

Abstract
Insulin lispro 200 U/mL (IL200) is a new strength formulation of insulin lispro (Humalog®, IL100), developed as an option for diabetic patients on higher daily mealtime insulin doses. This phase 1, open‐label, 2‐sequence, 4‐period crossover, randomized, 8‐hour euglycemic clamp study aimed to demonstrate the bioequivalence of IL200 and IL100 after subcutaneous administration of 20 U (U) to healthy subjects (n = 38). Pharmacokinetic (PK) and pharmacodynamic (PD) responses were similar in both formulations. All 90%CIs for the ratios of area under the concentration‐versus‐time curve from time zero to the time of the last measurable concentration (AUC0–tlast) and maximum observed drug concentration (Cmax), as well as the total glucose infused throughout the clamp (Gtot) and the maximum glucose infusion rate (Rmax), were contained within 0.80 and 1.25. Time of maximum observed drug concentration (tmax) was similar between formulations, with a median difference of 15 minutes and a 95%CI of the difference that included zero. Inter‐ and intrasubject variability estimates were similar for both formulations. Both formulations were well tolerated. IL200 was bioequivalent to IL100 after subcutaneous administration of 20‐U single doses, and PD responses were comparable between formulation strengths.
Keywords: bioequivalence, diabetes mellitus, insulin lispro, pharmacokinetic, pharmacodynamic
Insulin lispro 100 Units (U)/mL (Humalog®, IL100) is a rapid‐acting human insulin analogue indicated for the treatment of patients with diabetes mellitus who require insulin for the maintenance of normal glucose homeostasis. Insulin lispro was the first commercially available insulin analogue and has been proven to be well tolerated and comparable to human regular insulin in terms of lowering hemoglobin A1c.
Because of the correlation between insulin resistance and obesity,1 insulin requirements in patients with diabetes mellitus have increased. Worldwide, the prevalence of overweight and obese individuals combined rose by 27.5% for adults between 1980 and 2013. The number of overweight and obese individuals increased from 857 million in 1980 to 2.1 billion in 2013.2 In the United States, approximately 90% of patients with type 2 diabetes are considered overweight,3 and these patients can be expected to require progressively higher doses of insulin over the course of their disease.4 In a retrospective analysis of 18‐month data from 1739 adult US patients with type 2 diabetes, the average daily dose of the rapid‐acting prandial insulin aspart was 32 U/day.5 Consistently, a 6‐month efficacy study of 199 insulin‐dependent diabetes mellitus patients from the United Kingdom, Belgium, and the Netherlands showed the average daily insulin lispro dose was 37 U/day after 12 weeks of treatment.6 A study in 312 Asian type 2 diabetes patients used an average daily prandial dose of approximately 37 U/day for the entire population and 41 U/day for the insulin‐experienced population (n = 90) at the 24‐week end point.7
One approach for the management of patients with increased mealtime insulin requirements is the development of insulin products with higher strengths to address the challenges associated with the administration of higher doses of mealtime insulin. An increased strength formulation of Humalog may be beneficial for those patients who require higher mealtime insulin doses, as it allows for a greater number of units to be included in each injection device.
Humalog 200 U/mL is currently commercially available in Europe, presented in a prefilled pen injector (KwikPen®) device, which has been modified from the device used for the current 100 U/mL insulin lispro products. The modified prefilled pen contains twice as many units of insulin lispro (600 versus 300 U) in the same pen size as the currently marketed Humalog KwikPen.8 To accommodate the 600 U of the 200 U/mL strength within the same dimensions, the pen device mechanism was modified to allow variable dosing in 0.005‐mL increments (1 U = 0.005 mL compared with 0.010 mL in the 100 U/mL KwikPen).8 Patients dial the unit dose to be delivered in the same manner that they would for the traditional KwikPen, without needing to adjust for the difference in strength. The maximum single insulin lispro dose that can be delivered is 60 U, the same as in the Humalog 100 U/mL KwikPen device.9
The insulin lispro 200 U/mL formulation is based on the commercial Humalog 100 U/mL formulation and contains the same number of units in half the volume. Changes include an increase in insulin lispro strength from 100 to 200 U/mL, an increase in zinc concentration, and a change in buffering agent (trometamol instead of phosphate buffer).8 Zinc ions promote the association of insulin into hexamers, which are known to improve the stability of insulin.10, 11 Thus, the increase in the zinc/insulin ratio in the 200 U/mL formulation provides an improvement in the degradation rate. The tromethamine buffer reduced the potential for precipitation of phosphate salts (zinc phosphate) resulting from the increased zinc concentration.
The evaluation of the pharmacokinetic (PK) and pharmacodynamic (PD) properties of IL200 relative to IL100 is of interest, as demonstration of similar PK and PD profiles is considered the mainstay of proof of similar efficacy between the test and reference formulations.12 Therefore, this euglycemic clamp study was designed to demonstrate the bioequivalence of IL200 relative to IL100 based on area under the insulin concentration‐versus‐time curve from time zero to the time of the last measurable concentration (AUC0–tlast) and maximum observed drug concentration (Cmax) after subcutaneous administration of single 20‐U doses to healthy subjects. In addition, the PD responses were compared between formulations, and tolerability of both formulations was assessed in healthy subjects.
Materials and Methods
Study Design
This was a phase 1, open‐label, 2‐sequence, 4‐period crossover, randomized, replicate, 8‐hour euglycemic clamp study in healthy subjects13 (Clinicaltrials.gov Identifier: NCT01133392). Treatments were replicated such that each formulation was administered twice on different occasions, with a washout time between study periods that ranged from 4 to 7 days. Subjects were enrolled at a single center (Lilly‐National University of Singapore, Singapore). The study was approved by an ethical review board (National Healthcare Group, Domain Specific Review Board, Singapore), and all subjects provided written informed consent. All study procedures were carried out in compliance with the Declaration of Helsinki and Good Clinical Practices.
Subjects were randomly assigned to 1 of 2 dosing sequences (TRTR or RTRT). In each study period, subjects were dosed IL200 (test treatment [T]) on 2 occasions and IL100 (reference treatment [R]) on 2 occasions, after an approximately 8‐hour fast, and underwent a euglycemic clamp procedure.
Dosage and Administration
The dose of insulin lispro (20 U) was selected, as it is a clinically relevant mealtime dose of insulin lispro for patients with either type 1 or type 2 diabetes and was expected to result in measurable insulin concentrations. Insulin lispro formulations were administered subcutaneously into alternate lower abdominal quadrants at an approximately 90‐degree angle into a raised skinfold, using 3/10‐cc (0.3‐mL) insulin syringes with 29‐gauge, 12.7‐mm‐length (half‐inch‐length) needles.
Eligibility Criteria
Eligible subjects were healthy men and women between 21 and 50 years of age with a body mass index (BMI) between 18.5 and 29.9 kg/m2.
Exclusion criteria included systemic glucocorticoid use within 3 months prior to study entry, known allergies to insulin or its excipients, and a history or presence of comorbid conditions capable of significantly altering the absorption, metabolism, or elimination of drugs; of constituting a risk when taking the study medication; or of interfering with the interpretation of data. Subjects were also excluded if they smoked and if they had an elevated fasting venous blood glucose >6 mmol/L at screening.
Sample Size
The sample size for the present study was calculated based on an intrasubject variability of approximately 30% (% coefficient of variation) for area under the curve from time 0 to return to baseline of insulin concentration versus time, observed following administration of subcutaneous insulin lispro in a previous clamp study in a similar population.14 Thirty completers in a replicate treatment design were expected to provide at least 90% power to show the 90% confidence interval (CI) of the ratio of means for AUC0–tlast between the 2 formulations to be within the 0.80 and 1.25 bioequivalence limits.
Euglycemic Glucose Clamp Procedure
Prior to each clamp procedure, subjects were instructed to refrain from alcohol and vigorous exercise for 24 and 48 hours, respectively. Subjects fasted for approximately 8 hours before dosing and remained fasting until the end of the clamp. Each subject underwent a euglycemic clamp procedure7 on 4 separate visits. The time of insulin dosing was defined as time zero. A baseline blood glucose concentration was calculated for each subject by averaging the blood glucose concentrations taken at 10‐minute intervals for approximately 30 minutes prior to dosing. The target value for blood glucose concentrations during the clamp procedure was defined as 5 mg/dL (0.27 mmol/L) below the subject's fasting baseline blood glucose concentration. During the glucose clamp, the glucose infusion rate (GIR) was adjusted manually to maintain this predetermined target blood concentration for each individual subject. Glucose was infused as a 20% dextrose solution buffered to near neutral pH. Subjects underwent euglycemic clamps for up to 8 hours after dosing. Blood samples for PK evaluations were obtained up to 8 hours (480 minutes) postdose, and glucose concentrations, measured using the YSI glucose analyzer (YSI Life Sciences, Yellow Springs, Ohio) were obtained every 5 to 30 minutes throughout the clamping procedure. The clamp was discontinued if the GIR fell to 0 for at least 30 minutes after the clamp had been underway for at least 4 hours; PK sampling was continued in these instances. The GIR was documented throughout the procedure and was used to reflect the activity of insulin.
Tolerability was assessed throughout the study by monitoring adverse events (AEs), physical examinations, clinical laboratory tests, electrocardiograms, and vital signs.
Data Analyses
PK and PD analyses were performed on all subjects who completed at least 1 clamp procedure. Parameters were individually calculated for each subject based on actual collection times and are presented by summary statistics.
Bioanalytical
Blood samples were collected 10, 20, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes postdose to determine free (non‐autoantibody‐bound) immunoreactive insulin lispro‐specific concentrations for PK evaluations. Serum concentrations of insulin lispro were assayed using a validated competitive radioimmunoassay (RIA) method adapted from the Millipore Lispro Insulin RIA Kit (catalog LPI‐16K). This assay is specific for insulin lispro and demonstrates negligible cross‐reactivity with endogenous human insulin.15 Standards and controls were prepared in human serum using lispro insulin reference standards; standard concentrations ranged from 0.1 to 30.0 ng/mL. Standards, controls, and study samples were mixed with an equal volume of 25% polyethylene glycol prepared in 0.9% NaCl and placed on an ice bath for 25–30 minutes. Samples were centrifuged at 2°C–8°C at 3000 rpm for 25–30 minutes, and the supernatant was transferred into a clean tube. Duplicate 200‐µL aliquots of the sample supernatant were then processed in the RIA method per the kit instructions. The validated lower and upper limits of quantitation of the assay were 0.2 and 15 ng/mL, respectively. The assay was validated in accordance with regulatory guidelines.16 In validation experiments, interday precision and accuracy were within 13% (Eli Lilly and Company, data on file).
Pharmacokinetics
Serum concentrations were analyzed by conventional noncompartmental methods. PK parameters, including Cmax, time of maximum serum insulin concentration (tmax), AUC0–tlast, AUC from time zero to 8 hours (AUC0–8), and AUC from zero to infinity (AUC0– ∞) were estimated using WinNonlin Enterprise version 5.3 (Pharsight, Mountain View, California).
Concentrations that were below the quantitation limit at predose times were treated as zero; those postdose were treated as missing. Mean concentrations were calculated for all data points where at least two‐thirds of the subjects had available data from samples obtained within 10% of the nominal time.
Bioequivalence between insulin lispro 200 U/mL and insulin lispro 100 U/mL was analyzed using a linear mixed‐effects model where formulation (T or R), period, and sequence were included as fixed factors and subject as a random factor. The response variables (AUC0–tlast, AUC0–∞, AUC0–8, Cmax) were log‐transformed and analyzed separately. The difference in least‐squares (LS) mean estimates between formulations and the corresponding 90% confidence interval (CI) for the difference were back‐transformed to provide estimates of the ratio of geometric means and 90%CI for the ratio of means. Bioequivalence was concluded if the 90%CI for the ratio of IL200 to IL100 was completely contained within the limits of 0.80–1.25. The estimated difference in median time of occurrence of tmax was analyzed using the nonparametric Wilcoxon signed rank test.
Pharmacodynamics
Individual PD parameters were calculated from LOESS‐smoothed GIR‐versus‐time data. The LOESS smoothing technique (span = 0.1), was performed in S‐Plus® (version 7.0.6; Insightful Corp., Seattle, Washington). Values of zero GIR were included as such. Key parameters included total amount of glucose infused (Gtot) and maximum glucose infusion rate (Rmax). The following time PD parameters were also calculated: time of maximum glucose infusion rate (tRmax), times of half‐maximum GIR before and after Rmax (early and late tRmax50), time of the first change in GIR (tRonset), and time of last GIR (tRlast). Overall blood glucose data were plotted using a LOESS‐smoothing technique (span 0.1) in SigmaPlot for Windows (version 11.0), and summary blood glucose statistics are presented.
Comparison of key and time PD parameters between formulations was analyzed similarly as described for the PK parameters.
Results
Demographics
A total of 38 overtly healthy subjects, 36 male and 2 female, between ages 23 and 45 years (mean, 32.4 years), of whom 37 were Asian and 1 white, were randomly assigned to the treatments and received at least 1 dose of study drug. Subjects' weight and BMI ranged from 45.2 to 82.3 kg and from 19.0 to 25.9 kg/m2, respectively, with mean values of 66.57 kg and 22.57 kg/m2, respectively. Subjects were similar in age and BMI across sequence groups.
Pharmacokinetics
Mean insulin concentration‐versus‐time profiles were similar between formulations (Table 1 and Figure 1A). The similarity in PK profiles was confirmed by the ratio of LS means for AUC and Cmax values. Bioequivalence of IL200 relative to IL100 was demonstrated by the 90%CIs for the exposure ratios contained within 0.80–1.25. The tmax was also similar between formulations, with a median difference of 15 minutes and a 95%CI of the difference that included zero. Intra‐ and intersubject variability population estimates were similar for both formulations, with coefficients of variation of ≤14% and ≤15% for AUCs and ≤18% and ≤30% for Cmax, respectively.
Table 1.
Pharmacokinetic and Pharmacodynamic Results
| IL100 (R) | IL200 (T) | Ratio a or Difference b of LS Means | 90%CI (Ratio), 95%CI (Difference) | |
|---|---|---|---|---|
| N | 75 | 73 | ||
| PK Parameters | ||||
| AUC0–tlast (pmol · h/L) | 1940 (20) | 1920 (20) | 0.990 a | (0.948, 1.034) |
| 1980 c (368) d | 1960 c (390) d | |||
| Cmax (pmol/L) | 887 (34) | 819 (32) | 0.933 a | (0.897, 0.972) |
| 938 c (331) d | 860 c (280) d | |||
| tmax (h) e | 0.75 (0.50–3.00) | 1.00 (0.50–3.00) | 0.250 b | (0.000, 0.250) |
| t1/2 (h)c,d | 0.92 (0.25) | 0.83 (0.30) | — | — |
| AUC0–8 (pmol · h/L) | 2020 (19) | 2000 (19) | 0.994 a | (0.954, 1.036) |
| 2050 c (365) d | 2040 c (383) d | |||
| AUC0–∞ (pmol · h/L) | 2030 (19) | 2020 (19) | 0.993 a | (0.952, 1.036) |
| 2070 c (365) d | 2050 c (380) d | |||
| PD Parameters (%) | ||||
| Gtot (g) | 123 (30) | 125 (25) | 1.014 a | (0.961, 1.070) |
| Rmax (mg/min) | 539 (27) | 544 (23) | 1.005 a | (0.958, 1.054) |
| tRmax (h) | 2.00 (56) | 2.11 (49) | 0.100 b | (−0.400, 0.500) |
| Early tRmax50 (h) | 0.595 (29) | 0.568 (31) | −0.030 b | (−0.091, 0.007) |
| Late tRmax50 (h) | 4.34 (42) | 4.39 (37) | −0.036 b | (−0.156, 0.098) |
| tRonset (h) | 0.350 (43) | 0.337 (34) | −0.042 b | (−0.042, 0.042) |
| tRlast (h) | 7.12 (15) | 7.04 (14) | 0.000 b | (0.000, 0.000) |
Values are presented as geometric means and CV(%) unless otherwise indicated.
AUC0–t last, area under the concentration‐versus‐time curve from time zero to the last point with a measurable concentration; AUC0–8, area under the curve from time 0 to 8 hours; AUC0–∞, area under the curve from zero to infinity; CI, confidence interval; Cmax, maximum observed drug concentration; CV, coefficient of variation; GIR, glucose infusion rate; Gtot, total glucose infused throughout the clamp; h, hours; IL100, insulin lispro 100 U/mL; IL200, insulin lispro 200 U/mL; LS, least‐squares; N, number of subjects; PD, pharmacodynamics; PK, pharmacokinetic; R, reference treatment; Rmax, maximum glucose infusion rate; T, test treatment; tmax, time of maximum observed drug concentration; tRlast, time of the last nonzero GIR; tRmax, time of maximum glucose infusion rate; tRmax50, time of half‐maximum GIR before and after Rmax; tRonset, time of the first change in the nonzero GIR.
Ratio of LS means (T divided by R).
Median differences (T ‐ R).
Arithmetic mean.
Standard deviation.
Median (range).
Figure 1.
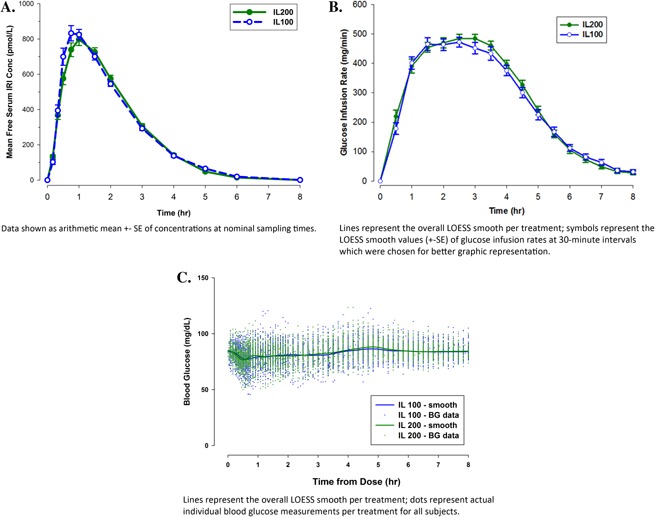
Mean free serum immunoreactive insulin (IRI) concentration versus time (A), mean glucose infusion rate (GIR) versus time (B), and overall LOESS smooth and individual blood glucose concentration data versus time for all subjects by treatment (C). BG, blood glucose; conc, concentration; h, hours; IL100, insulin lispro 100 U/mL; IL200, insulin lispro 200 U/mL; IRI, immunoreactive insulin; LOESS, locally weighted scatterplot smoothing.
Pharmacodynamics
Consistent with PK observations, administration of 20 U of IL200 or IL100 resulted in similar GIR‐versus‐time profiles and thus similar estimates for key PD parameters (Table 1 and Figure 1B). Median differences for PD time parameters were small (less than 6 minutes), and all 95%CIs encompassed zero. Consistent with PK observations, the intra‐ and intersubject variability population estimates were similar for both formulations, with coefficients of variation of ≤14% and <27% for Gtot and <18% and <24% for Rmax, respectively. Mean ± SE (CV%) blood glucose concentrations throughout the clamp procedure were were 81.9 ± 0.13 mg/dL (10.97%) for IL100 and 82.3 ± 0.13 mg/dL (10.76%) for IL200 (Figure 1C).
Tolerability
A total of 41 treatment‐emergent AEs were reported, with the most common AEs being catheter‐site hematoma (11 subjects) and procedural‐site reaction (5 subjects). In the opinion of the investigator, none of the AEs were deemed to be related to either insulin formulation (IL100 or IL200). The majority of AEs were mild in severity. Two AEs were considered moderate (1 catheter‐site hematoma resulting from each treatment). No subjects discontinued the study because of an AE. No clinically significant alterations in laboratory values, urinalysis values, or vital signs were identified. Results demonstrated both formulations to be well tolerated in healthy subjects, with no apparent differences with respect to tolerability between the 2 formulations.
Discussion
Data from this open‐label, 2‐sequence, 4‐period crossover, euglycemic clamp study demonstrated that insulin IL200 is bioequivalent to IL100 and that PD responses were also comparable between formulations. Inter‐ and intrasubject variability estimates were similar for both formulations. The flat shape and statistics of the blood glucose results confirm the clamp quality. In addition, both formulations were well tolerated in this study population.
The changes to the IL100 formulation, made to develop a new IL200 formulation, included increasing insulin lispro strength from 100 to 200 U/mL and changes to the zinc content and buffering agent of IL200. These changes did not affect the bioequivalence between the IL100 and IL200 formulations, which is due to the active substance (insulin lispro).8
The replicate crossover study design used in this study is a commonly used method to demonstrate bioequivalence, as it enables each subject to serve as his or her own control and to obtain repeated measures to improve the precision of the comparison.6 The treatment periods were separated by a 4‐ to 7‐day washout period to ensure that drug concentrations were below the lower limit of bioanalytical quantification in all subjects at the start of the subsequent period.
The subject population for bioequivalence studies is selected with the aim of permitting detection of differences between pharmaceutical products. To reduce variability not related to differences between products, bioequivalence studies are usually performed in healthy subjects, unless the drug's characteristics make this unfeasible. Insulin lispro has a well‐established safety profile. Therefore, a healthy subject population was used for this bioequivalence study, which would allow extrapolation of the results to populations for which insulin lispro is approved.6 Although the use of healthy subjects could be considered a study limitation as healthy subjects are not the target patient population, it does not impact the finding that the 2 formulations are bioequivalent. The BMI range in the population, which was chosen to facilitate study execution, would not influence the conclusions of this crossover study, in which subjects were their own controls.
A 20‐U dose of insulin lispro was selected for this study, as it is within the appropriate range of prandial doses for the treatment of diabetes in clinical practice and was anticipated to provide measurable insulin concentrations. Because IL200 and IL100 are bioequivalent, no difference in insulin PK would be expected between these formulations at other doses.
Therefore, the wide range of insulin dosing requirements among patients with diabetes can be delivered by using the 2 strengths of insulin lispro assessed in this study. The tolerability, efficacy, and PK properties of insulin lispro have already been established through an extensive clinical trial program, as well as its clinical use. Because this study has shown IL200 to be bioequivalent to IL100, the prior efficacy and safety established for Humalog would apply to the IL200 formulation.
Additionally, it is expected that IL200 may offer an increased benefit to patients requiring mealtime insulin doses higher than 20 U. Increasing the insulin strength from 100 to 200 U/mL in the same size pen device, thus doubling the number of units per pen, may allow patients to change insulin pens less frequently.
Conclusion
In summary, the IL200 formulation was bioequivalent to the IL100 formulation (Humalog) after subcutaneous administration of 20 U to healthy subjects, and PD responses, which are indicators of insulin effect, were similar for both formulations as well.
Declaration of Conflicting Interests
All authors are employees and stockholders of Eli Lilly and Company.
Funding
The study was supported by Eli Lilly and Company.
Acknowledgment
The authors acknowledge Aik Hoe Seah and Qun Lin (Eli Lilly and Company) for their statistical programming support, Sheila Corrigan (Eli Lilly and Company) for her expertise and review of this manuscript, and Stephanie Brillhart and Gina Moore (inVentiv Health Clinical) for their contributions as medical writers.
References
- 1. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000; 106:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ng M, Fleming T, Robinson M, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980‐2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014; 384:766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Obesity Society. Your weight and diabetes. 2010. www.obesity.org/resources‐for/your‐weight‐and‐diabetes.htm.
- 4. Watson L, Wilson BP, Alsop J, Kumar S. Weight and glycaemic control in type 2 diabetes: what is the outcome of insulin initiation? Diabetes Obes Metab. 2011; 13:823–831. [DOI] [PubMed] [Google Scholar]
- 5. Aagren M, Luo W, Moes E. Heathcare utilization changes in relation to treatment intensification with insulin aspart in patients with type 2 diabetes. Data from a large US managed‐care organization J Med Econom. 2010; 13(1)16–22. [DOI] [PubMed] [Google Scholar]
- 6. Holleman F, Schmitt H, Rottiers R, et al. Reduced frequency of severe hypoglycemia and coma in well‐controlled IDDM patients treated with insulin lispro. Diabetes Care. 1997; 20(12):1827–1832. [DOI] [PubMed] [Google Scholar]
- 7. Bebakar WM, Lim‐Abrahan MA, Jain AB, Seah D, Soewondo P. Safety and effectiveness of insulin aspart in type 2 diabetic patients: results from the ASEAN cohort of the A1chieve study. Diabetes Res Clin Pract. 2013;100(Suppl 1):S17–S23. [DOI] [PubMed] [Google Scholar]
- 8.European Medicines Agency. Humalog Assessment Report #EMEA/H/C/000088/X/0125. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Assessment_Report_‐_Variation/human/000088/WC500176634.pdf. Accessed March 19, 2015.
- 9.Eli Lilly and Company. Humalog KwikPen™ insulin lispro injection (rDNA origin): Instructions for use. http://pi.lilly.com/us/humalog‐kwikpen‐um.pdf. Accessed, March 19, 2015.
- 10. Dunn MF. Zinc‐ligand interactions modulate assembly and stability of the insulin hexamer—a review. Biometals. 2005; 18(4):295–303. [DOI] [PubMed] [Google Scholar]
- 11. Brange J, Langkjaer L. Chemical stability of insulin 3. Influence of excipients, formulation, and pH. Acta Pharm Nord. 1992; 4(3):149–158. [PubMed] [Google Scholar]
- 12.Committee for Medicinal Products for Human Use (CHMP). Guideline on the Investigation of Bioequivalence. European Medicines Agency. 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/ WC500070039. pdf. Accessed October 22, 2014.
- 13. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979; 237:E214–E223. [DOI] [PubMed] [Google Scholar]
- 14. Rave KM, Nosek L, de la Peña A. et al. Dose response of inhaled dry‐powder insulin and dose equivalence to subcutaneous insulin lispro Diabetes Care. 2005; 28(10)2400–2405. [DOI] [PubMed] [Google Scholar]
- 15.EMD Millipore. LisPro Insulin RIA [LPI‐16K]. http://www.emdmillipore.com/US/en/product/LisPro‐Insulin‐RIA,MM_NF‐LPI‐16K#anchor_TI. Accessed March 19, 2015.
- 16.US Department of Health and Human Services Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation. May 2001. http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf. Accessed March 19, 2015.