

Abstract
Human sirtuin 2 (SIRT2) is an NAD+-dependent deacetylase that primarily functions in the cytoplasm, where it can regulate α-tubulin acetylation levels. SIRT2 is linked to cancer progression, neurodegeneration, and infection with bacteria or viruses. However, the current knowledge about its interactions and the means through which it exerts its functions has remained limited. Here, we aimed to gain a better understanding of its cellular functions by characterizing SIRT2 subcellular localization, the identity and relative stability of its protein interactions, and its impact on the proteome of primary human fibroblasts. To assess the relative stability of SIRT2 interactions, we used immunoaffinity purification in conjunction with both label-free and metabolic labeling quantitative mass spectrometry. In addition to the expected associations with cytoskeleton proteins, including its known substrate TUBA1A, our results reveal that SIRT2 specifically interacts with proteins functioning in membrane trafficking, secretory processes, and transcriptional regulation. By quantifying their relative stability, we found most interactions to be transient, indicating a dynamic SIRT2 environment. We discover that SIRT2 localizes to the ER-Golgi intermediate compartment (ERGIC), and that this recruitment requires an intact ER-Golgi trafficking pathway. Further expanding these findings, we used microscopy and interaction assays to establish the interaction and coregulation of SIRT2 with liprin-β1 scaffolding protein (PPFiBP1), a protein with roles in focal adhesions disassembly. As SIRT2 functions may be accomplished via interactions, enzymatic activity, and transcriptional regulation, we next assessed the impact of SIRT2 levels on the cellular proteome. SIRT2 knockdown led to changes in the levels of proteins functioning in membrane trafficking, including some of its interaction partners. Altogether, our study expands the knowledge of SIRT2 cytoplasmic functions to define a previously unrecognized involvement in intracellular trafficking pathways, which may contribute to its roles in cellular homeostasis and human diseases.
Human sirtuin 2 (SIRT2)1 is one of seven NAD+-dependent deacylases (SIRT1–7) that were originally discovered as homologues of S. Cerevisiae Sir2 regulator of gene silencing in mating-type loci and telomeres (1). As the enzymatic activity of sirtuins is dependent on the presence of a major metabolic molecule, NAD+, these enzymes can act as sensors of intracellular energy states. Sirtuins are distributed throughout major intracellular compartments to deliver this information to nuclear (SIRT1, SIRT6), nucleolar (SIRT7), cytoplasmic (SIRT2), and mitochondrial (SIRT3, SIRT4, SIRT5) processes (2). The prominent enzymatic activity of sirtuins is thought to be deacetylation, i.e. removal of acetylation from lysine residues of protein substrates. However, studies from several research groups have demonstrated that sirtuins also possess other enzymatic activities. SIRT4 and SIRT6 can act as ADP-ribosyltransferases (3, 4), and more recently SIRT4 was identified as an efficient lipoamidase (5). SIRT2 and SIRT6 display demyristoylation activity (6, 7), whereas SIRT5 acts as demalonylase and desuccinylase (8). Given the wide range of histone and nonhistone substrates of sirtuins, these enzymes have been studied in the context of cancer, viral infection, neurological disorders, and lifespan (9–11). For instance, a recent study has demonstrated that sirtuins, including SIRT2, have broad-spectrum antiviral functions in primary human fibroblasts upon infection with several DNA or RNA viruses (11).
SIRT2 is primarily known as a cytoplasmic NAD+-dependent deacetylase. Previous studies reported that two SIRT2 isoforms are present in human cells, where isoform 1 represents the full-length protein, whereas isoform 2 results from alternative splicing and lacks the first 38 amino acids (12). Both isoforms are catalytically active, but differ in their tissue expression patterns. Isoform 1 is abundant in skeletal muscle, whereas isoform 2 is predominant in the brain (13). This suggests cell type-specific functions and contribution to different human diseases that remain to be fully understood. So far, most studies on SIRT2 functions within intracellular pathways have been focused on characterization of its substrates. The first identified substrate was α-tubulin, which is deacetylated by SIRT2 at Lys40 (14). This deacetylation process is thought to play a role in the progression of mitosis and in neuronal cell motility (15–18). SIRT2 can also regulate mitosis by relocating to chromatin, reducing global levels of acetylated histone H4 at Lys16, and thereby aiding in chromatin compaction (19–21). Another SIRT2 histone substrate, H3Lys18 was shown to regulate gene expression during bacterial infection with Listeria (22). The roles of SIRT2 were also explored in connection to metabolic diseases, in part via its deacetylation of ATP citrate lyase (ACLY) (23) and phosphoglycerate mutase (PGAM) (24). Overall, these studies of SIRT2 substrates indicate its involvement in a variety of intracellular processes.
In addition to regulating substrates, it is becoming evident that sirtuins and other histone deacetylases (HDACs) can also modulate protein functions through the formation of diverse protein-protein interactions. For instance, a global HDAC interactome study revealed previously undefined HDAC11 functions in mRNA splicing through its interaction with the SMN complex (25). Activity-dependent interactions of SIRT6 with the stress response factor G3BP1 pointed at its involvement in regulation of cellular stress responses (26, 27). So far, there is limited knowledge regarding SIRT2 protein–protein interactions, and it is mainly derived from either in vitro studies or analyses performed in immortalized cells. In a yeast two-hybrid screen, SIRT2 was found to interact with Homeobox Transcription Factor HOXA10 that could contribute to its role in cell cycle progression (28). Additionally, a large-scale study aimed at gaining insights into the broad interaction network of over 2000 human proteins was performed in HEK293T cells, and identified several associations when using FLAG-HA-tagged SIRT2 as one of the baits (29). However, the functional significance of these interactions requires further investigation, and there is a need to understand SIRT2 interactions in primary human cells.
Sirtuins can additionally impact cellular pathways through the ability of some of their substrates and interactions to regulate gene expression. For example, SIRT7 was found to play a role in RNA Pol I-dependent transcription through its interactions with the B-WICH chromatin remodeling complex (30). Both SIRT1 and SIRT2 were shown to modulate gene expression through deacetylation of several transcription factors, including nuclear factor κΒ (NFκΒ) (31, 32), p53 (33, 34), forkhead box protein O3 (FOXO3A) (35, 36), and coactivator β-catenin (37, 38). In addition, the neuroprotective effects of SIRT2 inhibition in Huntington's disease was associated with the SIRT2-dependent regulation of the SREBP-2 transcription factor trafficking from the endoplasmic reticulum to the nucleus (39). These studies highlight the importance of continuing the evaluation of sirtuin impact on gene expression and total protein levels.
The limited knowledge regarding the nonenzymatic roles of SIRT2 via either protein interactions or gene expression, as well as its likely context-dependent functions, may contribute to the different observations for SIRT2 functions in cancer progression. For instance, SIRT2 may act as a tumor suppressor, promoter, or both. In SIRT2 knockout mice, tumor development was observed to increase with age (40) or when challenged with mutagenic agents (41). It was also noted that SIRT2 expression is decreased in gliomas (42). On the other hand, disruption of SIRT2 activity was recently reported to have anticancer effects in nonsmall cell lung cancer (43), human colon carcinoma (44), and other cancer cell lines (45). In order to fill the gap in the current knowledge of SIRT2 biological functions, we need a better view of its protein interactions and impact on protein levels. Here, we addressed these questions by generating a refined network of SIRT2 protein interactions in primary human fibroblasts. We integrated label-free and metabolic labeling mass spectrometry to assess the relative stability of SIRT2 interactions. These analyses, in conjunction with microscopy and reciprocal isolation studies, led to the discovery of a previously unrecognized localization for SIRT2 at an intersection of intracellular trafficking routes and its interaction with proteins that function in these pathways. Specifically, we find SIRT2 to localize at the endoplasmic reticulum-Golgi intermediate compartment (ERGIC). This localization and its interaction with trafficked proteins, including the focal adhesion adaptor protein liprin-β1 (PPFiBP1), were dependent on the presence of an intact ER-Golgi trafficking pathway. Furthermore, we assessed the impact of SIRT2 knockdown on global protein levels, identifying an up-regulation of selected proteins in the endoplasmic reticulum and membrane organization pathways. Altogether, our results point to a role for SIRT2 in regulation of cellular trafficking through formation of protein interactions and regulation of protein levels.
EXPERIMENTAL PROCEDURES
Antibodies, Reagents, and Tissue Culture
The following primary antibodies and stains were used for Western blotting and/or confocal imaging: anti-SIRT2 (Abcam, Cambridge, MA), anti-PPFiBP1 (Abcam for Western blotting and Bethyl Laboratories for immunoaffinity purification), anti-ERGIC53 (Sigma Aldrich, Allentown, PA), DAPI (ThermoFisher Scientific, Waltham, MA). Anti-rabbit or anti-mouse light chain specific HRP-conjugated (Jackson ImmunoResearch, West Grove, PA) secondary antibodies were used for Western blotting. Alexa Fluor (Invitrogen Life Technology, Grand Island, NY) secondary antibodies were used for confocal imaging. ER tracker Red (BODIPY TR Glibenclamide, Life Technologies) and Golgi tracker red (BODIPY TR Ceramide) were used for confocal imaging of endoplasmic reticulum and golgi, respectively. Brefeldin A was obtained from Sigma and used at 3 μg/ml. Human MRC5 fibroblasts, HEK293T, and Phoenix cells were obtained from ATCC and grown in Dulbecco's Modified Eagle medium (DMEM, Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum at 37 °C and in 5% CO2. For the I-DIRT (30, 46) experiments, metabolic labeling was achieved by culturing MRC5 cells in SILAC DMEM (ThermoFisher Scientific, Waltham, MA) with the addition of “light” arginine and lysine (12C6) or “heavy” arginine and lysine (13C6) from Cambridge Isotope (Tewksbury, MA) and supplemented with dialyzed FBS (Gemini Bio-Products, West Sacramento, CA). Cell culture was allowed to proceed for five passages to ensure over 95% incorporation rate. Dulbecco's Phosphate-Buffered Saline (DPBS) buffer was obtained from ThermoFisher Scientific.
Generation of Cells Stably Expressing EGFP-FLAG and SIRT2-EGFP
EGFP open reading frame (ORF) was obtained from pEGFP-N1 (Clontech, Mountain View, CA) and inserted into pLXSN retroviral vector (Clontech, Mountain View, CA) alone or in fusion with FLAG at the 3′ end (EGFP-FLAG). SIRT2 isoform2-containing constructs were cloned from pCDNA3.1 plasmid (Addgene, Cambridge, MA) into pLXSN EGFP plasmid by PCR amplification of SIRT2 isoform-2 ORF. Phoenix cells were transfected with the resulting SIRT2-EGFP or EGFP-FLAG plasmid using Lipofectamin 2000 (Invitrogen Life Technology) in accordance with manufacturer's protocol. Generated retrovirus-containing media was filtered with 0.45 μm Acrodisc syringe filter (Pall, Port Washington, NY), supplemented with 4 μg/ml polybrene (Millipore, Billerica, MA) and used to transduce MRC5 fibroblasts. Cells stably expressing transduced construct were selected with 400 ng/ml G418 (ThermoFisher Scientific) for 7 days.
qPCR Analysis
Total mRNA was isolated with RNeasy mini kit (Qiagen, Manchester, UK). 1 μg of total mRNA was used for cDNA synthesis using RETROscript First Strand Synthesis Kit for RT-PCR (Ambion Life Technologies, ThermoFisher Scientific). Primers used for qPCR analysis were as followed: SIRT2: Fwd: 5′-ACCCGCTAAGCTGGATGAAAGAG-3′; Rev: 5′-AGTCTTCACACTTGGGCGTCAC-3′; β-actin: Fwd: 5′-TCCTCCTGAGCGCAAGTACTC-3′; Rev: 5′-CGGACTCGTCATACTCCTGCT-3′. ABI 7900 or ABI 384 thermocyclers (Applied Biosystems, Foster City, CA) were used for qPCR reaction and data collection. SDS 2.3 (Applied Biosystems) software was used for data processing.
Isolation of SIRT2 Protein Complexes and Sample Processing
SIRT2-EGFP and EGFP-FLAG immunoaffinity purification was performed as described previously (47). MRC5 cells stably expressing SIRT2-EGFP or EGFP-FLAG were washed with DPBS, resuspended in PVP-HEPES buffer (20 mm Na-HEPES, 1.2% polyvinylpyrrolidone (w/v) pH 7.4 with protease inhibitor mixture (Sigma Aldrich), and frozen as droplets in liquid nitrogen with frozen lysis buffer for cryogenic grinding. Lysis buffer was based on common TBT buffer components (200 mm K-HEPS pH7.4, 1.1 m KOac, 1% Tween-20(v/v)) with varying concentrations of NaCl and Triton X-100 used for optimization. Frozen pellets were ground using Retsch MM301 Mixer Mill (Retsch, Newtown, PA) at 30 Hz, 2.5 min cycle, 10 times. Cell powder was resuspended in additional optimized lysis buffer and subject to homogenization by Polytron (Kinematica, Bohemia, NY). Unsolubilized material was separated by centrifugation at 3400 × g, 4 °C. The soluble fraction was used as input for immunoaffinity purification using magnetic beads coated with epoxy groups and conjugated to anti-GFP antibody. The conjugation was performed using previously described method (48). After incubation with cell lysate for one hour at 4 °C, the beads were collected on a magnet and subjected to six washes in lysis buffer, followed by one wash in DPBS. Captured proteins were eluted with LDS sample buffer (247 mm Tris (pH 8.5), 2% LDS, 0.51 mm EDTA), boiled at 70 °C for 10min and left on TOMY shaker for 10min. The eluted proteins were analyzed by mass spectrometry or Western blotting. For mass spectrometry analysis, protein mixture was reduced with 20 mm TCEP, alkylated with 20 mm chloroacetamide and subjected to trypsin (Pierce) digestion in solution using filter-assisted sample preparation (FASP) method as described previously (49, 50). Briefly, protein was denatured in urea-containing buffer, captured on Amicon Ultra-0.5 centrifugal filters, 30 kDa NMWL (Millipore), and washed with 50 mm ammonium bicarbonate. Trypsin (ThermoFisher Scientific) was added on top of the filters at 1:50 (enzyme/substrate) ratio and digestion was carried overnight at 37 °C. Peptides were collected into spin columns and subjected to desalting on SDB-RPS StageTips as described below.
SIRT2 Deacetylation Activity Assay
Deacetylation activity assay was performed using SIRT2 Direct Fluorescent Screening Assay Kit (Cayman Chemicals, Ann Arbor, MI) according to the manufacturer's protocol. Briefly, SIRT2-EGFP and EGFP were isolated from human fibroblasts by immunoaffinity purification on magnetic beads. The bound proteins were incubated with 125 μm peptide substrate (Gln-Pro-Lys-Lys(ε-acetyl)-AMC) and 2 mm of the cofactor NAD+. As this deacetylation reaction is not performed with recombinant SIRT2, but rather with a lower amount of SIRT2 isolated (together with associated proteins) from cell culture, we optimized the incubation time, extending it to overnight incubation at 37 °C. The reaction was quenched with nicotinamide-containing developer. Fluorescence was measured using a SpectraMax Gemini XS microplate reader (Molecular Devices, Ramsey, MN).
Assessing the Relative Stability of Interactions Using I-DIRT
For I-DIRT-based analysis of interactions (46), equal amounts of metabolically labeled MRC5 fibroblasts wt (light) or SIRT2-EGFP expressing (heavy) were combined prior to cryogenic grinding. Ground cells were resuspended in stringent lysis buffer (TBT buffer containing 200 mm NaCl, 0.5% Triton, phosphatase inhibitor mixture 2 and 3, protease inhibitor mixture, and Benzonase (Pierce Universal Nuclease, ThermoFisher Scientific) and processed for IP as described above. An aliquot of heavy-labeled MRC5 was used to measure isotope label incorporation efficiency. Briefly, cells were lysed in 0.1 m ammonium bicarbonate, 2% deoxycholate with 20 mm TCEP, and 20 mm chloroacetamide, and the soluble fraction was collected. Approximately 50 μg of total protein was used for overnight trypsin (Pierce) digestion (1:50 enzyme/substrate) at 37 °C. After overnight incubation at 37 °C, equal volumes of ethyl acetate and 0.5%TFA were added to the sample, which was then subjected to centrifugation. The aqueous layer containing the peptides was recovered and subjected to desalting on SDB-RPS Stage Tips as described below. The peptide mixture was analyzed by mass spectrometry, as described below. I-DIRT scores were calculated for each interacting protein as Heavy/(Heavy+Light) isotopic ratio in ProteomeDiscoverer. In this formula, Heavy and Light denote average MS1 precursor peak areas of heavy and light isotopes, respectively. A minimum of two peptides per protein was used for quantification.
Generation of shRNA Expressing Cells
shRNA targeting SIRT2 (shSIRT2) or nontargeting (shCtrl) were obtained from Sigma (TRCN0000040221 and SHC202, respectively). HEK293T cells were transfected with the shRNA plasmids and helper plasmids (pMD2.G and pCMVΔR8.2, Addgene) using Lipofectamin2000 (Invitrogen) reagent according to manufacturer's protocol. Lentivirus-containing media was collected and used for transduction of MRC5 cells in the presence of 4 μg/ml polybrene (Millipore, Billerica, MA). Transduced cells were selected with 4 μg/ml puromycin (InvivoGen, San Diego, CA) for 7 days. shSIRT2 and shCtrl MRC5 cells were differentially labeled by stable isotope labeling as described above.
Sample Processing for the Assessment of Total Protein Levels
MRC5 cells stably expressing shSIRT2 or shCtrl and differentially labeled light or heavy, respectively, were collected by scraping into DPBS. Cell pellets were obtained by centrifugation and lysed in urea-based lysis buffer (25 mm HEPES pH8.0, 9 m urea, 1/100 (v/v) phosphatase inhibitor mixture II and III (Sigma), 1/100 (v/v) protease inhibitor mixture (Sigma), and 50 mm nicotinamide). Lysates were subjected to tip sonication (15W output, three bursts of 15 s each) and cooled on ice for 1min between each burst. Cell debris were pelleted by centrifugation at 20,000 × g for 15min at 4 °C. Protein concentration was measured by Bradford and equal amounts of total protein from each cell line were combined prior to protein digestion. Protein mixture was reduced and alkylated with 20 mm TCEP and 20 mm chloroacetamide, respectively. Reaction was quenched by the addition of 20 mm cysteine. Protein samples were digested by r-Lys-C (Promega) at 1:600 enzyme/substrate ratio for 7 h at room temperature. The digestion reaction was diluted threefold with 25 mm HEPES pH8.0 and trypsin (ThermoFisher Scientific) was added at 1:100 enzyme/substrate ratio. Trypsin digestion was carried overnight at 37 °C. Peptides were purified on Sep-Pak C18 column (WAT051910, Waters) according to manufacturer's protocol. Purified peptide mixture was subjected to desalting on SDB-RPS StageTips and sequential fractionation as described below.
Peptide Sample Preparation for Mass Spectrometry Analysis
Peptide mixture recovered after proteolytic digestion was acidified to 1%TFA and loaded onto StageTips assembled from low retention plastic tips (ThermoFisher Scientific) and 3 m SDB-RPS Empore Discs (Sigma-Aldrich). Loaded membrane was subjected to sequential washing (0.2% TFA) and elution (5% ammonium hydroxide and 80% acetonitrile) steps, as described previously (51). Isotopically labeled peptide samples from I-DIRT and shRNA experiments were subjected to sequential elution from StageTips using the following buffers: 0.1 m ammonium formate, 0.5% formic acid, 40% acetonitrile (elution 1), 0.15 m ammonium formate, 0.5% formic acid, 60% acetonitrile (elution 2), 5% ammonium hydroxide, and 80% acetonitrile (elution 3). Eluted peptides were concentrated by vacuum centrifugation and resuspended in 1% FA/2% acetonitrile to 9 μl for mass spectrometry analysis.
Mass Spectrometry Analysis
Approximately 4μl (for IP experiments) or 4 μg (for shRNA experiments) of peptide mixture prepared by StageTip was injected for nano-liquid chromatography/tandem mass spectrometry analysis (nLC-MS/MS) using Dionex Ultimate 3000 nanoRSLC (Dionex Corp., Sunnyvale, CA) with EASY-Spray source coupled to an LTQ-Orbitrap Velos (Thermo Fisher Scientific). Peptide mixture was first separated by reverse-phase chromatography on EASY-Spray column, 50 cm × 75 μm ID, PepMap RSLC C18, 2 μm (ThermoFisher Scientific) at a flow rate of 250 nL/min. For peptide mixture from IP experiments, 180 min 4% to 40%B linear acetonitrile gradient was applied (mobile phase A: 0.1% formic acid and 0.1% acetic acid in water; mobile phase B: 0.1% formic acid and 0.1% acetic acid in 97% acetonitrile). For shRNA experiment peptide mixture, 360 min 4% to 35%B gradient was applied. Mass spectrometer was set to data-dependent acquisition mode with FTMS preview scan disabled, enabled predictive automatic gain control, enabled dynamic exclusion that was optimized based on base peak width and other MS settings (52) (repeat count: 1, exclusion duration: 60), and enabled lock mass (mass list 371.101233). Other parameters included: FT MS1 target value of 1e6 and 500ms maximum injection time, ion trap MS2 target values of 1e4 and 100ms maximum injection time. One acquisition cycle comprised a single full-scan mass spectrum (m/z = 350.0–1700.0) in Orbitrap (r = 30,000 for regular IP, r = 60,000 for I-DIRT IP, or r = 100,000 for shRNA experiment at m/z = 400), followed by collision-induced dissociation on top 20 precursor ions in the dual-pressure linear ion trap with isolation width of 2.0 Th, normalized collision energy of 35 and activation time of 10ms.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD004212.
IP Experiments Mass Spectrometry Data Processing and Analysis
Raw data from MS analysis was extracted and subjected to search against UniProt Swiss-Prot sequence database (22,630 entries including human, herpesvirus, and common contaminants, downloaded in August, 2013) in Proteome Discoverer (v. 1.4.0.288, Thermo Fisher Scientific) by Sequest HT algorithm (v1.3, ThermoFisher Scientific). The following search criteria were used: full trypsin specificity, maximum 2 missed cleavage sites, precursor and fragment ion mass tolerance of 10 ppm and 0.5 Da, respectively; dynamic modifications: oxidation (+15.995 Da (M)), phospho (+79.966Da (S,T,Y)); static modifications: carbamidomethyl (+57.021 Da(C)). Peptide spectral match probabilities were calculated against decoy database by Percolator in Proteome Discoverer. Subsequent validation of protein identifications was performed in Scaffold (v. 4.4.8; Proteome Software, Inc., Portland, OR) using X!Tandem (53) and ProteinProphet (54) alogorithms. Additional variable modifications used in Scaffold search: Glu->pyro-Glu (−18.01), Ammonia-loss (−17.03), Gln->pyro-Glu (−17.03), deamidation (+0.98, N and Q), acetylation (+42.01, K and protein N terminus), carbamylation (+43.01, K and n). Peptide identifications were probabilistically validated using PeptideProphet (55) and corresponding protein probabilities were derived using ProteinProphet in Scaffold. To reduce the global protein and peptide false discover rate, probability filters were set to less than 1% and protein identification required at least two unique peptides. Common contaminant entries were removed and unweighted spectrum counts were exported for further analysis. Supplemental Tables S1 and S2 indicate number of unique peptides and sequence coverage, respectively, for each identified protein in label-free experiments. Supplemental Table S7 indicates number of unique peptides and sequence coverage for each identified protein in metabolic labeling experiment. Online SAINT (56) algorithm (www.crapome.org) was utilized to assign specificity scores to individual interactions based on spectral counts (supplemental Table S3). The following settings were used for probability score analysis: LowMode = 0, minFold = 0, Normalize = 1.
shRNA Experiments Mass Spectrometry Data Processing and Analysis
Raw data was searched against the UniProt Swiss-Prot sequence database (22630 entries, downloaded in August, 2013) using MaxQuant (v. 1.5.3.8) (57). The following group-specific parameters were used: standard type, multiplicity of 2 with Arg6 and Lys6 heavy labels, fully specific trypsin digestion with max. Two missed cleavages, precursor and fragment ion mass tolerance of 20ppm and 0.5Da, respectively, variable modifications: oxidation (M), acetyl (protein N-term), max. 5 modifications per peptide, requantify enabled, and “match from and to” type. The following global parameters were used: fixed modifications (carbamidomethyl, C), unique+razor peptides for quantification. Peptide-spectrum match, protein and site FDR were fixed at 1%. Other parameters were as preset. Supplemental Table S9 indicates number of unique peptides and sequence coverage for each identified protein.
MaxQuant output file containing information on identified protein groups, including normalized heavy/light (shCtrl/shSIRT2) isotopic ratios for each, was imported for further analysis into Perseus (v.1.5.2.4). For further processing, data from two replicates was first filtered to exclude potential contaminants and include at least one valid value in one of the replicates and ratio count>1 in both replicates. After that, each ratio was converted into shSIRT2/shCtrl and transformed into log2. For density estimation algorithm, shSIRT2/shCtrl normalized ratios were compared using P(x,y) distribution type. Gene Ontology Biological Process (GOBP) annotations were added in Perseus and 2D annotation enrichment algorithm (58) was applied to analyze overall changes in protein levels within each GOBP category.
Protein Network Analysis
SIRT2 interacting proteins that passed SAINT specificity score cutoff were loaded on string-db.org (59) for visual representation of existing evidence for known interactions present within isolated proteins with interaction score set to 0.4. For further analysis, STRING network was uploaded to Cytoscape software (cytoscape.org) (60) and attributes were added as indicated. Normalized spectral abundance factor (NSAF) (61) was used to calculate relative enrichment of protein within an IP. First, spectral abundance factor (SAF) was calculated by finding spectral counts to protein length (number of amino acids) ratio, then NSAF was calculated as the ratio of SAF to the sum of SAF across the IP. Total protein abundance values (PAX) were retrieved from integrated protein abundance database (PaxDb, pax-db.org) (62) and used to calculate NSAF/PAX ratio.
Reciprocal Isolations
MRC5 cells stably expressing SIRT2-EGFP were collected and washed with DPBS. Cells were lysed in IP buffer as described above. Soluble cell fraction was split equally between PPFiBP1 and control rabbit IgG IPs. Magnetic epoxy beads (Invitrogen) were conjugated with anti-PPFiBP1 antibodies (Bethyl Laboratories, Montgomery, TX) or IgG, and used for reciprocal isolations. Input and elution fractions were analyzed by Western blotting.
Confocal Microscopy
Cells grown on micro cover glass No1.5 (VWR, Radnor, PA) were washed in DPBS, fixed with 4% paraformaldehyde in PBS (ThermoFisher Scientific), and permeabilized with Triton X-100 (IBI Scientific, Peosta, IA). Samples were probed with indicated antibodies. DAPI was used for nuclear staining. Samples were mounted with Aqua-Poly/Mount medium (Polysciences, Inc., Warrington, PA) and imaged on LEICA SP5 confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL).
Experimental Design and Statistical Rationale
SIRT2-EGFP immunoaffinity purification (IP) experiments were performed from MRC5 fibroblasts in three biological replicates, two for label-free quantification and one for metabolic labeling (I-DIRT) quantification. Control EGFP IPs were performed in four biological replicates, and used in the label-free quantification experiment for assessment of nonspecific interactions using SAINT analysis (56). An additional of four control isolations were imported from the CRAPOME data set to give a total of eight controls for assessing the specificity of SIRT2 interactions. For I-DIRT score calculations, precursor peak intensities generated upon the analysis of metabolically labeled protein samples were utilized. Two biological replicates were used for quantification of total protein abundances in SILAC-labeled MRC5 cells stably expressing shCtrl or shSIRT2. Pearson's correlation coefficients were calculated from shCtrl/shSIRT2 SILAC ratios for correlation estimation between two replicate experiments.
RESULTS
Method Development for Characterizing the SIRT2 Protein Interaction Network in Human Fibroblasts
To characterize the SIRT2 protein interaction network, we selected to focus on primary human fibroblasts (MRC5). These cells can provide the first insights into SIRT2 interactions in normal, not immortalized cells. Furthermore, given the recent findings that SIRT2 acts in these MRC5 cells in antiviral defense against human cytomegalovirus, herpes simplex virus 1, and adenovirus infections (11), it is important to understand the specific functions of SIRT2 in fibroblasts. Given the lack of antibodies suitable for the efficient and specific immunoaffinity purification (IP) of endogenous SIRT2, we generated MRC5 cells that stably expressed SIRT2 isoform 2 tagged with EGFP at the C terminus (SIRT2-EGFP). This isoform was used to identify the first substrate of SIRT2, is functional, and is known to be critical in brain development (13). However, there is no current knowledge of its functions in human fibroblasts. In parallel, we generated control MRC5 cells expressing free EGFP. Given our prior success with using this strategy for characterizing the functions of other human sirtuins (5, 26, 30), we opted to use retrovirus transduction to avoid large levels of overexpression. In agreement with a moderate level of overexpression, we observed that SIRT2 mRNA levels were two times higher in the population of SIRT2-EGFP cells than in the wt or EGFP control MRC5 cells, indicating that at the mRNA level SIRT2-EGFP is present at relatively similar levels to endogenous SIRT2 (Fig. 1A). We also assessed the SIRT2 protein levels using Western blotting, and observed a moderate level of overexpression when compared with the endogenous SIRT2 isoform 2 (Fig. 1B). Next, we determined whether the tagged SIRT2-EGFP retains its enzymatic activity. Firstly, we assessed this by Western blotting by visualizing the levels of acetylated histone H4K16 (Fig. 1B), a known substrate of SIRT2 (21). As expected, we observed a decrease in substrate acetylation upon SIRT2-EGFP overexpression (compare H4Lys16ac in EGFP control and SIRT2-EGFP lanes), whereas the total level of H4 protein remained constant. Secondly, we isolated SIRT2-EGFP from fibroblasts and confirmed its ability to deacetylate in vitro a substrate p53 peptide by using a fluorometric assay (Fluor-de-Lys assay, Fig. 1C). Lastly, as the presence of EGFP allows for visualization by direct fluorescence microscopy (i.e. without the use of an antibody), we confirmed that SIRT2-EGFP localizes predominantly to the cytoplasm (Fig. 1D), similar to the endogenous SIRT2 (Fig. 1E). The control EGFP has a diffused localization throughout the cell (Fig. 1D). Altogether, these assays demonstrate that SIRT2-EGFP has the expected subcellular localization, retains its enzymatic activity, and is moderately overexpressed, providing the appropriate tool for studying SIRT2 interactions in human fibroblasts.
Fig. 1.

Experimental design for characterizing SIRT2 interactions in human fibroblasts. A, SIRT2-EGFP is localized to the cytoplasm. MRC5 cells stably expressing SIRT2-EGFP and EGFP were imaged by direct fluorescence microscopy, using DAPI as nuclear marker (blue). Bar size = 25 μm, 63Xmag. B, SIRT2-EGFP localizes similarly to endogenous SIRT2. MRC5 SIRT2-EGFP were imaged by immunofluorescence microscopy using anti-GFP (green) and anti-SIRT2 (red) antibodies, and DAPI nuclear stain (blue). Bar size = 25 μm, 63Xmag. Max Projection. C, SIRT2 mRNA levels in wt, EGFP, and SIRT2-EGFP MRC5 cells. SIRT2 levels were normalized to actin levels across all samples, n = 2. D, SIRT2 protein levels in EGFP and SIRT2-EGFP MRC5 cells. SIRT2-EGFP and endogenous SIRT2 were detected in MRC5 EGFP and SIRT2-EGFP whole-cell lysates using anti-SIRT2 antibody. Histone H4K16ac is a known SIRT2 substrate. Total histone H4 and actin are loading controls. E, SIRT2-EGFP construct is catalytically active. Deacetylase activity of SIRT2-EGFP and EGFP alone was measured upon IP from MRC5 cells using in vitro fluorometric assay. Western blotting using anti-GFP antibody shows equal isolation levels for each protein in the replicates (n = 3). F, Workflow using label-free and metabolic labeling approaches for identifying SIRT2 interactions, their specificity and relative stability. G, Workflow using metabolic labeling for determining changes in the cellular proteome upon shRNA-mediated knockdown of SIRT2.
For rigorous assessment of SIRT2 interactions, their specificity and relative stability, we next designed a mass spectrometry-based workflow that integrated label-free and metabolic labeling quantification of interactions (Fig. 1F). For label-free quantification, we performed IPs from MRC5 cells stably expressing either free EGFP or SIRT2-EGFP. The control EGFP cells were used to monitor nonspecific associations with the tag and the bead resin. For metabolic labeling, the SIRT2-EGFP and wild type MRC5 cells were differentially labeled with heavy (Lys6, Arg6) or light (Lys0, Arg0) amino acids in cell culture (SILAC (63)), respectively. The IPs were done after the 1:1 mixture of these two cell populations. For both approaches, cells were collected, frozen as pellets in liquid nitrogen, and lysed cryogenically as previously reported for studying protein complexes (64, 65). Next, ground cells were lysed in optimized IP buffer and the soluble fraction was used as an input for immunocapture using magnetic epoxy beads conjugated to high-affinity custom-made anti-GFP antibodies. The coisolated proteins were eluted, subjected to trypsin digestion, and the resulting peptides were analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). SAINT analysis (56, 66) was used to obtain interaction specificity scores based on peptide spectral counting. Additional controls from the CRAPome database (67) were used to further confirm the specificity of the observed SIRT2 interactions. Metabolic labeling-based scores were calculated from precursor peak intensities of heavy and light labeled peptides in SILAC protein mixture. Combined, SAINT and I-DIRT analyses provide information about the specificity and the relative stability of SIRT2 interactions, as detailed below.
SIRT2 Interacts With Proteins Involved in Membrane Trafficking, Cytoskeleton Organization, and Transcriptional Regulation
Following the optimization of the IP buffer for efficient isolation of SIRT2-EGFP, we confirmed that SIRT2-EGFP was soluble and not lost in the pellet fraction (Fig. 2A, left), and assessed the IP efficiency by Western blotting to ensure that the majority of SIRT2-EGFP was captured (compare IP fraction to input, flow through, FT, and bead fractions, Fig. 2A, right). The SIRT2 interaction network in primary fibroblasts was first assessed by label-free mass spectrometry, using two biological replicates for SIRT2-EGFP IPs and four biological replicates for control EGFP IPs (supplemental Tables S1 and S2). The SAINT algorithm (56) was used to determine the likely specificity of interaction for the coisolated proteins (supplemental Table S3). This method takes into account the number of spectral counts found for each prey in SIRT2-EGFP and EGFP IPs. Because SIRT2 is known to deacetylate abundant proteins, such as α-tubulin, characterizing its interactions has the challenges associated with studying a cytoskeleton-interacting protein. Such studies can suffer from both increased nonspecific associations and false assignment of a real interaction as a contaminant. To further address the specificity concern, we took a stringent approach by including four additional control isolations from the CRAPome database (67). These IPs were selected to represent anti-GFP isolations on magnetic beads, but from another cell type, transformed kidney HEK293 cells. This allowed us to remove common nonspecific associations to the GFP and bead resin. As a result of this SAINT analysis, the majority of the proteins found in the SIRT2-EGFP IP had low specificity scores (< 0.6). Some of these proteins included known SIRT2 substrates that are unfortunately abundant in cells and commonly found in control IPs in the CRAPome data set. For example, PGAM and ACLY are found in a quarter of the 411 IP experiments reported in the CRAPome, and their SAINT scores in our experiment were zero. (supplemental Table S4). Therefore, this increased filtering stringency is likely to remove real interactions. However, as a proteomic study focused on SIRT2 interactions has not been previously performed, and given the sparse knowledge regarding SIRT2 associations in primary human cells, we opted to focus only on the most likely specific interactions. Therefore, we only further examined the 57 proteins that had SAINT scores above 0.8 (when considering all eight control IPs).
Fig. 2.

SIRT2 interaction network in human fibroblasts. A, Isolation efficiency of SIRT2-EGFP from IP with optimized conditions. SIRT2-EGFP IP was performed from MRC5 cells with optimized lysis buffer using anti-GFP antibody conjugated to magnetic beads via epoxy group. Efficiency of isolation was demonstrated by Western blotting using anti-GFP antibody for detection of SIRT2-EGFP. Soluble - soluble fraction after lysis, Pellet - insoluble fraction, Input - IP input, FT - IP flow through, IP - IP elution, Beads - IP beads fraction after elution. B, Network of SIRT2-interacting partners. Interacting proteins were identified by nLC-MS/MS analysis of SIRT2-EGFP and EGFP control IPs as in Fig. 1F. SAINT algorithm was used to calculate specificity scores based on the number of spectral counts for each protein identified. Proteins passing a SAINT cut-off score of 0.8 were included in the network generated from string-db.com and Cytoscape software. Each node represents an interacting protein, where node color indicates protein enrichment in isolated fraction (NSAF) over its relative enrichment in the human proteome (PAX). Node shape indicates whether the protein is known to be acetylated (diamond) based on uniprot.org records. Protein clusters were assigned based on their known intracellular localizations and functions.
The SIRT2 interactions determined as likely specific were imported into STRING (string-db.org), and the interaction network was built using Cytoscape (Fig. 2B). Attributes were assigned to each node to represent enrichment index (node color) and known acetylation sites (node shape). The enrichment index for each interaction was calculated based on NSAF (normalized spectral abundance factor, (61)) normalized to protein abundance in a cell, as reported in the paxdb.org under H. Sapiens integrated cell line data set. The resulting network was arranged into clusters of proteins that share common pathways and/or intracellular localizations (Fig. 2B).
As expected, the identified interactions included known SIRT2 substrates, in particular α-tubulin (TUBA1A) (14) and NUDT21 (68). Additionally, this network pointed to a previously unrecognized association for SIRT2 with proteins involved in membrane trafficking, such as proteins localized to the Endoplasmic reticulum, trafficking vesicles, membranes, focal adhesions, and cytoskeletal proteins. Among membrane-bound proteins, RER1 (retention in endoplasmic reticulum sorting receptor (1) had the highest enrichment index value. RER1 plays an important role in membrane protein sorting by recognizing signals encoded in transmembrane domains of proteins that are destined for retrieval from Golgi to the ER (69). The SIRT2 network also contains other proteins that function in ER-Golgi and membrane-bound trafficking, such as coat proteins COPE and SEC13. SEC13 is part of the outer layer of COPII vesicles that deliver cargo from ER to Golgi (70). Collagens are among secreted proteins that are transported by COPII (71), and they were also present in SIRT2 IP in multiple isoforms (Fig. 2B). Finally, proteins that function in focal adhesion processes adjacent to plasma membrane, including liprin β-1 (PPFiBP1), ARF GTPase-activating protein GIT2, and Rho guanine nucleotide exchange factor (ARHGEF7), were also present among the SIRT2 interactions that passed the specificity criteria.
Proteins that function in transcription regulation, RNA processing, and translation (ribosomal proteins) were also found as likely specific interactions in SIRT2-EGFP IP (Fig. 2B). It was previously reported that FHL2 (four and a half LIM-2) promotes FOXO1 and SIRT1 interaction, which results in deacetylation of FOXO1 and inhibition of its transcription regulatory functions (72). It is tempting to speculate that this interaction can provide a spatial regulation of FHL2 via SIRT1 nuclear and SIRT2 cytoplasmic functions (73). In support of this prediction, when we performed a nuclear-cytoplasmic fractionation and isolated SIRT2-EGFP from the two fractions, we observed that FHL2 was detected in the SIRT2-EGFP IP from the cytoplasmic fraction (supplemental Table S5). Therefore, our results indicate that the SIRT2-FHL2 interaction takes place in the cytoplasm.
The identified SIRT2 interactions were not found as acetylated in our IP. However, some of these proteins (indicated with diamond shapes) have been reported to be acetylated in other cells, although not linked to SIRT2 functions. The absence of acetylation in our IP may be because of their low levels or to cell type-specific regulation. The presence of TUBA1A and NUDT21 in the SIRT2 IP suggests that other substrates may also be among the identified interactions. For example, β-tubulin (TUBB3) was recently reported to be acetylated at Lys252 by San acetyltransferase (74), but the enzyme responsible for its deacetylation remains unknown. Altogether, our SIRT2 interaction network suggests that one of the main roles for SIRT2 in normal human fibroblasts lies within intracellular trafficking pathways.
SIRT2 Interacts Transiently With Intracellular Trafficking Proteins
The association of SIRT2 with proteins functioning in membrane trafficking suggests that many of these associations are dynamic in nature. Therefore, we tested this hypothesis. We previously reported a mass spectrometry-based method for determining the relative stability for protein interactions (25). This approach integrates label-free SAINT analysis (56) with metabolic labeling using I-DIRT (Isotopic Differentiation of Interactions as Random or Targeted) (46), providing information about both interaction specificity and relative stability (25). To apply this method to studying SIRT2 interactions, we differentially labeled SIRT2-EGFP MRC5 and wild type MRC5 cells using either heavy or light Arg and Lys isotopes in cell culture, respectively (Fig. 1F). Following five passages, the incorporation of the labels was confirmed by mass spectrometry (supplemental Table S6). The two cell populations (SIRT2-GFP and wild type) were then mixed, cryogenically lysed, and used for IP via the GFP tag. Therefore, SIRT2 specific interactions would be expected to be predominantly heavy labeled, whereas nonspecific associations would derive equally from both cell types, being present as both light and heavy labeled. Furthermore, specific SIRT2 interactions that are fast-exchanging or dynamic in nature may also be detected as a combination of light and heavy peptides, as an on-off interaction can reform with proteins from either cell population. Therefore, the integration of the label-free SAINT approach allows the assessment of interaction specificity for these proteins, w the use of metabolic labeling provides information regarding their relative stability (Fig. 3A). As performing isolations from combined light and heavy cell populations has been shown to increase nonspecific background, for these analyses we used a more stringent lysis buffer. After nLC-MS/MS analysis, heavy/(light+heavy) ratios for each protein were quantified based on precursor peak intensities (supplemental Tables S7 and S8). For the proteins that passed the SAINT specificity threshold (>0.8), those that had high isotopic ratios (>0.7) were considered likely stable interactions, whereas those with 0.6–0.7 or at around 0.5 ratios were considered to be increasingly dynamic in their association with SIRT2. The proteins with very low isotopic score (<0.2) are likely to be environmental contaminants, as previously reported (25, 46). Therefore, by direct comparison of the label-free and metabolic-labeling experiments, we were able to assess whether SIRT2 interactions were specific and stable, nonspecific, or specific and transient (Fig. 3A).
Fig. 3.

Analysis of the relative stability of SIRT2 interactions. A, Combination of label-free and metabolic labeling MS-based approaches provides information on specificity and relative stability of SIRT2 interactions in human fibroblasts. B, Comparison of I-DIRT and SAINT scores from SIRT2-EGFP IP experiments. For the I-DIRT-based experiment, SIRT2-EGFP IP was performed upon mixing of MRC5 wt and MRC5 SIRT2-EGFP labeled light (Arg0, Lys0) and heavy (Arg6, Lys6), respectively. I-DIRT scores were calculated as a heavy/(heavy+light) ratio based on precursor peak intensity areas. Each dot represents an interacting protein identified in both experimental approaches. C, Table of interactions identified in SAINT and I-DIRT experiments color-coded based on I-DIRT ratio, as indicated in A. All shown interactions passed SAINT specificity score cutoff unless specified otherwise. Red outline indicates known SIRT2 substrates. D, Representative I-DIRT precursor peak intensity profiles for selected interacting proteins (SFRP1, PPFiBP1, SSR1).
Several proteins were observed with isotopic ratios indicative of relatively stable interactions. These included the known SIRT2 substrate TUBA1A (Fig. 3B–3C), as well as secreted frizzled-related protein 1 (SFRP1) (Fig. 3D). SFRP1 was not detected in the SAINT experiment, but uniquely identified in the I-DIRT experiment, possibly because of the increased lysis buffer stringency. SFRP1 is a secreted glycoprotein that functions as an antagonist of the Wnt signaling pathway (75). Another example of a protein involved in ER-Golgi trafficking, and identified only in the more stringent I-DIRT experiment, is reticulon 4 (RTN4) (Fig. 4A, supplemental Table S8). In support of our observation of the SIRT2 association with membrane trafficking pathways, the majority of the detected interactions had isotopic ratios reflective of transient associations (Fig. 3B–3C). Several proteins with high SAINT scores and intermediate isotopic ratios included another microtubule subunit protein, TUBB3 (SAINT = 0.95, I-DIRT = 0.65), focal adhesion proteins PPFiBP1 (SAINT = 1, I-DIRT = 0.63) and ARHGEF7 (SAINT = 1, I-DIRT = 0.69), ER translocon-associated protein SSR1 (SAINT = 1, I-DIRT = 0.63), and extracellular matrix protein COL18A1 (SAINT = 0.95, I-DIRT = 0.67). Many proteins with SAINT score above 0.8 were detected with isotopic ratios reflective of fast-exchanging interactions, including the known SIRT2 substrate NUDT21 (SAINT = 0.94, I-DIRT = 0.58) and the transcription regulator FHL2 (SAINT = 0.86, I-DIRT = 0.5). I-DIRT profiles of selected interacting partners identified as specific by label-free and/or metabolic labeling quantification are shown in Fig. 3D.
Fig. 4.

Mapping SIRT2 interactions to intracellular membrane trafficking pathways. A, SIRT2 interacting proteins are part of major secretory pathways and compartments. Schematic showing proteins identified as interacting partners by SIRT2-EGFP IP (orange) and their known intracellular localization sites. B–C, SIRT2-EGFP is juxtaposed to the nucleus, showing partial colocalization with ER and Golgi. Live MRC5 cells stably expressing SIRT2-EGFP (green) were imaged by confocal microscopy with ER-tracker (B, red) and Golgi-tracker (C, red) fluorescent stains. Bars - 25 μm (D) SIRT2-EGFP colocalizes with ERGIC. MRC5 cells stably expressing SIRT2-EGFP were imaged by immunofluorescence microscopy with anti-GFP (green) and anti-ERGIC53 (red) antibodies. E, Endogenous SIRT2 colocalizes with ERGIC. Wild type MRC5 cells were imaged by immunofluorescence microscopy with anti-SIRT2 (green) and anti-ERGIC53 (red) antibodies. Bars - 50 μm (F) SIRT2-EGFP localization was assessed with Brefeldin A treatment in MRC5 cells stably expressing SIRT2-EGFP. Immunofluorescence microscopy was performed using anti-EGFP (green) and anti-ERGIC53 (red) antibodies. Bars - 25 μm. DAPI is a nuclear marker in B–F.
Last, several known SIRT2 substrates that did not pass the SAINT specificity threshold were observed with isotopic ratios reflective of transient associations (Fig. 3B and 3C, bottom). These included isoforms of histones H3 and H4, ACLY, and PGAM1 (Fig. 3C). Altogether, this analysis of the specificity and relative stability of interactions highlighted the dynamic nature of SIRT2 associations with proteins functioning in intracellular trafficking pathways, such as SSR1, PPFiBP1, and SFRP1. Each of these proteins is targeted to a specific intracellular region, such as ER (SSR1), focal adhesions (PPFiBP1), or for an extracellular space (SFRP1). Therefore, our analysis of SIRT2 interaction stability suggests that this enzyme is able to function in the cell in membrane trafficking through both dynamic and stable associations with the pathway's cargo (PPFiBP1 and SFRP1) and regulatory proteins (SSR1).
SIRT2 Localizes at the Intersection of Intracellular Membrane Trafficking Routes
A large number of proteins specifically enriched in our SIRT2-EGFP IP experiments are known to localize to major intracellular secretory pathways (Fig. 4A). Among them are ER-bound proteins (ELOVL1, SSR1, and RTN4), proteins that function in vesicular trafficking (e.g. SEC13), Golgi proteins (e.g. RER1), and proteins transported to plasma membrane and/or secreted (e.g. PPFiBP1, SFRP1, CDC42, and IDE). Therefore, we next asked whether SIRT2 localizes to subcellular organelles that are known to regulate membrane trafficking. Imaging was performed in live fibroblasts by confocal microscopy, visualizing SIRT2-EGFP using direct fluorescence and organelles using ER or Golgi trackers. Our results indicated that SIRT2-EGFP does not have a perfect colocalization with the ER or Golgi, but rather localizes in close proximity to these organelles (Fig. 4B and 4C, respectively). Given this result, we next assessed whether SIRT2 localizes to the ER-Golgi intermediate compartment (ERGIC). This structure mediates trafficking from ER to Golgi and consists of tubular and vesicular membrane clusters enriched in ERGIC-53 marker protein (76). Using an anti-ERGIC53 antibody, we observed colocalization of SIRT2-EGFP with the ER-Golgi intermediate compartment (Fig. 4D). To further confirm this, we investigated endogenous SIRT2 in wild type fibroblasts, observing a similar colocalization with ERGIC53 (Fig. 4E). As these results provide the first evidence that SIRT2 is positioned at the ER-Golgi intermediate compartment, we next tested if this localization is dependent on an active trafficking between ER and Golgi. Treatment with Brefeldin A is known to disrupt ER-Golgi trafficking and the resulting protein secretion (77, 78), and, as expected, we observed this treatment to trigger the dissociation of ERGIC53 (Fig. 4F). Additionally, we found a similar dissociation for SIRT2-EGFP. Altogether, these results indicate that SIRT2 is trafficking between ER and Golgi, and is positioned to play a role in protein secretion and plasma membrane targeting.
Interaction and Microscopy Assays Demonstrate the Association of SIRT2 with Liprin-β1
One of the most prominent interactions identified in our SIRT2 interactome analysis, observed in both the label-free and metabolic labeling experiments, was that with PPFiBP1 (Fig. 3B), also known as liprin-β1. PPFiBP1 is in the family of liprins, which are scaffolding proteins originally identified in association with the receptor protein tyrosine phosphatase LAR (leukocyte common antigen-related) (79). Therefore, these proteins are thought to function in regulation of cell-matrix interactions through the formation of focal adhesions. These proteins were previously studied in the context of neuronal synapse formation (80) and tumor cell invasion regulation (81). Although PPFiBP1 was shown to be targeted by the metastasis-associated protein S100A4 (82) and to play a role as a mediator of lymphatic vessel integrity (83), the knowledge of its functions remains limited. Similar to other liprins, PPFiBP1 may play a role in plasma membrane-associated protein targeting through regulation of LAR cellular localization (79). Therefore, we investigated its localization relative to SIRT2-EGFP (Fig. 5A). By confocal microscopy, we observed PPFiBP1 signal in the proximity of plasma membrane. In addition, SIRT2 and PPFiBP1 seemed to colocalize at the region juxtaposed to the nucleus, suggesting that they are both localized to ERGIC. To further assess its association with SIRT2, we validated this interaction by reciprocal IP. Upon isolation of endogenous PPFiBP1, we observed that SIRT2-EGFP is present in PPFiBP1 IP, but not in the control IgG IP (Fig. 5B). In view of our findings that SIRT2 perinuclear localization is affected by the disruption of ER-Golgi trafficking, we next investigated whether PPFiBP1 also requires this active pathway. Interestingly, PPFiBP1 localization was dramatically changed upon Brefeldin A treatment (Fig. 5C). Perinuclear signal seemed to disappear, suggesting that this localization is dependent on an intact ER-Golgi trafficking, similar to ERGIC53 and SIRT2. Overall, these results suggest that SIRT2 can have a function in protein targeting or can itself be trafficked within this pathway through its interactions with ER and Golgi-associated proteins. For example, it can play a role in regulation of focal adhesion formation through its interactions with PPFiBP1.
Fig. 5.
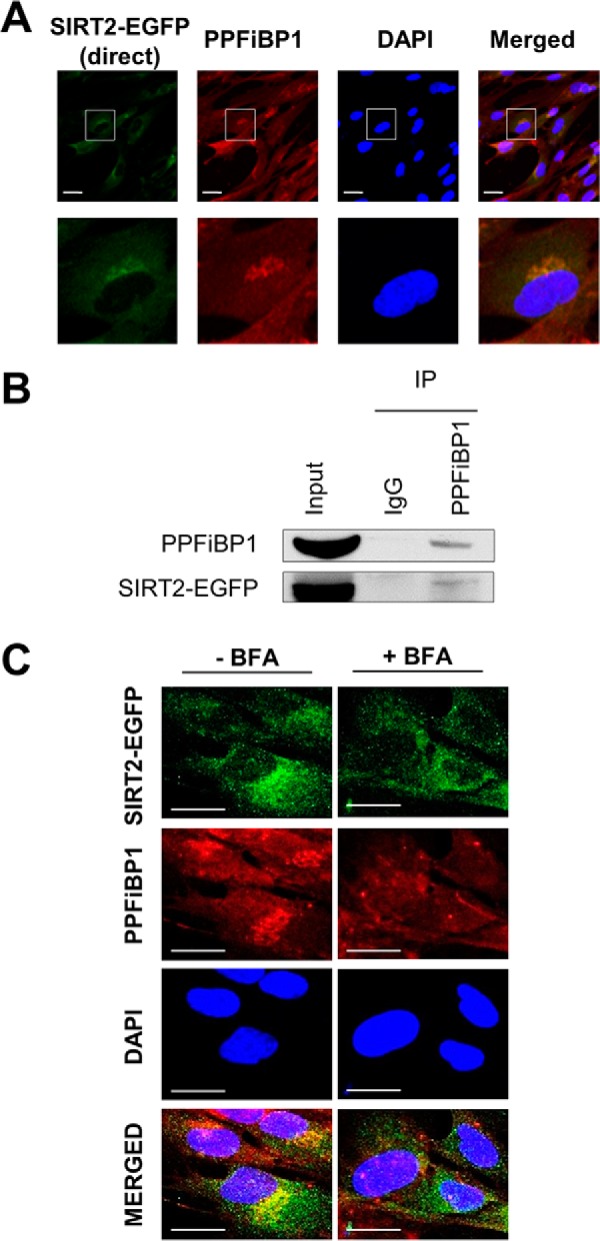
SIRT2 interacts and colocalizes with liprin-β1 (PPFiBP1). A, SIRT2 colocalizes with PPFiBP1. SIRT2-EGFP MRC5 cells were imaged by direct fluorescence (SIRT2-EGFP, green) or immunofluorescence microscopy (anti-PPFiBP1, red). Bars - 25 μm. B, PPFiBP1 reciprocally interacts with SIRT2. PPFiBP1 and IgG control IPs were performed in parallel. Western blotting analysis was done using anti-GFP antibody to detect SIRT2-EGFP. Input - 3% (v/v) of IP input fraction, IP - 30% (v/v) of IP elution fraction. C, PPFiBP1 localization was assessed with or without Brefeldin A treatment in MRC5 cells stably expressing SIRT2-EGFP. Immunofluorescence microscopy was performed using anti-EGFP (green) and anti-PPFiBP1 (red) antibodies. DAPI is a nuclear stain. Bars - 50 μm.
SIRT2 Down-regulation Increases Levels of Proteins With Functions in Membrane Trafficking
Our interactome and microscopy results indicated that SIRT2 is connected to proteins trafficked between the ER and Golgi or secreted to the plasma membrane (Figs. 2, 4). Furthermore, our SIRT2 interaction network includes proteins involved in transcriptional regulation (Figs. 2, 3). This is in agreement with its previously reported function in transcriptional regulation through deacetylation of several transcription factors (e.g. p53 (33), NFκB (31), FOXO3A (35)) and histones (H3Lys18ac (22)). Therefore, our results suggest that SIRT2 is positioned to function in ER-Golgi trafficking using multiple points of regulation: protein-protein interactions, post-translational modification of substrates, or by regulating gene expression. All of these SIRT2-mediated mechanisms could impact the levels of proteins that function within this pathway. Therefore, we next assessed whether SIRT2 levels can induce changes in total protein levels in the functional pathways identified from its interactions. For this, we generated fibroblasts (MRC5) stably expressing shRNA targeting SIRT2 (shSIRT2) or nontargeting shRNA (shCtrl). We measured SIRT2 mRNA levels by qPCR (Fig. 6A), which indicated ∼60% knockdown efficiency. We also validated the SIRT2 knockdown at the protein level, observing a similar decrease (Fig. 6B). We then differentially labeled shSIRT2 and shCtrl fibroblasts by SILAC with light or heavy Lys and Arg isotopes, respectively (Fig. 1G). The two cell types were then combined, lysed, and analyzed by LC-MS/MS. Analysis of protein levels based on their isotopic ratios revealed a good correlation between our two biological replicates (r = 0.82) (Fig. 6C, supplemental Table S10). The majority of proteins did not change their levels upon SIRT2 knockdown, illustrated by the local point density analysis in Fig. 6C. This was expected, indicating that SIRT2 does not globally impact the cellular proteome, but rather regulates specific cellular pathways.
Fig. 6.

SIRT2 impact on protein levels of membrane trafficking proteins in human fibroblasts. A, SIRT2 mRNA levels upon shRNA-mediated knockdown in MRC5 cells. MRC5 cells stably expressing nontargeting shRNA (shCtrl) or shSIRT2 were subjected to mRNA extraction and analysis of SIRT2 mRNA levels by qPCR. Β-Actin was used as endogenous control, n = 3. B, SIRT2 protein levels upon shRNA-mediated knockdown in MRC5 cells. Total endogenous SIRT2 levels were measured in MRC5 cells stably expressing shCtrl or shSIRT2 by Western blotting using anti-SIRT2 and anti-β-actin antibody as loading control. C, Changes in total protein levels upon SIRT2 KD in MRC5 cells from two replicate experiments. MRC5 shCtrl and shSIRT2 were differentially labeled by SILAC and total protein levels were assessed by mass spectrometry. Each dot represents quantified protein and its color indicates local point density. D, Table of GOBP categories with top annotation enrichment scores. Data set of changes in protein levels upon SIRT2 KD was analyzed in Perseus using 2D annotation enrichment algorithm based on two replicate experiments. GOBP enrichment scores were similar between replicates and one representative score from the two replicates is indicated. E, Changes in individual protein levels upon SIRT2 KD in MRC5 cells.
To gain a better understanding of the impact of SIRT2 levels on specific functional pathways in a cell, we bioinformatically analyzed the biological processes altered upon SIRT2 down-regulation. To do this, we utilized the 2D annotation enrichment tool (58) of the Perseus software for MaxQuant-based data analysis. Our analysis revealed that members of several Gene Ontology Biological Processes (GOBP) categories showed a tendency to increase or decrease in levels upon SIRT2 knockdown. Top categories with increased protein levels included hydrogen transport, ER organization, membrane docking, organelle fusion, and lipid transport (Fig. 4D, supplemental Table S11). As these categories are similar to the membrane trafficking clusters obtained in the SIRT2 interaction network analysis, we looked closer at the changes in individual protein levels within each category (Fig. 6E). Interestingly, we observed that several SIRT2-interacting proteins also changed in protein levels upon SIRT2 knockdown. One example is the twofold increase in SFRP1 levels (Fig. 6E and 3D), suggesting that one possible functional consequences of this SIRT2 interaction is regulation of protein levels or stability. Another example is RTN4, a protein that functions in ER organization (84), which was also identified in our I-DIRT experiment among the dynamic SIRT2 interactions (supplemental Table S8). The levels of RTN4 increased by 50% following SIRT2 knockdown. We also observed several decreases in protein levels, including the levels of extracellular matrix protein tenascin (TNC, Figs. 6E and 3B–3C). Overall, these results indicate that SIRT2 can function in membrane trafficking through protein-protein interactions, as well as through regulation of the levels of proteins within these pathways.
DISCUSSION
SIRT2 Role in Membrane Trafficking and Implications in Cellular Homeostasis and Human Disease
This study integrated microscopy, protein interaction, knockdown, and functional assays to investigate the roles of SIRT2 in primary human fibroblasts. Using label-free and metabolic labeling mass spectrometry-based quantification, we generated a network of SIRT2 interactions that indicated the involvement of SIRT2 within several specific intracellular processes. A distinctively large group of identified SIRT2 interactions has known functions in intracellular trafficking pathways, such as cytoskeletal organization, vesicular trafficking, localization to ER, plasma membrane and other cellular membranes. Among these, we identified the known SIRT2 substrate α-tubulin. The fact that SIRT2 deacetylates a major cytoskeletal protein is in agreement with our finding of its contribution to intracellular trafficking pathways, and highlights SIRT2 as a critical regulator of house-keeping functions in a cell. However, this also brings challenges to our analysis, where tubulin-associated proteins are abundantly present in the IP. To account for such associations, help reduce background and distinguish real interactions, we introduced a total of eight controls in our specificity assessment. Moreover, we designed and applied a metabolic labeling quantification approach, which in combination with label-free analysis, was used to confirm characterize the relative stability of SIRT2 interactions. This analysis also provided further experimental validation of SIRT2 interactions with proteins that function in membrane trafficking, such as α-tubulin, PPFiBP1, SSR1, and GIT2. Notably, I-DIRT-based analysis also was able to detect transient interactions of SIRT2 with its known substrates histone H3, histone H4, ACLY, and PGAM1 that received low SAINT specificity scores following label-free quantification. However, some previously reported SIRT2 interactions were not identified in our study. One possible reason for this difference is that our study represents one of the first attempts to characterize SIRT2 functions in normal cells. Most previous studies on SIRT2 interactions were performed in cancer cell lines or in other cell systems. For instance, the characterization of SIRT2-mediated deacetylation of p53 was performed either in the presence of p53 overexpressing plasmid in nonsmall cell lung carcinoma cell line or of etoposide treatment in melanoma cell line to increase p53 levels for detection (33). The SIRT2 interaction with FOXO3a was observed either in HEK293 cells transfected with FOXO3a and SIRT2-expressing plasmids or in brain lysates, where both proteins are abundantly expressed (35). NFκB and SIRT2 interaction was detected in THP-1 monocytes, a model system for studying immunity (31). We were not able to detect these known substrates in primary human fibroblasts, likely in part because of the absence of stimuli or the presence of cell type-specific functions.
In our study, the majority of the identified SIRT2 interactions had I-DIRT isotopic ratios that indicated transient associations. Given our finding that SIRT2 localizes to ERGIC, which is known to serve as a shuttling hub between ER and Golgi, this observation is expected. Moreover, we find that this SIRT2 localization is dependent on intact ER-Golgi pathway. Therefore, the transient nature of the SIRT2 interactions can also be driven by its dynamic association with these membrane trafficking organelles.
Our observation that SIRT2 distribution in the cytoplasm is tightly linked to ER-Golgi trafficking pathway also implies the existence of a previously unrecognized regulatory function for this sirtuin in membrane targeting. This function can be mediated through its substrates, such as α-tubulin. Significance of α-tubulin acetylation status in microtubule assembly remains largely unknown. It was observed as a consecutive event following microtubule stabilization, but it can also be present in other subpopulations of microtubules (85). Microtubule motor kinesin-1 was proposed to use the tubulin acetylation status as readout for its transport functions, most likely in combination with some other microtubule-associated proteins (86, 87). Because ER-Golgi transport is directed by microtubule-based motors (88), it is tempting to speculate that SIRT2 links tubulin and vesicular trafficking networks via its association with microtubules. Alternatively, other SIRT2 substrates or enzymatic functions can contribute to this role. For example, it has been proposed that SIRT2 has demyristoylation activity (7, 89). Fatty acid modification of proteins is closely linked with membrane targeting and protein-lipid interactions. For example, SIRT6-mediated demyristoylation was shown to regulate TNF-α secretion (6). Unfortunately, our knowledge on protein N(6)-myristoyllysine sites is still very limited. Whether SIRT2 interacting partners also possess myristoylation sites has to be further investigated.
What implication does SIRT2 role in membrane trafficking might have for its functions in disease? SIRT2 has recently emerged as a key player in neuronal processes, including glioma progression (9, 42) and neurodegeneration (10, 39). Because neurons rely on intercellular signaling through synaptic vesicle formation and trafficking, it is tempting to speculate that SIRT2 association with these diseases is partly provided by its role in protein secretion and other membrane-bound trafficking pathways. SIRT2 impact on viral infection (11, 90) might also be connected to intracellular trafficking. Viruses commonly use microtubule networks for capsid trafficking, genome delivery, and assembly (91). Our study provides an important groundwork for further investigation of this viral-host interaction mechanism.
SIRT2 Interaction with the Liprin Protein PPFiBP1
We demonstrated that SIRT2 associates and colocalizes with PPFiBP1, a member of liprin family of proteins known to interact with the receptor protein tyrosine phosphatase leukocyte common antigen-related (LAR) protein. Through interactions with other liprins, PPFiBP1 can play a role in focal adhesion disassembly (79). Our results confirm the localization of PPFiBP1 at the plasma membrane of primary human fibroblasts. We also discover that PPFiBP1 colocalizes with SIRT2 juxtaposed to the nucleus, and that this localization is affected when the ER-Golgi trafficking is disrupted. A large-scale study of the human acetylome in acute myeloid leukemia cell line reported that PPFiBP1can be acetylated at K322 (92). Interestingly, this lysine residue is within the N-terminal coiled coil region of PPFiBP1 known to mediate its homodimerization and possibly the formation of higher-order scaffolding structures (79). However, it is still not known what enzymes regulate this modification. Our observation that this largely uncharacterized protein associates with SIRT2 suggests a potential role for liprin-β1 in vesicular trafficking, such as the one previously observed for liprin-α in axonal transport (93).
SIRT2 Role in Regulation of Gene Expression
SIRT2 is positioned to regulate intracellular protein abundances at the levels of transcription, translation, and protein degradation. For instance, SIRT2 is known to deacetylate several transcription factors (p53 (33), NFκB (31), FOXO3a (35)), and histones (H3K18ac (22)). At the levels of pre-mRNA 3′-processing, SIRT2 can modulate the acetylation status of NUDT21 component of cleavage factor Im complex, potentially promoting its association with the complex (68). At the protein level, SIRT2 was demonstrated to negatively regulate autophagy-mediated degradation in neurons (94). In addition, it was shown that in the absence of SIRT2, Aurora-A levels are increased, possibly because of disregulation of anaphase-promoting complex/cyclosome (APC/C) ubiquitin ligase activity (40). In support of these previously proposed functions, we show that SIRT2 specifically interacts with proteins that function in transcription regulation, ribosomal processes, and RNA processing in human fibroblasts. In our study, three proteins with functions in transcription regulation passed our interaction specificity cutoff, including LIM domain protein FHL2. This protein is a transcriptional coactivator that is known to be induced by serum starvation (95). Moreover, FHL2, because of its association with focal adhesions, can transmit to the nucleus signals that originated at the cell membrane (96). Interestingly, its cytoplasmic localization is regulated by the same Crm1/exportin-dependent mechanism that has been demonstrated for SIRT2 (20, 95). Finally, FHL2 was shown to promote association of FOXO1 with SIRT1, which results in deacetylation and inhibition of the transcription factor (72). Altogether, these studies and our findings point at a possible localization-dependent regulation of FHL2 functions through association with SIRT1 and SIRT2 in the nucleus and the cytoplasm, respectively. As of note, such cooperation between SIRT1 and SIRT2 was previously described for their common substrate p53 (97).
To gain further insight into SIRT2-mediated gene expression events, we assessed total protein levels in human fibroblasts upon SIRT2 knockdown. Our analysis of SIRT2-dependent biological processes revealed up-regulation of proteins with functions in endoplasmic reticulum organization, membrane docking, organelle fusion, and lipid transport. Within the membrane docking category, ICAM1 and several RAB proteins showed almost twofold up-regulation in levels upon SIRT2 knockdown. RTN4 protein that is localized to ER and transiently associates with SIRT2, as demonstrated by our I-DIRT analysis, is also 50% up-regulated upon reduction in SIRT2 levels. This resident ER protein was demonstrated to be expressed in white matter of central nervous system and to play a role in inhibition of axonal regeneration (98). Interestingly, SIRT2 knockdown was previously demonstrated to impact neurite growth as well (17). Therefore, further exploration of a connection between RTN4 and SIRT2 in the context of neuron regeneration can provide insights into a cross-talk between their pathways. Several other SIRT2-interacting proteins were observed to change their protein levels upon SIRT2 knockdown. For example, the levels of SFRP1 protein that was identified as a relatively stable interacting partner by I-DIRT were increased twofold. SFRP1 is a secreted protein and is thought to function as an inhibitor of Wnt signaling pathway. Similar to SIRT2 (42), SFRP1 is down-regulated in several cancer types (99). Future studies can determine whether the SFRP1 interaction with SIRT2 and its up-regulation upon SIRT2 knockdown have a role in cancer. Together with our findings that SIRT2 interacts with membrane trafficking proteins, results of our analysis on changes in protein levels in response to SIRT2 knockdown, create a strong evidence for its function within intracellular trafficking pathways.
Perspective on SIRT2 Functions Via Protein Interactions and Enzymatic Activity
Two known SIRT2 substrates were observed in our interaction network, α-tubulin and NUDT21. However, many other interactions also have previously reported acetylation sites, including components of RNA processing machinery, ribosomal protein RRBP1, transcription regulator BUB3, cytoskeletal proteins, and focal adhesions protein PPFiBP1. Whether these sites can also be deacetylated by SIRT2 has to be further explored. Interestingly, in recent years, studies have emerged pointing at the existence of a cross-talk between different PTMs. Frequently, the same lysine residue is observed to be modified by different PTMs in different conditions, such as acetylation and succinylation (100). Emerging enzymatic activities of sirtuins also point at their ability to remove various groups from modified lysines, including desuccinylation/demalonylation by SIRT5 (8, 100), delipoylation by SIRT4 (5), and demyristoilation by SIRT2 (7) or SIRT6 (6). Several proteins in SIRT2 network have been identified in different screens as acetylated and/or succinylated at the same lysine residue, including Rho GDP-dissociation inhibitor ARHGDIA, glutathione S-transferase GSTP1, and glutaminase GLS. These sites can potentially serve as regulatory points of convergent sirtuin functions that would allow for immediate response to changes in intracellular metabolic state.
Although the majority of the current knowledge regarding SIRT2 is based on its enzymatic activities, gaining an understanding of its dynamic protein interactions within a cell is needed in order to fully define its functions. In recent years, it has become evident that sirtuins can modulate protein functions via formation of protein complexes, rather than only via enzymatic activity. For example, a function for SIRT6 in regulating stress granule formation was found following the identification of its interaction with Ras-GTPase-activating protein-binding protein 1 (G3BP1) (26, 27). Similarly, the interaction of SIRT7 with the transcription factor complex TFIIIC2 led to the identification of its role in Pol III-dependent transcription (101). The concept of acquiring multiple functions by formation of distinct protein complexes, rather than by deacetylation events, is well understood for the other classes of human deacetylases, histone deacetylases (HDACs)(25). The class IIa of HDACs (HDACs 4, 5, 7, and 9) is a prime example of this, as their functions are attained via transient interactions during their nuclear-cytoplasmic shuttling (102). The interaction between HDAC4 and the mutant huntingtin protein (mHTT) was shown to promote mHTT aggregation, pointing to a possible target for treatment of Huntington's disease (103, 104). As another example, the transient association between HDAC5 and Aurora B impacted the phosphorylation status of HDAC5, its association with the transcriptional regulatory complex NCoR, and its functions during cell cycle progression (50). Similarly, our finding that SIRT2 interacts with proteins involved in intracellular trafficking suggests that SIRT2 is positioned to regulate these biological processes via the formation of dynamic interactions.
In summary, our study provides the first insights into SIRT2 interactions in primary human fibroblasts, highlighting its role in cellular trafficking events. We observed that SIRT2 interacts with multiple intracellular trafficking proteins and showed that majority of its interactions are of transient nature. We confirmed its colocalization with ER-Golgi intermediate compartment and demonstrated that its cytoplasmic distribution is affected by the inhibition of the secretory pathway. We found and validated that SIRT2 interacts with the focal adhesion protein PPFiBP1 and demonstrated that the localization of this protein is also dependent on the ER-Golgi trafficking pathway. Finally, we observed up-regulation of proteins that function in ER organization upon SIRT2 knockdown. Given the prior sparse knowledge regarding SIRT2 associations, we expect our study to significantly aid in understanding its roles in health and disease. For example, our finding of a role of SIRT2 in membrane trafficking regulation can be further investigated in the context of a disease with altered vesicular transport, such as neurodegenerative diseases and viral infection.
Supplementary Material
Acknowledgments
We thank Dr. Yana Miteva for contributing to the EGFP control samples, Gary Laevsky and Christina DeCoste (Microscopy and Flow Cytometry Core Facilities, Princeton University), and Todd Greco and Joel Federspiel for technical support.
Footnotes
Author contributions: H.G.B. and I.M.C. designed research; H.G.B. performed research; I.M.C. contributed new reagents or analytic tools; H.G.B. and I.M.C. analyzed data; H.G.B. and I.M.C. wrote the paper.
* This work was supported by the National Institutes of Health, NIGMS grant GM114141 and NHBLI grant HL127640 to IMC, and an NSF graduate research fellowship DGE1148900 to HGB. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SIRT2
- sirtuin 2
- ACLY
- ATP citrate lyase
- EGFP
- enhanced Green Fluorescent Protein
- ER
- endoplasmic reticulum
- ERGIC
- ER-Golgi intermediate compartment
- FHL2
- four and a half LIM-2
- FOXO3A
- forkhead box protein O3
- GOBP
- gene ontology biological process
- HDAC
- histone deacetylase
- I-DIRT
- differentiation of interactions as random or targeted
- IP
- immunoaffinity purification
- MRC5
- human fibroblasts
- NAD+
- nicotinamide adenine dinucleotide
- NFκΒ
- nuclear factor kB
- NSAF
- normalized spectral abundance factor
- NUDT21
- cleavage and polyadenylation specificity factor subunit 5
- PAX
- protein abundance values
- PGAM
- phosphoglycerate mutase
- PPFiBP1
- liprin-beta-1
- RER1
- retention in endoplasmic reticulum sorting receptor 1
- RTN4
- reticulon 4
- SAINT
- significance analysis of interactome computational tool
- SFRP1
- secreted frizzled-related protein 1
- SILAC
- stable isotope labeling with amino acids in cell culture
- SSR1
- translocon-associated protein 1
- TUBA1A
- tubulin alpha-1A chain
- WT
- wild type.
REFERENCES
- 1. Frye R. A. (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798 [DOI] [PubMed] [Google Scholar]
- 2. Michishita E., Park J. Y., Burneskis J. M., Barrett J. C., and Horikawa I. (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liszt G., Ford E., Kurtev M., and Guarente L. (2005) Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 280, 21313–21320 [DOI] [PubMed] [Google Scholar]
- 4. Haigis M. C., Mostoslavsky R., Haigis K. M., Fahie K., Christodoulou D. C., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., Karow M., Blander G., Wolberger C., Prolla T. A., Weindruch R., Alt F. W., and Guarente L. (2006) SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954 [DOI] [PubMed] [Google Scholar]
- 5. Mathias R. A., Greco T. M., Oberstein A., Budayeva H. G., Chakrabarti R., Rowland E. A., Kang Y., Shenk T., and Cristea I. M. (2014) Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang H., Khan S., Wang Y., Charron G., He B., Sebastian C., Du J., Kim R., Ge E., Mostoslavsky R., Hang H. C., Hao Q., and Lin H. (2013) SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Teng Y. B., Jing H., Aramsangtienchai P., He B., Khan S., Hu J., Lin H., and Hao Q. (2015) Efficient demyristoylase activity of SIRT2 revealed by kinetic and structural studies. Sci. Rep. 5, 8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., and Lin H. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bosch-Presegue L., and Vaquero A. (2011) The dual role of sirtuins in cancer. Genes Cancer 2, 648–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chopra V., Quinti L., Kim J., Vollor L., Narayanan K. L., Edgerly C., Cipicchio P. M., Lauver M. A., Choi S. H., Silverman R. B., Ferrante R. J., Hersch S., and Kazantsev A. G. (2012) The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington's disease mouse models. Cell Rep. 2, 1492–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koyuncu E., Budayeva H. G., Miteva Y. V., Ricci D. P., Silhavy T. J., Shenk T., and Cristea I. M. (2014) Sirtuins are evolutionarily conserved viral restriction factors. MBio 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. North B. J., and Verdin E. (2007) Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem. 282, 19546–19555 [DOI] [PubMed] [Google Scholar]
- 13. Maxwell M. M., Tomkinson E. M., Nobles J., Wizeman J. W., Amore A. M., Quinti L., Chopra V., Hersch S. M., and Kazantsev A. G. (2011) The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum. Mol. Genet. 20, 3986–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. North B. J., Marshall B. L., Borra M. T., Denu J. M., and Verdin E. (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444 [DOI] [PubMed] [Google Scholar]
- 15. Dryden S. C., Nahhas F. A., Nowak J. E., Goustin A. S., and Tainsky M. A. (2003) Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 23, 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inoue T., Nakayama Y., Yamada H., Li Y. C., Yamaguchi S., Osaki M., Kurimasa A., Hiratsuka M., Katoh M., and Oshimura M. (2009) SIRT2 downregulation confers resistance to microtubule inhibitors by prolonging chronic mitotic arrest. Cell Cycle 8, 1279–1291 [DOI] [PubMed] [Google Scholar]
- 17. Pandithage R., Lilischkis R., Harting K., Wolf A., Jedamzik B., Luscher-Firzlaff J., Vervoorts J., Lasonder E., Kremmer E., Knoll B., and Luscher B. (2008) The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 180, 915–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Southwood C. M., Peppi M., Dryden S., Tainsky M. A., and Gow A. (2007) Microtubule deacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 32, 187–195 [DOI] [PubMed] [Google Scholar]
- 19. Inoue T., Hiratsuka M., Osaki M., Yamada H., Kishimoto I., Yamaguchi S., Nakano S., Katoh M., Ito H., and Oshimura M. (2007) SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 26, 945–957 [DOI] [PubMed] [Google Scholar]
- 20. North B. J., and Verdin E. (2007) Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2, e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vaquero A., Scher M. B., Lee D. H., Sutton A., Cheng H. L., Alt F. W., Serrano L., Sternglanz R., and Reinberg D. (2006) SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eskandarian H. A., Impens F., Nahori M. A., Soubigou G., Coppee J. Y., Cossart P., and Hamon M. A. (2013) A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 341, 1238858. [DOI] [PubMed] [Google Scholar]
- 23. Lin R., Tao R., Gao X., Li T., Zhou X., Guan K. L., Xiong Y., and Lei Q. Y. (2013) Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 51, 506–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsusaka T., Guo T., Yagura T., Inoue T., Yokode M., Inagaki N., and Kondoh H. (2014) Deacetylation of phosphoglycerate mutase in its distinct central region by SIRT2 down-regulates its enzymatic activity. Genes Cells 19, 766–777 [DOI] [PubMed] [Google Scholar]
- 25. Joshi P., Greco T. M., Guise A. J., Luo Y., Yu F., Nesvizhskii A. I., and Cristea I. M. (2013) The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miteva Y. V., and Cristea I. M. (2014) A proteomic perspective of Sirtuin 6 (SIRT6) phosphorylation and interactions and their dependence on its catalytic activity. Mol. Cell. Proteomics 13, 168–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simeoni F., Tasselli L., Tanaka S., Villanova L., Hayashi M., Kubota K., Isono F., Garcia B. A., Michishita-Kioi E., and Chua K. F. (2013) Proteomic analysis of the SIRT6 interactome: novel links to genome maintenance and cellular stress signaling. Sci. Rep. 3, 3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bae N. S., Swanson M. J., Vassilev A., and Howard B. H. (2004) Human histone deacetylase SIRT2 interacts with the homeobox transcription factor HOXA10. J. Biochem. 135, 695–700 [DOI] [PubMed] [Google Scholar]
- 29. Huttlin E. L., Ting L., Bruckner R. J., Gebreab F., Gygi M. P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K., Dong R., Guarani V., Vaites L. P., Ordureau A., Rad R., Erickson B. K., Wuhr M., Chick J., Zhai B., Kolippakkam D., Mintseris J., Obar R. A., Harris T., Artavanis-Tsakonas S., Sowa M. E., De Camilli P., Paulo J. A., Harper J. W., and Gygi S. P. (2015) The BioPlex Network: a systematic exploration of the human interactome. Cell 162, 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsai Y. C., Greco T. M., Boonmee A., Miteva Y., and Cristea I. M. (2012) Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol. Cell. Proteomics 11, 60–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rothgiesser K. M., Erener S., Waibel S., Luscher B., and Hottiger M. O. (2010) SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 123, 4251–4258 [DOI] [PubMed] [Google Scholar]
- 32. Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., and Mayo M. W. (2004) Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Leeuwen I. M., Higgins M., Campbell J., McCarthy A. R., Sachweh M. C., Navarro A. M., and Lain S. (2013) Modulation of p53 C-terminal acetylation by mdm2, p14ARF, and cytoplasmic SirT2. Mol. Cancer Ther. 12, 471–480 [DOI] [PubMed] [Google Scholar]
- 34. Vaziri H., Dessain S. K., Ng Eaton E., Imai S. I., Frye R. A., Pandita T. K., Guarente L., and Weinberg R. A. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 [DOI] [PubMed] [Google Scholar]
- 35. Wang F., Nguyen M., Qin F. X., and Tong Q. (2007) SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514 [DOI] [PubMed] [Google Scholar]
- 36. Wang F., Chan C. H., Chen K., Guan X., Lin H. K., and Tong Q. (2012) Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene 31, 1546–1557 [DOI] [PubMed] [Google Scholar]
- 37. Nguyen P., Lee S., Lorang-Leins D., Trepel J., and Smart D. K. (2014) SIRT2 interacts with beta-catenin to inhibit Wnt signaling output in response to radiation-induced stress. Mol. Cancer Res. 12, 1244–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Firestein R., Blander G., Michan S., Oberdoerffer P., Ogino S., Campbell J., Bhimavarapu A., Luikenhuis S., de Cabo R., Fuchs C., Hahn W. C., Guarente L. P., and Sinclair D. A. (2008) The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3, e2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luthi-Carter R., Taylor D. M., Pallos J., Lambert E., Amore A., Parker A., Moffitt H., Smith D. L., Runne H., Gokce O., Kuhn A., Xiang Z., Maxwell M. M., Reeves S. A., Bates G. P., Neri C., Thompson L. M., Marsh J. L., and Kazantsev A. G. (2010) SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 7927–7932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim H. S., Vassilopoulos A., Wang R. H., Lahusen T., Xiao Z., Xu X., Li C., Veenstra T. D., Li B., Yu H., Ji J., Wang X. W., Park S. H., Cha Y. I., Gius D., and Deng C. X. (2011) SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Serrano L., Martinez-Redondo P., Marazuela-Duque A., Vazquez B. N., Dooley S. J., Voigt P., Beck D. B., Kane-Goldsmith N., Tong Q., Rabanal R. M., Fondevila D., Munoz P., Kruger M., Tischfield J. A., and Vaquero A. (2013) The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 27, 639–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hiratsuka M., Inoue T., Toda T., Kimura N., Shirayoshi Y., Kamitani H., Watanabe T., Ohama E., Tahimic C. G., Kurimasa A., and Oshimura M. (2003) Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 309, 558–566 [DOI] [PubMed] [Google Scholar]
- 43. Hoffmann G., Breitenbucher F., Schuler M., and Ehrenhofer-Murray A. E. (2014) A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 289, 5208–5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheon M. G., Kim W., Choi M., and Kim J. E. (2015) AK-1, a specific SIRT2 inhibitor, induces cell cycle arrest by downregulating Snail in HCT116 human colon carcinoma cells. Cancer Lett. 356, 637–645 [DOI] [PubMed] [Google Scholar]
- 45. Jing H., Hu J., He B., Negron Abril Y. L., Stupinski J., Weiser K., Carbonaro M., Chiang Y. L., Southard T., Giannakakou P., Weiss R. S., and Lin H. (2016) A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 29, 297–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tackett A. J., DeGrasse J. A., Sekedat M. D., Oeffinger M., Rout M. P., and Chait B. T. (2005) I-DIRT, a general method for distinguishing between specific and nonspecific protein interactions. J. Proteome Res. 4, 1752–1756 [DOI] [PubMed] [Google Scholar]
- 47. Cristea I. M., Williams R., Chait B. T., and Rout M. P. (2005) Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics 4, 1933–1941 [DOI] [PubMed] [Google Scholar]
- 48. Cristea I. M., and Chait B. T. (2011) Conjugation of magnetic beads for immunopurification of protein complexes. Cold Spring Harb. Protoc. 2011, pdb prot5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wisniewski J. R., Zougman A., and Mann M. (2009) Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8, 5674–5678 [DOI] [PubMed] [Google Scholar]
- 50. Guise A. J., Greco T. M., Zhang I. Y., Yu F., and Cristea I. M. (2012) Aurora B-dependent regulation of class IIa histone deacetylases by mitotic nuclear localization signal phosphorylation. Mol. Cell. Proteomics 11, 1220–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Greco T. M., Guise A. J., and Cristea I. M. (2016) Determining the Composition and Stability of Protein Complexes Using an Integrated Label-Free and Stable Isotope Labeling Strategy. Methods Mol. Biol. 1410, 39–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y., Wen Z., Washburn M. P., and Florens L. (2009) Effect of dynamic exclusion duration on spectral count based quantitative proteomics. Anal. Chem. 81, 6317–6326 [DOI] [PubMed] [Google Scholar]
- 53. Craig R., and Beavis R. C. (2003) A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 17, 2310–2316 [DOI] [PubMed] [Google Scholar]
- 54. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 55. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 56. Choi H., Liu G., Mellacheruvu D., Tyers M., Gingras A. C., and Nesvizhskii A. I. (2012) Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Curr. Protoc. Bioinformatics Chapter 8, Unit8 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 58. Cox J., and Mann M. (2012) 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics 13, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jensen L. J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M., Bork P., and von Mering C. (2009) STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., and Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 62. Wang M., Herrmann C. J., Simonovic M., Szklarczyk D., and von Mering C. (2015) Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 15, 3163–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ong S. E., and Mann M. (2006) A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 1, 2650–2660 [DOI] [PubMed] [Google Scholar]
- 64. Budayeva H. G., and Cristea I. M. (2014) A mass spectrometry view of stable and transient protein interactions. Adv. Exp. Med. Biol. 806, 263–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Miteva Y. V., Budayeva H. G., and Cristea I. M. (2013) Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal. Chem. 85, 749–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Diner B. A., Li T., Greco T. M., Crow M. S., Fuesler J. A., Wang J., and Cristea I. M. (2015) The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol. Syst. Biol. 11, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., and Nesvizhskii A. I. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shimazu T., Horinouchi S., and Yoshida M. (2007) Multiple histone deacetylases and the CREB-binding protein regulate pre-mRNA 3′-end processing. J. Biol. Chem. 282, 4470–4478 [DOI] [PubMed] [Google Scholar]
- 69. Sato K., Sato M., and Nakano A. (2003) Rer1p, a retrieval receptor for ER membrane proteins, recognizes transmembrane domains in multiple modes. Mol. Biol. Cell 14, 3605–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lord C., Ferro-Novick S., and Miller E. A. (2013) The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the golgi. Cold Spring Harb. Perspect. Biol. 5, pii:a013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Townley A. K., Feng Y., Schmidt K., Carter D. A., Porter R., Verkade P., and Stephens D. J. (2008) Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J. Cell Sci. 121, 3025–3034 [DOI] [PubMed] [Google Scholar]
- 72. Yang Y., Hou H., Haller E. M., Nicosia S. V., and Bai W. (2005) Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 24, 1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jing E., Gesta S., and Kahn C. R. (2007) SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 6, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chu C. W., Hou F., Zhang J., Phu L., Loktev A. V., Kirkpatrick D. S., Jackson P. K., Zhao Y., and Zou H. (2011) A novel acetylation of beta-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol. Biol. Cell 22, 448–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Moon R. T., Brown J. D., Yang-Snyder J. A., and Miller J. R. (1997) Structurally related receptors and antagonists compete for secreted Wnt ligands. Cell 88, 725–728 [DOI] [PubMed] [Google Scholar]
- 76. Hauri H. P., Kappeler F., Andersson H., and Appenzeller C. (2000) ERGIC-53 and traffic in the secretory pathway. J. Cell Sci. 113, 587–596 [DOI] [PubMed] [Google Scholar]
- 77. Moorman N. J., Sharon-Friling R., Shenk T., and Cristea I. M. (2010) A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics 9, 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Breuza L., Halbeisen R., Jeno P., Otte S., Barlowe C., Hong W., and Hauri H. P. (2004) Proteomics of endoplasmic reticulum-Golgi intermediate compartment (ERGIC) membranes from brefeldin A-treated HepG2 cells identifies ERGIC-32, a new cycling protein that interacts with human Erv46. J. Biol. Chem. 279, 47242–47253 [DOI] [PubMed] [Google Scholar]
- 79. Serra-Pages C., Medley Q. G., Tang M., Hart A., and Streuli M. (1998) Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J. Biol. Chem. 273, 15611–15620 [DOI] [PubMed] [Google Scholar]
- 80. Astigarraga S., Hofmeyer K., Farajian R., and Treisman J. E. (2010) Three Drosophila liprins interact to control synapse formation. J. Neurosci. 30, 15358–15368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chiaretti S., Astro V., Chiricozzi E., and de Curtis I. (2016) Effects of the scaffold proteins liprin-alpha1, beta1 and beta2 on invasion by breast cancer cells. Biol. Cell 108, 65–75 [DOI] [PubMed] [Google Scholar]
- 82. Kriajevska M., Fischer-Larsen M., Moertz E., Vorm O., Tulchinsky E., Grigorian M., Ambartsumian N., and Lukanidin E. (2002) Liprin beta 1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1). J. Biol. Chem. 277, 5229–5235 [DOI] [PubMed] [Google Scholar]
- 83. Norrmen C., Vandevelde W., Ny A., Saharinen P., Gentile M., Haraldsen G., Puolakkainen P., Lukanidin E., Dewerchin M., Alitalo K., and Petrova T. V. (2010) Liprin (beta)1 is highly expressed in lymphatic vasculature and is important for lymphatic vessel integrity. Blood 115, 906–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. van de Velde H. J., Roebroek A. J., Senden N. H., Ramaekers F. C., and Van de Ven W. J. (1994) NSP-encoded reticulons, neuroendocrine proteins of a novel gene family associated with membranes of the endoplasmic reticulum. J. Cell Sci. 107 (Pt 9), 2403–2416 [DOI] [PubMed] [Google Scholar]
- 85. Perdiz D., Mackeh R., Pous C., and Baillet A. (2011) The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell Signal. 23, 763–771 [DOI] [PubMed] [Google Scholar]
- 86. Kaul N., Soppina V., and Verhey K. J. (2014) Effects of alpha-tubulin K40 acetylation and detyrosination on kinesin-1 motility in a purified system. Biophys. J. 106, 2636–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cai D., McEwen D. P., Martens J. R., Meyhofer E., and Verhey K. J. (2009) Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLos Biol. 7, e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stephens D. J. (2012) Functional coupling of microtubules to membranes- implications for membrane structure and dynamics. J. Cell Sci. 125, 2795–2804 [DOI] [PubMed] [Google Scholar]
- 89. Liu Z., Yang T., Li X., Peng T., Hang H. C., and Li X. D. (2015) Integrative chemical biology approaches for identification and characterization of “erasers” for fatty-acid-acylated lysine residues within proteins. Angew Chem. Int. Ed. Engl. 54, 1149–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Budayeva H. G., Rowland E. A., and Cristea I. M. (2016) Intricate Roles of Mammalian Sirtuins in Defense against Viral Pathogens. J. Virol. 90, 5–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dohner K., Nagel C. H., and Sodeik B. (2005) Viral stop-and-go along microtubules: taking a ride with dynein and kinesins. Trends Microbiol. 13, 320–327 [DOI] [PubMed] [Google Scholar]
- 92. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., and Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 93. Miller K. E., DeProto J., Kaufmann N., Patel B. N., Duckworth A., and Van Vactor D. (2005) Direct observation demonstrates that Liprin-alpha is required for trafficking of synaptic vesicles. Curr. Biol. 15, 684–689 [DOI] [PubMed] [Google Scholar]
- 94. Gal J., Bang Y., and Choi H. J. (2012) SIRT2 interferes with autophagy-mediated degradation of protein aggregates in neuronal cells under proteasome inhibition. Neurochem. Int. 61, 992–1000 [DOI] [PubMed] [Google Scholar]
- 95. Morlon A., and Sassone-Corsi P. (2003) The LIM-only protein FHL2 is a serum-inducible transcriptional coactivator of AP-1. Proc. Natl. Acad. Sci. U.S.A. 100, 3977–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wixler V., Geerts D., Laplantine E., Westhoff D., Smyth N., Aumailley M., Sonnenberg A., and Paulsson M. (2000) The LIM-only protein DRAL/FHL2 binds to the cytoplasmic domain of several alpha and beta integrin chains and is recruited to adhesion complexes. J. Biol. Chem. 275, 33669–33678 [DOI] [PubMed] [Google Scholar]
- 97. Peck B., Chen C. Y., Ho K. K., Di Fruscia P., Myatt S. S., Coombes R. C., Fuchter M. J., Hsiao C. D., and Lam E. W. (2010) SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 9, 844–855 [DOI] [PubMed] [Google Scholar]
- 98. GrandPre T., Nakamura F., Vartanian T., and Strittmatter S. M. (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403, 439–444 [DOI] [PubMed] [Google Scholar]
- 99. Kawano Y., and Kypta R. (2003) Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 116, 2627–2634 [DOI] [PubMed] [Google Scholar]
- 100. Rardin M. J., He W., Nishida Y., Newman J. C., Carrico C., Danielson S. R., Guo A., Gut P., Sahu A. K., Li B., Uppala R., Fitch M., Riiff T., Zhu L., Zhou J., Mulhern D., Stevens R. D., Ilkayeva O. R., Newgard C. B., Jacobson M. P., Hellerstein M., Goetzman E. S., Gibson B. W., and Verdin E. (2013) SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tsai Y. C., Greco T. M., and Cristea I. M. (2014) Sirtuin 7 plays a role in ribosome biogenesis and protein synthesis. Mol. Cell. Proteomics 13, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Greco T. M., Yu F., Guise A. J., and Cristea I. M. (2011) Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol. Cell. Proteomics 10, M110 004317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mielcarek M., Landles C., Weiss A., Bradaia A., Seredenina T., Inuabasi L., Osborne G. F., Wadel K., Touller C., Butler R., Robertson J., Franklin S. A., Smith D. L., Park L., Marks P. A., Wanker E. E., Olson E. N., Luthi-Carter R., van der Putten H., Beaumont V., and Bates G. P. (2013) HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLos Biol. 11, e1001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yue F., Li W., Zou J., Chen Q., Xu G., Huang H., Xu Z., Zhang S., Gallinari P., Wang F., McKeehan W. L., and Liu L. (2015) Blocking the association of HDAC4 with MAP1S accelerates autophagy clearance of mutant Huntingtin. Aging. 7, 839–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.