

Summary
Kawasaki disease (KD) is an acute systemic vasculitis of childhood that does not have a known cause or aetiology. The epidemiological features (existence of epidemics, community outbreaks and seasonality), unique age distribution and clinical symptoms and signs of KD suggest that the disease is caused by one or more infectious environmental triggers. However, KD is not transmitted person‐to‐person and does not occur in clusters within households, schools or nurseries. KD is a self‐limited illness that is not associated with the production of autoantibodies or the deposition of immune complexes, and it rarely recurs. Regarding the underlying pathophysiology of KD, innate immune activity (the inflammasome) is believed to play a role in the development of KD vasculitis, based on the results of studies with animal models and the clinical and laboratory findings of KD patients. Animal studies have demonstrated that innate immune pathogen‐associated molecular patterns (PAMPs) can cause vasculitis independently of acquired immunity and have provided valuable insights regarding the underlying mechanisms of this phenomenon. To validate this concept, we recently searched for KD‐specific PAMPs and identified such molecules with high specificity and sensitivity. These molecules have structures similar to those of microbe‐associated molecular patterns (MAMPs), as shown by liquid chromatography‐tandem mass spectrometry. We propose herein that KD is an innate immune disorder resulting from the exposure of a genetically predisposed individual to microbe‐derived innate immune stimulants and that it is not a typical infectious disease.
Keywords: Kawasaki disease, innate immunity, liquid chromatography‐mass spectrometry, pathogen‐associated molecular patterns
Introduction
Kawasaki disease (KD) is an acute self‐limiting systemic vasculitis of early childhood that was first described by Tomisaku Kawasaki in 1967 1. It affects predominantly the coronary arteries and causes coronary artery abnormalities in 25–30% of untreated patients 2. After the introduction of intravenous immunoglobulin (Ig), the incidence of coronary artery lesions (CALs) decreased to fewer than 5% 3. Nonetheless, KD is the most common cause of acquired childhood heart disease in developed countries 4, 5. The incidence of KD is still increasing, according to the most recent nationwide survey (2013–14) in Japan 6.
Although almost 50 years have passed since its initial description, the aetiology of KD remains unknown. KD is usually diagnosed by clinical symptoms and signs, because no specific diagnostic tests are available. The clinical and epidemiological features of KD suggest strongly that the disease results from the exposure of a genetically predisposed individual to an unidentified, possibly infectious environmental trigger 5, 7. This review will focus on the genetic, environmental and immunological aspects of KD in an attempt to elucidate its aetiology.
KD aetiology
Genetic background
Twin studies have revealed that the concordance rates of KD were 14·1% (11 of 78) and 13·3% (four of 30) for monozygotic and dizygotic twins, respectively, in Japan 8. In the United States, the concordance rate of KD for monozygotic twins was 25% (one out of four) 9. For a single‐gene disorder with complete penetrance, the expected concordance rate should be 100% for monozygotic twins, while it should be lower for dizygotic twins. For conditions that are determined completely by environmental factors, the concordance rates for monozygotic and dizygotic twins should be essentially equal and depend upon the shared environment in which the twins live. Thus, these twin studies suggest that environmental factors contribute more to the development of KD than genetic factors among individuals with the same ethnicity.
However, a genetic predisposition to KD has been proposed in various epidemiological studies. For example, the incidence of KD is highest in Asian populations, especially Japanese populations. The incidence among Japanese individuals is 10–15 times higher than that among Caucasians 10. The incidence of KD among Japanese American children in Hawaii is as high as that among Japanese children, while the incidence among Caucasian children in Hawaii is as low as that among Caucasian children in the continental United States 11, 12. The idea of genetic susceptibility to KD is supported further by the fact that a higher relative risk of KD exists within families 13.
Linkage analysis and genome‐wide association studies (GWASs) have identified KD susceptibility alleles for the following genes: ITPKC, CASP3, BLK, CD40, HLA and FCGR2A 14. Recently identified genetic variations of ORAI1 may explain the aforementioned Asian susceptibility to KD 15. Although inositol 1,4,5‐trisphosphate 3‐kinase C (ITPKC) was described initially as a negative regulator of T cell activation 16, ITPKC is a ubiquitous molecule that is present in innate and acquired immune cells, as well as endothelial cells. ITPKC, caspase‐3 (CASP3) and calcium release‐activated calcium channel protein 1 (ORAI1) may play important roles in KD development by regulating the calcium/nuclear factor of activated T cells (NFAT) pathway 14, 15, 16. The candidate gene approach showed that KD is linked to genetic variations (TGFB2, TGFBR2 and SMAD3) in the transforming growth factor beta (TGF‐β) signalling pathway that are important in inflammation and vascular remodelling 17. VEGFA, KDR and ANGPT1 have also been reported to be associated with KD. These data suggest that dysregulation of vascular endothelial growth factor (VEGF) and angiopoietins contributes to the disruption of vascular homeostasis in KD 18, 19.
Environmental factors
The clinical picture of KD supports the notion that microbes or infectious organisms are capable of triggering onset of the disease, as do the following facts: (a) children are affected mainly between 6 months and 5 years of age, and the peak age of disease onset coincides with the period during which children are most susceptible to common pathogens; (b) KD is characterized by an acute onset and follows a self‐limited clinical course; and (c) KD shows epidemics, community outbreaks and seasonality 3, 4, 5, 7.
Many microbes or microbe‐derived substances are believed to cause KD, including Rickettsia‐like agent, Propriobacterium acnes, Leptospira spp., Streptococcus sanguis, Staphylococcus aureus, Yersinia pseudotuberculosis, retroviruses, Epstein–Barr virus, cytomegalovirus, coronavirus, parvovirus B19, human bocavirus, undetermined RNA viruses and staphylococcal or streptococcal superantigens 20, 21. Rowley et al. detected cytoplasmic inclusion bodies containing virus‐like particles in the bronchial epithelium of a patient with acute KD, using synthetic antibodies 20, 22. However, no causative viruses have been identified thus far.
It is well known that approximately 10% of patients with Y. pseudotuberculosis infection develop Kawasaki syndrome in Japan 23 and that Kawasaki syndrome patients with known Y. pseudotuberculosis infection show a higher tendency to develop CALs 24. In addition, epidemiological data indicate that higher incidences of KD have been observed in populations at high risk for Y. pseudotuberculosis infection 25. Rodó et al. suggested that a wind‐borne environmental trigger induces KD 26, 27. However, no such agents have been identified 7, 28.
Regarding superantigens, Vβ‐restricted T cell expansion or activation in patients with KD has been observed by some researchers 29, 30. However, other research groups failed to detect such T cell expansion 31. Because only a small proportion of KD patients have shown Vβ‐restricted T cell activation 29, 30, 31, and there are no differences in KD symptoms or signs between patients with and without T cell activation (our unpublished observation), superantigen‐induced T cell activation may be an epiphenomenon rather than a necessity for the development of KD. Biofilms form as a result of interactions between microbes and the environment and are capable of producing large amounts of various bioactive molecules, including superantigens, through quorum‐sensing mechanisms 32. For example, when S. aureus was cultured in biofilms adhering to tampon sacs, the level of toxic shock syndrome (TSS) toxin‐1 in these bacteria was more than 1000‐fold higher than that in bacteria cultured via conventional methods 33. Although TSS caused by TSS toxin‐1 exhibits clinical features similar to those of KD 34, TSS is characterized by superantigen‐induced excessive T cell activation. In contrast, most cases of KD are characterized by T cell suppression, as well as endothelial cell/innate immune cell activation 35, 36. KD and TSS rarely develop in association with isolated sepsis or bacteraemia 37, in which bacteria grow under planktonic conditions. Thus, as is the case for TSS, the pathogenesis of KD may be evoked not by microbes themselves but by bioactive molecules that are produced by microbes under biofilm‐like conditions.
Immunological aspects
Innate immunity versus acquired immunity
The innate immune system has both cellular and humoral components. The cellular components include neutrophils, eosinophils, monocytes, macrophages, dendritic cells, γδT cells, natural killer cells and natural killer T cells 38. In addition, endothelial cells function as sentinel innate immune cells and detect foreign pathogens and endogenous danger signals in the bloodstream 39.
The following clinical and laboratory evidence suggests that the acute phase of KD is driven primarily by the innate immune system: (a) the absolute neutrophil and monocyte counts in the peripheral blood are increased 40; (b) the majority of the activated T lymphocytes in the peripheral blood are γδT cells 36; (c) the majority of the cells infiltrating the coronary arteries and skin lesions are macrophages 41, 42; (d) the levels of damage‐associated molecular patterns (DAMPs), such as S100 proteins and high mobility group box 1 (HMGB1), are elevated in the sera of KD patients during the acute phase 43, 44, 45, 46; (e) KD is sometimes associated with disorders characterized by hyperactive innate immunity, such as periodic fever, aphthous stomatitis, pharyngitis and adenitis (PFAPA) syndrome 47 and systemic juvenile idiopathic arthritis (JIA) 48, 49; and (f) KD patients have the highest recurrence rate within 12 months following the first episode 50, which may be attributed to the fact that the innate immune system lacks immunological memory.
KD is also regarded as a condition associated with acquired immune dysfunction that is characterized by (a) decreased absolute CD3+, CD4+ and CD8+ T cell counts in the peripheral blood 40; (b) marked suppression of T cell receptor/CD3‐induced T cell proliferation 35; (c) down‐regulation of T cell receptor and B cell receptor signalling pathways, as shown by microarray studies 36, 51, 52; and (d) suppression of regulatory T cells during the acute phase of the disease 53, 54. Whether T helper type 17 (Th17) cells contribute to the development of KD remains controversial, because a previous study showed that the levels of Th17 cells were only slightly elevated in the peripheral blood of KD patients 55, and inconsistent results were obtained by two other studies 56, 57. Based on currently available evidence KD is unlikely to have an autoimmune cause, as it is not associated with autoantibody production, resolves spontaneously and rarely recurs 5.
KD animal models
The clinical and laboratory features of KD suggest strongly that innate immunity plays a critical role in the development of coronary vasculitis in patients with KD. We therefore performed in‐vitro studies regarding the proinflammatory effects of innate immune ligands to corroborate this hypothesis, using human coronary artery endothelial cells (HCAECs). HCAECs have been shown to produce interleukin (IL)‐6 and IL‐8 when treated with ligands for Toll‐like receptors (TLR)−2 and −4 and nucleotide‐binding oligomerization domain‐containing proteins (NOD)1 and 2 58. To validate this finding in vivo, we injected various innate immune ligands into wild‐type C57Bl/6 mice and found that a NOD1 ligand, FK565, was a more potent inducer of coronary arteritis than any other ligand 58. Similar results were also obtained via oral administration of FK565, as shown in Fig. 1a 58. Using severe combined immunodeficient (SCID) mice, we demonstrated that NOD1 ligands induce coronary arteritis in the absence of functional T and B cells (Fig. 1c).
Figure 1.
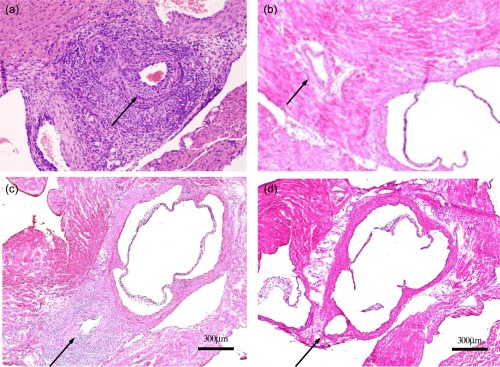
Nucleotide‐binding oligomerization domain‐containing protein (NOD1) ligand (FK565)‐induced coronary arteritis. (a) Administration of FK565 (100 μg/day p.o. for 2 weeks) induces coronary arteritis. This Kawasaki disease (KD) model is characterized by panarteritis with dense inflammatory cell infiltration involving neutrophils and macrophages, but is not associated with fibrinoid necrosis. This histopathology recapitulates the coronary artery lesions of KD. (b) Control solvent: no arteritis. (c) FK565‐induced coronary arteritis in a severe combined immunodeficient (SCID) mouse. This panel shows that FK565 also induces a milder form of coronary arteritis in SCID mice than that induced in wild‐type mice. Most of the infiltrating inflammatory cells are neutrophils and macrophages, as is the case in wild‐type mice. These data show that the coronary artery lesions of KD are mediated by the innate immune system (PAMPs) and develop independently of acquired immunity. (d) Absence of vasculitis in a NOD1‐knock‐out mouse from Nishio et al. 58.
FK565 is a synthetic acyltripeptide (heptanoyl‐γ‐D‐glutamyl‐meso‐diamino‐ pimelyl‐ D‐alanine) with a molecular weight (MW) of 502·6. By binding to NOD1, FK565, which harbours diaminopimelic acid within its structure, functions as a pathogen‐associated molecular pattern (PAMP). Diaminopimelic acid is also the active constituent of other environmental PAMPs. For example, dipeptide γ‐D‐glutamyl‐meso‐diaminopimelic acid (iE‐DAP, MW: 319·3), L‐alanyl‐γ‐D‐glutamyl‐meso‐diaminopimelic acid (MW: 390·39) and lauroyl‐γ‐D‐glutamyl‐meso‐diaminopimelic acid (MW: 501·61) are components of peptidoglycan in the cell walls of Gram‐negative and certain groups of Gram‐positive bacteria 58, 59.
Regarding the molecular and cellular pathophysiology of FK565‐induced coronary arteritis, we propose that NOD1 ligands activate proinflammatory signals in vascular endothelial cells, thereby producing large amounts of chemokines. In response to these chemokines, monocytes in the peripheral blood are recruited to FK565‐stimulated endothelial cells and differentiate subsequently into cardiac CD11c+ macrophages 60. Genetically manipulated mice lacking CD11c+ macrophages present with milder coronary vasculitis after administration of FK565, indicating that CD11c+ macrophages play a pivotal role in the pathogenesis of acute coronary arteritis (Fig. 2). We also verified that FK565 reproducibly induces acute coronary vasculitis in SCID and Rag1‐knock‐out mice 58, 60. These data provide new insights into the pathogenic mechanisms of vasculitis in humans and demonstrate that innate immunity (PAMPs) can cause vasculitis independently of acquired immunity.
Figure 2.
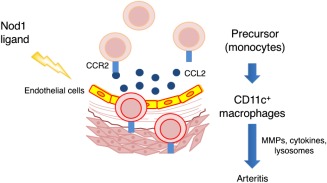
Schematic representation of the molecular and cellular mechanisms underlying nucleotide‐binding oligomerization domain‐containing protein (NOD1)‐induced arteritis. A NOD1 ligand, FK565, activates endothelial cells which produce large amounts of chemokines, including CCL2. In response to CCL2 and other chemokines, CCR2 (chemokine receptor)‐expressing precursor cells (monocytes) in the peripheral blood are recruited to FK565‐activated endothelial cells. This process subsequently induces the differentiation of cardiac CD11c+ macrophages, which play a pivotal role in the pathogenesis of acute coronary arteritis. MMP = matrix metalloproteinase.
To date, two other animal models of KD coronary arteritis have been established. These mouse models showed that crude microbe‐associated molecular patterns (MAMPs)/PAMPs from Lactobacillus caseii 61, 62, 63, 64, 65 and Candida albicans 66, 67, 68 induce acute coronary vasculitis (Table 1). Thus, these animal studies confirmed that stimulation of innate immunity with molecules such as NOD1 ligands induces vasculitis that mimics the coronary artery lesions of KD. In agreement with our study, an animal study using L. caseii cell wall extracts (LCWE) also showed that CD11c+ dendritic cells/macrophages and vascular stromal cells with cytokines (IL‐1α and β) are important in the pathogenesis of coronary arteritis 65. In addition, a recent study has demonstrated that the activation of endothelial Nlrp3 inflammasome, a key component of the innate immune system, may contribute to the development of coronary arteritis induced by LCWE 69.
Table 1.
Animal models of Kawasaki disease (KD)‐like vasculitis
| Microbe‐derived or synthetic substances | Innate immunity | Acquired immunity | Important cytokine/chemokine | |
|---|---|---|---|---|
| T cell | B cell | |||
|
LCWE (Lactobacillus caseii cell wall extract) 61, 62, 63, 64, 65, 69
Crude PAMP |
CD11c+ Macrophages/DC Vascular stromal cells TLR2/MyD88 Dectin‐1/Syk Nlrp3 inflammasome |
superantigen+++ | +/– |
IL‐1β IL‐1α TNF‐α |
|
CAWS (Candida albicans water‐ soluble fraction) 66, 67, 68
Crude PAMP |
Inflammatory monocytes |
++ Th17 Regulatory T cell |
+/– | IL‐6 |
|
NOD1 ligand (FK565) 58, 60
Pure PAMP (synthetic) |
Nod1 in endothelial cells CD11c+ macrophages |
+/– | +/– | CCL2 |
CCL = chemokine (C‐C motif) ligand; DC = dendritic cells; IL = interleukin; MyD = myeloid differentiation primary response; Nlrp3 = nucleotide‐binding, leucine‐rich repeat containing family pyrin domain containing 3; NOD1 = nucleotide‐binding oligomerization domain‐containing protein; TNF = tumour necrosis factor; Syk = spleen tyrosine kinase.
Searching for KD‐specific molecules in humans
A seminal study detected endothelial cell‐activating antigens in skin biopsy samples from KD patients 70. To confirm these findings, we searched for unknown ligands that may activate NOD1 or other vasculitis‐inducing pathways. We first prepared whole extracts and fractionated samples from the sera of KD patients and found that KD sera contain bioactive substances that induce production of IL‐6 and IL‐8 in HCAECs 71. However, no NOD1‐activating ligands were detected in these KD samples by cell‐based reporter systems or liquid chromatography‐mass spectrometry (LC‐MS) analysis 71. Thus, these results suggest that other innate immune receptor(s) may be associated with the development of KD vasculitis. Alternatively, the sensitivities of cell‐based reporter system and LC‐MS analysis may not be high enough to detect NOD1 ligands in KD serum samples.
To exclude the possibility of cytokine and chemokine contamination in the aqueous fractions of sera from KD patients, we analysed the lipophilic fractions of the above‐mentioned 117 samples. We detected novel molecules between 227·1 m/z and 1487·8 m/z with a specificity of 100% and a low sensitivity ranging from 9·3 to 48·8% 71. We defined them as ‘KD‐specific molecules’. Then, we investigated whether these KD‐specific molecules had structures similar to those of MAMPs from Y. pseudotuberculosis 23, 24, 25 and airborne bacteria 26, 27, using liquid chromatography‐tandem mass spectrometry (LC‐MS/MS). We used various types of culture media, as well as different temperatures, durations of shaking and incubation and supplemental nutrients, to stimulate bacterial growth. Lipid extracts from three bacterial culture components (cell, supernatant and biofilm) were subjected to LC‐MS/MS analysis 71. The serum KD‐specific molecules showed MS/MS fragmentation patterns that were similar to those of MAMPs in the biofilm extracts from Y. pseudotuberculosis and airborne bacteria; however, these patterns were not similar to those of MAMPs in the other two extracts (cell and supernatant). Production of MAMPs similar to serum KD‐specific molecules was enhanced markedly under biofilm‐forming conditions in the presence of butter 71. HCAEC‐stimulatory activities (IL‐6 and IL‐8 production from HCAECs) also tended to be much higher in the biofilm extracts from Y. pseudotuberculosis and airborne bacteria in cultures supplemented with butter, as shown in Fig. 3.
Figure 3.
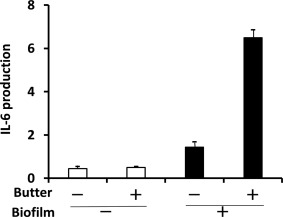
Human coronary artery endothelial cells (HCAECs)‐stimulatory activities of biofilm extracts from Yersinia pseudotuberculosis. HCAECs‐stimulatory activities of Y. pseudotuberculosis extracts were measured by interleukin (IL)‐6 production from HCAECs. Y. pseudotuberculosis extracts were prepared from culture supernatants (□) or biofilms (▪) of Y. pseudotuberculosis cultured in the presence (+) or absence (–) of butter. Medium alone, ethyl acetate alone or ethyl acetate extract from glass slides cultured in the absence of microbes was used as a negative control. FK565 (10 µg/ml) was used as a positive control. Data are expressed as the fold change in induction of IL‐6 production compared to positive control levels. Modified from the data of Kusuda et al. 71.
More recently, we used modified extraction and analysis methods in a nationwide collaborative study. KD‐specific molecules were detected in the sera of affected patients with a specificity of 100% and a sensitivity of almost 100% (Nakashima et al. 2016, manuscript in preparation). In this study, we confirmed that KD‐specific molecules possessed structures similar to those of MAMPs found in biofilm extracts from Y. pseudotuberculosis and airborne bacteria (Nakashima et al. 2016, manuscript in preparation).
A comprehensive view of KD pathogenesis in the context of systemic vasculitides
The epidemiological features (existence of epidemics, community outbreaks and seasonality) of KD suggest that the disease is caused by one or more infectious environmental triggers 5, 7, 72. Among other systemic vasculitides, only IgA vasculitis exhibits seasonality in the absence of outbreaks or epidemics 73, as shown in Table 2. The incidence of KD, as well as those of allergies and non‐infectious inflammatory bowel disease, has increased 6, 74, 75, while those of infectious diseases have decreased continuously in Japan. These facts suggest that KD is not a typical infectious disease.
Table 2.
Kawasaki disease (KD) pathogenic features in light of those of other systemic vasculitides
| KD | Other systemic vasculitides | |
|---|---|---|
| Epidemiological features | Epidemics +, community outbreaks +seasonality + |
No epidemics, no outbreaks usually no seasonality (IgA vasculitis: seasonality +) |
| Clinical and laboratory features |
Age: mostly infants abrupt onset acute infection‐like symptoms self‐limited/no recurrence in most cases Autoantibodies: usually absent immune complexes: usually absent association with innate immune disorders, PFAPA syndrome and systemic JIA |
Common in older age acute‐chronic onset constitutional symptoms chronic and/or recurrent (IgA vasculitis: usually self‐limited) autoantibodies: some + immune complexes: some + no association with PFAPA syndrome and systemic JIA |
| Pathophysiological features |
Inhibited T cell receptor signalling pathway Inflammasome involvement + hypercytokinaemia (IL‐1, IL‐6, TNF‐α, etc.) Detection of possible PAMPs in sera |
none Inflammasome involvement: some + hypercytokinaemia: some +no data |
IL = interleukin; JIA = juvenile idiopathic arthritis; PAMPs = pathogen‐associated molecular patterns; PFAPA syndrome = periodic fever, aphthous stomatitis, pharyngitis and adenitis syndrome; TNF = tumour necrosis factor.
The unique age distribution (more than 80% of cases occur between the ages of 6 months and 4 years) of KD is reminiscent of paediatric infectious diseases 3, 4, 5, 7. However, as KD is not transmitted person‐to‐person and does not occur in clusters within households, schools or nurseries, it does not possess the characteristics of an infectious disease 76. Conversely, the peak age of IgA vasculitis onset is between 4 and 6 years, and those of the onset of other systemic vasculitides with possible autoimmune aetiologies are much higher 77.
The clinical symptoms and signs (fever, injection of the eyes or oropharynx, rash and cervical lymphadenopathy) of KD mimic those of acute infections, while those of other systemic vasculitides usually do not 77. Innate immune disorders, such as PFAPA syndrome 78, and superantigen‐induced diseases, such as TSS, have clinical features that are indistinguishable from those of infectious diseases. As superantigen‐induced αβT cell activation is usually not observed in most patients with KD 30, 31, 36, it is possible that KD is an innate immune disorder.
KD is a self‐limited illness that is not characterized by autoantibody production or immune complex deposition and it rarely recurs, which suggests that it is unlikely to be an autoimmune disease 5. In contrast, large‐vessel vasculitides are considered PAMP (TLR‐ligands)‐triggered T cell‐mediated autoimmune disorders 79, 80, 81. The immunopathogenic process of polyarteritis nodosa (PAN), a medium‐vessel vasculitis, is associated with both innate and acquired immunity, although the exact pathogenic mechanisms remain unknown 82, 83, 84. The small vessel vasculitides (SVVs) comprise antineutrophil cytoplasmic antibody (ANCA)‐associated vasculitis (AAV) and immune complex SVVs. In AAV, ANCAs seem to play an indispensable role in the development of vasculitis by activating primed neutrophils and monocytes, triggering a subsequent inflammatory amplification loop in the vessel wall 85, 86. Among immune complex SVVs, IgA vasculitis is considered to be a predominantly IgA‐mediated immune disorder 73, 79.
The genetic background of KD differs from that of other systemic vasculitic syndromes. GWASs have demonstrated an association between the genes (ITPKC, CASP3 and ORAI1) of the calcium/NFAT pathway and KD 14, 15, 16; however, these genes do not appear to be associated with other vasculitic syndromes 87. Only the variant alleles of FCGR2A 88 have been linked to susceptibility to KD and Takayasu's arteritis 87.
Regarding the pathophysiology of KD, T cell suppression 35, and down‐regulation of T cell receptor and B cell receptor signalling pathways 36, 51, 52 in KD have not been documented in other systemic vasculitides. Furthermore, animal models 65, 69, as well as the clinical and laboratory features (increased serum IL‐1β levels, IL‐1 signalling pathway up‐regulation and anti‐IL‐1β treatment effectiveness) of KD 52, 89, 90, suggest that the inflammasome (a key component of the innate immune system) is associated with the development of KD vasculitis. Inflammasome activation may also be associated with the development of other systemic vasculitides, including autoinflammatory disease‐associated systemic vasculitis and Behçet disease 91, 92. Serum levels of a variety of cytokines are elevated in KD 3 as well as in several other systemic vasculitides 77, 85. In addition, we have identified possible PAMPs in KD sera with high specificity and sensitivity by LC‐MS/MS (Nakashima et al. 2016, manuscript in preparation). Further study is necessary to identify such molecules in other systemic vasculitides linked closely to infections. KD is also associated with innate immune disorders (PFAPA syndrome and systemic JIA) 47, 48, 49; however, no other systemic vasculitides have been linked to these innate immune disorders.
In contrast to the animal models of other systemic vasculitides, our KD animal model has provided new insights regarding the mechanisms underlying the disease and shown that PAMPs associated with innate immunity can cause vasculitis independently of acquired immunity 58, 60. The possible presence of PAMPs and DAMPs, such as S100 proteins and HMGB1, in KD patient sera 43, 44, 45, 46, 70 support the hypothesis that PAMPs/MAMPs, together with DAMPs, activate endothelial and immune cells co‐operatively through innate immune pattern recognition receptors (PRRs), as shown in Fig. 4. Recruitment of immune cells to activated endothelial cells and destruction of vascular structures lead to the development of KD vasculitis and aneurysms. These molecular scenarios may be even more prominent in genetically predisposed individuals. Although vasculitis can occur independently of T cell‐ and B cell‐mediated immunity, acquired immunity is undoubtedly associated with the vasculitides mediated by the innate immune system in humans. Regarding late KD vasculopathy 93 and the premature development of atherosclerosis in patients with a prior history of KD 94, further studies are necessary to elucidate the mechanisms underlying these phenomena. They may result from persistent exposure to small amounts of vasculitis‐inducing molecules produced by endogenous microbes or from acquired immunity‐mediated vascular inflammation 95, 96.
Figure 4.
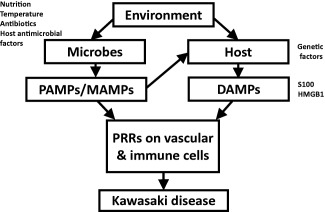
Innate immune‐mediated vasculitis as a pathogenic model for Kawasaki disease (KD). Microbes produce massive amounts of pathogen‐associated molecular patterns /microbe‐associated molecular patterns (PAMPs/MAMPs) under certain biofilm‐like conditions. PAMPs/MAMPs induce damage‐associated molecular pattern (DAMP) [S100 proteins and high mobility group box 1 (HMGB1)] production by and release from host cells. These molecules activate endothelial and immune cells co‐operatively through innate immune pattern recognition receptors (PRRs). Recruitment of immune cells to activated endothelial cells and destruction of vascular structures result in the development of KD vasculitis and aneurysms. These molecular scenarios may be even more prominent in genetically predisposed individuals.
Conclusions
Based on the results of epidemiological, clinical, laboratory and animal studies, we have concluded that KD is not an infectious disease but an innate immune disorder. We propose that KD results from the exposure of a genetically predisposed individual to PAMPs from microbes growing under biofilm‐like conditions, as well as DAMPs.
Disclosure
The authors declare that there are no disclosures.
Acknowledgements
This work was supported by a Grant for Research on Allergic Diseases and Immunology (Elucidation of the pathogenesis and pathophysiology of Kawasaki disease and the development of a novel therapy: Principal Investigator T. H.) from the Japan Agency for Medical Research and Development; a Health and Labour Sciences Research Grant (to T. H.) from the Ministry of Health, Labour and Welfare of Japan; a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to T. H.); and a grant for Kawasaki Disease Research from the Japan Blood Products Organization (to Y. N.).
References
- 1. Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi 1967; 16:178–222. [PubMed] [Google Scholar]
- 2. Kato H, Koike S, Yamamoto M, Ito Y, Yano E. Coronary aneurysms in infants and young children with acute febrile mucocutaneous lymph node syndrome. J Pediatr 1975; 86:892–8. [DOI] [PubMed] [Google Scholar]
- 3. Galeotti C, Bayry J, Kone‐Paut I, Kaveri SV. Kawasaki disease: aetiopathogenesis and therapeutic utility of intravenous immunoglobulin. Autoimmun Rev 2010; 9:441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Newburger JW, Takahashi M, Gerber MA et al Diagnosis, treatment, and long‐term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation 2004; 110:2747–71. [DOI] [PubMed] [Google Scholar]
- 5. Jamieson N, Singh‐Grewal D. Kawasaki disease: a clinician's update. Int J Pediatr 2013; 2013:645391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yashiro M, Makino N, Nakamura Y, Yanagawa H, Kawasaki T. Results of the 23rd nationwide survey on Kawasaki disease – the highest number of patients ever. In: Proceedings of the 35th annual meetings of the Japanese Society of Kawasaki Disease, 2015:52.
- 7. Kim KY, Kim DS. Recent advances in Kawasaki disease. Yonsei Med J 2016; 57:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harada F, Sada M, Kamiya T, Yanase Y, Kawasaki T, Sasazuki T. Genetic analysis of Kawasaki syndrome. Am J Hum Genet 1986; 39:537–9. [PMC free article] [PubMed] [Google Scholar]
- 9. Kottek A, Shimizu C, Burns JC. Kawasaki disease in monozygotic twins. Pediatr Infect Dis J 2011; 30:1114–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Uehara R, Belay ED. Epidemiology of Kawasaki disease in Asia, Europe, and the United States. J Epidemiol 2012; 22:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997–2007. Pediatr Infect Dis J 2010; 29:483–8. [DOI] [PubMed] [Google Scholar]
- 12. Holman RC, Christensen KY, Belay ED et al Racial/ethnic differences in the incidence of Kawasaki syndrome among children in Hawaii. Hawaii Med J 2010; 69:194–7. [PMC free article] [PubMed] [Google Scholar]
- 13. Fujita Y, Nakamura Y, Sakata K et al Kawasaki disease in families. Pediatrics 1989; 84:666–9. [PubMed] [Google Scholar]
- 14. Onouchi Y. Genetics of Kawasaki disease: what we know and don't know. Circ J 2012; 76:1581–6. [DOI] [PubMed] [Google Scholar]
- 15. Onouchi Y, Fukazawa R, Yamamura K et al Variations in ORAI1 gene associated with Kawasaki disease. PLOS ONE 2016; 11: e0145486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Onouchi Y, Gunji T, Burns JC et al ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet 2008; 40:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shimizu C, Jain S, Davila S et al Transforming growth factor‐beta signaling pathway in patients with Kawasaki disease. Circ Cardiovasc Genet 2011; 4:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kariyazono H, Ohno T, Khajoee V et al Association of vascular endothelial growth factor (VEGF) and VEGF receptor gene polymorphisms with coronary artery lesions of Kawasaki disease. Pediatr Res 2004; 56:953–9. [DOI] [PubMed] [Google Scholar]
- 19. Breunis WB, Davila S, Shimizu C et al Disruption of vascular homeostasis in patients with Kawasaki disease: involvement of vascular endothelial growth factor and angiopoietins. Arthritis Rheum 2012; 64:306–15. [DOI] [PubMed] [Google Scholar]
- 20. Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of Kawasaki disease – cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol 2008; 6:394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pinna GS1, Kafetzis DA, Tselkas OI, Skevaki CL. Kawasaki disease: an overview. Curr Opin Infect Dis 2008; 21:263–70. [DOI] [PubMed] [Google Scholar]
- 22. Rowley AH, Baker SC, Shulman ST et al Cytoplasmic inclusion bodies are detected by synthetic antibody in ciliated bronchial epithelium during acute Kawasaki disease. J Infect Dis 2005; 192:1757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sato K, Ouchi K, Taki M. Yersinia pseudotuberculosis infection in children, resembling Izumi fever and Kawasaki syndrome. Pediatr Infect Dis 1983; 2:123–6. [DOI] [PubMed] [Google Scholar]
- 24. Tahara M, Baba K, Waki K, Arakaki Y. Analysis of Kawasaki disease showing elevated antibody titers of Yersinia pseudotuberculosis . Acta Paediatr 2006; 95:1661–4. [DOI] [PubMed] [Google Scholar]
- 25. Vincent P, Salo E, Skurnik M, Fukushima H, Simonet M. Similarities of Kawasaki disease and Yersinia pseudotuberculosis infection epidemiology. Pediatr Infect Dis J 2007; 26:629–31. [DOI] [PubMed] [Google Scholar]
- 26. Rodó X, Ballester J, Cayan D. Association of Kawasaki disease with tropospheric wind patterns. Sci Rep 2011; 1:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodó X, Curcoll R, Robinson M et al Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proc Natl Acad Sci USA 2014; 111:7952–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shulman ST, Rowley AH. Kawasaki disease: insights into pathogenesis and approaches to treatment. Nat Rev Rheumatol 2015; 11:475–82. [DOI] [PubMed] [Google Scholar]
- 29. Abe J, Kotzin BL, Meissner C et al Characterization of T cell repertoire changes in acute Kawasaki disease. J Exp Med 1993; 177:791–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Curtis N, Zheng R, Lamb JR, Levin M. Evidence for a superantigen mediated process in Kawasaki disease. Arch Dis Child 1995; 72:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pietra BA, De Inocencio J, Giannini EH, Hirsch R. TCR V beta family repertoire and T cell activation markers in Kawasaki disease. J Immunol 1994; 153:1881–8. [PubMed] [Google Scholar]
- 32. Aparna MS1, Yadav S. Biofilms: microbes and disease. Braz J Infect Dis 2008; 12:526–30. [DOI] [PubMed] [Google Scholar]
- 33. Schlievert PM, Peterson ML. Glycerol monolaurate antibacterial activity in broth and biofilm cultures. PLOS ONE 2012; 7:e40350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dominguez SR, Friedman K, Seewald R, Anderson MS, Willis L, Glodé MP. Kawasaki disease in a pediatric intensive care unit: a case–control study. Pediatrics 2008; 122:e786–90. [DOI] [PubMed] [Google Scholar]
- 35. Kuijpers TW, Wiegman A, van Lier RAW et al Kawasaki disease: a maturational defect in immune responsiveness. J Infect Dis 1999; 180:1869–77. [DOI] [PubMed] [Google Scholar]
- 36. Ikeda K, Yamaguchi K, Tanaka T et al Unique activation status of peripheral blood mononuclear cells at acute phase of Kawasaki disease. Clin Exp Immunol 2010; 160:246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li Z, Peres AG, Damian AC, Madrenas J. Immunomodulation and disease tolerance to Staphylococcus aureus . Pathogens 2015; 4:793–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002; 20:197–216. [DOI] [PubMed] [Google Scholar]
- 39. Mai J, Virtue A, Shen J, Wang H, Yang X‐F. An evolving new paradigm: endothelial cells – conditional innate immune cells. J Hematol Oncol 2013; 6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Furukawa S, Matsubara T, Yabuta K. Mononuclear cell subsets and coronary artery lesions in Kawasaki disease. Arch Dis Child 1992; 67:706–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takahashi K, Oharaseki T, Yokouchi Y et al Kawasaki disease as a systemic vasculitis in childhood. Ann Vasc Dis 2010; 3:173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sato N, Sagawa K, Sasaguri Y et al Immunopathology and cytokine detection in the skin lesions of patients with Kawasaki disease. J Pediatr 1993; 122:198–203. [DOI] [PubMed] [Google Scholar]
- 43. Foell D, Ichida F, Vogl T et al S100A12 (EN‐RAGE) in monitoring Kawasaki disease. Lancet 2003; 361:1270–2. [DOI] [PubMed] [Google Scholar]
- 44. Ye F, Foell D, Hirono KI et al Neutrophil‐derived S100A12 is profoundly upregulated in the early stage of acute Kawasaki disease. Am J Cardiol 2004; 94:840–4. [DOI] [PubMed] [Google Scholar]
- 45. Ebihara T, Endo R, Kikuta H et al Differential gene expression of S100 protein family in leukocytes from patients with Kawasaki disease. Eur J Pediatr 2006; 164:427–31. [DOI] [PubMed] [Google Scholar]
- 46. Hoshina T, Kusuhara K, Ikeda K et al High mobility group box 1 (HMGB1) and macrophage migration inhibitory factor (MIF) in Kawasaki disease. Scand J Rheumatol 2008; 37:445–9. [DOI] [PubMed] [Google Scholar]
- 47. Broderick L, Tremoulet AH, Burns JC et al Recurrent fever syndromes in patients after recovery from Kawasaki syndrome. Pediatrics 2011; 127:e489–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Komatsu H, Tateno A. Failure to distinguish systemic‐onset juvenile idiopathic arthritis from incomplete Kawasaki disease in an infant. J Paediatr Child Health 2007; 43:707–9. [DOI] [PubMed] [Google Scholar]
- 49. Dong S, Bout‐Tabaku S, Texter K, Jaggi P. Diagnosis of systemic‐onset juvenile idiopathic arthritis after treatment for presumed Kawasaki disease. J Pediatr 2015; 166:1283–8. [DOI] [PubMed] [Google Scholar]
- 50. Hirata S, Nakamura Y, Yanagawa H. Incidence rate of recurrent Kawasaki disease and related risk factors: from the results of nationwide surveys of Kawasaki disease in Japan. Acta Paediatr 2001; 90:40–4. [DOI] [PubMed] [Google Scholar]
- 51. Ling XB, Lau K, Kanegaye JT et al A diagnostic algorithm combining clinical and molecular data distinguishes Kawasaki disease from other febrile illnesses. BMC Medicine 2011; 9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoang LT, Shimizu C, Ling L et al Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med 2014; 6:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Furuno K, Yuge T, Kusuhara K et al CD25+CD4+ regulatory T cells in patients with Kawasaki disease. J Pediatr 2004; 145:385–90. [DOI] [PubMed] [Google Scholar]
- 54. Olivito B, Taddio A, Simonini G et al Defective FOXP3 expression in patients with acute Kawasaki disease and restoration by intravenous immunoglobulin therapy. Clin Exp Rheumatol 2010; 28:93–7. [PubMed] [Google Scholar]
- 55. Jia S, Li C, Wang G, Yang J, Zu Y. The T helper type 17/regulatory T cell imbalance in patients with acute Kawasaki disease. Clin Exp Immunol 2010; 162:131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rasouli M, Heidari B, Kalani M. Downregulation of Th17 cells and the related cytokines with treatment in Kawasaki disease. Immunol Lett 2014; 162:269–75. [DOI] [PubMed] [Google Scholar]
- 57. Guo MM, Tseng WN, Ko CH, Pan HM, Hsieh KS, Kuo HC. Th17‐ and Treg‐related cytokine and mRNA expression are associated with acute and resolving Kawasaki disease. Allergy 2015; 70:310–8. [DOI] [PubMed] [Google Scholar]
- 58. Nishio H, Kanno S, Onoyama S et al NOD1 ligands induce site‐specific vascular inflammation. Arterioscler Thromb Vasc Biol 2011; 31:1093–9. [DOI] [PubMed] [Google Scholar]
- 59. Tukhvatulin AI, Logunov DY, Gitlin II et al An in vitro and in vivo study of the ability of NOD1 ligands to activate the transcriptional factor NF‐kB. Acta Naturae 2011; 3:77–84. [PMC free article] [PubMed] [Google Scholar]
- 60. Motomura Y, Kanno S, Asano K et al Identification of pathogenic cardiac CD11c+ macrophages in NOD1‐mediated acute coronary arteritis. Arterioscler Thromb Vasc Biol 2015; 35:1423–33. [DOI] [PubMed] [Google Scholar]
- 61. Duong TT1, Silverman ED, Bissessar MV, Yeung RS. Superantigenic activity is responsible for induction of coronary arteritis in mice: an animal model of Kawasaki disease. Int Immunol 2003; 15:79–89. [DOI] [PubMed] [Google Scholar]
- 62. Rosenkranz ME, Schulte DJ, Agle LM et al TLR2 and MyD88 contribute to Lactobacillus casei extract‐induced focal coronary arteritis in a mouse model of Kawasaki disease. Circulation 2005; 112:2966–73. [DOI] [PubMed] [Google Scholar]
- 63. Hui‐Yuen JS, Duong TT, Yeung RS. TNF‐alpha is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. J Immunol 2006; 176:6294–301. [DOI] [PubMed] [Google Scholar]
- 64. Lin IC, Suen JL, Huang SK et al Dectin‐1/Syk signaling is involved in Lactobacillus casei cell wall extract‐induced mouse model of Kawasaki disease. Immunobiology 2013; 218:201–12. [DOI] [PubMed] [Google Scholar]
- 65. Lee Y, Wakita D, Dagvadorj J et al IL‐1 signaling is critically required in stromal cells in Kawasaki disease vasculitis mouse model: role of both IL‐1α and IL‐1β. Arterioscler Thromb Vasc Biol 2015; 35:2605–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Murata H, Naoe S. Experimental Candida‐induced arteritis in mice‐relation to arteritis in Kawasaki disease. Prog Clin Biol Res 1987; 250:523. [PubMed] [Google Scholar]
- 67. Nagi‐Miura N, Harada T, Shinohara H et al Lethal and severe coronary arteritis in DBA/2 mice induced by fungal pathogen, CAWS, Candida albicans water‐soluble fraction. Atherosclerosis 2006; 186:310–20. [DOI] [PubMed] [Google Scholar]
- 68. Martinez HG, Quinones MP, Jimenez F et al Important role of CCR2 in a murine model of coronary vasculitis. BMC Immunol 2012; 13:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen Y, Li X, Boini KM et al Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta 2015; 1853:396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Leung DY, Cotran RS, Kurt‐Jones E, Burns JC, Newburger JW, Pober JS. Endothelial cell activation and high interleukin‐1 secretion in the pathogenesis of acute Kawasaki disease. Lancet 1989; 2:1298–302. [DOI] [PubMed] [Google Scholar]
- 71. Kusuda T, Nakashima Y, Murata K et al Kawasaki disease‐specific molecules in the sera are linked to microbe‐associated molecular patterns in the biofilms. PLOS ONE 2014; 9:e113054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Scott DGI, Bacon P. Epidemiology of systemic vasculitis In: Sharma A, ed. Textbook of systemic vasculitis. London: The Health Sciences Publisher, 2015:3–9. [Google Scholar]
- 73. Brogan P, Bagga A. Leukocytoclastic vasculitis: Henoch–Schönlein purpura and hypersensitivity vasculitis In: Petty RE, Laxer RM, Lindsley CB, eds. Textbook of pediatric rheumatology. Philadelphia, PA: Elsevier, 2016:452–61. [Google Scholar]
- 74. Futamura M, Ohya Y, Akashi M et al Age‐related prevalence of allergic diseases in Tokyo schoolchildren. Allergol Int 2011; 60:509–15. [DOI] [PubMed] [Google Scholar]
- 75. Molodecky NA, Soon IS, Doreen M et al Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012; 142:46–54. [DOI] [PubMed] [Google Scholar]
- 76. Lloyd AJ, Walker C, Wilkinso M. Kawasaki disease: is it caused by an infectious agent? Br J Biomed Sci 2001; 58:122–8. [PubMed] [Google Scholar]
- 77. Weiss PF. Pediatric vasculitis. Pediatr Clin North Am 2012; 59:407–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Stojanov S, Lapidus S, Chitkara P et al Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL‐1 blockade. Proc Natl Acad Sci USA 2011; 108:7148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Misra DP, Wakhlu A, Agarwal V. Pathogenesis of vasculitis In: Sharma A, ed. Textbook of systemic vasculitis. London: The Health Sciences Publisher, 2015:40–8. [Google Scholar]
- 80. Pryshchep O, Ma‐Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel‐specific Toll‐like receptor profiles in human medium and large arteries. Circulation 2008; 118:1276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Deng J, Ma‐Krupa W, Gewirtz AT, Younge BR, Goronzy JJ, Weyand CM. Toll‐like receptors 4 and 5 induce distinct types of vasculitis. Circ Res 2009; 104:488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dillon MJ, Eleftheriou D, Brogan PA. Medium‐size‐vessel vasculitis. Pediatr Nephrol 2010; 25:1641–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Prajs K, Bobrowska‐Snarska D, Skała M, Brzosko M. Polyarteritis nodosa and Sjögren's syndrome: overlap syndrome. Rheumatol Int 2012; 32:4019–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Eleftheriou D, Ozen S. Polyarteritis nodosa In: Petty RE, Laxer RM, Lindsley CB, eds. Textbook of pediatric rheumatology. Philadelphia, PA: Elsevier, 2016:462–6. [Google Scholar]
- 85. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody‐mediated disease. Nat Rev Rheumatol 2014; 10:463–73. [DOI] [PubMed] [Google Scholar]
- 86. Cabral DA, Morishita K. Antineutrophil cytoplasmic antibody associated vasculitis In: Petty RE, Laxer RM, Lindsley CB, eds. Textbook of pediatric rheumatology. Philadelphia, PA: Elsevier, 2016:462–6. [Google Scholar]
- 87. Mathew J, Ramya J. Genetics of vasculitis In: Sharma A, ed. Textbook of systemic vasculitis. London: The Health Sciences Publisher, 2015:10–5. [Google Scholar]
- 88. Khor CC, Davila S, Breunis WB et al Genome‐wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet 2011; 43:1241–6. [DOI] [PubMed] [Google Scholar]
- 89. Maury CP, Salo E, Pelkonen P. Circulating interleukin‐1 beta in patients with Kawasaki disease. N Engl J Med 1988; 319:1670–1. [DOI] [PubMed] [Google Scholar]
- 90. Campbell AJ, Burns JC. Adjunctive therapies for Kawasaki disease. J Infect 2016; 72 Suppl:S1–5. [DOI] [PubMed] [Google Scholar]
- 91. Ramirez GA, Maugeri N, Sabbadini MG, Rovere‐Querini P, Manfredi AA. Intravascular immunity as a key to systemic vasculitis: a work in progress, gaining momentum. Clin Exp Immunol 2014; 175:150–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kim EH, Park MJ, Park S, Lee ES. Increased expression of the NLRP3 inflammasome components in patients with Behçet's disease. J Inflamm (Lond) 2015; 12:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shah V, Christov G, Mukasa T et al Cardiovascular status after Kawasaki disease in the UK. Heart 2015; 0:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Noto N, Okada T. Premature atherosclerosis long after Kawasaki disease: In: Atiq M, ed. Recent advances in cardiovascular risk factors. Shanghai: InTech, 2012. ISBN: 978‐953‐51‐0321‐9. Available at: http://www.intechopen.com/books/recent-advances-in-cardiovascular-risk-factors/prematureatherosclerosis-long-after-kawasaki-disease. Last accessed: June 4, 2016. [Google Scholar]
- 95. Chen S, Lee Y Crother TR, et al Marked acceleration of atherosclerosis after Lactobacillus casei‐induced coronary arteritis in a mouse model of Kawasaki disease. Arterioscler Thromb Vasc Biol 2012; 32:e60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kanno S, Nishio H, Tanaka T et al Activation of an innate immune receptor, Nod1, accelerates atherogenesis in Apoe–/– mice. J Immunol 2015; 194:773–80. [DOI] [PubMed] [Google Scholar]