

Abstract
A stereocontrolled first total synthesis of muraymycin D1 (1) has been achieved. The synthetic route is highly stereoselective, featuring 1) selective β-ribosylation of the C2-methylated amino ribose, 2) selective Strecker reaction, 3) ring-opening reaction of a diastereomeric mixture of a diaminolactone to synthesize muraymycidine (epi-capreomycidine). The acid-cleavable protecting groups for secondary alcohol and uridine ureido nitrogen are applied for simultaneous deprotections with the Boc and tBu groups. Muraymycin D1 (1) and its amide derivatives (2 and 3) exhibited growth inhibitory activity against Mycobacterium tuberculosis (MIC50 1.56–6.25 μg/mL) and strong enzyme inhibitory activities against the bacterial phosphotransferases (MurX and WecA) (IC50 0.096−0.69 μM).
Graphical abstract
INTRODUCTION
Muraymycins belong to aminoribosyl-uridyl peptides that were isolated from Streptomyces spp. by McDonald et al.1 To date, 19 muraymycin congeners (muraymycin A1–5, B1–7, C1–4, D1–3) have been isolated. Their structural diversity is observed in the lipid moiety (R2) and the appended C5′-aminoribose unit (R1) (Figure 1). Muraymycin A1 is one of the most active members of this family and showed bactericidal activity against both Gram-positive and Gram-negative bacteria. Notably, muraymycin A1 demonstrated efficacy in the Staphylococcus aureus infected mice models (ED50 1.1 mg/kg).1 The muraymycins are structurally related to the other uridyl peptide antibiotics such as the liposidomycins, mureidomycins, pacidamycins, and tunicamycin.2 This class of natural products is reported to exhibit strong inhibitory activities against translocase I (MraY/MurX), essential peptidoglycan biosynthesis enzymes that catalyze the formation of lipid I from Park’s nucleotide (UDP-MurNAc-pentapeptide) with polyprenyl phosphate.3 Besides muraymycin A1, in vitro properties of the other muraymycin congeners have been poorly characterized. The difficulties in isolating the muraymycins in their pure form via reverse-phase HPLC as well as inaccessibility of the muraymycin-producing strain preclude biological evaluation. Recently, muraymycin D2 (R1, R2 = H in Figure 1) was reported to show no significant antibacterial activity, even though it has strong MraY enzyme inhibitory activity (IC50 0.01 μM).4 In addition, some structure-activity relationship (SAR) studies were also described based on the structure of muraymycins.5 Muraymycin D1 (1) is synthetically more challenging than other members of the muraymycin D series. Because 1 lacks only the lipophilic side chain appended in the L-leucine moiety of muraymycin A1, achievement of synthesis of 1 will make a promising step toward the total synthesis of muraymycin A1. Therefore, we desired to establish an efficient synthesis of 1 and thoroughly evaluate the efficacy of 1 in vitro. Several groups have reported synthetic efforts on muraymycins including a total synthesis of muraymycin D2.4,6 Although remarkable accomplishments have been documented in the reported syntheses, more efficient strategies that minimize generations of diastereomers and protecting group manipulations will accelerate the development of new analogs for multi-drug resistant (MDR) bacterial infections. Herein, we report a highly stereocontrolled total synthesis of muraymycin D1 (1), its amide analogs (2 and 3), and their evaluation against the bacterial phosphotransferases.
Figure 1.
Structures of representative muraymycins
RESULTS AND DISCUSSION
Our retrosynthesis for muraymycin D1 (1) is illustrated in Scheme 1; the challenging synthetic outcomes are highlighted in the structure of 1. Muraymycin D1 is retrosynthetically divided into the left- and right-half segments. We envisioned that the 3-aminopropyl amino acid moiety (C6′,7′-positions) of 1 could be constructed via Strecker reaction of the aldehyde 8 with the mono-protected 1,3-diaminopropane in the presence of an appropriate CN source. We have extensively studied ribosylations via non-anchimeric assistance of the C2-position and found that β-selective ribosylations can be achieved when the ribose-donors possess a bulky ester group at the C3″-postion.7 The muraymycins are vulnerable to strong bases and give rise to complex mixtures upon exposure. In order to achieve facile deprotection of the acyl group under acidic conditions, we planned to introduce the 3,3-dimethyl-5-(triisopropylsilyloxy)pentanoate protecting group for the alcohol at the C3″-position of the amino ribose (see 9).7 Construction of the R-configuration at C5′-position relies on Carreira’s asymmetric alkynation.8 Presence of (2S,3S)-muraymycidine (epi-capreomycidine) is one of the characteristics of the muraymycins.9 We have previously investigated lactone-opening reactions to synthesize 2S,3S-ureido-muraymycidine 7 through a diastero-mixture of 13.10 In addition, a unique selective-deprotection method to remove the 2-(trimethylsilyl)ethanol group of 7 followed by capturing the carboxylate using the polymer-supported fluoride (PS-F) is applied to facilitate the synthesis of the left-half segment (the ureido-tripeptide carboxylic acid).11 Coupling of the right- and left-half segments, global deprotections of all acid labile groups including monomethoxytetrachlorodiphenylmethoxymethyl (MTPM)12, followed by hydrolysis of the amide group are envisioned to furnish 1 in a single step.
Scheme 1.
Retrosynthetic analysis of muraymycin D1
The synthesis of 1 commenced with the left-half segment 7 (Scheme 2). We previously reported a scalable synthesis of (2R,3S)- and (2S,3S)-diaminolactones from (2S)-2-amino γ-butyrolactone.10 Extensive studies of the opening of 13 with a wide range of amino acids revealed that the undesired 2R-configuration of 13 is completely epimerized to the desired (2S,3S)-13 by treatment with 2(1H)-pyridinone at 70 °C. Interestingly, nucleophilic attacks of (2R,3S)-13 with C-protected amino acids did not take place, while (2S,3S)-13 underwent thermal amide-forming reaction. Taking advantage of these observations, a one-pot epimerization/lactone-opening reaction with the hydrazide 14 gave rise to the dipeptide 15. The overall yield of the transformation from 13 to 15 was determined to be >80% after acetylation of the primary alcohol of 15. Although the phenylhydrazide could serve as an appropriate C-protecting group to accomplish the synthesis of the left-half segment 23, deprotection of the phenylhydrazide group in 22 required multiple time-consuming purifications via reverse-phase HPLC (CH3OH-0.1% TFA = 50 : 50) to provide 23 in its pure form. In order to facilitate the synthesis of 23, we revised the orthogonal protection strategy. The hydrazide group of 16 was converted to the trimethylsilylethyl (TMSE) ester 17 in 95% yield by using N-bromosuccinimide (NBS)/NaHCO3 in anhydrous CH2Cl2. Hydrogenations of 17 provided the free-amine, which was then subjected to the urea-forming reaction with the imidazole-carboxamido derivative 18 to furnish 19 in 65% overall yield. The Boc group of 19 was removed with 4N HCl, and the generated HCl-amine salt was coupled with N,N′-di-tert-butoxycarbonyl-S-methyl isothiourea in the presence of Et3N and HgCl2 to afford 20 in 75% overall yield.13 [tBu2Sn(OH)Cl]2-catalyzed deacetylation14 of 20 followed by tosylation of the primary alcohol provided the intermediate 21 which subsequently underwent intramolecular cyclization and concomitant deprotection of the Boc group of the imino-N, yielding the ureido-muraymycidine tripeptide 7 in 85% overall yield.
Scheme 2.
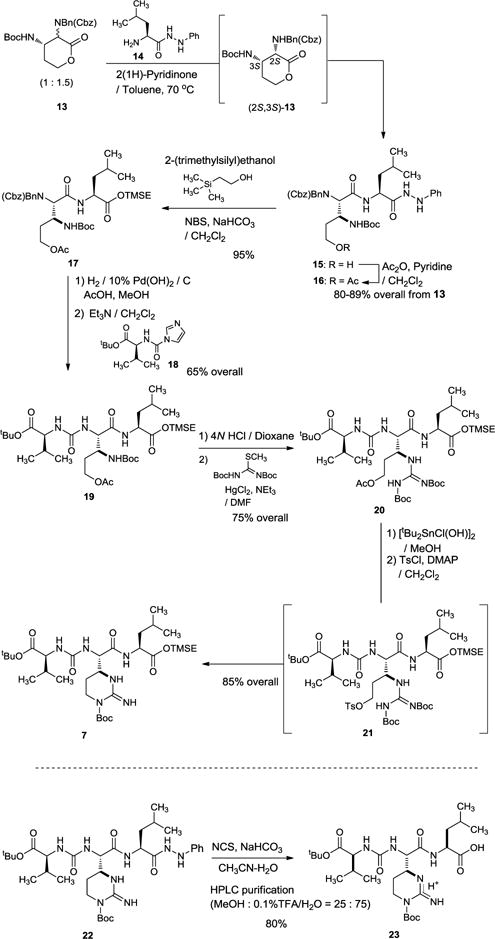
Synthesis of the left-half segment
We have introduced the monomethoxytetrachlorodiphenylmethoxymethyl (MTPM) protecting group because it has significant advantages over ordinal protecting groups (e.g. BOM) for uridine ureido nitrogen; the MTPM group is stable under hydrogenation conditions and to a wide range of acids, but it can be deprotected by solvolytic cleavage with 30% TFA.12,15 The MTPM-protected uridine 1115 was subjected to a modified Swern oxidation to provide the corresponding aldehyde in quantitative yield which was then subjected to Carreira’s asymmetric alkynation reaction using (+)-N-methylephedrine,8 yielding the (S)-propargyl alcohol 10 in 80% yield with S/R = >98:2 selectivity. Without the chiral controller, the 1,2-addition of the zinc acetylide species provided a mixture of the propargyl alcohols in 75% yield with S/R = 1.7:1.0 selectivity (Scheme 3). The stereochemistry of the secondary alcohol of 10 generated via Carreira’s alkynation was unequivocally determined by the advanced Mosher’s method.16 NIS-AgBF4 promoted ribosylation of 10 with the thioglycoside 9 furnished the β-glycoside 24 exclusively in 91% isolated yield.17 It is worth mentioning that the ribosylation demonstrated with 9 is an unusual observation in that the C2-ether-protected ribose donor provided β-glycoside without contamination of the α-glycoside. This observation may be attributable to C3-acyl group participation in the oxocarbenium ion transition state, leading exclusively to β-ribosylation of 9.7 The azido group of 24 was reduced with Zn metal in the presence of aq. NH4Cl, and the generated free-amine was protected with (Boc)2O to furnish 25 in 90% overall yield. The alkyne moiety of 25 was converted to the aldehyde via a standard three step procedure including partial reduction with a Lindlar’s catalyst, osmylation, and oxidative cleavage with Pb(OAc)4. The crude aldehyde 8 was subjected to a thiourea-catalyzed Strecker reaction18 with the Cbz-protected 1,3-diaminopropane to provide the desired aminonitrile 26 in 60% overall yield from 25. In this reaction, the undesired (R)-diastereomer was not observed by LC-MS, TLC or 1H-NMR analyses of the reaction mixture; thus, selectivity of the Strecker reaction of 8 was determined to be >25/1. Later, we realized that same transformation (from 8 to 26) could be catalyzed by MgSO4, producing 26 with the yield and S/R-selectivity comparable to the reaction catalyzed by the thiourea. The nitrile group of 26 was hydrated with HgCl2-aldoxime to furnish the amide 6 in 70% yield. The stereogenic center (C6′) generated via the Strecker reaction (8→26) was confirmed by the 1H-NMR analyses of 6 where the value of the coupling constant between H-5′ and H-6′ (J5′,6′ = 3.4 Hz) was in good agreement with the reported J value for (2S,3S)-2-amino-3,4-dihydroxybutyric acid derivatives (J = 3.2–8.0 Hz) and the muraymycin D2 synthetic intermediate (J = 3.5 Hz).1a,4,19,20 The Cbz group of 6 was removed under hydrogenation conditions21 followed by treatment with 1N HCl to provide the diamine HCl salts 27 in quantitative yield.
Scheme 3.
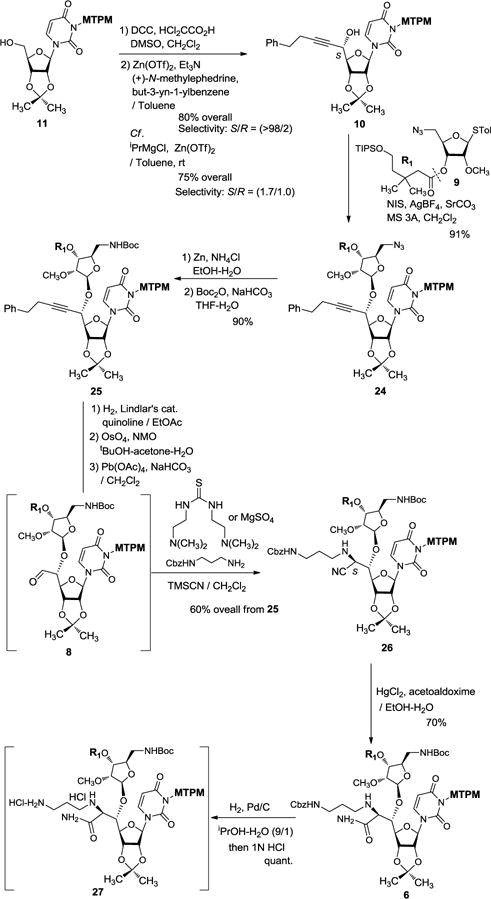
Synthesis of the right-half segment
The 2-(trimethylsilyl)ethanol (TMSE) group of the left-half segment 7 was selectively cleaved with PS-F11 to furnish the PS-ammonium salt 28 (Scheme 4). The purity of 28 was determined to be >92% by 1H-NMR and HPLC analyses of the protonated form 23 (see Scheme 2). Decomplexation of the PS-ammonium complex 28 was not observed under neutral conditions; conveniently, 28 could be dissociated under the peptide-forming reaction conditions (Glyceroacetonide-Oxyma (GOx, 29), EDCI, NaHCO3 in DMF-H2O),22 and the coupling reaction with 27 was complete in 3h to afford the protected muraymycin D1, 4 with >90% purity after water work-up and filtration. Global deprotection of 4 to form muraymycin D1 (1) was performed in two steps in a one-pot procedure with 84% overall yield; the MTPM, Boc, tBu and acyl (R1) groups were first removed via 30% TFA/CH2Cl2, then addition of 50% TFA/H2O removed the acetonide and amide groups leading to 1. Similarly, muraymycin D1-amide (2) was synthesized via treatment with 50% TFA/CH2Cl2 followed by addition of H2O. Primary amide-formation of the C-terminus of 2 was accomplished via our standard coupling conditions (29, EDCI, NaHCO3 in DMF-H2O)22 with excess NH4Cl to give rise to 3 in 75% yield. Muraymycin D1 and its amide analogs were synthesized in their pure forms as determined by C18 reverse-phase HPLC analyses (retention time of 1, 2, and 3: 10.0, 17.5, and 18.0 min, respectively; solvent system: MeOH: 0.1% TFA/H2O = 25 : 75, flow rate : 2.0 mL/min, UV: 254 nm).23
Scheme 4.
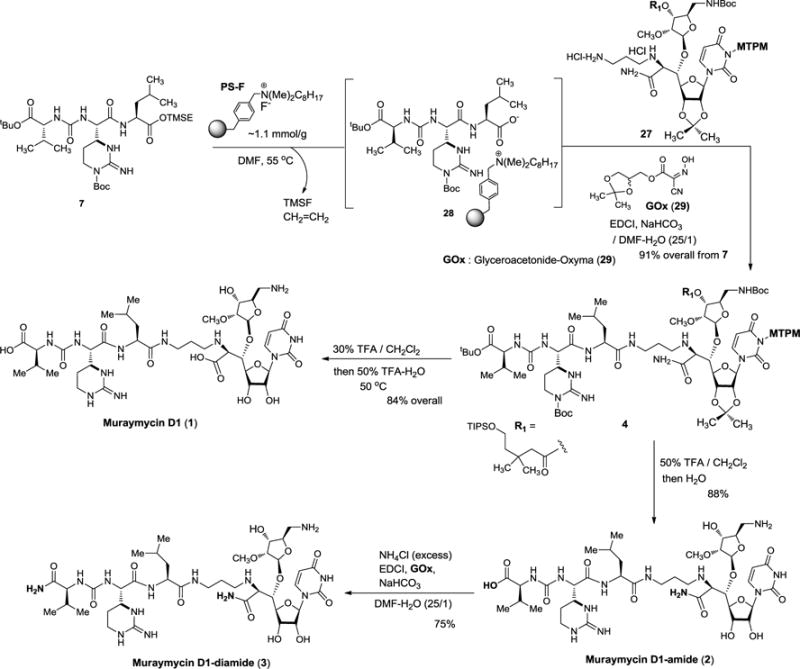
Synthesis of muraymycin D1 and its amide analogs
Antibacterial activity of some muraymycins is believed to be solely due to inhibition of MraY/MurX. The other bacterial phosphotransferase, polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA), has never been investigated as a potential mechanism of action for the muraymycins. WecA is an essential enzyme for the growth of M. tuberculosis. Inhibition of WecA blocks the entire biosynthesis of essential cell wall components of M. tuberculosis in both replicating and non-replicating states, making this enzyme a target for development of novel TB drugs.24 The synthetic molecules (1, 2, and 3) were evaluated in MurX and WecA assays (Table 1). Muraymycin D1 (1) and muraymycin D1-amide (2) exhibited equal enzyme inhibitory activities against the bacterial phosphotransferases (MurX and WecA) and their IC50 values were in low μM range. Inhibition of WecA enzyme of muraymycin D1-diamide (3) was ~10 times greater than that of 1 and 2. Extensive bacterial growth inhibitory assays of 1, 2, and 3 against Gram-positive and -negative bacteria including Mycobacterium spp. revealed that 1–3 exhibited bacteriostatic activity against M. tuberculosis; the MIC50 values are comparable to UT-01320 (a selective WecA inhibitor)24,25, capuramycin (a selective MurX inhibitor), and tunicamycin (a non-selective phosphotransferase inhibitor). However, 1–3 did not show antibacterial activity against Gram-negative bacteria (E. coli, K. pneumoniae, P. aeruginosa and A. baumannii) and Gram-positive bacteria (S. aureus, C. difficile, and E. faecium) even at >100 μg/mL concentrations. Unlike tunicamycin, 1–3 did not exhibit cytotoxicity against mammalian cells such as Vero cells even at 300 μg/mL concentration (see SI).
Table 1.
Bacterial phosphotransferase activities and MICs against M. tuberculosis
| Compound | WecA inhibition IC50 (μM)a |
MurX inhibition IC50 (μM)a |
M. tuberculosis growth inhibition [MIC50 (μg/mL)]b |
|---|---|---|---|
| Muraymycin D1 (1) | 0.69 | 0.011 | 1.56 |
| Muraymycin D1-amide (2) | 0.66 | 0.011 | 1.56 |
| Muraymycin D1-diamide (3) | 0.070 | 0.0096 | 6.25 |
| Tunicamycin | 0.15 | 3.38 | 3.13 |
| Capuramycin | – | 0.22 | 6.25 |
| UT-01320 | 0.060 | – | 1.56 |
CONCLUSIONS
In summary, a highly stereocontrolled first total synthesis of muraymycin D1 (1) has been achieved from the reported intermediates.10,12 The principal features of this synthesis include 1) stereoselective synthesis of the ureido-muraymycidine tripeptide, 2) β-selective glycosylation of the C2-methyl ether of the amino-ribose, and 3) syn-selective Strecker reaction to construct the 3-aminopropyl α-amino acid moiety in a single step. The acid-cleavable protecting groups introduced here allowed us to accomplish the synthesis of 1 with a minimum number of protecting group manipulations. Primary amide-formation of the free carboxylic acid of 2 could be achieved via a GOx/EDCI based coupling condition in H2O-containing solvents without protections of the amino and alcohol groups. We have demonstrated that the amide derivatives of 1 do not diminish MurX enzyme inhibitory activity. Muraymycin D1 and its amide derivatives are also effective in inhibiting WecA enzyme activity at low concentrations. Muraymycin D1-diamide (3) shows significantly greater inhibition of the WecA enzyme than its natural form. To date, only a few investigational TB drugs such as UT-01320 and CPZEN-45 have been reported to inhibit the WecA enzymes at low concentrations.25,26 Although the activity of muraymycin A1 has been evaluated in vitro and in vivo,1a the antibacterial activity of the other muraymycins (B, C and D) has not been thoroughly investigated. Interestingly, we have identified that muraymycin D1 shows strong bacteriostatic activity against M. tuberculosis by targeting both MurX and WecA enzymes. Amide derivatives of muraymycins can be purified readily via conventional methods without the need for HPLC purification. These chemical properties will facilitate the discovery of new muraymycin analogs. Application of the synthetic strategies presented here continues for the synthesis of muraymycin A1 and its analogs in our laboratory. Efficacy of muraymycin congeners against non-replicating M. tuberculosis will be reported elsewhere.
Supplementary Material
Acknowledgments
The National Institutes of Health is greatly acknowledged for financial support of this work (AI084411 and GM114611). MK thanks University of Tennessee for generous financial support (CORNET Award). NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant. The following reagent was obtained through BEI Resources, NIAID, NIH: M. tuberculosis, Strain H37Rv and Gamma-Irradiated M. tuberculosis, NR-14819. We gratefully acknowledge Dr. William Clemons (California Institute of Technology) for useful discussions of the WecA assays.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publication website at http://pubs.acs.org.
Complete experimental details, compound characterization data, and biological evaluation and data (PDF)
Notes
The authors declare no competing financial interests.
REFERENCES AND NOTES
- 1.(a) McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. J Am Chem Soc. 2002;124:10260–10261. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]; (b) McDonald LA, Barbieri LR, Carter GT, Kruppa G, Feng X, Lotvin JA, Siegel MM. Anal Chem. 2003;75:2730–2739. doi: 10.1021/ac0264731. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wiegmann D, Koppermann S, Wirth M, Niro G, Leyerer K, Ducho C. Beilstein J Org Chem. 2016;12:769–795. doi: 10.3762/bjoc.12.77. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Winn M, Goss RJ, Kimura K, Bugg TD. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]; (c) Kimura K, Bugg TD. Nat Prod Rep. 2003;20:252–273. doi: 10.1039/b202149h. [DOI] [PubMed] [Google Scholar]; (d) Knapp S. Chem Rev. 1995;95:1859–1876. [Google Scholar]
- 3.(a) Chung BC, Mashalidis EH, Tanino T, Kim M, Matsuda A, Hong J, Ichikawa S, Lee S-Y. Nature. 2016;533:557–560. doi: 10.1038/nature17636. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Niu G, Tan H. Trends Microbiol. 2015;23:110–119. doi: 10.1016/j.tim.2014.10.007. [DOI] [PubMed] [Google Scholar]; (c) Tanino T, Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A, Oyama H, Ichikawa S, Matsuda A. J Med Chem. 2011;54:8421–8439. doi: 10.1021/jm200906r. [DOI] [PubMed] [Google Scholar]; (d) Dini C. Curr Top Med Chem. 2005;5:1221–1236. doi: 10.2174/156802605774463042. [DOI] [PubMed] [Google Scholar]; (e) Yamashita A, Norton E, Petersen PJ, Rasmussen BA, Singh G, Yang Y, Mansour TS, Ho DM. Bioorg Med Chem Lett. 2003;13:3345–3350. doi: 10.1016/s0960-894x(03)00671-1. [DOI] [PubMed] [Google Scholar]; (f) Yamashita A, Norton EB, Williamson RT, Ho DM, Sinishtaj S, Mansour TS. Org Lett. 2003;5:3305–3308. doi: 10.1021/ol030085j. [DOI] [PubMed] [Google Scholar]
- 4.Tanino T, Ichikawa S, Shiro M, Matsuda A. J Org Chem. 2010;75:1366–1377. doi: 10.1021/jo9027193. [DOI] [PubMed] [Google Scholar]
- 5.(a) Takeoka Y, Tanino T, Sekiguchi M, Yonezawa S, Sakagami M, Takahashi F, Togame H, Tanaka Y, Takemoto H, Ichikawa S, Matsuda A. ACS Med Chem Lett. 2014;5:556–560. doi: 10.1021/ml5000096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hirano S, Ichikawa S, Matsuda A. J Org Chem. 2008;73:569–577. doi: 10.1021/jo702264e. [DOI] [PubMed] [Google Scholar]
- 6.(a) Spork AP, Büschleb M, Ries O, Wiegmann D, Boettcher S, Mihalyi A, Bugg TDH, Ducho C. Chem Eur J. 2014;20:15292–15297. doi: 10.1002/chem.201404775. [DOI] [PubMed] [Google Scholar]; (b) Tanino T, Ichikawa S, Matsuda A. Org Lett. 2011;13:4028–4031. doi: 10.1021/ol201527k. [DOI] [PubMed] [Google Scholar]; (c) Spork AP, Koppermann S, Dittrich B, Herbst-Irmer R, Ducho C. Tetrahedron: Asymmetry. 2010;21:763–766. [Google Scholar]; (d) Tanino T, Hirano S, Ichikawa S, Matsuda A. Nucleic Acids Symp Ser. 2008;52:557–558. doi: 10.1093/nass/nrn282. [DOI] [PubMed] [Google Scholar]; (e) Hirano S, Ichikawa S, Matsuda A. Tetrahedron. 2007;63:2798–2804. [Google Scholar]; (f) Srabia F, Martín-Ortiz L. Tetrahedron. 2005;61:11850–11865. [Google Scholar]; (g) Yamashita A, Norton EB, Ho DM, Sinishtaj S, Mansour TS. Org Lett. 2003;5:3305–3308. doi: 10.1021/ol030085j. [DOI] [PubMed] [Google Scholar]
- 7.Kurosu M, Li K. J Org Chem. 2008;73:9767–9770. doi: 10.1021/jo801408x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frantz DE, Fassler R, Carreira EM. J Am Chem Soc. 2000;122:1806–1807. [Google Scholar]
- 9.(a) DeMong DE, Williams RM. Tetrahedron Lett. 2001;42:3529–3532. [Google Scholar]; (b) Nomoto S, Teshima T, Wakamiya T, Shiba T. Tetrahedron. 1978;34:921–92. [Google Scholar]; (c) Nomoto S, Teshima T, Wakamiya T, Shiba T. J Antibiot. 1977;30:955–959. doi: 10.7164/antibiotics.30.955. [DOI] [PubMed] [Google Scholar]
- 10.Aleiwi BA, Schneider CM, Kurosu M. J Org Chem. 2012;77:3859–3867. doi: 10.1021/jo300205b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurosu M, Crick DC. Tetrahedron Lett. 2006;47:5325–5328. [Google Scholar]
- 12.Wang Y, Kurosu M. Tetrahedron. 2012;68:4797–4803. doi: 10.1016/j.tet.2012.03.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeMong DE, Williams RM. J Am Chem Soc. 2003;125:8561–8565. doi: 10.1021/ja0351241. [DOI] [PubMed] [Google Scholar]
- 14.Orita A, Hamada Y, Nakano T, Toyoshima S, Otera J. Chem Eur J. 2001;7:3321–3327. doi: 10.1002/1521-3765(20010803)7:15<3321::aid-chem3321>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Siricilla S, Aleiwi BA, Kurosu M. Chem Eur J. 2013;19:13847–13858. doi: 10.1002/chem.201302389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–519. [Google Scholar]; b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Org Chem. 1991;56:1296–1298. [Google Scholar]
- 17.(a) Kurosu M, Li K, Crick DC. Org Lett. 2009;11:2393–2397. doi: 10.1021/ol900458w. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ribosylation of the 5′-glycyl derivatives of uridine from other groups;; (b) Nakamura H, Yoshida T, Tsukano C, Takemoto Y. Org Lett. 2016;18:2300–2303. doi: 10.1021/acs.orglett.6b00943. [DOI] [PubMed] [Google Scholar]; (c) Nakamura H, Tsukano C, Yasui M, Yokouchi S, Igarashi M, Takemoto Y. Angew Chem Int Ed Engl. 2015;54:3136–3139. doi: 10.1002/anie.201411954. [DOI] [PubMed] [Google Scholar]; (d) Gopinath P, Wang L, Abe H, Ravi G, Masuda T, Watanabe T, Shibasaki M. Org Lett. 2014;16:3364–3367. doi: 10.1021/ol501397b. [DOI] [PubMed] [Google Scholar]; (e) Hirano S, Ichikawa S, Matsuda A. J Org Chem. 2007;72:9936–9946. doi: 10.1021/jo701699h. [DOI] [PubMed] [Google Scholar]; (f) Hirano S, Ichikawa S, Matsuda A. Angew Chem Int Ed. 2005;44:1854–1856. doi: 10.1002/anie.200462439. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Liu X, Feng X. Chem Rev. 2011;111:6947–6983. doi: 10.1021/cr200057t. [DOI] [PubMed] [Google Scholar]
- 19.(a) Spork AP, Ducho C. Synlett. 2013;24:343–346. [Google Scholar]; (b) Cativiela C, Díaz-de-Villegas MD, Gálvez JA, García JI. Tetrahedron. 1996;52:9563–9574. [Google Scholar]
- 20.Syn-selective Strecker reaction of 8 was also supported by an accomplishment of the streocontrolled synthesis of (+)-FR-900493, whose structure was well-characterized by 1H-NMR.6e Mitachi, K.; Kurosu, M. unpublished data.
- 21.Aleiwi BA, Kurosu M. Tetrahedron Lett. 2012;53:3758–3762. doi: 10.1016/j.tetlet.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q, Wang Y, Kurosu M. Org Lett. 2012;14:3372–3375. doi: 10.1021/ol3013556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The compounds 1, 2, and 3 were isolated as their TFA salts.
- 24.(a) Siricilla S, Mitachi K, Skorupinska-Tudek K, Swiezewska E, Kurosu M. Anal Biochem. 2014;461:36–45. doi: 10.1016/j.ab.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mitachi K, Siricilla S, Yang D, Kong Y, Skorupinska-Tudek K, Swiezewska E, Kurosu M. Anal Biochem. 2016;512:78–90. doi: 10.1016/j.ab.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siricilla S, Mitachi K, Wan B, Franzblau SG, Kurosu M. J Antibiot. 2015;68:271–278. doi: 10.1038/ja.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishizaki Y, Hayashi C, Inoue K, Igarashi M, Takahashi Y, Pujari V, Crick DC, Brennan PJ, Nomoto A. J Biol Chem. 2013;288:30309–30319. doi: 10.1074/jbc.M113.492173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.