

Abstract
Whole-genome sequencing is a useful approach for identification of chemical-induced lesions, but previous applications involved tedious genetic mapping to pinpoint the causative mutations. We propose that saturation mutagenesis under low mutagenic loads, followed by whole-genome sequencing, should allow direct implication of genes by identifying multiple independent alleles of each relevant gene. We tested the hypothesis by performing three genetic screens with chemical mutagenesis in the social soil amoeba Dictyostelium discoideum. Through genome sequencing, we successfully identified mutant genes with multiple alleles in near-saturation screens, including resistance to intense illumination and strong suppressors of defects in an allorecognition pathway. We tested the causality of the mutations by comparison to published data and by direct complementation tests, finding both dominant and recessive causative mutations. Therefore, our strategy provides a cost- and time-efficient approach to gene discovery by integrating chemical mutagenesis and whole-genome sequencing. The method should be applicable to many microbial systems, and it is expected to revolutionize the field of functional genomics in Dictyostelium by greatly expanding the mutation spectrum relative to other common mutagenesis methods.
The social amoeba, Dictyostelium discoideum, is a eukaryote that grows as a unicellular organism and develops as a multicellular organism. It is used in a wide array of biomedical and basic biology research (Annesley and Fisher 2009; Williams 2010). Early genetic studies in Dictyostelium utilized chemical mutagens or UV irradiation to generate mutants. Although it is possible to map causative mutations to linkage groups by parasexual genetics, most of the target genes have not been identified because recombination is very limited under these conditions (Loomis 1987). Restriction enzyme-mediated integration (REMI) mutagenesis, which allows researchers to tag and clone the target genes in forward genetic screens, replaced chemical mutagenesis in the 1990s (Kuspa and Loomis 1992). Since then, REMI has been the workhorse of gene discovery in Dictyostelium. Other techniques, including gene silencing by antisense RNA (Spann et al. 1996) and by RNA interference (RNAi) (Kuhlmann et al. 2006), have been developed, too, but they are not commonly used in Dictyostelium research.
We wanted to resurrect the use of chemical mutagenesis and integrate it with whole-genome sequencing in forward genetic screens for three major reasons. First, chemical mutagenesis generates a broad spectrum of mutant alleles. The mutations can generate gain-of-function, temperature sensitive, and null alleles. They can also generate subtle variations that would facilitate the study of protein structure and function (Loomis 1987). Secondly, Dictyostelium has a very gene-rich, haploid genome of 34 Mb (Eichinger et al. 2005), which is readily amenable to high-throughput sequencing technologies, especially with the low cost afforded by sample multiplexing. Lastly, we found that REMI suppressors are rare and weak in previous screens for suppressors of the tgrC1-defective phenotypes (Li et al. 2015; Wang and Shaulsky 2015), and we wanted a method that would generate a variety of stronger suppressor mutations.
The major challenge in chemical mutagenesis stems from the large number of mutations induced per genome, which requires implementation of tedious genetic mapping techniques to locate the causative mutation to a defined genomic region (Sarin et al. 2008; Haelterman et al. 2014). Here, we report on a method for identification of chemical-induced mutations in Dictyostelium. First, we limited the number of mutations per genome by titrating the dose of the chemical mutagen. Next, we identified multiple mutant strains with common phenotypes. We then utilized sample multiplexing to reduce the whole-genome sequencing costs and developed a data analysis pipeline to identify, filter, and annotate mutations at a genomic scale. We implicated the target genes by recovering multiple mutant alleles and successfully identified and validated the causative mutations, thereby bypassing the need for genetic mapping. This chemical mutagenesis-based gene discovery pipeline is a significant addition to Dictyostelium genetics and is additionally applicable to other organisms.
Results
Genetic screens with chemical mutagenesis span a wide range of relevant target sizes
We performed genetic screens to test the limits of the system, one in which the number of possible genes (relevant target size) was small and one in which it was large. We induced mutations by exposing Dictyostelium cells to N-methyl-N′-nitro-N-nitrosoguanidine (NTG). In the first screen, we selected for resistance to light-induced cell death. The predicted targets were genes that encode biosynthetic components of light-sensitive chromophores (e.g., flavins and porphyrins) (Godley et al. 2005), so we presumed a small target size even though this phenotype has not been investigated in Dictyostelium before. Figure 1A shows that wild-type cells grow well on bacteria in the dark but fail to grow under intense illumination. We isolated six mutants (four shown, Fig. 1A) that grow both in the dark and under intense light. The mutants grow slightly better in the dark, suggesting that the mutations do not completely protect them from the lethal effects of intense light.
Figure 1.

Genetic screens with chemical mutagenesis. (A) We compared the viability of the wild-type AX4 and the light-resistant (Lr#) mutants. We spotted 5 to 250 Dictyostelium cells on nutrient agar in association with bacteria. We incubated the plates in darkness (Dark) or under intense illumination (Light) for 4–5 d. Opaque circles are thick bacterial lawns (e.g., AX4, Light, 250) and dark plaques within the lawns are clearings that indicate amoebae growth. Whenever the amoebae cleared the bacterial lawns, starved and developed, a mass of fruiting bodies is evident as opaque spots and protrusions over the plaque background (e.g., Lr1, 250). (B,C) We inoculated the indicated Dictyostelium strains in association with bacteria (bac) on nutrient agar and incubated for 4–5 d. Cells (AX4 and NTG#) in the center of the plaque starved and developed into mature fruiting bodies with a sorus (ss) atop a stalk (st). Aggregation-less (aggless#) mutants did not form aggregates. tgrB1AX4tgrC1QS38 cells arrested at the loose mound (lm) stage. Vegetative (veg) Dictyostelium cells resided in the translucent periphery of the plaque. Insets are magnified (3×) for visual clarity. The black arrowheads indicate inoculation spots. Scale bar = 2 mm. (D) We measured the survival rate (%, y-axis) over time (minutes, x-axis) during mutagenesis of two strains, AX4 (n = 13) and tgrB1AX4tgrC1QS38 (n = 4). (E) We measured the frequency (%, y-axis) of mutants with the desired phenotypes and plotted against the negative logarithm of survival (%, x-axis) in the genetic screens for aggregation defects (n = 5) and for suppression of the tgrB1-tgrC1 mismatch phenotype (n = 2).
In the second set of experiments, we performed a screen for an aggregation-less phenotype. Dictyostelium aggregation has been studied extensively, and the networks that regulate it contain 100–150 genes (Swaney et al. 2010), a rather large relevant target size. When amoebae grow on bacteria, they consume the bacteria and grow outwardly, forming an expanding circular plaque in the bacterial lawns around them. Cells in the center of the plaque starve as the bacteria are consumed, while cells at the edge of the plaque continue to grow. Starving wild-type cells then aggregate and develop into fruiting bodies (Fig. 1B). We isolated 14 mutants (five shown, Fig. 1B) that grew and cleared the bacterial lawns but remained flat even upon prolonged starvation, indicating failure to aggregate.
In the third set of experiments, we sought genes in a novel pathway, combining a selection with a visual screen for suppression of the tgrB1-C1 mismatch phenotypes. TgrB1 and TgrC1 are cell-surface adhesion proteins that form heterophilic interactions in trans (Chen et al. 2013). Cells carrying a mismatched allelic pair of the tgrB1 and tgrC1 genes exhibit impaired cell-cell recognition and are unable to complete development (Benabentos et al. 2009; Hirose et al. 2011, 2015). The tgrB1AX4tgrC1QS38 cells carry a mismatched pair of alleles, so their development is arrested at the loose mound stage and they do not form spores (Fig. 1C; Hirose et al. 2011). Previous REMI mutagenesis screens yielded small numbers of weak suppressors (Li et al. 2015; Wang and Shaulsky 2015), so we did not know if it was even possible to generate strong suppressors, let alone the relevant target size. We mutated these cells, allowed the population to develop, and selected for spores by detergent treatment. We plated the survivors at low density on nutrient agar in association with bacteria and screened for mutants that formed fruiting bodies. We recovered 63 mutants that fulfilled these criteria (five shown, Fig. 1C), indicating that it is possible to obtain a large number of strong suppressors of the tgrB1-C1 mismatch phenotype.
The three genetic screens verify previous reports that chemical mutagenesis can be used for identification of aggregation-less mutants (Williams and Newell 1976) and show that new phenotypes can be detected as well.
Low levels of chemical mutagenesis produce enough mutants for screening
We wanted to find conditions that would produce enough mutant strains for screening and selection while keeping a low number of mutations in each genome. Previous studies used conditions that resulted in low survival rates (Loomis 1987). It is hard to evaluate the number of mutations in each genome in those studies, but it was estimated at several hundred to several thousand. To evaluate the relationship between the number of mutations per genome and the number of mutants in the population, we measured the relationship between the duration of NTG treatment and the survival rate, as well as the relationship between survival rate and frequency of mutants with defined phenotypes. We found that survival was inversely correlated with the duration of NTG treatment (Fig. 1D) and that the frequency of mutants was inversely correlated with survival (Fig. 1E). We also found that it is possible to generate enough mutants for selections and screens even with high survival rates, suggesting that it would be possible to identify the causative mutations by whole-genome sequencing.
Whole-genome sequencing reveals a small number of variants per genome
We used high-throughput sequencing to analyze the genomes of the two parental strains (AX4 and tgrB1AX4tgrC1QS38) and the 83 mutants found in the three screens. We multiplexed the samples for cost effectiveness such that 16–24 strains were grouped in each sequencing lane. We obtained 15.8 ± 3.6 (mean ± standard deviation [SD]) million reads per genome and were able to map 99% of the reads to the reference genome with a mean fold-coverage of 19.4 ± 5.2 (mean ± SD) over the entire genome. The coverage of individual chromosomes is shown in Supplemental Figure S1A. To identify mutations, we performed multisample variant calling on single nucleotide variants (SNVs) and indels (McKenna et al. 2010). To remove false positive variants, we imposed three filters: read depth, allele frequency, and pre-existing variations in the parental strain. These filters removed 99% of the raw SNVs and indels (Table 1), leaving 16.3–29.7 SNVs and 0.1–1.0 indels per genome. The number of variants per genome was negatively correlated with survival (Fig. 2A). More specifically, survival rates of 30%, 10%, and 0.1% corresponded to an average of 13, 21, and 34 variants per genome, respectively. Since ∼30% of the entire genome and ∼10% of the coding exons did not pass the read depth filter and were therefore excluded from SNP calling (Supplemental Fig. S1B), we probably underestimated the number of NTG-induced mutations per genome. Nevertheless, this relatively small number of mutations per genome suggested that it would be possible to identify causative mutations by comparing the genome sequences of strains with similar phenotypes. Figure 2A also shows a diminishing returns relationship between survival and the number of variants per genome, suggesting that reducing the survival rate to 0.1% or below does not contribute significantly to mutant discovery.
Table 1.
Filtering variants found in whole-genome sequencing
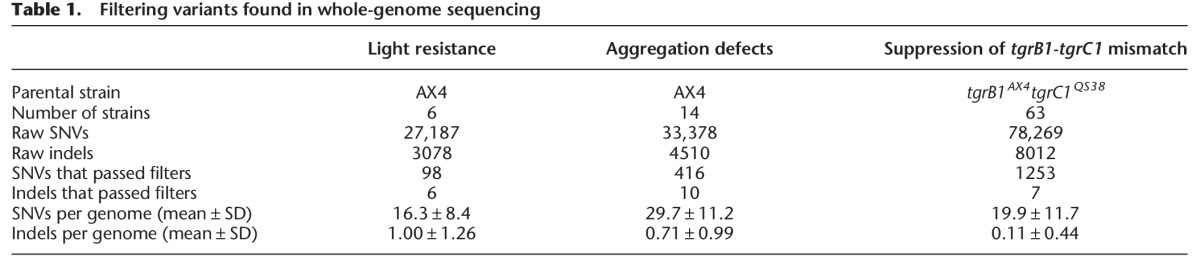
Figure 2.

Properties of the chemically induced mutations. (A) We plotted the number of mutations (single nucleotide variants and indels) per genome (y-axis) against the negative logarithm of survival (%, x-axis). Dots represent means and whiskers represent 95% confidence intervals. Sample sizes are indicated next to each whisker, and the framed legend describes the experiments. (B) The bars show the proportion of filtered single nucleotide variants (SNVs) that are G > A or T > C transitions in individual experiments (white) and the other substitutions (gray). Percentages of G > A or T > C transitions are indicated inside the bars. The total numbers of filtered SNVs are indicated on the right. Sample size: light resistance (n = 6), aggregation defects (n = 14), and tgrB1-C1 mismatch (n = 63). (C) SNVs were classified into eight groups based on the locations and types of substitution mutations (missense, synonymous, nonsense, intergenic, intronic [excluding splice sites], splice donor site, splice acceptor site, and NCG [noncoding gene], as indicated on the right). Titles above the pie charts describe the experiments. The average number of SNVs per genome is indicated on the bottom. The percentages of individual mutation types are indicated next to individual sectors.
Most SNVs cause missense mutations
Most of the NTG-induced SNVs we found are G > A or T > C transitions (G > A:T > C ratio = 19:1), consistent with the mutagenic action of NTG (Fig. 2B; Lucchesi et al. 1986; Ohta 2000). We classified the SNVs into eight categories (Fig. 2C). The majority of SNVs occurred in coding exons, which is expected because coding exons contain 71.7% of the G-C base pairs in the Dictyostelium genome (Eichinger et al. 2005). Statistical testing indicates significant enrichment of exonic mutations in two experiments (exact binomial test: 77.4% of the 416 SNVs in the aggregation defects experiment, P value < 0.01; 86.5% of the 1253 SNVs in the suppression of tgrC1-B1 mismatch experiment, P value < 0.001). Moreover, the Dictyostelium codon usage bias predicts that 76.6% of the exonic G > A transitions would result in missense mutations. Indeed, our results show that the predominant type of exonic mutations is missense, with significant enrichment in two experiments (exact binomial test: 82.0% of the 322 exonic mutations in the aggregation defects experiment, P value = 0.02; 82.0% of the 1084 exonic mutations in the suppression of tgrC1-B1 mismatch experiment, P value < 0.001). Nonsense mutations occurred at 3.2%–7.1%, with a frequent occurrence in the glutamine codon (CAA > TAA, 60%). Figure 3 shows the locations of the mutations, suggesting that NTG-induced mutations are randomly distributed throughout the genome (Kolmogorov-Smirnov test for uniformity: light resistance, P value = 0.2314; aggregation defects, P value = 0.2545; suppression of tgrC1-B1 mismatch, P value = 0.1757). We conclude that NTG-induced mutations are distributed nearly randomly throughout the genome and are most likely to modify protein-coding genes.
Figure 3.
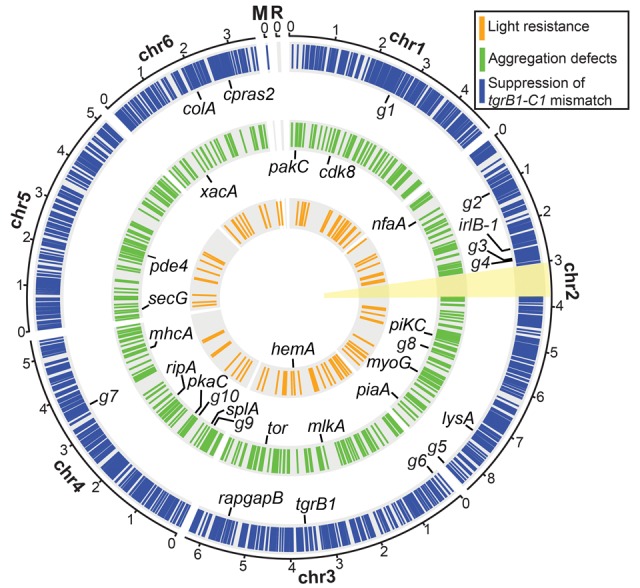
The genomic distribution of chemically induced mutations. The circular plot summarizes the distribution of chemically induced mutations in the six chromosomal (Chr 1–6), mitochondrial (M), and ribosomal (R) DNAs. Colored bars on the three concentric circles represent the mutations found in the three genetic screens as indicated in the legend: suppression of the tgrB1-C1 mismatch (blue, n = 63 strains), aggregation defects (green, n = 14 strains), and light resistance (orange, n = 6 strains). The candidate genes in the individual screens are labeled inside the respective circles, and the black lines indicate their chromosomal positions. Abbreviations: g1, DDB_G0270488; g2, DDB_G0271904; g3, DDB_G0273247; g4, DDB_G0273385; g5, DDB_G0277749; g6, DDB_G0277997; g7, DDB_G0285705; g8, DDB_G0275861; g9, DDB_G0283339; g10, DDB_G0283893. The yellow sector (around 3 o'clock) indicates the second copy of the Chromosome 2 duplication (Chr 2: 3016083…3768654), which was masked during the alignment of sequencing reads. The full list of mutations is detailed in Supplemental Data 1–3 and the candidate genes in Supplemental Table S1. The scale outside the circles shows positions in million base pairs (Mb).
Multiple mutations in the ALA synthase gene, hemA, in light-resistant mutants
To identify phenotype-causing mutations, we looked for genes that were mutated independently in multiple strains with common phenotypes. We recovered six light-resistant strains (Table 1) and found that hemA was mutated independently in four of them (Fig. 3; Supplemental Table S1). None of the other genes mutated in this screen was common to two or more strains.
hemA encodes the D. discoideum 5-aminolevulinate (ALA) synthase. ALA is a precursor in heme biosynthesis (Ajioka et al. 2006) and intense illumination damages cells by causing ALA-induced porphyrin accumulation and intra-cellular ROS production (Godley et al. 2005). ALA is photosensitive and its derivatives are used as photosensitizers in photodynamic therapy (Dolmans et al. 2003). Our finding of multiple mutations only in hemA and the known role of ALA in photosensitivity implicate the mutations in hemA as the cause of the light-resistance phenotype. The two strains that do not carry mutations in hemA suggest that other genes are also involved. Most importantly, these results demonstrate that we can reach near-saturation and identify recurrent mutations in a genetic screen over a small relevant target size.
Known aggregation genes found in a screen for aggregation defects
Dictyostelium aggregation involves more than 110 genes (Swaney et al. 2010), so we did not expect to reach saturation with only 14 mutants (Table 1). Nevertheless, we did expect to find mutations in some of the known genes among the aggregation-less mutants. Indeed, we identified one variant each in 13 aggregation-related genes (Supplemental Table S1). We also identified two or more variants each in five other genes (tor, splA, DDB_G0275861, DDB_G0283339, and DDB_G0283893) (Fig. 3; Supplemental Table S1). Only one of them, tor, has been previously implicated in aggregation (Lee et al. 2005; Swaney et al. 2010). Among the other four genes, splA and DDB_G0283893 are likely false positives. splA is dispensable for aggregation (Nuckolls et al. 1996), and the two mutant strains that carry splA mutations also harbor mutations in known aggregation-related genes that could account for the observed phenotypes. Moreover, as larger genes are more likely to be mutated at random, a statistical test indicates that the two mutations in DDB_G0283893 were likely to occur by chance (binomial distribution, P value = 0.058). In summary, we identified 14 known aggregation-related genes in the screen for aggregation defects, supporting our expectation and suggesting that it is possible to identify recurrent mutations in a genetic screen over a large relevant target size. If we were studying an unknown pathway, we would need to achieve saturation to identify most of the genes, but even our limited screen has implicated two new genes as potential aggregation-related genes.
Multiple alleles reveal 13 suppressors of the tgrB1-C1 mismatch phenotype
We recovered 63 mutants that suppressed the morphological and sporulation defects of the tgrB1-C1 mismatch phenotype (Table 1). Many of the mutants were nearly indistinguishable from the wild type (Fig. 4A). The sequencing results revealed 79 genes that were mutated ≥2 times. Gene Ontology (GO) enrichment analysis of these genes suggests that six (htt, lmpB, lysA, talA, tgrB1, and rapgapB) are involved in cell-cell adhesion (P value = 0.0001, FDR = 0.0265). Thirteen genes were mutated ≥3 times (Fig. 3; Supplemental Table S1). Indeed, we identified 19 mutations in rapgapB and nine mutations in tgrB1, suggesting that our screen has been carried to near-saturation. These results suggest that our method is useful in implicating genes in a genetic screen over a moderate relevant target size.
Figure 4.
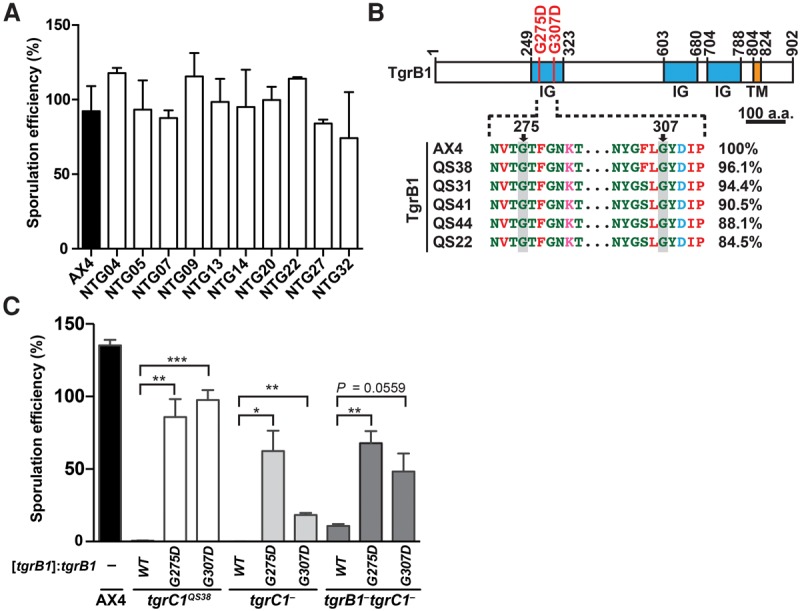
Dominant causative mutations in tgrB1 identified in the screen for suppression of the tgrB1-C1 mismatch phenotype. (A) We developed cells for 72 h on a nitrocellulose filter and counted the fraction (%) of cells that formed spores. Strain names are indicated under the x-axis: AX4 cells (black bar) and 10 NTG-mutagenized mutants (white bars) isolated from the screen for suppression of the tgrB1-C1 mismatch phenotype. The bar graph represents the mean ± standard deviation (SD) of three independent replicates. (B) (Top) TgrB1 contains three immunoglobulin-like domains (IG, blue) and one transmembrane domain (TM, orange). Numbers above the chart represent amino acid positions. Scale bar = 100 aa. Recurrent missense mutations (G275D and G307D) are indicated as red bars. (Bottom) Partial protein sequence alignment of the polymorphic tgrB1 alleles from AX4 and five wild isolates as indicated. The pairwise sequence identity (%) compared to TgrB1AX4 is indicated on the right. (C) We expressed three different alleles (tgrB1WT, tgrB1G275D, or tgrB1G307D, as indicated immediately below the x-axis) driven by the native promoter, in three tgrC1-defective mutants (tgrC1QS38, tgrC1−, and tgrB1−tgrC1−, as indicated on the bottom). We allowed the cells to develop and measured their sporulation efficiencies as the proportion of cells (%) that formed spores (y-axis). Bars represent the means ± SD of four independent replicates. Welch's unequal variances t-test (two-tailed): (*) P value < 0.05; (**) P value < 0.01; (***) P value < 0.005.
So far, we have relied on multiple alleles and on published results to validate our screen results. The highest numbers of alleles were found in rapgapB and tgrB1, and we know that TgrB1 and TgrC1 interact to regulate development (Hirose et al. 2015) and that rapgapB encodes a RapA GTPase regulatory protein B that regulates cell-cell and cell-substrate adhesion (Parkinson et al. 2009). This knowledge and the availability of molecular tools allowed us to perform direct causality tests of these mutations.
Lack of correlation between mutations and fine morphological phenotypes
We hypothesized that the mutants’ morphological phenotypes would be instructive in analyzing the underlying mutations. We therefore analyzed the morphologies of 25 tgrB1-C1-mismatch-suppressor strains at seven time points over 36 h of development. The genotypes and phenotypes did not seem to correlate when visualized by multidimensional scaling (Supplemental Fig. S2), and analysis by multiple linear regressions found no significant association either (mutations in rapgapB, P value = 0.3128; mutations in tgrB1, P value = 0.5462; mutations in any of the 13 suppressor genes, P value = 0.0920). We conclude that fine morphological phenotyping did not help to group mutants with similar suppressor mutations in this context.
Dominant causative mutations in tgrB1
Most of the mutations in tgrB1 restored sporulation efficiency to near wild-type levels (Fig. 4A). Two mutant alleles, tgrB1G275D and tgrB1G307D, independently arose twice and three times, respectively, in our screen (Supplemental Table S1). Both mutations (G275D and G307D) occurred in the invariable part of the first immunoglobulin-like domain in the extracellular region of the TgrB1 protein (Fig. 4B). We hypothesized that tgrB1G275D and tgrB1G307D are gain-of-function alleles because deletion of tgrB1 confers only weak suppression of the sporulation defect in tgrC1− cells (Benabentos et al. 2009). We therefore tested whether expressing tgrB1G275D or tgrB1G307D driven by the native tgrB1 promoter would suppress the phenotypic defects of three mutants: the parental mismatch strain tgrB1AX4tgrC1QS38, the single-gene deletion strain tgrC1−, and the double-gene deletion strain tgrB1−tgrC1− (Fig. 4C). We found that transformation with either of the two mutant tgrB1 alleles strongly suppressed the sporulation defects, whereas the wild-type allele tgrB1WT did not (Fig. 4C). These results suggest that the G275D and G307D mutations in tgrB1 were indeed the causative mutations in the original strains and that both alleles are dominant.
Recessive causative mutations in rapgapB
To characterize the rapgapB alleles, we tried to complement the original suppressor mutant strains by transformation with wild-type rapgapB. We transformed the rapgapBWT allele, driven by its native promoter, into mutants carrying the mutated rapgapB (Fig. 5A,B). We grew the transformants in association with bacteria on nutrient agar and counted plaques that lost the suppression phenotype (Fig. 5B). We found that the majority of plaques in NTG82 (70%, rapgapBM1I) and NTG14 (90%, rapgapBG382D) exhibited a loose-aggregate phenotype, which is the tgrB1-C1 mismatch phenotype. This finding indicates that M1I and G382D are recessive causative mutations of the phenotypic suppression. In contrast, transformation with rapgapBWT did not affect the suppression phenotype of NTG09 (0%, rapgapBG191D) and NTG64 (0%, rapgapBQ44*) (Fig. 5B). A likely explanation is that these two variants are dominant causative alleles, but it is possible that other mutations in these genomes caused the phenotypic suppression.
Figure 5.

Recessive causative mutations in rapgapB observed in the screen for suppression of the tgrB1-C1 mismatch phenotype. (A) Schematic representation of RapGAPB. The blue rectangle indicates the Rap GTPase-activating protein (RapGAP) domain. Numbers above the chart represent amino acid positions. Scale bar = 100 aa. Four of the 17 amino acid substitutions observed in the screen are indicated in red (M1I, Q44*, G191D, and G382D). (B) We performed a complementation test by introducing the rapgapBWT allele driven by the endogenous promoter ([rapgapB]:rapgapBWT) into chemically mutagenized strains carrying the indicated rapgapB mutations. We grew the transformants in association with bacteria on nutrient agar and measured the proportion of colonies that lost the suppression phenotype (%, red text). We photographed the morphology of the mutants with or without the rapgapBWT expression construct, as indicated below the line of each panel. The names of the individual strains (NTG#) and the respective rapgapB mutations are indicated above the line in each panel. Insets are magnified (3×). Scale bar = 1 mm.
Altogether, the genetic analyses of tgrB1 and rapgapB support the hypothesis that multiple independent alleles implicate specific genes in this type of genetic screen. They also illustrate the power of the method to generate a broad spectrum of mutations.
Discussion
Our findings show that chemical mutagenesis followed by whole-genome sequencing can reveal a broad spectrum of causative mutations in genetic screens. The key technical feature is the low level of mutagenesis, with 10%–50% survival and 20–30 SNVs per genome. The key conceptual feature is the implication of a single common gene by multiple independent mutations. Mutations outside of the implicated gene help to distinguish independent mutants from siblings. We used complementation with expression vectors to validate our conclusions. This step is not necessary in future screens, but complementation is a useful tool for analyzing the genetic nature of the mutated alleles.
Previous chemical mutagenesis studies were done mostly at high levels of mutagenesis and at ∼0.1% survival. These rates were interpreted as an indication that mutagenesis resulted in an average of seven lethal hits per genome (Loomis 1978, 1987). We have observed a diminishing returns relationship between the killing rate and the number of SNVs per genome, suggesting that lethality in previous studies was compounded by toxicity. Regardless, the conditions we used are suitable for large-scale screens. With 20 genic mutations per genome, the odds of mutating any one of the 12,000 genes in one Dictyostelium genome are 1:600. A typical mutagenesis reaction produces approximately 107 mutants, enough to saturate almost any gene. Remarkably, the number of SNVs per genome is rather homogeneous, which makes the downstream analysis rather convenient.
High-throughput sequencing was not available in the 1970s, and genetic mapping in Dictyostelium is difficult, so most mutations were never characterized in molecular detail. The presumed large number of mutations per genome also resulted in confounding effects, with a 50% chance that secondary mutations affected the studied phenotype (Loomis 1987). Therefore, researchers had to perform genetic crosses and to study several mutants in order to associate genotypes with phenotypes (Newell 1978). Here, we implicated genes by identifying multiple independent alleles in genome sequencing data without genetic mapping. Another observation in previous studies was that most mutations were recessive (Loomis 1987). Our findings indicate that chemical mutagenesis produces dominant alleles as well.
Some analyses of chemical mutagenesis data were based on the assumption that mutations occur at random (Loomis 1987). These analyses were remarkably accurate despite our finding that NTG mutagenesis is biased toward exons of coding genes. This bias is a combined result of the skewed nucleotide content of the Dictyostelium genome (Eichinger et al. 2005) and the preference of NTG to alkylate guanine residues (Ohta 2000). Nevertheless, NTG mutagenesis seems to be unbiased at the genome level, as all the genes appear to be equally susceptible. The bias to coding exons is convenient because mutations in these sequences are somewhat simpler to interpret. Compared to other mutagenesis methods, such as REMI (Kuspa and Loomis 1992), antisense RNA (Spann et al. 1996), and RNA interference (Kuhlmann et al. 2006), our method provides a broader spectrum of mutations and opens the field to exploration of modifications that are not necessarily null or loss-of-function mutations. For example, NTG mutagenesis can generate temperature sensitive alleles (Loomis 1969; Liwerant and Pereira Da Silva 1975), and we found strong suppressors of the tgrB1-tgrC1 mismatch phenotype, which were not found by REMI screens (Li et al. 2015; Wang and Shaulsky 2015). Some mutations, such as the dominant alleles of tgrB1, cannot be produced by REMI. The mutation spectrum could be broadened further by using UV irradiation and other chemicals that have been tested in Dictyostelium (Liwerant and Pereira Da Silva 1975). According to the central repository of Dictyostelium genomic data (dictyBase, April 2016 [Fey et al. 2013]), ∼11% of the Dictyostelium genes have been mutated by REMI or by methods such as homologous recombination. Most of these mutations conferred loss-of-function. Using the method described here is expected to increase the number of mutated genes and the variety of available mutations, opening the field of functional genomics in Dictyostelium to new exploration.
Important considerations in genetic screens are the relevant target size and saturation. Target size is a function of the number of genes that participate in a biological process and the size of each gene. We tested our method on three phenotypes: Resistance to light was presumed to have a small target size (approximately 1–5 genes), defective aggregation was known to have a large target size (approximately 100 genes), and suppression of the tgrB1-tgrC1 mismatch had an unknown target size that turned out to be in the middle (approximately 10–20 genes). As expected, saturation was achieved in the small and middle size screens and not in the large size one. Our method relies on saturation because we can only implicate genes that are independently mutated several times. There is no theoretical way to predict the relevant target size, but chemical mutagenesis can be used to estimate the number of genes required in any biological process (Loomis 1987).
In terms of cost and efficiency, our method relies on economies of scale. Generating mutants is inexpensive and fast, whereas the screen or selection phase depends on the experimental design. In the sequencing phase, we used 16- to 24-fold multiplexing, generating an average of 20-fold sequence coverage. We discovered ∼90% of the mutations in the coding regions in our screens (Supplemental Fig. S1B), and we estimate that doubling the multiplexing would allow discovery of ∼80%. This compromise may be acceptable depending on the application. The commercial cost of sequencing in the United States on January 2016 was roughly $2500–$4000 per lane. This cost can be considerably lower through institutional core services, and it is likely to decline in the future. We note that next-generation sequencing data management and analysis can be expensive, too, but the cost can be reduced through use of shared institutional resources or public free cloud platforms, such as Galaxy (https://usegalaxy.org). Our method is therefore accessible to laboratories with modest budgets.
The large number of mutants generated by NTG mutagenesis and the advantage of working with a microbial system makes the method applicable to genetic screens as well as selections. Each approach has advantages and disadvantages (Shuman and Silhavy 2003), but our findings indicate that either approach, or a combination thereof, can be used. Genetic suppression is a powerful tool in Dictyostelium (Shaulsky et al. 1996), and our method is suitable for this type of investigation as well. More importantly, the ease of NTG mutagenesis makes it applicable to nonaxenic strains (Loomis 1987), where transformation-based methods are possible but somewhat difficult (Veltman et al. 2014). Our method could also be readily adapted to any haploid organism with a small, sequenced genome, including many amoebae, fungi, and bacteria.
Methods
Genetic screens by chemical mutagenesis
We performed chemical mutagenesis as described (Loomis 1987) with minor modifications. The stock solution of N-methyl-N′-nitro-N-nitrosoguanidine (NTG, Pfaltz&Bauer) was prepared in DMSO at 10 mg/mL and stored as 100 µL aliquots for one-time use at −80°C. We harvested exponentially growing Dictyostelium cells and washed once with KK2 buffer (16.3 mM KH2PO4 and 3.7 mM K2HPO4). We mutagenized 108 cells in 1 mL of KK2 buffer containing 500 µg/mL of NTG for 10, 20, 30, 40, or 60 min. Afterward, we collected the cells by centrifugation at 500g for 30 sec and resuspended in 10 mL HL5 medium. We estimated the survival rate by plating 100 to 10,000 cells on SM plates in association with Klebsiella aerogenes. We plated the rest of the mutagenized cells on SM plates with bacteria at a density of 103 to 107 cells per 10-cm plate. For aggregation defects, we inspected plaques under a stereomicroscope for strains that did not form streams and mounds. For light resistance, we selected for mutagenized AX4 cells that grew and developed under constant exposure to 5000 to 10,000 lux of light (Mastech light meter) illuminated with compact fluorescent light (CFL) bulbs (GE 100-Watt daylight 6500K). Ten separate pools of 106 mutagenized cells were selected in this case, and we isolated only one stable mutant from each population to avoid siblings. For the suppression of the tgrB1-C1 mismatch, we grew mutagenized tgrB1AX4tgrC1QS38 cells on SM plates with bacteria and allowed them to develop for 3 d after clearing the plate. We collected the cells and treated them with detergent (0.1% NP-40 and 10 mM EDTA) to kill nonspore cells. We plated the surviving spores on fresh SM plates with bacteria, followed by screening for fruiting body-forming plaques.
Genomic DNA sequencing
We prepared genomic DNA (gDNA) as described (Santhanam et al. 2015). Briefly, we harvested 108–109 cells grown in HL5 medium, followed by washing once with KK2 buffer. We lysed the cells in 10 mL of nuclei buffer (40 mM Tris-HCl pH 7.8, 1.5% sucrose, 0.1 mM EDTA, 6 mM MgCl2, 40 mM KCl, 5 mM DTT, and 0.4% NP40) on ice for 10 min. We collected the nuclei by centrifuging at 20,000g for 10 min. We incubated the nuclei in 500 µL of STE/SDS/Protease K solution (450 µL STE solution [10 mM Tris-HCl pH 8.0, 10 mM EDTA, and 400 mM NaCl], 25 µL 20% SDS, 15 µL ddH2O, and 10 µL of 10 mg/mL Proteinase K) at 60°C for 1 h. We performed phenol/chloroform extraction and ethanol precipitation and repeated the process once more after treating the gDNA with RNase A. For library preparation, we sheared 1 µg of gDNA in 100 µL of ddH2O in a Covaris microTUBE (#520052) by Covaris S220 Focused-ultrasonicator (60 sec, 10% duty cycle, 200 cycles per burst, 14W acoustic power, 7°C). The rest of the library preparation was performed as described for cDNA library preparation (Miranda et al. 2013). We quantified individual gDNA libraries by quantitative PCR to determine the PCR cycle number for enriching and barcoding with indexing primers (Meyer and Kircher 2010; Miranda et al. 2013). We multiplexed 16–24 barcoded samples on one lane for 100-bp paired-end sequencing on the Illumina HiSeq 2000 platform.
Sequence read alignment and variant detection
We demultiplexed the sequencing reads based on the barcodes and mapped the processed reads to the reference Dictyostelium AX4 genome by the BWA-MEM (Li and Durbin 2009) algorithm (version 0.7.5a) using default parameters. For all of the analyses, we masked one copy of the 750-kb duplication on Chromosome 2 (position 3016083 to 3768654) in the reference genome (Eichinger et al. 2005). For strains carrying the foreign tgrC1QS38 allele, we replaced the tgrC1AX4 allele with the tgrC1QS38 allele in the reference genome. After read alignment, we used Picard tools (version 1.119) to mark and remove duplicates (paired-end reads that carried identical sequence information and most likely arose at the indexing PCR step of the library preparation). We evaluated the quality statistics of the mapped reads with Qualimap (version 2.1.1) (García-Alcalde et al. 2012). Subsequently, we detected single nucleotide variants and indels by performing multisample variant calling with the Genome Analysis Toolkit's Unified Genotyper (GATK version 3.4–46) (McKenna et al. 2010). In variant calling, we considered the duplication on Chromosome 2 as a diploid genome and the rest of the chromosomal regions as a haploid genome. All of the raw reads, aligned reads, and VCF files can be accessed on dictyExpress (https://dictyexpress.research.bcm.edu/genboard/#/chemical-mutagenesis/). We created the circular plot in Figure 3 with the R package OmicCircos (Hu et al. 2014).
Filtering variants
We applied several filters to the raw SNVs and indels called by GATK. First, a locus must be covered by at least five sequencing reads. Secondly, a locus in the haploid Dictyostelium genome can only carry one allele at a time. We considered an allele at a given locus to be the one supported by ≥80% of the reads. If there was no allele that passed the threshold, we discarded the locus. Third, a locus on the Chromosome 2 duplication can carry two alleles. We considered the locus to be heterozygous if the two alleles were both supported by 40% to 60% of the reads. Lastly, we filtered the variants detected in the chemically mutagenized strains against the pre-existing variants in the relevant parental strain. The full list of filtered variants in this study is summarized in Supplemental Data 1–3. R codes for variant filtering can be found on the GitHub repository (https://github.com/chenglinli/chemical_mutagenesis) and in Supplemental File 1.
Genetic constructs
To construct the rapgapBWT expression vector, we PCR-amplified a 3-kb DNA fragment including the rapgapB gene (1.6 kb), 5′ promoter region (0.8 kb), and 3′ region (0.6 kb) with the following primers: 5′-ATTATCTTTGTGGGTTTAGTTGTG-3′ and 5′-TGATGCTGAACCTCTTGATATG-3′. We cloned the 3-kb fragment into the pCR-Blunt II-TOPO vector (ThermoFisher Scientific), followed by subcloning into the EcoRV and BamHI sites of the pLPBLP vector (Faix et al. 2004). The tgrB1WT expression construct, in which expression was driven by the native promoter, was kindly provided by S. Hirose. We replaced the wild-type allele tgrB1AX4 with the mutated alleles tgrB1G275D and tgrB1G307D by cutting and pasting PCR-amplified mutant tgrB1 alleles with EcoNI and AgeI. The primers for amplifying the mutant tgrB1 alleles were 5′-AAATGGATACAAATGGAG-3′ and 5′-TTACAGTCATATTCTTAACACC-3′.
Phenotyping
To group the Dictyostelium strains based on their developmental morphology, we developed individual strains on a 1.5% nonnutrient KK2 agar containing 500 µL/mL streptomycin. We took pictures of the developmental morphology from above under a stereomicroscope at seven time points (8, 10, 12, 16, 20, 24, and 36 h). We scored the morphology at each time point based on the predominant developmental structures. The scores for individual structures were 1 for ripples, 2 for streaming, 3 for loose aggregates, 4 for tight aggregates, 5 for tipped mounds, 6 for slugs and fingers, 7 for Mexican hats, 8 for early culmination, 9 for mid-culmination, and 10 for fruiting bodies. We calculated the pairwise distances between the strains by aligning the time series with the dynamic time warping (DTW) algorithm (Sakoe and Chiba 1978), while computing the distance between the two morphological states as the absolute difference between the scores for their corresponding predominant structures. The time-shift penalty for DTW was set to 0.2. In addition to morphology, we quantified the sorus size of individual strains after growth and development on SM plates in association with Klebsiella aerogenes, but this information was not included in the distance calculation. To measure sporulation efficiency, we developed a known number of cells for 48 h, collected spores, and counted them with phase contrast microscopy. Sporulation efficiency was calculated as the fraction (%) of cells that became spores.
Data access
The whole-genome sequence data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP073746.
Supplementary Material
Acknowledgments
We thank R. Sucgang and A. Kuspa for helpful suggestions and insightful discussions, A. Kuspa and R. Gomer for critical review of the manuscript, S. Hirose for the tgrB1WT expression construct, and M. Katoh-Kurasawa for technical guidance with whole-genome sequencing. This work was supported by National Institutes of Health (NIH) grants R01 GM084992 and P01 HD03691, and A.N.W. was partly supported by grant T32 GM008307 from the NIH; C.F.L. was partly supported by the Burroughs Wellcome Fund through the Houston Laboratory and Population Sciences Training Program in Gene-Environment.
Author contributions: C.F.L. and G.S. conceived and designed the experiments. C.F.L. performed the experiments and analyzed the data. B.S. participated in the development of the genome analysis code, A.N.W. photographed the developmental structures, and B.Z. developed and implemented the dynamic time warping algorithm for morphological phenotyping. C.F.L. and G.S. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.205682.116.
References
- Ajioka RS, Phillips JD, Kushner JP. 2006. Biosynthesis of heme in mammals. Biochim Biophys Acta 1763: 723–736. [DOI] [PubMed] [Google Scholar]
- Annesley SJ, Fisher PR. 2009. Dictyostelium discoideum—a model for many reasons. Mol Cell Biochem 329: 73–91. [DOI] [PubMed] [Google Scholar]
- Benabentos R, Hirose S, Sucgang R, Curk T, Katoh M, Ostrowski EA, Strassmann JE, Queller DC, Zupan B, Shaulsky G, et al. 2009. Polymorphic members of the lag gene family mediate kin discrimination in Dictyostelium. Curr Biol 19: 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Wang J, Xu X, Wu X, Piao R, Siu C-H. 2013. TgrC1 mediates cell–cell adhesion by interacting with TgrB1 via mutual IPT/TIG domains during development of Dictyostelium discoideum. Biochem J 452: 259–269. [DOI] [PubMed] [Google Scholar]
- Dolmans DEJGJ, Fukumura D, Jain RK. 2003. Timeline: Photodynamic therapy for cancer. Nat Rev Cancer 3: 380–387. [DOI] [PubMed] [Google Scholar]
- Eichinger L, Pachebat JA, Glöckner G, Rajandream M, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, et al. 2005. The genome of the social amoeba Dictyostelium discoideum. Nature 435: 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faix J, Kreppel L, Shaulsky G, Schleicher M, Kimmel AR. 2004. A rapid and efficient method to generate multiple gene disruptions in Dictyostelium discoideum using a single selectable marker and the Cre-loxP system. Nucleic Acids Res 32: e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey P, Dodson RJ, Basu S, Chisholm RL. 2013. One stop shop for everything Dictyostelium: dictyBase and the Dicty Stock Center in 2012. Methods Mol Biol 983: 59–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, Dopazo J, Meyer TF, Conesa A. 2012. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28: 2678–2679. [DOI] [PubMed] [Google Scholar]
- Godley BF, Shamsi FA, Liang F-Q, Jarrett SG, Davies S, Boulton M. 2005. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J Biol Chem 280: 21061–21066. [DOI] [PubMed] [Google Scholar]
- Haelterman NA, Jiang L, Li Y, Bayat V, Sandoval H, Ugur B, Tan KL, Zhang K, Bei D, Xiong B, et al. 2014. Large-scale identification of chemically induced mutations in Drosophila melanogaster. Genome Res 24: 1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S, Benabentos R, Ho H-I, Kuspa A, Shaulsky G. 2011. Self-recognition in social amoebae is mediated by allelic pairs of tiger genes. Science 333: 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S, Santhanam B, Katoh-Kurosawa M, Shaulsky G, Kuspa A. 2015. Allorecognition, via TgrB1 and TgrC1, mediates the transition from unicellularity to multicellularity in the social amoeba Dictyostelium discoideum. Development 142: 3561–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Yan C, Hsu C, Chen Q, Niu K, Komatsoulis GA, Meerzaman D. 2014. OmicCircos: a simple-to-use R package for the circular visualization of multidimensional omics data. Cancer Inform 13: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann M, Popova B, Nellen W. 2006. RNA interference and antisense-mediated gene silencing in Dictyostelium. In Dictyostelium discoideum protocols (ed. Eichinger L, Rivero F), Vol. 346, pp. 211–226. Humana Press, New Jersey. [DOI] [PubMed] [Google Scholar]
- Kuspa A, Loomis WF. 1992. Tagging developmental genes in Dictyostelium by restriction enzyme-mediated integration of plasmid DNA. Proc Natl Acad Sci 89: 8803–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Comer FI, Sasaki A, McLeod IX, Duong Y, Okumura K, Yates JR, Parent CA, Firtel RA. 2005. TOR complex 2 integrates cell movement during chemotaxis and signal relay in Dictyostelium. Mol Biol Cell 16: 4572–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C-LF, Chen G, Webb AN, Shaulsky G. 2015. Altered N-glycosylation modulates TgrB1- and TgrC1-mediated development but not allorecognition in Dictyostelium. J Cell Sci 128: 3990–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liwerant IJ, Pereira Da Silva LH. 1975. Comparative mutagenic effects of ethyl methanesulfonate, N-methyl-N′-nitro-N-nitrosoguanidine, ultraviolet radiation and caffeine on Dictyostelium discoideum. Mutat Res 33: 135–146. [DOI] [PubMed] [Google Scholar]
- Loomis WF. 1969. Temperature-sensitive mutants of Dictyostelium discoideum. J Bacteriol 99: 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis WF. 1978. The number of developmental genes in Dictyostelium. Birth Defects Orig Artic Ser 14: 497–505. [PubMed] [Google Scholar]
- Loomis WF. 1987. Chapter 3 - Genetic tools for Dictyostelium discoideum. In Methods in cell biology (ed. Spudich JA), Vol. 28, pp. 31–65. Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Lucchesi P, Carraway M, Marinus MG. 1986. Analysis of forward mutations induced by N-methyl-N′-nitro-N-nitrosoguanidine in the bacteriophage P22 mnt repressor gene. J Bacteriol 166: 34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc 2010: ppdb.prot5448. [DOI] [PubMed] [Google Scholar]
- Miranda ER, Rot G, Toplak M, Santhanam B, Curk T, Shaulsky G, Zupan B. 2013. Transcriptional profiling of Dictyostelium with RNA sequencing. Methods Mol Biol 983: 139–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell PC. 1978. Genetics of the cellular slime molds. Annu Rev Genet 12: 69–93. [DOI] [PubMed] [Google Scholar]
- Nuckolls GH, Osherov N, Loomis WF, Spudich JA. 1996. The Dictyostelium dual-specificity kinase splA is essential for spore differentiation. Development 122: 3295–3305. [DOI] [PubMed] [Google Scholar]
- Ohta T. 2000. A comparison of mutation spectra detected by the Escherichia coli Lac+ reversion assay and the Salmonella typhimurium His+ reversion assay. Mutagenesis 15: 317–323. [DOI] [PubMed] [Google Scholar]
- Parkinson K, Bolourani P, Traynor D, Aldren NL, Kay RR, Weeks G, Thompson CRL. 2009. Regulation of Rap1 activity is required for differential adhesion, cell-type patterning and morphogenesis in Dictyostelium. J Cell Sci 122: 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakoe H, Chiba S. 1978. Dynamic programming algorithm optimization for spoken word recognition. IEEE Trans Acoust ASSP 26: 43–49. [Google Scholar]
- Santhanam B, Cai H, Devreotes PN, Shaulsky G, Katoh-Kurasawa M. 2015. The GATA transcription factor GtaC regulates early developmental gene expression dynamics in Dictyostelium. Nat Commun 6: 7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S, Prabhu S, O'Meara MM, Pe'er I, Hobert O. 2008. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods 5: 865–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulsky G, Escalante R, Loomis WF. 1996. Developmental signal transduction pathways uncovered by genetic suppressors. Proc Natl Acad Sci 93: 15260–15265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman HA, Silhavy TJ. 2003. Microbial genetics: The art and design of genetic screens: Escherichia coli. Nat Rev Genet 4: 419–431. [DOI] [PubMed] [Google Scholar]
- Spann TP, Brock DA, Lindsey DF, Wood SA, Gomer RH. 1996. Mutagenesis and gene identification in Dictyostelium by shotgun antisense. Proc Natl Acad Sci 93: 5003–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney KF, Huang C-H, Devreotes PN. 2010. Eukaryotic chemotaxis: a network of signaling pathways controls motility, directional sensing, and polarity. Annu Rev Biophys 39: 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman DM, Lemieux MG, Knecht DA, Insall RH. 2014. PIP3-dependent macropinocytosis is incompatible with chemotaxis. J Cell Biol 204: 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shaulsky G. 2015. TgrC1 has distinct functions in Dictyostelium development and allorecognition. PLoS One 10: e0124270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JG. 2010. Dictyostelium finds new roles to model. Genetics 185: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KL, Newell PC. 1976. A genetic study of aggregation in the cellular slime mould Dictyostelium discoideum using complementation analysis. Genetics 82: 287–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.