

ABSTRACT
Leber congenital amaurosis (LCA) is a severe autosomal‐recessive retinal dystrophy leading to congenital blindness. A recently identified LCA gene is NMNAT1, located in the LCA9 locus. Although most mutations in blindness genes are coding variations, there is accumulating evidence for hidden noncoding defects or structural variations (SVs). The starting point of this study was an LCA9‐associated consanguineous family in which no coding mutations were found in the LCA9 region. Exploring the untranslated regions of NMNAT1 revealed a novel homozygous 5′UTR variant, c.‐70A>T. Moreover, an adjacent 5′UTR variant, c.‐69C>T, was identified in a second consanguineous family displaying a similar phenotype. Both 5′UTR variants resulted in decreased NMNAT1 mRNA abundance in patients’ lymphocytes, and caused decreased luciferase activity in human retinal pigment epithelial RPE‐1 cells. Second, we unraveled pseudohomozygosity of a coding NMNAT1 mutation in two unrelated LCA patients by the identification of two distinct heterozygous partial NMNAT1 deletions. Molecular characterization of the breakpoint junctions revealed a complex Alu‐rich genomic architecture. Our study uncovered hidden genetic variation in NMNAT1‐associated LCA and emphasized a shift from coding to noncoding regulatory mutations and repeat‐mediated SVs in the molecular pathogenesis of heterogeneous recessive disorders such as hereditary blindness.
Keywords: Leber congenital amaurosis, LCA9, NMNAT1, 5′UTR variants, Alu‐mediated deletions
Introduction
Leber congenital amaurosis (LCA; MIM #204000) is the earliest and most severe inherited retinal dystrophy, causing profound visual deficiency, nystagmus, and an undetectable or severely reduced electroretinogram (ERG) in the 1st year of life [Leber, 1869; Franceschetti and Dieterle, 1954]. The disease may develop as a cone‐rod (type I) or rod‐cone dystrophy (type II), with associated features further impairing visual function [Hanein et al., 2004]. So far, 19 disease genes have been associated with autosomal‐recessive LCA (RetNet). The encoded proteins function in retina‐specific as well as general processes [den Hollander et al., 2008]. In recent years, genetic causes have been found in up to 70% of cases, facilitated by massively parallel sequencing (MPS) technologies such as targeted resequencing of linkage intervals or whole‐exome sequencing (WES) [Neveling et al., 2013]. Although most mutations underlying LCA are located in coding regions, there is accumulating evidence for genetic defects in noncoding regions such as deep intronic mutations or regulatory mutations in enhancers, promoters, or untranslated regions (UTRs). A striking example is a deep intronic mutation of CEP290, accounting for 15% of congenital blindness in Europe [Coppieters et al., 2010].
In 2012, four groups independently identified the gene encoding nicotinamide nucleotide adenylyltransferase 1 (NMNAT1; MIM #608700) as a new LCA disease gene through WES [Chiang et al., 2012; Falk et al., 2012; Koenekoop et al., 2012; Perrault et al., 2012]. Interestingly, all studies reported severe phenotypes with macular coloboma or atrophic macular lesions in almost all cases, representing one of the strongest genotype–phenotype correlations ever observed in LCA. Subsequent sequencing of the NMNAT1 coding region in 740 additional prescreened LCA cases revealed homozygous or compound heterozygous mutations in 45 probands (6.1%) from different origins [Chiang et al., 2012; Falk et al., 2012; Koenekoop et al., 2012; Perrault et al., 2012]. The majority of NMNAT1 mutations causing LCA are missense changes, predicted to alter protein structure and/or abolish function based on the NMNAT1 crystal structure [Zhou et al., 2002], and in vivo as well as in vitro enzymatic activity assays [Chiang et al., 2012; Falk et al., 2012; Koenekoop et al., 2012; Perrault et al., 2012; Siemiatkowska et al., 2014]. Until now, only five nonsense, three frameshift, and one splice‐site mutation have been identified. In addition, the segregation of a read‐through mutation in the original LCA9 family sustained the elucidation of the last remaining LCA locus [Keen et al., 2003; Koenekoop et al., 2012]. Thus far, only coding NMNAT1 mutations have been reported.
NMNAT1, which is localized to the nucleus, catalyzes NAD+ synthesis in the last step of a salvage synthesis pathway that recycles nicotinamide back to NAD+. Both a fusion protein (Ube4b/Nmnat1) from a spontaneous mouse model [Coleman and Freeman, 2010] and NMNAT1 overexpression protect axons from degeneration [Zhu et al., 2013; Fang et al., 2014], opening therapeutic perspectives for retinal neurodegeneration.
Here, we aimed to identify the hidden genetic defect in a LCA9‐associated family. The absence of a NMNAT1 coding mutation prompted us to explore the UTRs, revealing a novel 5′UTR variant, c.‐70A>T. Subsequently, a second adjacent 5′UTR variant, c.‐69C>T, was identified in an unrelated family with LCA and macular involvement. Functional studies of both 5′UTR variants suggest a loss‐of‐function effect. Second, we aimed at unraveling the pseudohomozygosity of a coding NMNAT1 mutation in two unrelated patients with a similar form of LCA, leading to the identification of two distinct heterozygous partial deletions of NMNAT1. Our study uncovers, for the first time, hidden genetic variation and a complex molecular pathogenesis of NMNAT1‐associated LCA and is exemplary for congenital blindness and for other heterogeneous autosomal‐recessive diseases in general.
Materials and Methods
Ethics Statement
This study was conducted following the tenets of the Declaration of Helsinki and ethical approval was given by the local ethics committee (Ghent University Hospital).
Patients
This study included 101 consenting subjects initially diagnosed with LCA (74) or early‐onset retinal dystrophy (EORD) (27) and referred for molecular testing by an ophthalmologist and/or geneticist. All affected individuals were subjected to standard ophthalmologic evaluation, among which visual acuity measurement, fundoscopy, ERG, infrared, visual field assessment, and autofluorescence imaging.
Genomic DNA (gDNA) was extracted from leukocytes using the Gentra Puregene Blood Kit (QIAGEN Benelux BV, Antwerp, Belgium), whereas RNA was extracted from leukocytes using the RNeasy Mini kit and the RNase‐Free DNase set (Qiagen), followed by cDNA synthesis (iScript cDNA Synthesis kit; Bio‐Rad Laboratories NV, Temse, Belgium). A selection of lymphocyte cultures were treated with puromycin to block nonsense‐mediated decay. If available, parents and/or siblings were also genotyped. Most probands previously underwent at least LCA chip analysis, testing 344 to 780 mutations in eight to 15 LCA and EORD genes (LCA chip Versions 2004–2012; Asper Ophthalmics, Tartu, Estonia, Estonia) (http://www.asperbio.com).
Identity‐by‐Descent Mapping in F1
At the onset of this study, gDNA was available of two affected cases (IV:1 and IV:2) of F1, originating from Niger. These individuals underwent genome‐wide identity‐by‐descent (IBD) mapping using Affymetrix GeneChip Human Mapping 250K Nsp arrays according to manufacturer protocols (DNAVision, Charleroi, Belgium). Single‐nucleotide polymorphism (SNP) genotypes were analyzed with PLINK [Purcell et al., 2007]. A healthy sibling (IV:3) was born later and underwent SNP genotyping later in the course of this study.
MPS in F1
The four largest IBD regions common between both oldest affected siblings of F1 (IV:1 and IV:2) were selected for MPS using the NimbleGen Sequence Capture 385K array. The array design was based on the RefSeq (NCBI), AceView (NCBI), and RNAgene (UCSC) resources and included all coding exons, 50 bp upstream intronic sequence, 20 bp downstream intronic sequence, UTRs (RefSeq), and 500 bp upstream genomic region (putative promoter, RefSeq) (NCBI36/hg18). Two samples enriched with this method, one of which was the proband of F1, were sequenced on a Roche, Vilvoorde, Belgium GS FLX Titanium run. The CLC Genomics Workbench (version 6.5; CLC bio, Aarhus, Denmark) was employed for read mapping against the human genome reference sequence (NCBI, GRCh37.p5), postmapping duplicate read removal, coverage analysis, and quality‐based variant detection.
NMNAT1 Sequencing on gDNA and cDNA Level in F1 and F2
All probands underwent gDNA sequencing of exon 1, all coding regions and intron–exon boundaries of NMNAT1. cDNA sequencing was carried out using intron‐spanning amplicons in all individuals of F1 and F2. PCR primers were designed with Primer3Plus [Untergasser et al., 2007]. Sanger sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, ThermoFisher Scientific, Waltham, MA) on a 3730xl DNA Analyzer (Applied Biosystems).
Evaluation of Sequence Changes
Mutation nomenclature uses numbering with the A of the initiation codon ATG as +1 (NM_022787.3) (http://www.hgvs.org/mutnomen). All variants and individuals have been submitted to the following locus‐specific database: http://www.lovd.nl/NMNAT1. In silico evaluation of novel NMNAT1 variants was done with the Alamut mutation interpretation software v.2.3 (Interactive Biosoftware, Rouen, France). Segregation analysis of disease alleles was performed if possible, whereas gDNA obtained from unrelated healthy individuals of Belgian origin (>173) was used as a control panel.
NMNAT1 and LZIC mRNA Quantification
Intron‐spanning primer pairs for NMNAT1 and LZIC mRNA quantification were designed using PrimerXL (http://www.primerxl.org). qPCR‐based mRNA quantification was performed as previously described [Derveaux et al., 2010]. At least two reference genes were used for data normalization. Data analysis was performed with qbase+ software (Biogazelle, Ghent, Belgium). P‐values were calculated in qbase+ using a two‐sided two‐sample t‐test with unequal variances. Six healthy controls were included in which no mutations were identified following Sanger sequencing of exon 1, all coding regions, and intron–exon boundaries of NMNAT1.
Bisulfite Sequencing
Bisulfite conversion of DNA was performed using the EZ DNA Methylation kit and the Human Methylated and Non‐Methylated DNA Set (Zymo Research, Freiburg, Germany), followed by Sanger sequencing and analysis in SeqScape v2.5 (Applied Biosystems).
Luciferase Assays
Three different luciferase constructs were used: one for the NMNAT1 promoter and two for the NMNAT1 5′UTR. For the promoter construct, we used the commercially available NMNAT1 pLightSwitch_Prom reporter vector, which was mutated to generate the c.‐70A>T (F1) and c.‐69C>T (F2) mutant constructs (SwitchGear Genomics, La Hulpe, Belgium). For the 5′UTR constructs, wild‐type and mutant DNA oligos (Integrated DNA Technologies, BVBA, Leuven, Belgium) were cloned into the pGL3 reporter vectors pGL3‐prom and pGL3‐basic (Promega Benelux BV, Leiden, The Netherlands), the latter containing a minimal CMV promoter sequence.
RPE‐1 (human retinal pigment epithelial) cells were used to perform functional analysis. They were grown in DMEM‐F12 medium, supplemented with 10% FBS and 1% penicillin/streptomycin (ThermoFisher Scientific, Waltham, MA Gibco, ThermoFisher Scientific, Waltham, MA). RPE‐1 cells were seeded 16 hr prior to transfection at a density of 4.104 cells/cm2. Each experiment was performed in six replicates in 96‐well culture plates. Cells were transfected with 100 ng of DNA per well using fuGENE‐HD (SwitchGear Genomics). Forty‐eight hours after transfection, cells were washed with PBS before lysis and firefly luciferase and renilla luciferase activities were assessed by the Dual‐Luciferase Reporter Assay System (Promega). Luciferase results are reported as relative light units (RLU, i.e., the ratio of the Renilla luciferase and firefly luciferase activities). Each value is the mean of six independent experiments, and standard error bar represent the standard error of the mean.
Copy‐Number Variation Analysis
For each of the five exons, a qPCR amplicon was designed (http://www.primerxl.org). Identified deletions were further delineated at the 5′ and 3′ end using additional qPCR amplicons. qPCR‐based copy‐number variation (CNV) analysis was performed as previously described, using two reference genes for normalization of the relative quantities [D'Haene et al., 2010]. Data analysis was performed with qbase+ software (Biogazelle). The junction products were amplified with the iProof High‐Fidelity DNA Polymerase (Bio‐Rad), followed by Sanger sequencing using internal sequencing primers.
Bioinformatic Analysis of the Deletion Junctions
Bioinformatic analysis of the deletions junctions was performed as previously described [Verdin et al., 2013].
In short, known repetitive elements intersecting the breakpoints were identified using the RepeatMasker track in the UCSC genome browser, followed by BLAST2 (NCBI) analysis to determine the percentage of sequence identity between the different elements. The presence of DNA sequences leading to non‐B DNA conformations in the breakpoints was examined using the following tools: RepeatAround [Goios et al., 2006], QGRS Mapper [Kikin et al., 2006], and nBMST [Cer et al., 2012]. Finally, the presence of 40 previously described sequence motifs was analyzed with Fuzznuc [Rice et al., 2000; Abeysinghe et al., 2003].
Results
First NMNAT1 5′UTR Mutation in LCA9‐Linked Family
The starting point of this study was a consanguineous family from Niger (F1) in which all affected individuals suffered from severe LCA with progressive macular involvement. Genome‐wide IBD mapping using SNP arrays in the two oldest affected individuals of F1, IV:1 and IV:2, revealed four common regions larger than 3 Mb (Supp. Table S1). Two of these could be partially excluded later when the healthy sibling (IV:3) was born, as the latter child was homozygous for the same haplotype. Three regions remained in total, together harboring 22.9 Mb and 321 genes. Interestingly, the largest region contained the LCA9 locus (Supp. Fig. S1) [Keen et al., 2003].
MPS of the IBD regions was performed at the time when only IV:1 and IV:2 were available, and included capturing and resequencing of all coding exons, intron–exon boundaries, UTRs, and putative promoters of the four common IBD regions (Supp. Table S1). Of the 1.6 Mb target region, 87.5% was covered by the NimbleGen Sequence Capture 385K platform design considering an offset of 100 bp. MPS generated 539,559 reads, of which 463,208 (86%) unique reads were aligned against the human genome reference sequence (NCBI, GRCh37.p5), resulting in a minimal coverage of 10x and 20x for 93% and 85%, respectively, of the target regions. In total, 6,153 variants were detected with a coverage and variant allele frequency equal to or above 5x and 70%, respectively. Variants were prioritized based on their predicted effect on the protein (nonsense/frameshift > missense > silent) or splicing, and subsequently subjected to segregation analysis in the family and control samples. However, this approach did not allow the identification of the causal defect.
Following the identification of NMNAT1 as the causal disease gene for the LCA9 locus [Koenekoop et al., 2012], MPS data were revisited and subjected to an in‐depth analysis of the five exons of NMNAT1, including the UTRs. Supp. Table S2 lists all NMNAT1 variants identified in the proband of F1 through MPS. Of the nine variants, four were located in an intron, one upstream of the gene, one in the 5′UTR and three in the 3′UTR. Two variants were novel, with only one predicted to affect splicing, namely, c.‐70A>T (Supp. Fig. S2). This variant is located in exon 1, which is part of the 5′UTR, and segregates with the disease in the family (Fig. 1).
Figure 1.
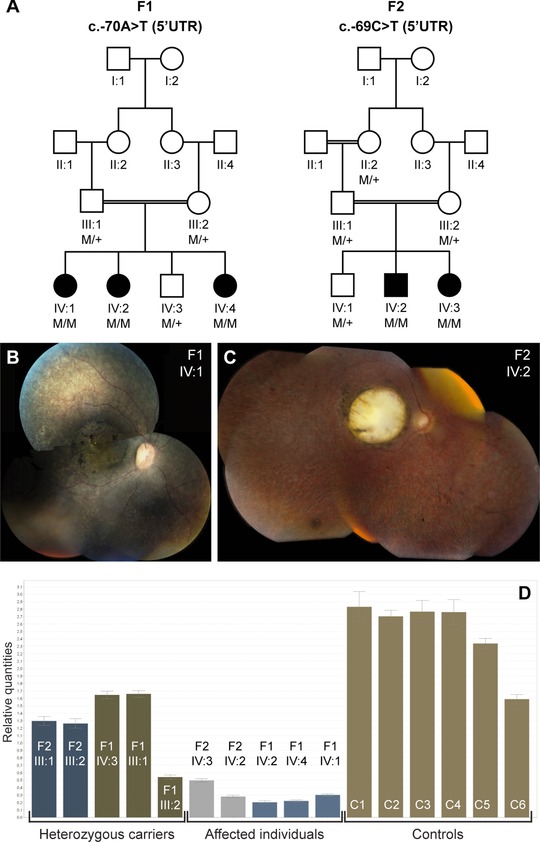
Segregation analysis, associated phenotype, and mRNA quantification of the NMNAT1 5′UTR mutations. A: Segregation analysis of the c.‐70A>T mutation in F1 and the c.‐69C>T mutation in F2. B: Fundus picture of right eye of female patient IV:1 (F1) at age 7 years; note area of atrophy of outer retinal layers with some intraretinal pigment migration and highlighted luteal pigment in macula; mottled aspect of peripheral retinal pigment epithelium illustrates disease in outer retinal layers alternating with anatomically better preserved spots. C: Fundus picture of right eye of male patient IV:2 (F2) at age 21 years; note large area of total retinal and choroidal atrophy in macular area; this area was smaller with atrophy limited only to outer retinal layers but not choroid at age 7 years (data not shown); also note grayish area of outer retinal atrophy in retinal periphery, with limited intraretinal pigment migration. D: qPCR quantification of NMNAT1 mRNA abundance on lymphocyte cDNA of all available family members of F1 and F2, and six healthy controls. The abundance of NMNAT1 mRNA was significantly lower in the affected individuals of F1 in comparison with the controls (P = 5,473E‐5). No significant difference was observed however in the affected individuals of F2 in comparison with controls (P = 0.072) (unpaired t‐test). Abbreviations used: C, control; F, family; M, mutant allele, +, wild‐type allele; UTR, untranslated region.
Second Adjacent NMNAT1 5′UTR Mutation in Unrelated Family
Sanger sequencing of the 5′UTR, all coding regions and intron–exon boundaries of NMNAT1 in 98 additional LCA (71) and EORD (27) probands revealed potential pathogenic coding variants in five LCA families (Supp. Table S3). Biallelic coding mutations were found in F5, F6, and F7. In the probands of F8 and F9, however, only a single heterozygous variant was identified. Unfortunately, no phenotype data are available for these two patients. In both individuals, Sanger sequencing of the 5′UTR did not reveal a second mutation. For F8, no DNA was available anymore to perform CNV analysis. In F9, qPCR analysis did not reveal a CNV, pointing to other mutation mechanisms in NMNAT1 or to the involvement of another retinal disease gene.
Interestingly, a homozygous 5′UTR variant, c.‐69C>T, was found in F2, an unrelated consanguineous family from Morocco (Table 1). This variant is located one nucleotide downstream of c.‐70A>T; however, unlike c.‐70A>T, it is not predicted to affect splicing (data not shown). The c.‐69C>T variant was the only NMNAT1 variant identified in the proband of F2 and segregates with disease. Similar to F1, the affected individuals of F2 display LCA with progressive macular involvement, which is in agreement with the typical NMNAT1‐associated phenotype (Fig. 1).
Table 1.
NMNAT1 5′UTR Mutations and Copy‐Number Variations Explaining Hidden Genetic Variation in This Study
| Allele 1 | Allele 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Family | Exon | cDNA | Protein | Reference | Exon/intron | cDNA | Protein | Reference |
| F1 | E1 (5′UTR) | c.‐70A>T | p.? | Novel | E1 (5′UTR) | c.‐70A>T | p.? | Novel |
| F2 | E1 (5′UTR) | c.‐69C>T | p.? | Novel | E1 (5′UTR) | c.‐69C>T | p.? | Novel |
| F3 | E5 | c.709C>T | p.Arg237Cys | Falk et al. (2012); Perrault et al. (2012) | NMNAT1 partial deletion (g.10039763_10056271del) (hg19) | Novel | ||
| F4 | E5 | c.709C>T | p.Arg237Cys | Falk et al. (2012) Perrault et al. (2012) | NMNAT1 partial deletion (g.10038285_10043034del) (hg19) | Novel | ||
Abbreviations used: F, family; E, exon; UTR, untranslated region.
Functional Assessment of c.‐70A>T and c.‐69C>T
NMNAT1 cDNA sequencing and quantification
Subsequent Sanger sequencing was performed on lymphocyte‐derived cDNA from all available family members of F1 and F2. As c.‐70A>T was predicted to affect splicing, lymphocyte cultures of F1 were treated with and without puromycine, a protein synthesis inhibitor used to prevent nonsense‐mediated mRNA decay. No splicing defects were identified. However, haplotype analysis at the cDNA level of three NMNAT1 variants previously found at the gDNA level revealed allele‐specific expression in the parents and the healthy sib of F1 (Supp. Fig. S3). This result was obtained in cells treated with and without puromycine, suggesting that the genetic defect has an effect prior to translation. In F2, cDNA was only available from untreated lymphocyte cultures. As in F1, cDNA sequencing showed allele‐specific expression in both parents. In both families, superposition of low sequence traces could be seen, representing residual mutant mRNA (Supp. Fig. S3).
In order to quantify NMNAT1 mRNA abundance, qPCR analysis was performed on cDNA of all available family members of F1 and F2, and six healthy controls. This revealed a lower mRNA abundance in the affected, homozygous individuals in comparison with the healthy controls (Fig. 1D).
LZIC mRNA quantification in F1
The NMNAT1 gene is transcribed from the forward DNA strand. NMNAT1 mutations, c.‐70A>T and c.‐69C>T, are located 133 and 134 bp downstream of the 5′UTR of the LZIC gene (UCSC genes), which is transcribed from the negative DNA strand and thus located in a head‐to‐head orientation with NMNAT1 (Fig. 2B). The LZIC protein is required for neuronal survival in zebrafish [Clements and Kimelman, 2005]. Therefore, an effect on LZIC mRNA abundance was assessed using qPCR analysis, which revealed no difference in mRNA abundance between the affected individuals of F1 and six healthy control individuals (Supp. Fig. S4).
Figure 2.
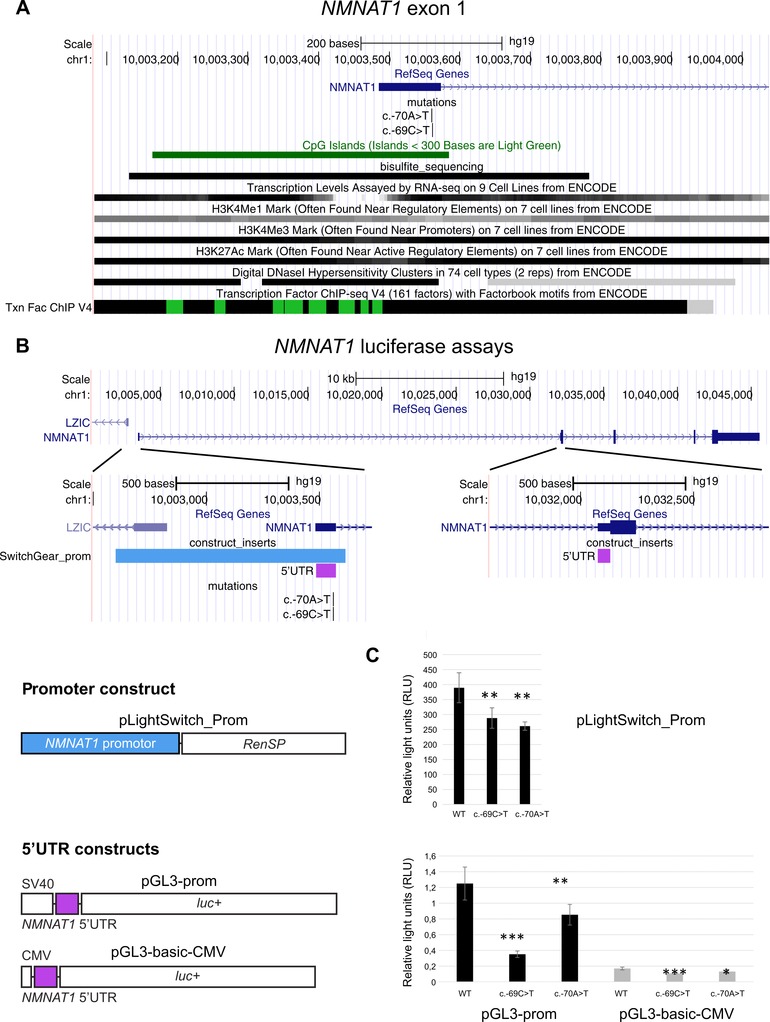
Location and functional analysis of the NMNAT1 5′UTR mutations, c.‐70A>T and c.‐69C>T. A: Overview of a selection of the ENCODE Integrated Regulation tracks aligning with exon 1. The bisulfite_sequencing custom track shows the region covered by the bisulfite sequencing performed in F1. B: Overview of the NMNAT1 gene locus and luciferase reporter constructs. The NMNAT1 5′UTR consists of exon 1 and part of exon 2, which are separated from each other by a large intron (28 kb). The promoter construct contains the NMNAT1 promoter upstream of a Renilla luciferase gene. The 5′UTR constructs contain an SV40/CMV promoter, followed by the NMNAT1 5′UTR and a firefly luciferase gene. C: Results luciferase assays. Upper panel: transcriptional activities of WT and mutated NMNAT1 promoter. Luciferase assays were performed in RPE‐1 cells. Relative lights units (RLU) correspond to the ratio of the activity of the Renilla reporter (RenSP) over that of the firefly luciferase reporter (internal control of transfection efficiency, pGL3‐CMV‐empty). Lower panel: transcriptional activities of WT and mutated NMNAT1 5′UTR. RLU correspond to the ratio of the activity of the firefly luciferase reporter over that of the Renilla reporter (internal control of transfection efficiency, pRL‐RSV). Statistical significance in Student's t‐tests: n.s., P.0.05; *: P,0.05; **, P,0.01, ***, P,0.001.
In silico prediction of the effects of the 5′UTR mutations
Both 5′UTR mutations are located in exon 1, which is part of a transcription unit of NMNAT1 according to several ENCODE‐integrated regulation tracks. In addition, both mutations are located in a CpG island encompassing exon 1 (Fig. 2A). 5′UTRs mutations can cause aberrant mRNA instability or translational regulation through different mechanisms. We performed an in silico evaluation of the mutated and wild‐type sequences that revealed a potential loss of several transcription factor binding sites (Supp. Table S4), potential disruption of secondary structures (Supp. Fig. S5), or location in G‐quadruplexes (Supp. Table S5).
Bisulfite sequencing
To determine whether the 5′UTR variants might affect methylation of the CpG island, we performed Sanger sequencing of bisulfite‐converted gDNA of all family members of F1, three control individuals, and one methylated and one nonmethylated control sample (Fig. 2A). No difference was seen in the methylation status of F1 in comparison with the healthy controls (data not shown). In all individuals, each evaluated CpG nucleotide was unmethylated.
Luciferase assays
Figure 2B shows an overview of the NMNAT1 genomic locus. The NMNAT1 gene contains five exons. The 5′UTR encompasses exon 1 and part of exon 2. The first two exons are separated by a large intron of 28 kb. To study the potential effects of the mutations on the regulation of NMNAT1 expression or on the stability/translatability of its transcripts, we created three different luciferase constructs: one for the putative NMNAT1 promoter and two for the NMNAT1 5′UTR. The promoter construct contained 1,020 bp of sequence harboring exon 1 and an upstream region, followed by the Renilla reporter gene RenSP (SwitchGear Genomics). In the two different 5′UTR constructs, the 5′UTR, consisting of exon 1 and part of exon 2, was cloned downstream of either an SV40 or CMV promoter, followed by the firefly luciferase gene (pGL3‐prom and pGL3‐basic reporter vectors; Promega) (Fig. 2B).
Figure 2C shows the expression of these constructs in human retinal pigment epithelial RPE‐1 cells. First, we assessed the activity of the putative NMNAT1 promoter. The two mutated forms show a decrease in luciferase activity (25% for c.‐69>T and 33% for c.‐70A>T), suggesting that a single mutation is able to reduce the promoter activity. Second, the WT or mutated 5′UTR were cloned as described above with a strong promoter (SV40 or CMV). Again, in both constructs, there is a significant decrease in luciferase activity with the two mutated forms, c.‐69C>T and c.‐70A>T, relative to the wild type. With pGL3‐prom, containing a SV40 promoter, luciferase activity is reduced with 72% for c.‐69C>T and 32 % for c.‐70A>T. With pGL3‐CMV, luciferase activity is reduced with 36% for c.‐69C>T and 22 % for c.‐70A>T, respectively.
Structural Variations of NMNAT1
In two Japanese families (F3 and F4), pseudohomozygosity for the same coding NMNAT1 mutation c.709C>T p.(Arg237Cys) [Falk et al., 2012; Perrault et al., 2012] was demonstrated [Suzuki et al., 2014], suggesting hemizygosity due to a heterozygous deletion (Table 1; Supp. Fig. S6A). Both probands showed LCA with macular degeneration in both eyes at the time of diagnosis (age 3 months). In the proband of F4, the macular degeneration was progressed at the age of 4 months (Supp. Table S6).
Subsequently, CNV screening was performed using five qPCR assays, one for each exon. This revealed a heterozygous deletion of the amplicons for exon 4 and 5 in the proband of F3 and of the amplicon for exon 4 in the proband of F4 (Supp. Fig. S6B). Figure 3 shows the delineation of both deletions using PCR walking with additional qPCR amplicons. Subsequently, the junction product was generated using long‐range PCR and underwent Sanger sequencing, revealing partial gene deletions of 16.5 kb (F3) and 4.8 kb (F4), respectively. Both CNVs segregated with disease in the families (Supp. Fig. S6C).
Figure 3.
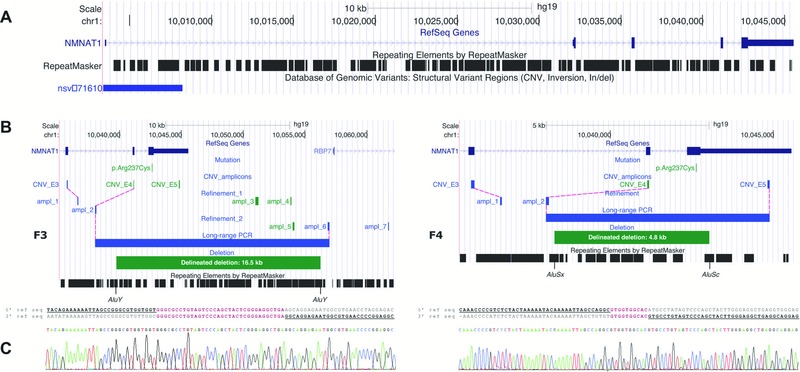
Molecular characterization of two distinct heterozygous NMNAT1 deletions. A: Overview of the NMNAT1 genomic locus, which is scattered with Alu repeats. The Database of Genomic Variants contains one duplication. B: Refinement and delineation of both heterozygous deletions at the nucleotide level. Deleted amplicons are indicated in green; amplicons with a normal copy number are indicated in blue. In F3, two subsequent refinement rounds with seven additional qPCR amplicons in total (ampl_1–ampl_7) were needed to refine the deletion at both ends to a region of 13.0–18.7 kb. Subsequent long‐range PCR and Sanger sequencing revealed a deletion of 16.5 kb. In F4, refinement at the 5′ end (ampl_1 and ampl_2) delineated the deletion to a region of 1.5–6.7 kb. At the 3′ end, the deletion breakpoint appeared to be located between the p.Arg237Cys mutation and the forward primer of the amplicon CNV_E5 (3′ UTR), as this amplicon was not deleted according to qPCR results (Supp. Fig. S6B). Subsequent long‐range PCR and Sanger sequencing revealed a deletion of 4.8 kb. For both deletions, the breakpoints regions are located in Alu repeats. C: Alignment of the junction product with the 5′ and 3′ reference sequence revealed microhomology regions of 34 and 10 bp for F3 and F4, respectively.
Bioinformatics Study of the Breakpoint Junctions
The breakpoints of the partial NMNAT1 deletions were subjected to an extensive bioinformatic analysis to explore the local genomic architecture and potential underlying mechanisms (Supp. Table S7). The deletion junctions displayed microhomology of 34 bp (F3) and 10 bp (F4) between their breakpoints (Fig. 3C). Based on these observations, microhomology mediated break‐induced replication is a potential underlying mechanism. Interestingly, Alu elements were observed at all breakpoints, consisting of AluY‐AluY (F3) and AluSx‐AluSc (F4) combinations. Blast2 analysis showed a sequence identity of 92% (F3) and 88% (F4) between the combined Alu repeats. Because of this similarity of the Alu elements involved and the relatively short distance separating them, single‐strand annealing is also possible, although this is a minor mechanism in the formation of structural variations (SVs) [Hastings et al., 2009].
Discussion
NMNAT1 is one of the most recently identified LCA genes (LCA9). Thus far, eight studies listed 45 distinct mutations, all located in the coding region or intron–exon boundaries of NMNAT1 [Chiang et al., 2012; Falk et al., 2012; Koenekoop et al., 2012; Perrault et al., 2012; Corton et al., 2013; Coppieters et al., 2014; Jin et al., 2014; Siemiatkowska et al., 2014]. Here, we demonstrate for the first time that both regulatory 5′UTR variations and SVs of NMNAT1 add to the complex molecular pathogenesis of LCA.
The pathogenicity of the 5′UTR mutations, c.‐70A>T and c.‐69C>T, identified in two unrelated families with a typical NMNAT1‐associated phenotype, has been substantiated by several pieces of evidence: (1) segregation analysis, (2) allele‐specific expression at the cDNA level, (3) decreased mRNA abundance in patients’ lymphocytes compared with healthy controls, and (4) decreased luciferase activity of mutated constructs in retinal RPE‐1 cells compared with wild‐type constructs (Figs. 1 and 2; Supp. Fig. S3). These results suggest a loss‐of‐function effect of both 5′UTR mutations, which is in line with the known NMNAT1 mutation spectrum. Apart from a few nonsense and frameshift mutations, most mutations are missense changes. For a selection of these, in vivo and/or in vitro functional assays were performed, demonstrating reduced enzymatic activity [Falk et al., 2012; Koenekoop et al., 2012]. In addition, we excluded an effect on expression of a neighboring gene, LZIC (Supp. Fig. S4), and methylation changes of an overlapping CpG island. The exact molecular consequences of both 5′UTR mutations on mRNA stability or translation efficiency remain to be studied. As both mutations are adjacent, an effect on secondary structure formation or disruption of a motif representing a DNA/RNA protein‐binding site seems plausible (Supp. Tables S4 and S5; Supp. Fig. S5).
Interestingly, the occurrence of cis‐acting variants affecting NMNAT1 expression levels has recently been put forward in another context. Based on the high population frequency of p.Glu257Lys, the most common NMNAT1 mutation, and nonpenetrance of this mutation in a family with intellectual disability, a recent study hypothesizes that p.Glu257Lys is a hypomorphic variant only causing LCA in combination with more severe NMNAT1 mutations, or in combination with trans‐ or cis‐acting variants resulting in reduced transcription. Moreover, two different haplotypes in NMNAT1 homozygotes for p.Glu257Lys suggest the presence of cis‐acting factors influencing expression [Siemiatkowska et al., 2014].
Thus far, only 40%–70% of the retinal dystrophy cases can be attributed to mutations in the known disease genes. Until now, it was presumed that the remaining cases were caused by mutations in novel disease genes. The widely applied high‐throughput sequencing technologies did not yield the expected number of novel, frequently mutated genes, however. This underscores the importance of a much larger number than expected of less prevalent disease genes, or, more likely, of hidden mutations in already known disease genes. These mutations might be balanced or unbalanced SVs, deep intronic mutations, silent variants affecting splicing, or regulatory mutations in promoters, UTRs, and enhancers. Thus far, only a handful of mutations affecting regulatory regions have been identified in retinal dystrophies. A few examples are a homozygous 1.6 kb deletion encompassing 1 kb of the promoter region and the first noncoding exon of LCA5 in autosomal‐recessive LCA [den Hollander et al., 2007], two distinct heterozygous substitutions in the 3′UTR of CA4 in three families with autosomal‐dominant/sporadic retinitis pigmentosa [Yang et al., 2005; Tian et al., 2010], a heterozygous substitution in the 5′UTR of PRPF31 in autosomal‐dominant retinitis pigmentosa [Rose et al., 2012], and a heterozygous 18‐bp deletion crossing the transcription start site of PRPF4 in autosomal‐dominant retinitis pigmentosa [Chen et al., 2014]. In most of these studies, decreased mRNA abundance in patients’ blood cells and/or decreased luciferase activity in vitro was shown, in agreement with our findings.
Both the identification and interpretation of these categories of mutations is extremely challenging. We initially failed to identify the causal mutation in the largest IBD interval of family F1, despite the inclusion of UTRs and putative promoter regions in our custom enrichment design. Similarly, despite the inclusion of UTRs in several commercial WES designs [Clark et al., 2011], only a few UTR mutations in novel genes have been identified and functionally validated thus far using this approach [Cho et al., 2012; Semler et al., 2012; Williamson et al., 2014]. The main reason for this is the tremendous number of variants that should undergo data analysis if UTRs would be kept within a standard variant‐filtering pipeline. In order to facilitate this process, novel high‐throughput evaluation tools are needed to evaluate this category of noncoding variants as well.
In the second part of this study, we identified two different partial deletions of the NMNAT1 gene following CNV analysis in two families with pseudohomozygosity of a coding NMNAT1 mutation (Supp. Fig. S6). Both deletions were delineated at the nucleotide level, and a bioinformatics study revealed Alu‐combinations at the breakpoints, possibly predisposing to the deletions (Fig. 3; Supp. Table S7). So far, SVs have been detected in a selection of other known retinal dystrophy genes, such as PCDH15 and EYS [Le Guedard et al., 2007; Pieras et al., 2011]. It can even be expected that the occurrence of heterozygous CNVs will be higher than initially anticipated, mainly because of the current lack of proper detection methods. For most retinal dystrophy genes, PCR or capture‐based MPS enrichment platforms followed by regular paired‐end sequencing have now been developed. It is, however, still a challenge to perform CNV analysis solely based on coverage calculations. Mate‐pair sequencing or whole‐genome sequencing are more appropriate MPS methods for this purpose.
Conclusion
This study is the first to identify 5′UTR single‐base mutations in NMNAT1‐related LCA, and autosomal‐recessive blindness in general. In addition, we uncovered two distinct heterozygous CNVs in NMNAT1, further expanding the complex mutation spectrum of this gene. Hence, it can be anticipated that the high number of patients with hidden genetic variation in which, for instance, only a single heterozygous mutation has been found, can be explained by regulatory mutations or SVs of NMNAT1, requiring additional strategies apart from resequencing of the NMNAT1 coding region. Finally, our study illustrates a paradigm shift from coding mutations to noncoding, regulatory mutations and repeat‐mediated SVs in the molecular pathogenesis of unexplained cases of highly heterogeneous recessive disorders such as hereditary blindness.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Material
Acknowledgments
We are most grateful to the families who participated in this study. Brecht Crombez and Ilse Coene are thanked for their expert technical assistance. Greta Van der Cruyssen, Petra Vermassen, Wouter Steyaert, and Claudia Carvalho are acknowledged for their advice. F.C. and H.V. are recipient of a postdoctoral fellowship of the FWO. M.B. is recipient of a PhD fellowship of the FWO. E.D.B. and B.P.L. are Senior Clinical Investigators of the FWO.
Disclosure statement: The authors have nothing to disclose.
Contract grant sponsors: Research Foundation Flanders (FWO) (FWO 3G079711, FWO/KAN/1520913N); Ghent University Special Research Fund (BOF15/GOA/011); Belspo IAP project P7/43 (Belgian Medical Genomics Initiative: BeMGI).
Communicated by Daniel Schorderet
References
- Abeysinghe SS, Chuzhanova N, Krawczak M, Ball EV, Cooper DN. 2003. Translocation and gross deletion breakpoints in human inherited disease and cancer I: nucleotide composition and recombination‐associated motifs. Hum Mutat 22:229–244. [DOI] [PubMed] [Google Scholar]
- Cer RZ, Bruce KH, Donohue DE, Temiz NA, Mudunuri US, Yi M, Volfovsky N, Bacolla A, Luke BT, Collins JR, Stephens RM. 2012. Searching for non‐B DNA‐forming motifs using nBMST (non‐B DNA motif search tool). Curr Protoc Hum Genet Chapter 18:Unit 18.17. 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liu Y, Sheng X, Tam PO, Zhao K, Rong W, Liu X, Pan X, Chen LJ, Zhao Q, Vollrath D, Pang CP, et al. 2014. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum Mol Genet 23:2926–2939. [DOI] [PubMed] [Google Scholar]
- Chiang PW, Wang J, Chen Y, Fu Q, Zhong J, Yi X, Wu R, Gan H, Shi Y, Barnett C, Wheaton D, Day M, et al. 2012. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat Genet 44:972–974. [DOI] [PubMed] [Google Scholar]
- Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, Lee G, Kim HN, Lee HR, Eom HH, Lee ZH, Kim OH, et al. 2012. A single recurrent mutation in the 5′‐UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet 91:343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Chen R, Lam HY, Karczewski KJ, Euskirchen G, Butte AJ, Snyder M. 2011. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol 29:908–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements WK, Kimelman D. 2005. LZIC regulates neuronal survival during zebrafish development. Dev Biol 283:322–334. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. 2010. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci 33:245–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters F, Lefever S, Leroy BP, De Baere E. 2010. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat 31:1097–1108. [DOI] [PubMed] [Google Scholar]
- Coppieters F, Van Schil K, Bauwens M, Verdin H, De Jaegher A, Syx D, Sante T, Lefever S, Abdelmoula NB, Depasse F, Casteels I, de Ravel T, et al. 2014. Identity‐by‐descent‐guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet Med 6:671–680. [DOI] [PubMed] [Google Scholar]
- Corton M, Nishiguchi KM, Avila‐Fernandez A, Nikopoulos K, Riveiro‐Alvarez R, Tatu SD, Ayuso C, Rivolta C. 2013. Exome sequencing of index patients with retinal dystrophies as a tool for molecular diagnosis. PLoS One 8:e65574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Haene B, Vandesompele J, Hellemans J. 2010. Accurate and objective copy number profiling using real‐time quantitative PCR. Methods 50:262–270. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Koenekoop RK, Mohamed MD, Arts HH, Boldt K, Towns KV, Sedmak T, Beer M, Nagel‐Wolfrum K, McKibbin M, Dharmaraj S, Lopez I, et al. 2007. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat Genet 39:889–895. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. 2008. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 27:391–419. [DOI] [PubMed] [Google Scholar]
- Derveaux S, Vandesompele J, Hellemans J. 2010. How to do successful gene expression analysis using real‐time PCR. Methods 50:227–230. [DOI] [PubMed] [Google Scholar]
- Falk MJ, Zhang Q, Nakamaru‐Ogiso E, Kannabiran C, Fonseca‐Kelly Z, Chakarova C, Audo I, Mackay DS, Zeitz C, Borman AD, Staniszewska M, Shukla R, et al. 2012. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet 44:1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C, Decker H, Banker G. 2014. Axonal transport plays a crucial role in mediating the axon‐protective effects of NmNAT. Neurobiol Dis 68C:78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschetti A, Dieterle P. 1954. [Diagnostic and prognostic importance of the electroretinogram in tapetoretinal degeneration with reduction of the visual field and hemeralopia.]. Confin Neurol 14:184–186. [PubMed] [Google Scholar]
- Goios A, Meirinhos J, Rocha R, Lopes R, Amorim A, Pereira L. 2006. RepeatAround: a software tool for finding and visualizing repeats in circular genomes and its application to a human mtDNA database. Mitochondrion 6:218–224. [DOI] [PubMed] [Google Scholar]
- Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P, Dollfus H, Hamel C, Lopponen T, Munier F, Santos L, et al. 2004. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype‐phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 23:306–317. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. 2009. Mechanisms of change in gene copy number. Nat Rev Genet 10:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Qu LH, Meng XH, Xu HW, Yin ZQ. 2014. Detecting genetic variations in hereditary retinal dystrophies with next‐generation sequencing technology. Mol Vis 20:553–560. [PMC free article] [PubMed] [Google Scholar]
- Keen TJ, Mohamed MD, McKibbin M, Rashid Y, Jafri H, Maumenee IH, Inglehearn CF. 2003. Identification of a locus (LCA9) for Leber's congenital amaurosis on chromosome 1p36. Eur J Hum Genet 11:420–423. [DOI] [PubMed] [Google Scholar]
- Kikin O, D'Antonio L, Bagga PS. 2006. QGRS mapper: a web‐based server for predicting G‐quadruplexes in nucleotide sequences. Nucleic Acids Res 34:W676–W682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, Chen Y, Li Y, Fishman GA, Genead M, Schwartzentruber J, Solanki N, et al. 2012. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet 44:1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber T. 1869. Uber retinitis pigmentosa und angeborene amaurose. Von Graefe's Arch. Ophthalmology 15:25. [Google Scholar]
- Le Guedard S, Faugere V, Malcolm S, Claustres M, Roux AF. 2007. Large genomic rearrangements within the PCDH15 gene are a significant cause of USH1F syndrome. Mol Vis 13:102–107. [PMC free article] [PubMed] [Google Scholar]
- Neveling K, den Hollander AI, Cremers FP, Collin RW. 2013. Identification and analysis of inherited retinal disease genes. Methods Mol Biol 935:3–23. [DOI] [PubMed] [Google Scholar]
- Perrault I, Hanein S, Zanlonghi X, Serre V, Nicouleau M, Defoort‐Delhemmes S, Delphin N, Fares‐Taie L, Gerber S, Xerri O, Edelson C, Goldenberg A, et al. 2012. Mutations in NMNAT1 cause Leber congenital amaurosis with early‐onset severe macular and optic atrophy. Nat Genet 44:975–977. [DOI] [PubMed] [Google Scholar]
- Pieras JI, Barragan I, Borrego S, Audo I, Gonzalez‐Del Pozo M, Bernal S, Baiget M, Zeitz C, Bhattacharya SS, Antinolo G. 2011. Copy‐number variations in EYS: a significant event in the appearance of arRP. Invest Ophthalmol Vis Sci 52:5625–5631. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. 2007. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “RetNet.” Retrieved from http://www.sph.uth.tmc.edu/RetNet/.
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277. [DOI] [PubMed] [Google Scholar]
- Rose AM, Shah AZ, Waseem NH, Chakarova CF, Alfano G, Coussa RG, Ajlan R, Koenekoop RK, Bhattacharya SS. 2012. Expression of PRPF31 and TFPT: regulation in health and retinal disease. Hum Mol Genet 21:4126–4137. [DOI] [PubMed] [Google Scholar]
- Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, Schoenau E, Bohlander SK, et al. 2012. A mutation in the 5′‐UTR of IFITM5 creates an in‐frame start codon and causes autosomal‐dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet 91:349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemiatkowska AM, Schuurs‐Hoeijmakers JH, Bosch DG, Boonstra FN, Riemslag FC, Ruiter M, de Vries BB, den Hollander AI, Collin RW, Cremers FP. 2014. Nonpenetrance of the most frequent autosomal recessive Leber congenital amaurosis mutation in NMNAT1. JAMA Ophthalmol 132:1002–1004. [DOI] [PubMed] [Google Scholar]
- Siemiatkowska AM, van den Born LI, van Genderen MM, Bertelsen M, Zobor D, Rohrschneider K, van Huet RA, Nurohmah S, Klevering BJ, Kohl S, Faradz SM, Rosenberg T, et al. 2014. Novel compound heterozygous NMNAT1 variants associated with Leber congenital amaurosis. Mol Vis 20:753–759. [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Fujimaki T, Yanagawa A, Arai E, Fujiki K, Wada Y, Murakami A. 2014. A novel exon 17 deletion mutation of RPGRIP1 gene in two siblings with Leber congenital amaurosis. Jpn J Ophthalmol 58:528–535. [DOI] [PubMed] [Google Scholar]
- Tian Y, Tang L, Cui J, Zhu X. 2010. Screening for the carbonic anhydrase IV gene mutations in Chinese retinitis pigmentosa patients. Curr Eye Res 35:440–444. [DOI] [PubMed] [Google Scholar]
- Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. 2007. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin H, D'Haene B, Beysen D, Novikova Y, Menten B, Sante T, Lapunzina P, Nevado J, Carvalho CM, Lupski JR, De Baere E. 2013. Microhomology‐mediated mechanisms underlie non‐recurrent disease‐causing microdeletions of the FOXL2 gene or its regulatory domain. PLoS Genet 9:e1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson KA, Rainger J, Floyd JA, Ansari M, Meynert A, Aldridge KV, Rainger JK, Anderson CA, Moore AT, Hurles ME, Clarke A, van Heyningen V, et al. 2014. Heterozygous loss‐of‐function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am J Hum Genet 94:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Alvarez BV, Chakarova C, Jiang L, Karan G, Frederick JM, Zhao Y, Sauve Y, Li X, Zrenner E, Wissinger B, Hollander AI, et al. 2005. Mutant carbonic anhydrase 4 impairs pH regulation and causes retinal photoreceptor degeneration. Hum Mol Genet 14:255–265. [DOI] [PubMed] [Google Scholar]
- Zhou T, Kurnasov O, Tomchick DR, Binns DD, Grishin NV, Marquez VE, Osterman AL, Zhang H. 2002. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J Biol Chem 277:13148–13154. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Zhang L, Sasaki Y, Milbrandt J, Gidday JM. 2013. Protection of mouse retinal ganglion cell axons and soma from glaucomatous and ischemic injury by cytoplasmic overexpression of Nmnat1. Invest Ophthalmol Vis Sci 54:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Material