tg/hCD1Tg/Rag2−/− mice were stimulated with 5 μg/mL anti-CD3 (eBioscience, 16-0031-86), 2 μg/mL anti-CD28 (Bioxcell, BE00155), and 20 ng/mL IL-12 (Peprotech, 210–12) for 48 h. The cells were “rested” for 48 h in 70% RPMI-10 medium and 30% supplemented Mischell Dutton media with 20 U/mL IL-2 (Peprotech, 212–12) (CTL medium) and cultured for an additional 3 d in 100% CTL medium. At this point cells were used for intra-tumor injections.
Intra-tumor injection of HJ1 T cells in RMA-S tumor model
hCD1Tg (CD45.1) mice were inoculated subcutaneously with 5 × 106 RMA-S or RMA-S/CD1b cells 4 d before HJ1 T cell transfer. On day 0, in vitro expanded 2–4 × 106 HJ1 T cells were injected into tumors with or without 40 μg CpG in a volume of 100 μL PBS. Six days later, HJ1 T cells were injected into the tumors for the second time. Two perpendicular diameters of the tumor mass were measured on specified days, and tumor volumes were calculated as: π/6 × length × width2.
Statistical analysis
Statistical analyses were performed using Prism 6 software (GraphPad). The statistical significance of differences between experimental groups was analyzed using a Student's t test. A p value of <0 .05 was considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the NIH tetramer core facility for purified CD1b protein, Dr. Hak-Jong Choi for assistance on CD1b plate bound assays and Dr. Sebastian Joyce (Vanderbilt University) for providing 2C12 NKT cell hybridoma.
Funding
This work is supported by NIH grant R01AI057460 and R21AI117238 to C.-R.W.
References
- 1.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14:1014-22; PMID:24048123; http://dx.doi.org/ 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011; 29:235-71; PMID:21219185; http://dx.doi.org/ 10.1146/annurev-immunol-031210-101324 [DOI] [PubMed] [Google Scholar]
- 3.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14:135-46; PMID:24457417; http://dx.doi.org/ 10.1038/nrc3670 [DOI] [PubMed] [Google Scholar]
- 4.Van Der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, Chapiro J, Van Den Eynde BJ, Brasseur F, Boon T. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev 2002; 188:51-64; PMID:12445281; http://dx.doi.org/ 10.1034/j.1600-065X.2002.18806.x [DOI] [PubMed] [Google Scholar]
- 5.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 2013; 6:1353-63; PMID:24203995; http://dx.doi.org/ 10.1242/dmm.011338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Podo F. Tumour phospholipid metabolism. NMR Biomed 1999; 12:413-39; PMID:10654290; http://dx.doi.org/ 10.1002/(SICI)1099-1492(199911)12:7%3c413::AID-NBM587%3e3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- 7.Roos DS, Choppin PW. Tumorigenicity of cell lines with altered lipid composition. Proc Natl Acad Sci U S A 1984; 81:7622-6; PMID:6594705; http://dx.doi.org/ 10.1073/pnas.81.23.7622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barral DC, Brenner MB. CD1 antigen presentation: how it works. Nat Rev Immunol 2007; 7:929-41; PMID:18037897; http://dx.doi.org/ 10.1038/nri2191 [DOI] [PubMed] [Google Scholar]
- 9.Salio M, Cerundolo V. Regulation of lipid specific and vitamin specific non-MHC restricted T cells by antigen presenting cells and their therapeutic potentials. Front Immunol 2015; 6:388; PMID:26284072; http://dx.doi.org/ 10.3389/fimmu.2015.00388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macho-Fernandez E, Brigl M. The Extended Family of CD1d-Restricted NKT Cells: Sifting through a Mixed Bag of TCRs, Antigens, and Functions. Front Immunol 2015; 6:362; PMID:26284062; http://dx.doi.org/ 10.3389/fimmu.2015.00362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dascher CC, Brenner MB. Evolutionary constraints on CD1 structure: insights from comparative genomic analysis. Trends Immunol 2003; 24:412-8; PMID:12909453; http://dx.doi.org/ 10.1016/S1471-4906(03)00179-0 [DOI] [PubMed] [Google Scholar]
- 12.Smyth MJ, Crowe NY, Pellicci DG, Kyparissoudis K, Kelly JM, Takeda K, Yagita H, Godfrey DI. Sequential production of interferon-gamma by NK1.1(+) T cells and natural killer cells is essential for the antimetastatic effect of α-galactosylceramide. Blood 2002; 99:1259-66; PMID:11830474; http://dx.doi.org/ 10.1182/blood.V99.4.1259 [DOI] [PubMed] [Google Scholar]
- 13.Berzofsky JA, Terabe M. NKT cells in tumor immunity: opposing subsets define a new immunoregulatory axis. J Immunol 2008; 180:3627-35; PMID:18322166; http://dx.doi.org/ 10.4049/jimmunol.180.6.3627 [DOI] [PubMed] [Google Scholar]
- 14.Pilones KA, Aryankalayil J, Demaria S. Invariant NKT cells as novel targets for immunotherapy in solid tumors. Clin Dev Immunol 2012; 2012:720803; PMID:23118781; http://dx.doi.org/ 10.1155/2012/720803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagnara D, Ibatici A, Corselli M, Sessarego N, Tenca C, De Santanna A, Mazzarello A, Daga A, Corvo R, De Rossi G et al.. Adoptive immunotherapy mediated by ex vivo expanded natural killer T cells against CD1d-expressing lymphoid neoplasms. Haematologica 2009; 94:967-74; PMID:19454494; http://dx.doi.org/ 10.3324/haematol.2008.001339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renukaradhya GJ, Sriram V, Du W, Gervay-Hague J, Van Kaer L, Brutkiewicz RR. Inhibition of antitumor immunity by invariant natural killer T cells in a T-cell lymphoma model in vivo. Int J Cancer 2006; 118:3045-53; PMID:16395717; http://dx.doi.org/ 10.1002/ijc.21764 [DOI] [PubMed] [Google Scholar]
- 17.Bjordahl RL, Gapin L, Marrack P, Refaeli Y. iNKT cells suppress the CD8+ T cell response to a murine Burkitt's-like B cell lymphoma. PLoS One 2012; 7:e42635; PMID:22880059; http://dx.doi.org/ 10.1371/journal.pone.0042635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, Godfrey DI, Ostrand-Rosenberg S, Smyth MJ, Berzofsky JA. A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med 2005; 202:1627-33; PMID:16365146; http://dx.doi.org/ 10.1084/jem.20051381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, Carbone DP, Paul WE, Berzofsky JA. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol 2000; 1:515-20; PMID:11101874; http://dx.doi.org/ 10.1038/82771 [DOI] [PubMed] [Google Scholar]
- 20.Ambrosino E, Terabe M, Halder RC, Peng J, Takaku S, Miyake S, Yamamura T, Kumar V, Berzofsky JA. Cross-regulation between type I and type II NKT cells in regulating tumor immunity: a new immunoregulatory axis. J Immunol 2007; 179:5126-36; PMID:17911598; http://dx.doi.org/ 10.4049/jimmunol.179.8.5126 [DOI] [PubMed] [Google Scholar]
- 21.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B et al.. Transforming growth factor-β production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med 2003; 198:1741-52; PMID:14657224; http://dx.doi.org/ 10.1084/jem.20022227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao J, Weng X, Bagchi S, Wang CR. Polyclonal type II natural killer T cells require PLZF and SAP for their development and contribute to CpG-mediated antitumor response. Proc Natl Acad Sci U S A 2014; 111:2674-9; PMID:24550295; http://dx.doi.org/ 10.1073/pnas.1323845111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lepore M, de Lalla C, Gundimeda SR, Gsellinger H, Consonni M, Garavaglia C, Sansano S, Piccolo F, Scelfo A, Haussinger D et al.. A novel self-lipid antigen targets human T cells against CD1c(+) leukemias. J Exp Med 2014; 211:1363-77; PMID:24935257; http://dx.doi.org/ 10.1084/jem.20140410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vincent MS, Xiong X, Grant EP, Peng W, Brenner MB. CD1a-, b-, and c-restricted TCRs recognize both self and foreign antigens. J Immunol 2005; 175:6344-51; PMID:16272286; http://dx.doi.org/ 10.4049/jimmunol.175.10.6344 [DOI] [PubMed] [Google Scholar]
- 25.Shamshiev A, Donda A, Carena I, Mori L, Kappos L, De Libero G. Self glycolipids as T-cell autoantigens. Eur J Immunol 1999; 29:1667-75; PMID:10359121; http://dx.doi.org/ 10.1002/(SICI)1521-4141(199905)29:05%3c1667::AID-IMMU1667%3e3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- 26.de Jong A, Pena-Cruz V, Cheng TY, Clark RA, Van Rhijn I, Moody DB. CD1a-autoreactive T cells are a normal component of the human alphabeta T cell repertoire. Nat Immunol 2010; 11:1102-9; PMID:21037579; http://dx.doi.org/ 10.1038/ni.1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Rhijn I, Kasmar A, de Jong A, Gras S, Bhati M, Doorenspleet ME, de Vries N, Godfrey DI, Altman JD, de Jager W et al.. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat Immunol 2013; 14:706-13; PMID:23727893; http://dx.doi.org/ 10.1038/ni.2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shamshiev A, Donda A, Prigozy TI, Mori L, Chigorno V, Benedict CA, Kappos L, Sonnino S, Kronenberg M, De Libero G. The alphabeta T cell response to self-glycolipids shows a novel mechanism of CD1b loading and a requirement for complex oligosaccharides. Immunity 2000; 13:255-64; PMID:10981968; http://dx.doi.org/ 10.1016/S1074-7613(00)00025-X [DOI] [PubMed] [Google Scholar]
- 29.Shamshiev A, Gober HJ, Donda A, Mazorra Z, Mori L, De Libero G. Presentation of the same glycolipid by different CD1 molecules. J Exp Med 2002; 195:1013-21; PMID:11956292; http://dx.doi.org/ 10.1084/jem.20011963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Lalla C, Lepore M, Piccolo FM, Rinaldi A, Scelfo A, Garavaglia C, Mori L, De Libero G, Dellabona P, Casorati G. High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol 2011; 41:602-10; PMID:21246542; http://dx.doi.org/ 10.1002/eji.201041211 [DOI] [PubMed] [Google Scholar]
- 31.de Jong A, Cheng TY, Huang S, Gras S, Birkinshaw RW, Kasmar AG, Van Rhijn I, Pena-Cruz V, Ruan DT, Altman JD et al.. CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Immunol 2014; 15:177-85; PMID:24362891; http://dx.doi.org/ 10.1038/ni.2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Rhijn I, van Berlo T, Hilmenyuk T, Cheng TY, Wolf BJ, Tatituri RV, Uldrich AP, Napolitani G, Cerundolo V, Altman JD et al.. Human autoreactive T cells recognize CD1b and phospholipids. Proc Natl Acad Sci U S A 2016; 113:380-5; PMID:26621732; http://dx.doi.org/ 10.1073/pnas.1520947112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mansour S, Tocheva AS, Cave-Ayland C, Machelett MM, Sander B, Lissin NM, Molloy PE, Baird MS, Stubs G, Schroder NW et al.. Cholesteryl esters stabilize human CD1c conformations for recognition by self-reactive T cells. Proc Natl Acad Sci U S A 2016; 113:E1266-75; PMID:26884207; http://dx.doi.org/ 10.1073/pnas.1519246113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Felio K, Nguyen H, Dascher CC, Choi HJ, Li S, Zimmer MI, Colmone A, Moody DB, Brenner MB, Wang CR. CD1-restricted adaptive immune responses to Mycobacteria in human group 1 CD1 transgenic mice. J Exp Med 2009; 206:2497-509; PMID:19808251; http://dx.doi.org/ 10.1084/jem.20090898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li S, Choi HJ, Felio K, Wang CR. Autoreactive CD1b-restricted T cells: a new innate-like T-cell population that contributes to immunity against infection. Blood 2011; 118:3870-8; PMID:21860021; http://dx.doi.org/ 10.1182/blood-2011-03-341941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Facciotti F, Ramanjaneyulu GS, Lepore M, Sansano S, Cavallari M, Kistowska M, Forss-Petter S, Ni G, Colone A, Singhal A et al.. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat Immunol 2012; 13:474-80; PMID:22426352; http://dx.doi.org/ 10.1038/ni.2245 [DOI] [PubMed] [Google Scholar]
- 37.Daly PF, Zugmaier G, Sandler D, Carpen M, Myers CE, Cohen JS. Regulation of the cytidine phospholipid pathways in human cancer cells and effects of 1-β-D-arabinofuranosylcytosine: a noninvasive 31P nuclear magnetic resonance study. Cancer Res 1990; 50:552-7; PMID:2153442 [PubMed] [Google Scholar]
- 38.Jahrsdorfer B, Weiner GJ. CpG oligodeoxynucleotides as immunotherapy in cancer. Update Cancer Ther 2008; 3:27-32; PMID:19255607; http://dx.doi.org/ 10.1016/j.uct.2007.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol 2007; 5:405-17; PMID:17487145; http://dx.doi.org/ 10.1038/nrmicro1657 [DOI] [PubMed] [Google Scholar]
- 40.Crowe NY, Smyth MJ, Godfrey DI. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J Exp Med 2002; 196:119-27; PMID:12093876; http://dx.doi.org/ 10.1084/jem.20020092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nair S, Boddupalli CS, Verma R, Liu J, Yang R, Pastores GM, Mistry PK, Dhodapkar MV. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood 2015; 125:1256-71; PMID:25499455; http://dx.doi.org/ 10.1182/blood-2014-09-600270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brennan PJ, Tatituri RV, Brigl M, Kim EY, Tuli A, Sanderson JP, Gadola SD, Hsu FF, Besra GS, Brenner MB. Invariant natural killer T cells recognize lipid self-antigen induced by microbial danger signals. Nat Immunol 2011; 12:1202-11; PMID:22037601; http://dx.doi.org/ 10.1038/ni.2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brennan PJ, Tatituri RV, Heiss C, Watts GF, Hsu FF, Veerapen N, Cox LR, Azadi P, Besra GS, Brenner MB. Activation of iNKT cells by a distinct constituent of the endogenous glucosylceramide fraction. Proc Natl Acad Sci U S A 2014; 111:13433-8; PMID:25197085; http://dx.doi.org/ 10.1073/pnas.1415357111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, Zhao M, Self K, Teyton A, Everett C et al.. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian α-linked glycosylceramides. Immunity 2014; 41:543-54; PMID:25367571; http://dx.doi.org/ 10.1016/j.immuni.2014.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gumperz JE, Roy C, Makowska A, Lum D, Sugita M, Podrebarac T, Koezuka Y, Porcelli SA, Cardell S, Brenner MB et al.. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity 2000; 12:211-21; PMID:10714687; http://dx.doi.org/ 10.1016/S1074-7613(00)80174-0 [DOI] [PubMed] [Google Scholar]
- 46.Lasek W, Zagozdzon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother 2014; 63:419-35; PMID:24514955; http://dx.doi.org/ 10.1007/s00262-014-1523-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Townsend A, Ohlen C, Bastin J, Ljunggren HG, Foster L, Karre K. Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature 1989; 340:443-8; PMID:2666863; http://dx.doi.org/ 10.1038/340443a0 [DOI] [PubMed] [Google Scholar]
- 48.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957; 226:497-509; PMID:13428781 [PubMed] [Google Scholar]
- 49.Canaday DH. Production of CD4(+) and CD8(+) T cell hybridomas. Methods Mol Biol 2013; 960:297-307; PMID:23329495; http://dx.doi.org/ 10.1007/978-1-62703-218-6_22 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.