

Abstract
The fidelity of DNA replication together with repair mechanisms ensure that the genetic material is properly copied from one generation to another. However, on extremely rare occasions when damages to DNA or replication errors are not repaired, germline mutations can be transmitted to the next generation. Because of the rarity of these events, studying the rate at which new mutations arise across organisms has been a great challenge, especially in multicellular nonmodel organisms with large genomes. We sequenced the genomes of 11 birds from a three-generation pedigree of the collared flycatcher (Ficedula albicollis) and used highly stringent bioinformatic criteria for mutation detection and used several procedures to validate mutations, including following the stable inheritance of new mutations to subsequent generations. We identified 55 de novo mutations with a 10-fold enrichment of mutations at CpG sites and with only a modest male mutation bias. The estimated rate of mutation per site per generation was 4.6 × 10−9, which corresponds to 2.3 × 10−9 mutations per site per year. Compared to mammals, this is similar to mouse but about half of that reported for humans, which may be due to the higher frequency of male mutations in humans. We confirm that mutation rate scales positively with genome size and that there is a strong negative relationship between mutation rate and effective population size, in line with the drift-barrier hypothesis. Our study illustrates that it should be feasible to obtain direct estimates of the rate of mutation in essentially any organism from which family material can be obtained.
The rate of mutation is one of the most central parameters in evolutionary and population genetics (Lynch 2010a) but is notoriously difficult to accurately estimate. The primary difficulty lies in that mutation rates are extremely low (Drake et al. 1998; Lynch 2010a), and detecting mutations when they arise has therefore constituted a formidable task, especially in eukaryotes with large genomes. In classical genetics, the approach has been to derive locus-specific rate estimates based on observable phenotypes in crosses or pedigrees (Stadler 1930; Schalet 1960; Russell and Russell 1996). However, unless the question of interest is the rate of mutation by which penetrant phenotypes arise, a representative estimate of the per-locus (or per-nucleotide) mutation rate can only be obtained by such methods if the strength of selection for recessive or dominant phenotypes can concurrently be estimated. In practice, such rate estimates are therefore only gross approximations. Phenotypic effects have also been exploited for mutation rate estimation in mutation accumulation experiments (the Bateman-Muller-Mukai method [Muller 1928; Bateman 1959; Mukai 1964] sensu Drake and colleagues [Drake et al. 1998]), in which phenotypic effects are recorded at the end of the experiment after line propagation.
A shift in the way mutation rate could be estimated came about with DNA sequencing technology and by the appreciation that the rate of neutral sequence divergence is equal to the rate of mutation (Kimura 1968). If orthologous, neutral sequences of two species are aligned and compared, it is relatively straightforward in principle to estimate divergence and from there indirectly estimate the rate of mutation (Kondrashov and Crow 1993; Nachman and Crowell 2000; Keightley 2012). However, there are a number of caveats associated with this by now widely used phylogenetic approach. Some of these caveats are related to methodology (e.g., sequence alignment, models applied for taking multiple mutations into account), and others to the underlying assumption of selective neutrality of the sequences analyzed and to the scaling parameters needed to convert divergence estimates to mutation rate estimates (e.g., divergence time and generation time). It has subsequently turned out that the phylogenetic approach can give substantially different mutation rate estimates compared with approaches that rely on more direct ways of detecting mutations (Shendure and Akey 2015).
Next-generation sequencing has provided novel means for direct detection of germline mutations by allowing for comparisons of genome sequences between subsequent generations (Scally and Durbin 2012). One way of benefitting from the power of genomic resequencing for mutation rate estimation is to employ mutation accumulation lines (e.g., Ossowski et al. 2010), in which mutations have built up over generations, but this approach is bound to be limited to organisms in which controlled line propagation in the laboratory is feasible. Of more general applicability is the possibility to sequence multiple individuals of pedigreed families, including parent–offspring trios or larger two- or three-generation pedigrees. A suite of such studies has recently been reported for humans (Awadalla et al. 2010; Roach et al. 2010; Conrad et al. 2011; Kong et al. 2012; Michaelson et al. 2012; Samocha et al. 2014; Francioli et al. 2015), revealing an estimated rate of point mutation in human of ≈1 × 10−8 per nucleotide site and generation. By the identification of large numbers of de novo mutations, this has also revealed the spectrum of mutations (Lynch 2010b; Shendure and Akey 2015). The rate of human mutation is among the highest observed across the relatively limited number of eukaryotic species so far investigated, spanning a range of at least two orders of magnitude of variation with unicellular organisms in the lower end (Lynch 2010a).
Pedigree sequencing should in principle be applicable to any organism (Keightley et al. 2014, 2015; Venn et al. 2014) and thus has the potential to unveil the rate of mutations in organisms in which such estimates previously have been out of reach, like for the vast majority of all vertebrates. Here we estimate the rate of spontaneously arising mutations in an avian species—the collared flycatcher (Ficedula albicollis)—a small (≈15 g) migratory songbird breeding in Europe and wintering in sub-Saharan Africa. This is a short-lived species in which most individuals start breeding at the age of 1 yr, with ≈50% of breeders in the population being 1-yr-old individuals and with a generation time of 2 yr (Brommer et al. 2004). Because mutation rate is hypothesized to scale with genome size (Lynch 2010a), as well as with the effective population size (Ne) (Sung et al. 2012a), mutation data from birds would be valuable for further testing of this hypothesis given that the size of avian genomes is intermediate (≈1 Gb) to that of mammals (several Gb) and of other animals and plants for which mutation rate estimates are available (≈100–200 Mb). Moreover, alternative means for translating observed sequence divergence into divergence times would act complementary to calibration points based on fossil records for molecular dating in phylogenetic analysis. The collared flycatcher has been subject to detailed genomic investigation, and there is a 1.123-Gb genome assembly with a super-scaffold N50 of 20.2 Mb and with 93.4% of the assembly anchored, ordered, and oriented to chromosomes via a high-density genetic linkage map (Ellegren et al. 2012; Kawakami et al. 2014; Smeds et al. 2015). Levels of genetic diversity in this species are intermediate to that observed in humans and Drosophila melanogaster, with a mean nucleotide diversity (π) of ≈4 × 10−3 in different populations (Burri et al. 2015). We performed deep resequencing of 11 members of a three-generation flycatcher pedigree and detected de novo mutations by applying highly stringent filtering criteria and independent validation by genotyping and by following the inheritance of mutations in the pedigree.
Results
Identification of de novo mutations
A complete three-generation pedigree (paternal and maternal grandparents, father, mother, and five offspring) (Fig. 1) was resequenced at a mean coverage of 42× with Illumina technology and 2 × 100-bp paired-end reads. A total of 11.7 million SNPs, before filtering, was detected to segregate in the pedigree, which is in line with expectations from the level of nucleotide diversity previously seen in the population (Ellegren et al. 2012). We carefully screened F1 and F2 individuals for novel sequence variants, applying highly stringent filtering criteria (coverage, quality, unambiguous presence/absence of reads across the pedigree), and identified 55 heterozygous positions in F1 and/or F2 birds (two to 13 per individual) that were homozygous for the reference allele in all grandparents. These positions (reported in Supplemental Table S1) represent sites of putative de novo mutation events, and none of the sites had previously been detected as segregating in the study population, based on resequencing data of more than 100 individuals.
Figure 1.
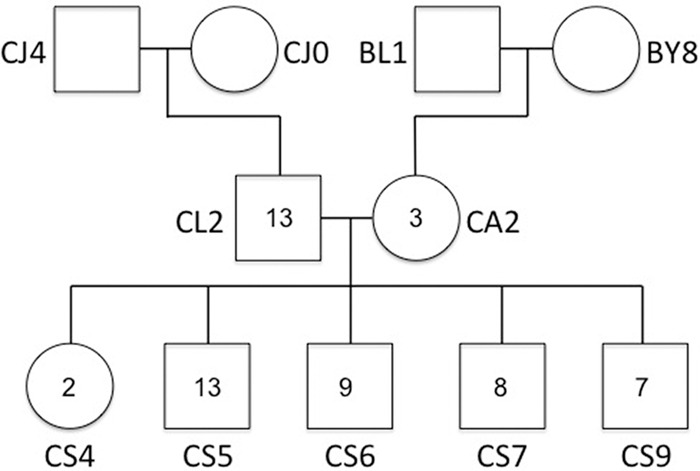
Collared flycatcher pedigree used for mutation detection by whole-genome resequencing. The number of de novo mutations detected in each F1 and F2 offspring is shown within individual symbols.
Sixteen mutations were detected in one (but never both) of the F1 parents, and 15 of these mutations were transmitted to F2 offspring with a distribution of the number of offspring showing the mutant allele closely following binomial expectations (χ2 = 3.8, d.f. = 5, P = 0.58) (Fig. 2). Note that one mutant out of 16 not being transmitted to any of five offspring has a probability of 0.5 and is thus entirely plausible. These observations confirm germline origin and stable Mendelian inheritance of newly arisen mutations. The remaining 36 mutation events were detected in single F2 offspring showing an alternative allele not detected in the P and F1 generations. The proportions of mutations detected in the F1 (16/55 = 29.1%) and F2 generations (70.9%) were in perfect agreement with the proportions of meiosis scored in the P (4/14 = 28.6%) and F1 (10/14 = 71.4%) generations.
Figure 2.
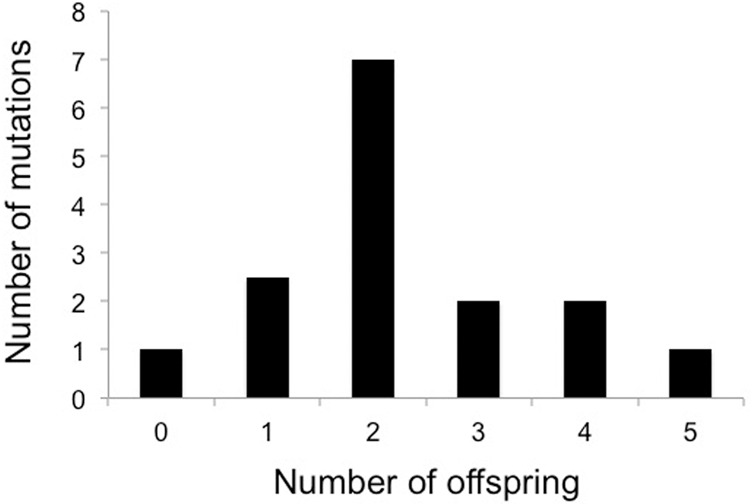
Distribution of the number of F2 offspring to which mutations originating in the F1 generation were transmitted.
Validation of de novo mutations
In addition to confirmation by inheritance, we sought to validate mutations by SNP genotyping. Thirty-two assays met the recommended Illumina GoldenGate assay quality criteria for a high likelihood of assay conversion and valid genotyping (design score, >0.6). After genotyping of the pedigree and 20 unrelated birds from the population, we obtained unambiguous genotype calls from 31 assays. All of these confirmed the mutation event: A heterozygous genotype was only seen in those individuals from the pedigree in which the mutation was detected by sequencing, while all other individuals of the pedigree as well as of the population sample were homozygous for the reference allele. For another 12 mutation events, the assay did not meet recommended quality criteria (design score, 0–0.6), yet genotyping was attempted and confirmed the mutation in 11 cases. In summary, given the multiple lines of evidence supporting the validity of the detected mutations, we consider the false-positive rate as very low, and the analyses presented below are therefore based on the whole set of detected mutation events. Given the depth of coverage, we also consider the rate of false negatives as very low.
Characteristics of de novo mutations
There were 33 mutations in intergenic regions, 21 in introns, and one in coding sequence. The flycatcher genome consists of 67.5% intergenic DNA, 30.4% introns, and 2.1% coding sequence. The distribution of observed mutation events among the three sequence categories does not differ from expectation based on the genomic frequency of the respective categories (χ2 = 1.57, P = 0.45). The single coding sequence mutation was a nonsynonymous change in the carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 5 gene (CHST5). The distribution of mutations among chromosomes did not deviate from expectations from the size distribution of chromosomes (χ2 = 18.7, d.f. = 18, P = 0.41).
The transition/transversion ratio was 2.67 (95% confidence interval, CI = 1.56–5.44), which is similar to what is typically seen in molecular evolutionary data as well as in screens for de novo mutations in humans (Kong et al. 2012). Seven mutations were C>T or G>A transitions at CpG sites. Based on the frequency of CpG sites in the avian genome (Mugal et al. 2015) and assuming a uniform mutation rate, only 0.51 CpG mutations would have been expected (P < 10−6, binomial test), indicating an approximately 10-fold increase in the rate of mutation at CpG sites due to cytosine methylation and deamination. In total, there were 17 A:T>G:C and 30 G:C>A:T mutations (Table 1), confirming a mutation pressure in the direction of A+T previously seen in both eukaryotes (Lynch 2010b) and prokaryotes (Hershberg and Petrov 2010).
Table 1.
Direction of 55 de novo mutations in collared flycatcher

There was only a modest excess of paternally derived mutations with 18 mutations traceable to originate from the male germline and 15 from the female germline; the parent-of-origin was determined by reads or read-pairs spanning the mutation and a nearby heterozygous site unique to one of the parents. This corresponds to a male-to-female mutation rate ratio of 1.20 (95% CI = 0.60–2.51). It should be noted that the sequenced male of the F1 generation (in which most mutations arose) was only 1 yr old, potentially reducing the paternal excess of mutations in this material because the male mutation rate generally increases with age (Crow 2000). On a related note, it is intriguing that the two female F1 and F2 offspring both had fewer mutations detected (two and three, respectively) than all five male offspring (7–13). It is difficult to see a biologically plausible explanation to this observation.
Mutation rate estimates
With 55 mutations observed in 14 transmissions, there was a mean of 3.9 mutation events per meiosis. The amount of sequence available for screening after filtering ranged between 844.9–852.8 Mb, with a mean of 848.3 Mb, approximately representing 80% of the genome (see Methods; parts not included were either repetitive or did not meet filter criteria for coverage and quality). The rate of mutation can thus be estimated to be 4.6 × 10−9 (95% CI = 3.4 × 10−9–5.9 × 10−9, assuming a Poisson distribution) per site per meiosis, which is the same as per site per generation or per site per haploid genome. Based on a long-term field study of more than 1400 breeding attempts recorded during 20 yr, Brommer et al. (2004) reported the generation time of the (female) collared flycatcher to be 1.8 yr. This should be adjusted to 2.0 yr by taking differences in reproductive output among age classes into account. Based on these data, the estimated rate of mutation per site per year in the flycatcher is 2.3 × 10−9 (95% CI = 1.7 × 10−9–3.0 × 10−9).
With a detailed mutation rate estimate available, we can provide rule of thumb figures for how neutral sequence divergence scales with divergence time in bird lineages with similar generation times as in flycatchers. Specifically, with a mutation rate of 2.3 × 10−9 per site per year, 1% sequence divergence at neutral sites would correspond to a divergence time of 4.3 Myr; 5% divergence, to 21.7 Myr (without correction for multiple hits).
Discussion
Lynch (2010a) stated “… to be fully reliable, future molecular investigations with a goal of interpreting evolutionary mechanisms should take advantage of direct estimates of mutation rates.” Indeed, mutation rate estimates are necessary to many applications of population genetics, phylogenetics, and molecular evolution (Lynch 2010a). For example, the population genetic parameter θ predicts the amount of genetic diversity in a population, and based on mutation rate data, estimates of θ can be used to infer selection and demography. In phylogenetics, mutation rate estimates are required to convert branch lengths into time units (molecular dating).
The current knowledge on the rate of mutation in the nuclear genome of birds is limited. The existing rate estimates (e.g., Axelsson et al. 2004) have relied on the phylogenetic approach and have been limited by at least two sources of uncertainty. First, as is inherent to such analyses, selecting a genomic category of selectively neutral sequences is nontrivial. For instance, just as in other organisms (Chamary et al. 2006), there is some evidence that fourfold degenerate sites—often taken to represent a neutral reference—may evolve under constraint (Künstner et al. 2011), and avian intergenic regions and introns contain a wealth of conserved regulatory elements (Zhang et al. 2014; Lowe et al. 2015). Inherent problems associated with such analyses also include taking multiple hits into account and obtaining reliable alignments. Second, the fossil record of birds is not extensive, and fossil calibration points are necessary to estimate mutation rates from divergence data.
Based on direct observations of 55 mutation events in a three-generation pedigree of flycatchers, we estimated the spontaneous rate of germline mutation to be 4.6 × 10−9 (3.4 × 10−9–5.9 × 10−9) per site per generation or 2.3 × 10−9 (1.7 × 10−9–3.0 × 10−9) per site per year. This is very similar to a point estimate based on divergence at fourfold degenerate sites in the lineage leading to zebra finch (2.2 × 10−9 per site per year) (Nam et al. 2010), which like flycatchers belongs to the order Passeriformes. It is somewhat higher than point estimates obtained for Galliformes, including chicken and turkey, based on intronic divergence (1.3 × 10−9) (Axelsson et al. 2004) and divergence at fourfold degenerate sites (1.9 × 10−9) (Nam et al. 2010). It thus seems that the fossil-calibrated and sequence divergence–based mutation rate estimates of birds agree well with the direct approach applied herein; however, the fact that these previous estimates were likely associated with large variances makes inference less conclusive.
The rate of synonymous substitution (dS) has been found to vary significantly among bird lineages (Galtier et al. 2009; Lanfear et al. 2010; Nabholz et al. 2011), suggestive of an underlying variation in the rate of mutation. Weber et al. (2014) found a negative correlation between dS and body size among 48 bird species, with body size representing a proxy for generation time (i.e., assuming longer generation times in larger birds). Their results hence support the generation-time hypothesis for rate of molecular evolution in birds since short-lived species are likely to undergo more generations per time unit, and thereby be exposed to more opportunities for germline mutation, than long-lived species. This assumes that most mutations are arising in replicating DNA. We may expect the generation time of most birds to be in the range 2–10 yr (De Magalhäes and Costa 2009). If the per-generation mutation rate is constant across bird species, this would lead to a predicted fivefold rate variation per year among avian lineages, with the estimate in flycatcher (2.3 × 10−9) representing the upper end. However, the per-generation mutation rate may not necessarily be constant across species, and additional pedigree-based studies directly estimating the rate of mutation in different avian groups will be needed to resolve this matter.
How does our estimate of the mutation rate in flycatchers (4.6 × 10−9) compare to direct estimates of the rate of mutation in other organisms? Studies of pedigreed families from different human populations have reported rates of 1.0 × 10−8–1.4 × 10−8 per site per generation (Awadalla et al. 2010; Roach et al. 2010; Conrad et al. 2011; Kong et al. 2012). Similarly, a rate of 1.2 × 10−8 has been obtained for chimpanzee (Venn et al. 2014). The ≈2.5 times higher rate in hominids than in at least one avian taxon indicates that the fidelity of germline DNA replication and efficiency of repair are lower among the former. However, this may not necessarily be the case on a per-cell division basis. The rate of germline mutation in humans and chimpanzee shows a pronounced male bias and is strongly influenced by father's age (Kong et al. 2012; Francioli et al. 2015), most likely due to the accumulating number of mitotic cell divisions in male germline during life. A 20-yr-old man is expected to transmit about 40 new mutations to each child, whereas the number doubles at the age of 40 (Kong et al. 2012), approximately corresponding to male-to-female mutation rate ratios (α) of three and six, respectively. The male mutation bias in birds is less pronounced (Ellegren 2007), which we confirmed in this study. The higher per-generation rate of mutation in humans and chimpanzees compared with the flycatcher may thus at least in part be explained by a larger fraction of paternally derived mutations in the former species. Specifically, assuming that the human mutation rate of 1.2 × 10−8 is driven by a mean α of four (cf. Kong et al. 2012), halving α would give a rate of 6.6 × 10−9, and if there were no male excess, the rate would only be 4.5 × 10−9, i.e., essentially identical to our avian estimate. A strong paternal age effect in man may also explain why the per-generation mutation rate in mice (5.4 × 10−9) (Uchimura et al. 2015), in which α is about two (Sandstedt and Tucker 2005), is more similar to our estimate for birds than to the estimates for humans and chimpanzee. If annual rates are considered, the estimate we obtained for flycatchers (2.3 × 10−9 per site per year) is much higher than that in humans (4.4 × 10−10, assuming a generation time of 25 yr) but much less than in mice (1.1 × 10−8, conservatively assuming a generation time of 0.5 yr).
Table 2 summarizes direct estimates of the rate of mutation from studies using sequencing of pedigrees or mutation accumulation lines in other organisms. Besides the above-mentioned work on humans and chimpanzee, the most extensive pedigree sequencing study is the report of Yang et al. (2015) on selfing lines of Arabidopsis thaliana and rice and on honey bee. There are also two insect studies with a limited number of mutations detected in D. melanogaster and Heliconius melpomene families (Keightley et al. 2014, 2015). A positive scaling of mutation rate with genome size has previously been reported for eukaryotes (Lynch 2010a), which we confirm based on new data from this and other recent studies (r = 0.538) (Fig. 3A). This does not necessarily imply a causal effect of genome size on the rate of mutation (per nucleotide) since such effect may come from factors that covary with genome size. Specifically, it has been suggested that mutation rate evolution conforms to the drift-barrier hypothesis (Lynch 2010a, 2011; Sung et al. 2012a), which postulates that at some point the selective advantage of mutation rate modifiers further reducing the incidence of deleterious mutations becomes smaller than the power of genetic drift. The hypothesis leads to the prediction that mutation rate should scale negatively with Ne as the extent of genetic drift is a direction function of Ne. We used reported levels of nucleotide diversity (π) to solve Ne from the formula θ = 4Neµ (where µ is the mutation rate and by replacing θ with π) to analyze the relationship between mutation rate (again based on direct estimates) and Ne. There is a strong negative exponential relationship between the two parameters (µ = Ne−0.46, r = 0.88), in support of the drift-barrier hypothesis (Fig. 3B).
Table 2.
Summary of direct estimates of the germline mutation rate in different organisms based on pedigree sequencing (PS) or sequencing of mutation accumulation lines (MA)
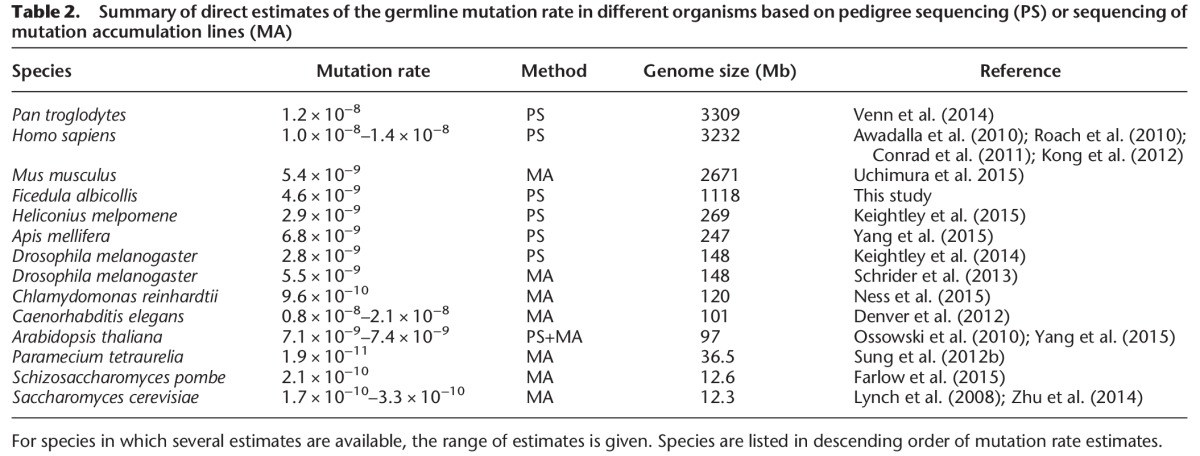
Figure 3.
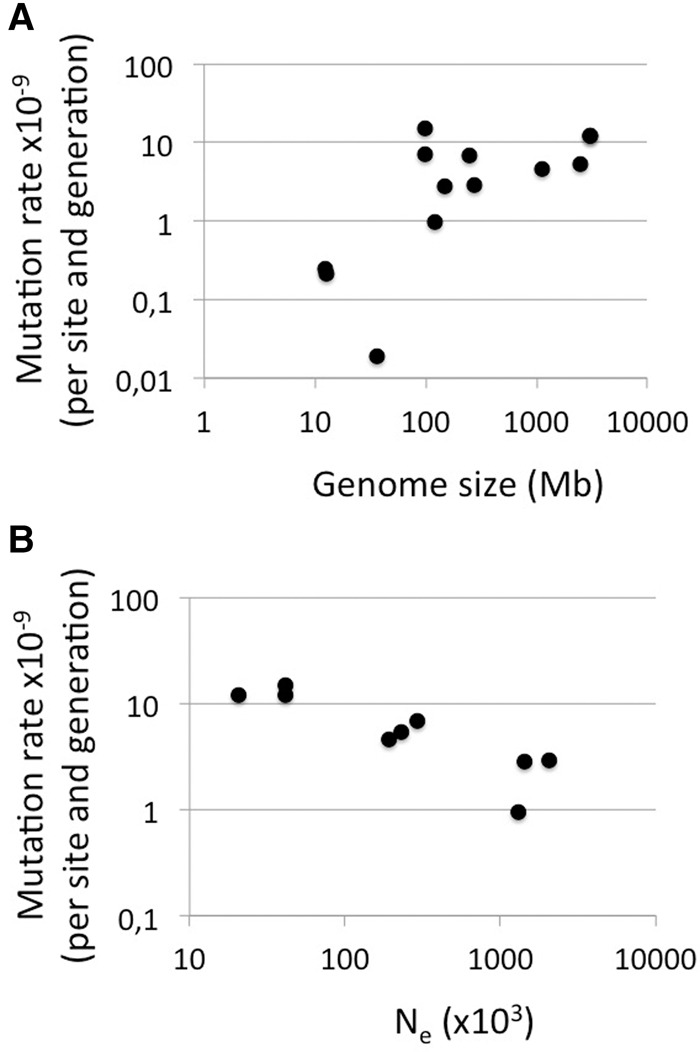
Relationship between mutation rate (per nucleotide and generation) and genome size (A) and effective population size (Ne) (B). Mutation rate estimates were taken from Table 2 and the references cited therein. Genome size is the length of assembled genome sequence as available at http://www.ncbi.nlm.nih.gov. For B, the species are in order of increasing Ne: Homo sapiens (π used to estimate Ne from the International SNP Map Working Group 2001), Pan troglodytes (The Chimpanzee Sequencing and Analysis Consortium 2005), Caenorhabditis elegans (Cutter et al. 2009), Ficedula albicollis (Burri et al. 2015), Mus musculus (Lindblad-Toh et al. 2000), Apis mellifera (Wallberg et al. 2014), Chlamydomonas reinhardtii (Flowers et al. 2015), Drosophila melanogaster (Andolfatto 2001), and Heliconius melpomene (Keightley et al. 2015).
We judge the false-positive rate in our approach to be very low. In addition to the use of a stringent bioinformatic pipeline, including final manual inspection, this conclusion was supported by validation by independent genotyping, by the fact that sites of mutation events were monomorphic in large population samples, and by the fact that stable inheritance was confirmed for mutations first appearing in the F1 generation. However, measures to ensure a low false-positive rate may in theory come with the price of false negatives. Using a very similar bioinformatic pipeline for mutation detection as in our study, Keightley et al. (2014) addressed the rate of false negatives by adding “synthetic” mutations to read data from a D. melanogaster pedigree consisting of 14 individuals. The pipeline detected 99.4% of all callable synthetic mutations, suggesting that the rate of false negatives was negligible. We find no reason to expect that the false-negative rate should be significantly different in our study. One methodological aspect that is worth acknowledging is that we filtered both sites that were called heterozygous in the P generation and candidate mutations in the F1 or F2 generations that corresponded to known segregating alleles in the population. In theory, heterozygous/segregating sites could represent mutational hotspots, potentially leading us to underestimate the rate of mutation. However, heterozygosity in collared flycatcher is <0.25% (Burri et al. 2015) so polymorphic sites only constitute a minor fraction of the genome.
This study focused on the rate of point mutation. There are obviously several other types of mutations, including short insertions-deletions, larger structural mutations (e.g., inversions and transpositions), and copy number mutations in tandem repetitive DNAs, like minisatellites and microsatellites. Of these, the highest rate is expected for hypermutable tandem repeat sequences. In birds, tetranucleotide repeat microsatellites with a mutation rate greater than 1 × 10−2 have been reported (Primmer et al. 1998; Beck et al. 2003), although loci with such high rates are probably rare in the genome. An interesting possibility for future research will be to use whole-genome sequence information from pedigrees combined with efficient algorithms for repeat profiling to obtain genome-wide estimates of microsatellite mutation rates (Gymrek et al. 2012). Another aspect worth mentioning is that our filtering criteria would eliminate possible de novo mutations for which the parent is mosaic.
To summarize, extrapolating from a mean of 3.9 new mutations found in screening ≈80% of the genome, there are about five new point mutations in the 1.1 billion-bp genome of every flycatcher, clearly indicating the needle-in-the-haystack challenge of finding de novo mutations. Yet, with careful processing of sequence data, our study demonstrates the feasibility of estimating the spontaneous rate of germline mutation in nonmodel species. We foresee that the approach taken herein should be applicable to essentially any species from which DNA samples of families can be collected and that a suite of direct mutation rate estimates should thus become available in the near future. This will aid in further elucidating the determinants and constraints of mutation rate evolution.
Methods
Samples and sequencing
Blood samples from 11 collared flycatchers from a three-generation pedigree (Supplemental Table S2) were collected on Öland, Sweden, as part of a long-term study following the breeding biology of the population. DNA was extracted by a standard proteinase K digestion/phenol-chloroform purification protocol, and each individual was sequenced to ≈40× coverage (Supplemental Table S2) on an Illumina HiSeq instrument with paired-end 100-bp reads and an approximate library insert size of 450 bp. The reads were aligned to the collared flycatcher reference genome FicAlb1.5 (GenBank Accession GCA_000247815.2) with BWA 0.7.5a (Li and Durbin 2009), deduplicated, recalibrated, and cleaned with GATK 3.2.2 (DePristo et al. 2011).
Variant calling
Variants were called with GATKs HaplotypeCaller and GenotypeGVCFs (version 3.3.0). We did not perform variant quality recalibration (VQSR) according to the best practices since de novo mutations, if not transmitted between generations, should only occur in single individuals and are therefore more likely to be filtered out as low-quality variants. Instead we applied an extensive set of hard filters to increase the likelihood of only calling true variants. Repetitive regions were masked with a combination of RepeatMasker v3.2.9 (Smit et al. 1996–2010) and a flycatcher-specific repeat library (Smeds et al. 2015), Tandem Repeats Finder v4.07 (Benson 1999), and a custom Perl script to remove any homopolymers >10 bp that were not already masked. Then each site had to pass GATKs CallableLoci level and genotype quality (GQ) had to be at least 30. Since we only considered single-nucleotide variants (SNV) in this study, all called insertions and deletions (indels) were masked; there were 2.4 million indels segregating in the pedigree.
A hard coverage threshold of 15 was used to minimize false variant calls due to insufficient read data (89.2%–90.7% of the genome met this criterion). This represents a very stringent per-site coverage filter and was considered important as to reduce the initial frequency of false positives before further quality control as described below. After the filtering we were left with 845–853 Mb sequence per individual (∼80% of the genome and basically the same 80% among all of the individuals).
Detection of de novo mutations
Screening for new mutations represents a challenging task and has to be treated with the utmost care (see, e.g., the useful discussion by Keightley et al. 2014). Roach et al. (2010) noted “… most apparent aberrations in allele inheritance will be due to errors in the data and not to mutation.” We applied extremely stringent filtering in attempts to minimize the false-discovery rate. For each individual in the F1 and F2 generations, heterozygous positions were extracted from the background and had to meet the following criteria to be considered as potential de novo mutations:
No alternative reads in any of the parents (making parental mosaicism unlikely),
No other individuals in the same or the previous generation(s) are heterozygous or homozygous for the alternative allele,
At least 25% of the reads support the alternative allele,
Does not overlap with known SNPs from genomic resequencing of more than 100 birds from the same population (Burri et al. 2015; Kardos et al. 2016), and
Both parents are homozygous for the reference allele.
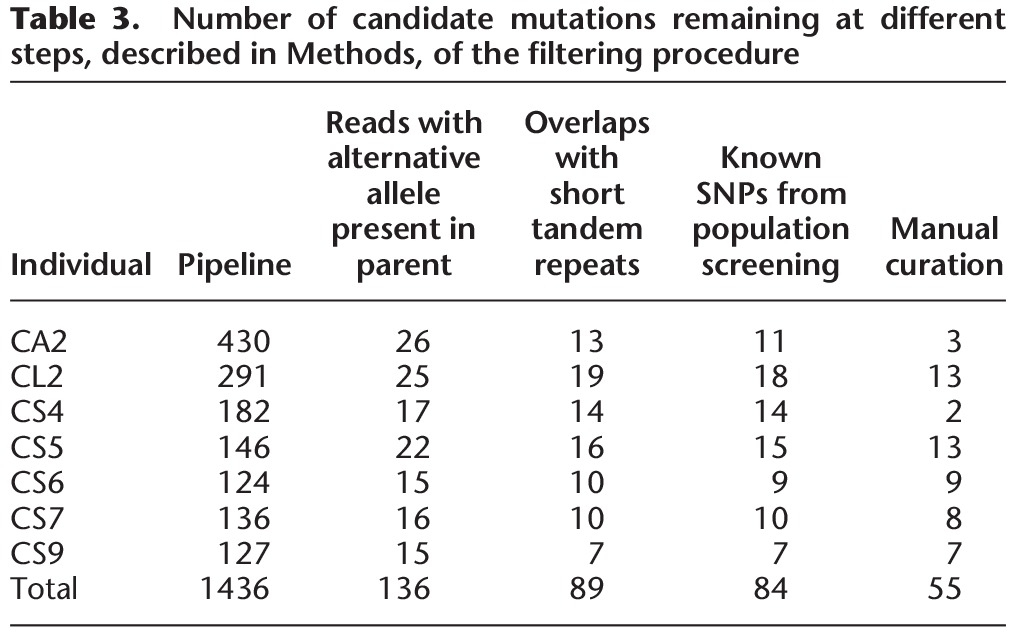
To manually curate potentially mutated positions, we used the SAMtools mpileup of BAM files and, similar to Keightley et al. (2014), the Integrated Genomics Viewer (IGV) (Thorvaldsdóttir et al. 2013). The latter was particularly valuable for detection of mapping errors and insertions or deletions associated with candidate mutations. The type of false positives detected in this way are well described by the examples shown in the supplemental figures S1 through S4 by Keightley et al. (2014). In Table 3 the number of candidate mutations remaining after each filtering step is provided, and we suggest that reporting this should be standard in this type of study.
Table 3.
Number of candidate mutations remaining at different steps, described in Methods, of the filtering procedure
SNP genotyping
We extracted flanking sequences for all mutation events and attempted to convert each event into a single-nucleotide polymorphism assay using the Illumina GoldenGate platform. We used the “design score,” a proprietary algorithm provided by the manufacturer, to assess the conversion rate. Eleven events failed assay design. Genotyping was performed at the SNP & SEQ Technology Platform, Uppsala University (http://molmed.medsci. uu.se/SNP+SEQ+Technology+Platform/). Genotype data are provided in Supplemental Table S3.
Data access
Raw sequence reads and all variant data from this study have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under accession numbers ERX1326426 and ERZ312631, respectively.
Supplementary Material
Acknowledgments
We thank Michael Lynch and Carina Mugal for helpful discussions. Financial support was obtained from the European Research Council (AdG 249976), Knut and Alice Wallenberg Foundation, and the Swedish Research Council (2007–8731, 2010–5650, and 2013–8271).
Author contributions: L.S. performed all bioinformatic analyses. L.S. and H.E. analyzed the data. H.E. conceived of and designed the study and wrote the paper. A.Q. contributed with flycatcher family.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.204669.116.
Freely available online through the Genome Research Open Access option.
References
- Andolfatto P. 2001. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol Biol Evol 18: 279–290. [DOI] [PubMed] [Google Scholar]
- Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR, Côté M, Henrion E, Spiegelman D, Tarabeux J, et al. 2010. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet 87: 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, Smith NGC, Sundström H, Berlin S, Ellegren H. 2004. Male-biased mutation rate and divergence in autosomal, Z-linked and W-linked introns of chicken and turkey. Mol Biol Evol 21: 1538–1547. [DOI] [PubMed] [Google Scholar]
- Bateman AJ. 1959. The viability of near-normal irradiated chromosomes. Int J Radiat Biol Relat Stud Phys Chem Med 1: 170–180. [Google Scholar]
- Beck NR, Double MC, Cockburn A. 2003. Microsatellite evolution at two hypervariable loci revealed by extensive avian pedigrees. Mol Biol Evol 20: 54–61. [DOI] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brommer JE, Gustafsson L, Pietiäinen H, Merilä J. 2004. Single-generation estimates of individual fitness as proxies for long-term genetic contribution. Am Nat 163: 505–517. [DOI] [PubMed] [Google Scholar]
- Burri R, Nater A, Kawakami T, Mugal CF, Olason PI, Smeds L, Suh A, Dutoit L, Bureš S, Garamszegi LZ, et al. 2015. Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res 25: 1656–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. 2006. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet 7: 98–108. [DOI] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87. [DOI] [PubMed] [Google Scholar]
- Conrad D, Keebler J, DePristo M, Lindsay S, Zhang Y, Casals F, Idaghdour Y, Hartl C, Torroja C, Garimella K, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF. 2000. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1: 40–47. [DOI] [PubMed] [Google Scholar]
- Cutter AD, Dey A, Murray RL. 2009. Evolution of the Caenorhabditis elegans genome. Mol Biol Evol 26: 1199–1234. [DOI] [PubMed] [Google Scholar]
- De Magalhäes JP, Costa J. 2009. A database of vertebrate longevity records and their relation to other life-history traits. J Evol Biol 22: 1770–1774. [DOI] [PubMed] [Google Scholar]
- Denver DR, Wilhelm LJ, Howe DK, Gafner K, Dolan PC, Baer CF. 2012. Variation in base-substitution mutation in experimental and natural lineages of Caenorhabditis nematodes. Genome Biol Evol 4: 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. 2007. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc R Soc Lond B Biol Sci 274: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Smeds L, Burri R, Olason PI, Backstrom N, Kawakami T, Kunstner A, Makinen H, Nadachowska-Brzyska K, Qvarnstrom A, et al. 2012. The genomic landscape of species divergence in Ficedula flycatchers. Nature 491: 756–760. [DOI] [PubMed] [Google Scholar]
- Farlow A, Long H, Arnoux S, Sung W, Doak TG, Nordborg M, Lynch M. 2015. The spontaneous mutation rate in the fission yeast Schizosaccharomyces pombe. Genetics 201: 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers JM, Hazzouri KM, Pham GM, Rosas U, Bahmani T, Khraiwesh B, Nelson DR, Jijakli K, Abdrabu R, Harris EH, et al. 2015. Whole-genome resequencing reveals extensive natural variation in the model green alga Chlamydomonas reinhardtii. Plant Cell 27: 2353–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I; Genome of the Netherlands Consortium, van Duijn CM, Swertz M, Wijmenga C, et al. 2015. Genome-wide patterns and properties of de novo mutations in humans. Nat Genet 47: 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Blier PU, Nabholz B. 2009. Inverse relationship between longevity and evolutionary rate of mitochondrial proteins in mammals and birds. Mitochondrion 9: 51–57. [DOI] [PubMed] [Google Scholar]
- Gymrek M, Golan D, Rosset S, Erlich Y. 2012. lobSTR: a short tandem repeat profiler for personal genomes. Genome Res 22: 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. 2010. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet 6: e1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International SNP Map Working Group. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928–933. [DOI] [PubMed] [Google Scholar]
- Kardos M, Husby A, McFarlane E, Qvarnstrom A, Ellegren H. 2016. Whole genome resequencing of extreme phenotypes in collared flycatchers highlights the difficulty of detecting quantitative trait loci in natural populations. Mol Ecol Resour 16: 727–741. [DOI] [PubMed] [Google Scholar]
- Kawakami T, Smeds L, Backström N, Husby A, Qvarnström A, Mugal CF, Olason P, Ellegren H. 2014. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol Ecol 23: 4035–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD. 2012. Rates and fitness consequences of new mutations in humans. Genetics 190: 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Ness RW, Halligan DL, Haddrill PR. 2014. Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full-sib family. Genetics 196: 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Pinharanda A, Ness RW, Simpson F, Dasmahapatra KK, Mallet J, Davey JW, Jiggins CD. 2015. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol Biol Evol 32: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1968. Evolutionary rate at the molecular level. Nature 217: 624–626. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS, Crow JF. 1993. A molecular approach to estimating the human deleterious mutation rate. Hum Mutat 2: 229–234. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Künstner A, Nabholz B, Ellegren H. 2011. Significant selective constraint at fourfold degenerate sites in the avian genome and its consequence for detection of positive selection. Genome Biol Evol 3: 1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Ho SYW, Love D, Bromham L. 2010. Mutation rate is linked to diversification in birds. Proc Natl Acad Sci 107: 20423–20428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Winchester E, Daly MJ, Wang DG, Hirschhorn JN, Laviolette J-P, Ardlie K, Reich DE, Robinson E, Sklar P, et al. 2000. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet 24: 381–386. [DOI] [PubMed] [Google Scholar]
- Lowe CB, Clarke JA, Baker AJ, Haussler D, Edwards SV. 2015. Feather development genes and associated regulatory innovation predate the origin of Dinosauria. Mol Biol Evol 32: 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010a. Evolution of the mutation rate. Trends Genet 26: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010b. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci 107: 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2011. The lower bound to the evolution of mutation rates. Genome Biol Evol 3: 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci 105: 9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson Jacob J, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. 2012. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151: 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugal CF, Arndt PF, Holm L, Ellegren H. 2015. Evolutionary consequences of DNA methylation on the GC content in vertebrate genomes. G3 (Bethesda) 5: 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T. 1964. The genetic structure of natural populations of Drosophila melanogaster. I. Spontaneous mutation rate of polygenes controlling viability. Genetics 50: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1928. The measurement of gene mutation rate in Drosophila, its high variability, and its dependence upon temperature. Genetics 13: 279–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabholz B, Künstner A, Wang R, Jarvis ED, Ellegren H. 2011. Dynamic evolution of base composition: causes and consequences in avian phylogenomics. Mol Biol Evol 28: 2197–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. 2000. Estimate of the mutation rate per nucleotide in humans. Genetics 156: 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam K, Mugal C, Nabholz B, Schielzeth H, Wolf J, Backstrom N, Kunstner A, Balakrishnan C, Heger A, Ponting C, et al. 2010. Molecular evolution of genes in avian genomes. Genome Biol 11: R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Vasanthakrishnan RB, Colegrave N, Keightley PD. 2015. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii. Genome Res 25: 1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327: 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primmer CR, Saino N, Moller AP, Ellegren H. 1998. Unraveling the processes of microsatellite evolution through analysis of germ line mutations in barn swallows Hirundo rustica. Mol Biol Evol 15: 1047–1054. [Google Scholar]
- Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, et al. 2010. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328: 636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell LB, Russell WL. 1996. Spontaneous mutations recovered as mosaics in the mouse specific-locus test. Proc Natl Acad Sci 93: 13072–13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnstrom K, Mallick S, Kirby A, et al. 2014. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46: 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstedt S, Tucker P. 2005. Male-driven evolution in closely related species of the mouse genus Mus. J Mol Evol 61: 138–144. [DOI] [PubMed] [Google Scholar]
- Scally A, Durbin R. 2012. Revising the human mutation rate: implications for understanding human evolution. Nat Rev Genet 13: 745–753. [DOI] [PubMed] [Google Scholar]
- Schalet AP. 1960. “A study of spontaneous visible mutations in Drosophila melanogaster.” PhD thesis, Indiana University, Bloomington, IN. [Google Scholar]
- Schrider DR, Houle D, Lynch M, Hahn MW. 2013. Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster. Genetics 194: 937–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Akey JM. 2015. The origins, determinants, and consequences of human mutations. Science 349: 1478–1483. [DOI] [PubMed] [Google Scholar]
- Smeds L, Warmuth V, Bolivar P, Uebbing S, Burri R, Suh A, Nater A, Bures S, Garamszegi LZ, Hogner S, et al. 2015. Evolutionary analysis of the female-specific avian W chromosome. Nat Commun 6: 7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 1996–2010. RepeatMasker Open-3.0. http://www.repeatmasker.org/.
- Stadler LJ. 1930. The frequency of mutation of specific genes in maize. Anat Rec 47: 381. [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. 2012a. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci 109: 18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Tucker AE, Doak TG, Choi E, Thomas WK, Lynch M. 2012b. Extraordinary genome stability in the ciliate Paramecium tetraurelia. Proc Natl Acad Sci 109: 19339–19344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura A, Higuchi M, Minakuchi Y, Ohno M, Toyoda A, Fujiyama A, Miura I, Wakana S, Nishino J, Yagi T. 2015. Germline mutation rates and the long-term phenotypic effects of mutation accumulation in wild-type laboratory mice and mutator mice. Genome Res 25: 1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venn O, Turner I, Mathieson I, de Groot N, Bontrop R, McVean G. 2014. Strong male bias drives germline mutation in chimpanzees. Science 344: 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg A, Han F, Wellhagen G, Dahle B, Kawata M, Haddad N, Simoes ZLP, Allsopp MH, Kandemir I, De la Rua P, et al. 2014. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat Genet 46: 1081–1088. [DOI] [PubMed] [Google Scholar]
- Weber C, Nabholz B, Romiguier J, Ellegren H. 2014. Kr/Kc but not dN/dS correlates positively with body mass in birds, raising implications for inferring lineage-specific selection. Genome Biol 15: 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang L, Huang J, Zhang X, Yuan Y, Chen J-Q, Hurst LD, Tian D. 2015. Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature 523: 463–467. [DOI] [PubMed] [Google Scholar]
- Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, Storz JF, Antunes A, Greenwold MJ, Meredith RW, et al. 2014. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346: 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci 111: E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.