

Abstract
The connection of microbial biosynthetic gene clusters to the small molecule metabolites they encode is central to the discovery and characterization of new metabolic pathways with ecological and pharmacological potential. With increasing microbial genome sequence information being deposited into publicly available databases, it is clear that microbes have the coding capacity for many more biologically active small molecules than previously realized. Of increasing interest are the small molecules encoded by the human microbiome, as these metabolites likely mediate a variety of currently uncharacterized human-microbe interactions that influence health and disease. In this mini-review, we describe the ongoing biosynthetic, structural, and functional characterizations of the genotoxic colibactin pathway in gut bacteria as a thematic example of linking biosynthetic gene clusters to their metabolites. We also highlight other natural products that are produced through analogous biosynthetic logic and comment on some current disconnects between bioinformatics predictions and experimental structural characterizations. Lastly, we describe the use of pathway-targeted molecular networking as a tool to characterize secondary metabolic pathways within complex metabolomes and to aid in downstream metabolite structural elucidation efforts.
Keywords: Biosynthesis, Colibactin, Pathway-Targeted Molecular Networking
1. INTRODUCTION
Secondary metabolites have long served as inspirational structural and functional scaffolds for the development of new-in-class pharmaceuticals [1-2]. A longstanding era of secondary metabolite discovery followed the discovery of penicillin in the 1920s, and by the 1990s, approximately 80% of commercial drugs were natural products or natural product derivatives [3-4]. This percentage has decreased over the last few decades due to the expansion of combinatorial synthetic methods and an increase in the rediscovery rates of natural products through traditional discovery campaigns. However, with the continued expansion of microbial genome and metagenome sequence information, a resurgence in interdisciplinary academic and industrial natural product discovery campaigns is well underway [5-11]. Several major challenges exist for the discovery of new microbial natural product-derived drug leads, such as: 1) our inability to culture the majority of microbes from environmental samples (e.g., “the great plate count anomaly”) [12-17]; 2) our general lack of robust tools to broadly activate bioactive small molecule production from diverse “silent” pathways in the microbes (or heterologous expression hosts) that we can readily cultivate in the lab [18-21]; and 3) our inefficiencies in quickly identifying and dereplicating unknown metabolites from expressed pathways with often unpredictable structural and functional properties [22]. As a result, there is a continued general need for the development of interdisciplinary approaches to link “orphan” biosynthetic gene clusters to the bioactive small molecules they produce.
Multipartite animal-microbe symbioses have provided rich sources of novel bacterial small molecules that naturally function in animal environments, enhancing their pharmacological potential [23-28]. By understanding the ecological niche, new small molecules can be stimulated, discovered, and investigated with overarching ecological and functional contexts. Indeed, since the turn of the century, we have come to appreciate humans and all other animals as being “superorganisms” [29]. Our resident microbes, the human microbiota, is one such source that has emerged as a prominent player in regulating both human health and disease, and thus its metabolite coding capacity, the human microbiome, is a rich reservoir of potential clinically-relevant small molecules [30-32]. The microbiota affects the host through various mechanisms including the exchange of nutrients, regulation of the immune system, protection from pathogens, and metabolism of indigestible compounds [26, 33-34]. Because of the importance of this symbiotic relationship, dysregulation of the microbiota communities (dysbiosis) has been correlated with the onset of serious health issues, including obesity, diabetes, inflammatory bowel diseases, and cancers [35-40].
Small molecule metabolites regularly mediate host-microbe interactions. And despite the ecological importance of microbial natural products, we know very little about the structures and functional roles of these compounds and how they affect human health. (Meta)genomics-guided approaches have started to shed light on the extent of this question. Sequencing of the human microbiota [41-42] revealed that human-associated bacteria encode for a wide diversity of biosynthetic gene clusters, with over 3,000 biosynthetic gene clusters being widely distributed among the sequenced microbiota of healthy individuals [30]. Much of the chemical diversity encompassed by the small molecule products of these gene clusters is found in bacteria that are associated with the oral and gut cavities. The majority of these compounds have not yet been characterized.
One of the more heavily studied biosynthetic gene clusters from the microbiome is the colibactin pathway [43]. The colibactin gene cluster is a ~55 kb biosynthetic gene cluster that produces a family of polyketide-nonribosomal peptide hybrid molecules. This gene cluster is found among the Enterobacteriaceae, including Escherichia coli, Citrobacter koseri, Klebsiella pneumoniae, and Enterobacter aerogenes [44]. Additionally, the gene cluster has been discovered in the microbiota of infected coral [45] and of honeybees exhibiting an intestinal scab phenotype [46-47]. Bacteria expressing the pathway induce DNA double strand breaks and cause genomic instability of mammalian cells [48-49]. The presence of this gene cluster is epidemiologically associated with long-term persistence in the host [50]. Under inflammatory conditions, such as in inflammatory bowel disease (IBD), Enterobacteriaceae members containing this gene cluster proliferate [51]. As a result of the cytotoxicity exhibited by the small molecules from this pathway, the colibactin pathway has been directly linked to colorectal tumorogenesis in colitis mouse models [38-39, 52]. However, other strains containing the colibactin cluster, such as E. coli Nissle 1917, paradoxically have also been demonstrated to exhibit probiotic effects for patients with ulcerative colitis [53]. Gaining mechanistic insights for these functional disconnects remain the subjects of ongoing investigations. Mechanistic understanding of the phenotypes exhibited by this pathway have been hindered by the lack of colibactin structural information. Fortunately, structural and small molecule functional data are starting to emerge, providing new vantage points to experimentally elucidate the mechanistic underpinnings for the various colibactin pathway functions [54-61]. In this mini-review, we focus on the colibactin pathway as a central thematic example of linking biosynthetic gene clusters to the small molecules they produce and draw connections to other pathways invoking related biosynthetic logic. We highlight “pathway-targeted” molecular networking as one approach to more finely map expressed secondary metabolic pathways within complex metabolomes to aid in secondary metabolite identification and characterization [62]. Lastly, we discuss a few of the disconnects between secondary metabolite structure and biosynthetic predictions as illustrative examples for the continued need of enzymological characterizations of orphan biosynthetic gene clusters [63].
2. Genomics-guided secondary metabolite discovery
The “structure first,” then hunt for its responsible gene cluster paradigm, is transitioning to “sequence first,” then hunt for the many possible products encoded in the (meta)genomic information. Genes-to-molecules discovery approaches inherently reduce rediscovery rates of known metabolites, as novel gene clusters, i.e., novel “biosynthetic codes,” are selected as the genetic source materials. Briefly, natural products of the polyketide and the nonribosomal peptide families, for example, are often biosynthesized according to a biosynthetic code [64-66]. Many of these biosynthetic systems follow a “co-linearity” rule, in which the organization of domains dictates the order of biosynthetic operations in the pathway. Because of this logic and the wealth of mechanistic enzymology knowledge in this area, it is possible to predict with some accuracy the possible core structure(s) of assembly line-derived polyketides and nonribosomal peptides [64-66]. New bioinformatic programs such as antiSMASH [67-70] and ClusterFinder [71] have integrated a variety of existing bioinformatic algorithms and have largely automated the process of finding novel secondary metabolite gene clusters and predicting possible core structures. However, many nonlinear and iterative pathways among other confounding factors, such as hypothetical proteins serving as novel biocatalysts, necessitate a continued need for “biosynthetic code breaking.” Indeed, about half of the genes in the human microbiome are listed as hypotheticals and are completely unknown [72]. A portion of these genes will undoubtedly contribute to novel small molecule metabolism. Additionally, many putative secondary metabolite pathways are emerging in genome databases that contain proteins with no significant similarity to previously reported pathways and are not detected in algorithms that rely on currently known pathways as inputs, raising genome-guided opportunities for the discovery of new small molecule classes.
3. Pathway-targeted molecular networking
When comparing experimental conditions, A versus B, metabolomics groups often make functional claims based on the responses of molecules that can be mapped to established external and internal databases. Without question, many functional insights have been gained from these approaches, but what about all of the product ions that are not found in any current database? In secondary metabolite discovery operations, mapping small molecules to known databases most often fails, as novel metabolites by definition have not yet been characterized. Foundational approaches are emerging that are beginning to address this key bottleneck in microbial secondary metabolism [73]. Specifically, molecular networking techniques enhance diverse investigations of secondary metabolite discovery, regulation, and their functional roles by interconnecting structurally related molecules in silico [74]. This unbiased approach scores tandem MS (MS2) spectra based on small molecule fragmentation similarities. The molecules are then represented in a molecular network as interconnected nodes based on fragmentation relationships [74]. Using this method, an individual node, or “molecular feature” (MoF) groups with similar MoFs, forming structurally related clusters, or “molecular families.” Molecular networking has found many recent uses in investigating metabolic responses from individual microorganisms to complex cell-to-cell interactions. For example, coupling nanospray desorption electrospray ionization (nanoDESI) MS with molecular networking, the metabolic status of living microbial colonies has been characterized [74]. By growing the colony next to a competing species, the metabolic response of the microbial colony can be assessed. From the MS/MS network, small molecules that are stimulated by the challenge can be grouped into general families of metabolites. Consequently, if a novel molecule is not yet in a database but falls within a known molecular family in the network, structural information can more readily be proposed. These foundational methods accelerate natural product dereplication approaches [22], facilitate the study of intraspecies, interspecies, and even interkingdom interactions [75-76], and significantly aid in the decoding of “orphan” biosynthetic gene clusters and the “cryptic” small molecules they produce [77].
When collecting high-resolution tandem MS data in untargeted fragmentation modes, the subsequent molecular networks generated from this data are enriched in the more abundant molecules – primary metabolites, media components, and abundant secondary metabolites – and often lack less abundant molecules encoded by the pathway of interest. To address this roadblock, we conducted a modified “pathway-targeted” molecular networking strategy for the colibactin pathway that ties into the existing untargeted networking frameworks and resources [56]. In our modified strategy, we first collect high-resolution untargeted metabolomics data (MS) in isogenic wildtype and mutant strains to identify parent ions dependent on the presence of the secondary metabolic pathway, which can be cleanly deleted or inserted into a heterologous host without significant effects to cell growth. Then, we run a tandem MS experiment focusing on the fragmentation of only those unique pathway-dependent features for molecular networking. Multiple tandem MS datasets can be pooled if needed to enhance overall pathway-targeted network coverage. To illustrate the output of this approach, Fig. (1) compares an untargeted and a pathway-targeted molecular network from E. coli heterologously expressing an example secondary metabolic pathway. We recently applied this approach to the bacterial colibactin pathway found in select strains of E. coli and elsewhere [56-57, 62]. The result was a focused network map of the colibactin pathway that contained critically important, lower abundance ions that were missed in untargeted fragmentation modes. Because pathway intermediates and products inherently share structural similarities, the networks from freshly prepared organic extracts greatly facilitated the structural predictions of colibactin pathway-dependent molecules in the network, some of which were unstable and decomposed during chemical processing, relative to a handful of stable pathway-dependent reference molecules extensively characterized by NMR and/or synthesis. We have found this approach to be more generally applicable in conducting detailed systems-level biosynthetic analyses of secondary metabolic pathways, such as determining genetic mutation consequences at the metabolic network level (pinpointing bottlenecks in secondary metabolism) and assessing NRPS amino acid substrate specificities at the pathway level using a combination of system-wide isotopic labeling studies and pathway-targeted molecular networking.
Fig. (1).
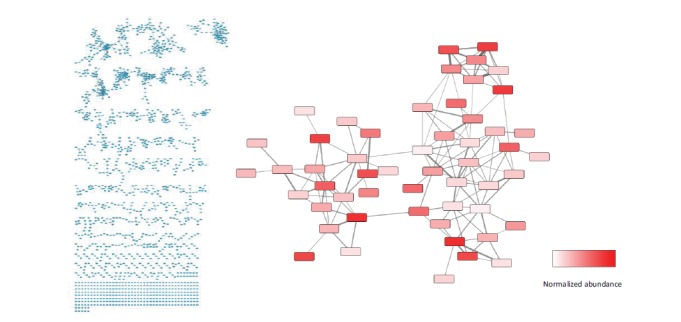
Pathway-targeted molecular networking of gene clusters links genes to the molecules they produce.
While pathway-targeted molecular networking has the potential to aid in natural product discovery efforts, there are a few limitations that must be considered. Pathway-targeted comparative metabolomics requires the ability to acquire spectra from both a producing and non-producing strain (comparison of a functionally expressed pathway versus a strain lacking a functional secondary metabolic pathway). Thus, a successful experiment necessitates that the pathway is not “silent” under laboratory growth conditions and that the producing host is genetically tractable, or that the pathway can be transferred and functionally expressed in a heterologous host. Despite these limitations, pathway-targeted molecular networking is a useful tool in metabolite discovery efforts and can be coupled to emerging synthetic biology techniques for pathway activation. We discuss two biosynthetic events found in the colibactin pathway – hydrolytic maturation of secondary metabolites and the incorporation of unexpected amino acids – and how pathway-targeted molecular networking has and continues to aid in the elucidation of diverse structures from this important pathway.
4. Hydrolytic maturation of secondary metabolites
More and more examples have emerged where sequencing of a biosynthetic gene cluster leads to a predicted biosynthetic code that does not match its expected product. One important mechanism underlying this disconnect is in the hydrolytic maturation of nonribosomal peptides and hybrids thereof, in which a larger precursor is cleaved to form smaller constituents [78]. A growing number of natural products fall into this category, including colibactin. Analogous proteolytic events occur during the maturation of many ribosomally synthesized and post-translationally modified peptides (RiPPs) from larger precursor peptides [79]. An important nonribosomal peptide example is in the biosynthesis of the prototypical monocyclic β-lactam antibiotic family, the nocardicins, which are broad spectrum antibacterials first described in the 1970s [80]. When Townsend and co-workers reported the biosynthetic gene cluster for nocardicin in 2004 through a structure-guided sequencing approach, they noted that the gene cluster encoded five modules for the predicted production of a pentapetide, but nocardicin A consisted of only a modified tripeptide sequence [81]. The group proposed that either the pathway contained inactive modules, which was the favored mechanism at the time, or engaged in proteolytic processing to explain the discrepancy between the number of modules in the biosynthetic gene cluster and the number of amino acids in the final structure of the mature antibiotic [81]. Because a candidate protease was not found in the gene cluster, experimental support for the less favored proteolytic mechanism came later through a model protease cleavage assay [82] and biochemical analysis of individual catalytic domains [83-84].
Hydrolytic processing of polyketide synthase/nonri-bosomal peptide synthetase (PKS/NRPS) hybrid products was proposed as the primary route for zwittermicin biosynthesis [85]. The structure of zwittermicin was first described in 1994 [86], but it was not until the gene cluster was identified in 2009 that it was proposed that zwittermicin is one of two major metabolites cleaved from a larger precursor molecule [85]. This precursor, prezwittermicin A, was proposed to be capped with an N-terminal N-acyl-D-asparagine. A transmembrane peptidase, ZmaM, which is encoded in the gene cluster, was then proposed to cleave the N-acyl-D-asparagine during export from the cell, releasing mature zwittermicin A. However, the first strong complementary experimental evidence for this maturation mechanism was presented in two related pathways, small molecule structures of “prexenocoumacins” from the xenocoumacin antibiotic pathway [87] and shortly thereafter an X-ray crystal structure of a peptidase from the colibactin genotoxin pathway like those found in the zwittermicin and xenocoumacin pathways [88]. From the xenocoumacin pathway, a transmembrane peptidase XcnG was required to produce the active xenocoumacins. Deletion of this peptidase led to the characterization of a family of prexenocoumacins that are capped with an N-acyl-D-asparagine. The capped prexenocoumacins exhibited no detectable bioactivity. Due to the differences in antibiotic activity between prexenocoumacins and xenocoumacins, the maturation event was described as a “pro-drug activation mechanism” [87]. The complementary X-ray structure and biochemical analysis of the related peptidase ClbP in the colibactin pathway, showed that the transmembrane peptidase was necessary for genotoxicity and was located on the inner membrane facing the periplasm, supporting the cleavage of “precolibactins” in the periplasm during export [88]. The authors noted the key similarities between the colibactin pathway and the xenocoumacin/zwittermicin pathways, but the structures of precolibactins remained unknown [89]. ClbP-dependent N-acyl-D-Asparagines, e.g. N-myristoyl-D-Asn and ClbP precursor analogs, from the colibactin pathway were identified later in accordance with the above biosynthetic logic [54-56]. The function of this N-terminal cap is likely to protect the producing strain from genotoxicity. However, the liberated N-myristoyl-D-asparagine has also been shown to have biological activities in vitro, including weak bacterial growth inhibitory effects against Gram-positive bacteria and antagonistic activities against the serotonin-7 receptor and the dopamine-5 transporter [56]. Speculatively, these activities may contribute to bacteria-bacteria and/or host-bacteria interactions in the gut [56].
An untargeted network of a single E. coli organic extract (left) contains over 1500 molecular features (MoFs). Features are clustered based on the similarity of MS/MS spectra. Endogenous metabolites, media components, as well as metabolites of interest are included in this network. A targeted analysis fragmenting gene cluster-dependent features returns approximately 50 MoFs (right) with higher pathway coverage. The production of each of these features is dependent on the presence of a gene cluster of interest. Nodes are colored according to average ionization intensity, with white nodes being present at low abundance and black nodes being present at high abundance.
A variety of other NRPS/PKS molecules are enzymatically hydrolyzed during maturation. The didemnins and zeamines contain polypeptides that are removed during maturation. Didemnin B is a cyclic depsipeptide that has been investigated for use as an anticancer agent [90]. Didemnin X and Y contain the core structure of the active Didemnin B with an additional N-terminal β-hydroxyl-polyglutamine cap [90]. These derivatives were isolated with the didmenin gene cluster in hand, enabling a comparative gene cluster – small molecule structural outcome correlation. Zeamine is a PKS/NRPS hybrid molecule that was originally isolated from Dickeya zeae. Pre-zeamine was isolated from Serratia plymuthica and contains a C-terminal pentapeptide that is proposed to be cleaved by a peptidase in the gene cluster [91]. Pyoverdine is a fluorescent siderophore produced by Psuedomonas aeruginosa that is required for virulence [92-93]. Periplasmic proteins are responsible for the maturation of the chromophore as well as cleavage of a myristoyl moiety [94-95]. In vitro reconstitution of the saframycin biosynthetic pathway revealed that it is synthesized with an N-terminal long-chain acyl group [96]. Amicoumacin [97], a compound from B. subtilis that is structurally related to xenocoumacin, also employs a hydrolytic maturation strategy in which an N-terminal acyl-asparagine or acyl-glutamine is removed upon activation [98-99]. As with xenocoumacin, the acylated compound is inactive, while the mature compound is a potent antibiotic. Fig. (2) summarizes currently reported NRPS and NRPS/PKS-derived structures that undergo enzymatic hydrolytic maturation. For a dedicated review of NRPS/PKS products that mature via enzy-
Fig. (2).

Known NRPS/PKS products that undergo enzymatic hydrolytic maturation.
The active cleaved metabolite is shown in black and the leader structural features, which may also have biological activity, are shown in grey. The cleaved bond is bolded. For precolibactin A, the proposed thiazoline and thiazole order and its stereochemistry were predicted by bioinformatics, tandem MS, and isotopic labeling studies, and further experimental evidence is needed. matic hydrolysis, the reader is also directed to a recent review by Bode and co-workers [78].
Hydrolytic maturation of secondary metabolites has been well established when extending beyond NRPS/PKS assembly lines. For example, cofactors used in primary metabolism can be incorporated into secondary metabolites and hydrolyzed into smaller structural units. We provide three striking examples: thienamycin, gliotoxin, and lincomycin (Fig. 3). In contrast to the examples discussed above, these compounds retain part of the building blocks incorporated during biosynthesis, rather than using hydrolyzable motifs encoded in thiotemplate assembly line biosynthesis. In thienamycin biosynthesis, coenzyme A (CoA) is used as a source for cysteamine [100]. Notably, the phosphopantethiene arms between CoA and PKS/NRPS carrier proteins are shared, and polar CoA analogs produced inside of the cell typically remain sequestered in the intracellular environment. Two hydrolases associated with the thienamycin biosynthetic gene cluster are responsible for the stepwise hydrolysis of CoA to pantetheine [100]. A third hydrolase processes the carbapenem-pantetheine adduct into thienamycin, which is exported for antimicrobial defense/signaling. In other selected examples, redox-relevant cofactors such as glutathione and mycothiol are also used as sulfur donors in the biosynthesis of secondary metabolites. Gliotoxin, an epidithiodiketopiperazine produced by the fungus Aspergillus fumigatus, is a nonribosomal peptide virulence factor [101-102]. Deletion of individual genes in the gliotoxin cluster revealed that glutathione serves as an unusual sulfur donor [103]. A bisglutathione conjugate is initially formed after synthesis of the core. Subsequent hydrolyses truncate the adduct to a biscysteine conjugate. Cleavage of the C-S bond and oxidation to the disulfide result in mature gliotoxin. An analogous mechanism has been observed in the biosynthesis of the saccharide antibiotic lincomycin A produced by Gram-positive Streptomyces [104]. A mycothiol conjugate is hydrolyzed to release an N-acyl-cysteine conjugate. C-S bond cleavage and methylation follow to produce the final antibiotic. It is intriguing that the latter two examples exploit general cellular toxin detoxification strategies, the thiol nucleophiles glutathione and mycothiol, which typically neutralize electrophilic toxins, for channeling potentially toxic antibiotic intermediates.
Fig. (3).
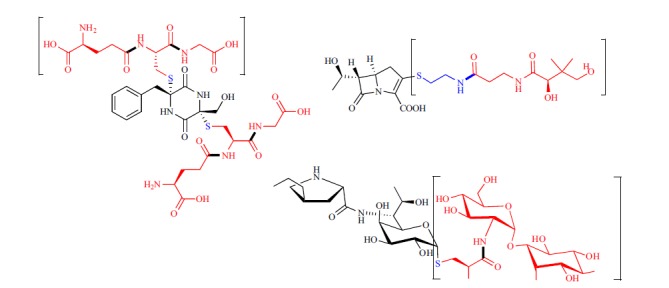
Hydrolytic maturation events are commonly used to introduce structural features into secondary metabolites. Three striking examples are shown. Atoms in blue are retained in the mature structures, while red atoms are excised. Sites of amide bond hydrolysis are bolded. Metabolites involved in primary metabolism that play a role in the biosynthesis of these compounds are bracketed.
With genome sequence information now complementing structural characterization efforts, diverse maturation strategies are now emerging as more prominent routes for nonribosomal peptide processing. Moving forward, we expect more related nonribosomal peptide maturation strategies to emerge. With further mechanistic characterizations and a better understanding of the processing enzymes, the disconnection between expected structures and bioinformatics predictions will begin to close. Additionally, current bioinformatics approaches, which rely on homology to known systems or machine learning algorithms trained on known systems, still fail to identify putative “atypical” gene clusters. Because microbial biosynthetic gene clusters represent chemical traits and are well known to transfer from one organism to another via horizontal gene transfer [105], genome synteny analyses – examining the co-localization of genetic loci in phylogenetically-related organisms – continue to provide a promising route for the discovery of “atypical” natural product enzymes and pathways [28, 106]. The continued characterization of atypical secondary metabolic pathways, new biosynthetic enzymes, and new maturation strategies, and their subsequent integration into online bioinformatics programs, such as antiSMASH, is needed.
5. Nonribosomal peptide building blocks
NRPSs can sample from about 500 amino acid substrates, which provide a good variety of potential monomer building blocks for secondary metabolite structural diversification [66]. The continued characterization of these building blocks and their associated biosynthetic enzymes will aid in both nonribosomal, and more increasingly, in ribosomal peptide/protein engineering. While the gene cluster for colibactin was described in 2006, the recently proposed structure of precolibactin A, which accounts for a majority of the enzymatic domains in the biosynthetic pathway, contains an unexpected cyclopropane moiety [57]. This moiety was similarly found in a smaller precolibactin shunt product, where the cyclopropane was proposed to be derived from the amino acid building block 1-aminocyclopropane-1-carboxylic acid (ACC) [57-59]. In plants, ACC is an intermediate in the production of ethylene, a signaling hormone [107]. ACC is biosyntheized from S-adenosylmethionine in a PLP-dependent manner. In the proposed mechanism, nucleophilic displacement liberates methylthioadenosine and forms the three-membered ring [108]. This mechanism is paralleled in the synthesis of coronatine, albeit with a different leaving group [109]. Coronamic acid is a cyclopropyl containing intermediate that is produced through a cryptic halogenation event. A carrier protein-tethered isoleucine is chlorinated at the γ-carbon by an α-ketogluterate non-heme Fe2+-dependent oxygenase. Deprotonation of the α-carbon yields the enolate, which then displaces chloride, forming the cyclopropane moiety. Other variations of this mechanism are found in the biosynthesis of, for example, kutzneride 2 and curacin A [108].
None of the previously described mechanisms for the biosynthesis of cyclopropane substructures readily appear to be encoded in the colibactin gene cluster. The colibactin gene cluster lacks homologous PLP-dependent or Fe2+-dependent oxygenases. Isotope labeling studies indicate that the four carbons (aminobutyryl) are derived from methionine [57-58], which was also previously observed in the biosynthesis of the cytotrienin cyclopropyl moiety [110]. Since ACC is derived from methionine in bacteria, it is possible that free ACC is loaded onto the carrier protein; however, feeding studies with free deuterated ACC showed no detectable incorporation of the free amino acid in bacterial cell cultures [57]. One NRPS module with an unusual architecture is involved in the production of the spirobicyclic structural feature [57-58]. This protein, ClbH, contains an additional adenylation domain. In contrast to canonical NRPS modules that contain a condensation domain, (C), followed by an adenlyation domain, (A), and then a thiolation domain, (T), this particular protein has the architecture A-C-A-T. The second A-domain was speculatively proposed to potentially convert a carrier protein-tethered Met into SAM for cyclization [58]. Exactly how isotopically labeled Met is processed to the ACC-derived feature in colibactin remains a subject of current investigation. It is not without precedent, however, where NRPS domains directly catalyze the formation of ring-strained units, such as in the biosynthesis of the β-lactam nocardicin [84]. There, the condensation domain catalyzes the condensation with the upstream intermediate as well as β-lactam formation [84].
More recent studies on ClbH indicate that the A1 protein domain fragment activates serine in vitro as predicted by bioinformatics [111]. Free-standing carrier protein ClbE accepts this substrate in vitro, which is further oxidized to α-aminomalonate (detected as its decarboxylation product) by isolated dehydrogenases ClbD and ClbF. ClbG and ClbO, a discrete acyltransferase and PKS module, respectively, which are currently unaccounted for in colibactin biosynthesis, might be available for incorporation of this rare extender unit [111]. α-aminomalonate extender units can also be found in the zwittermicin [112], guadinomine [113], and lumiquinone antibiotics [114]. These genes are necessary for bacterial cells harboring the colibactin pathway to initiate mammalian cell DNA damage, suggesting that functional (pre)colibactin derivatives in the bacteria-host interaction may incorporate an α-aminomalonate substrate [111]. (Pre)Colibactins with this structural unit have not yet been described. Additionally, peptidase ClbL remains to be included in the colibactin biosynthetic model. However, suggestions have been made for ClbL’s potential involvement in a second cleavage event based on gene deletion analysis [58].
Many cyclopropane-containing compounds exhibit toxicity through covalent modifications of DNA. Release of the ring strain contained in cyclopropanes can contribute to their reactivity. Oftentimes, the formation of aromaticity accompanies ring opening for these irreversible reactions. Nucleophilic positions on guanine or adenine can attack the ring, particularly when the ring is positioned in conjugation with an α-β-unsaturated carbonyl, leading to DNA alkylation. For example, in duocarmycin, upon binding DNA, a conformational change positions the cyclopropane for attack by the N3 of adenine [108]. This leads to DNA alkylation and subsequent cytotoxicity. In the case of colibactin, an analogous alkylation reaction can occur (Fig. 4a). However, the proposed alkylated ring-opened intermediate contains a Michael acceptor (or an analogous conjugated iminium acceptor) (Fig. 4b). This could allow the second strand of the duplex DNA to attack, forming an interstrand crosslink between the two strands of DNA [57]. A model precolibactin shunt product, which served as a mimic of the open chain product, was shown to exhibit weak DNA interstrand crosslinking activity in in vitro assays, supporting this notion. Based on this data, we proposed several possible modes of action for colibactin toxicity (open chain and closed chain molecules), which are shown in (Fig. 4ab) [57]. The Balskus group identified the same model compound and subjected it to a ClbP protease cleavage assay in Lysogeny Broth growth conditions (LB, 10 g/L NaCl). A mass consistent with Cl- addition to the cyclopropyl moiety of the closed-chain structure was detected. Based on this data, they similarly proposed a colibactin activity consistent with DNA alkylation (Fig. 4a, bottom) [59]. In isotopically-labeled minimal media, we did not detect Cl- adducts, but we could see masses consistent with model colibactin cleavage products in both the open-chain (primary amine) and closed-chain (imine) forms, from freshly prepared organic extracts [57]. Alkylation and crosslinking activities can result in mutagenesis, activation of apoptotic pathways, and downstream DNA double strand breaks [115].
Fig. (4).
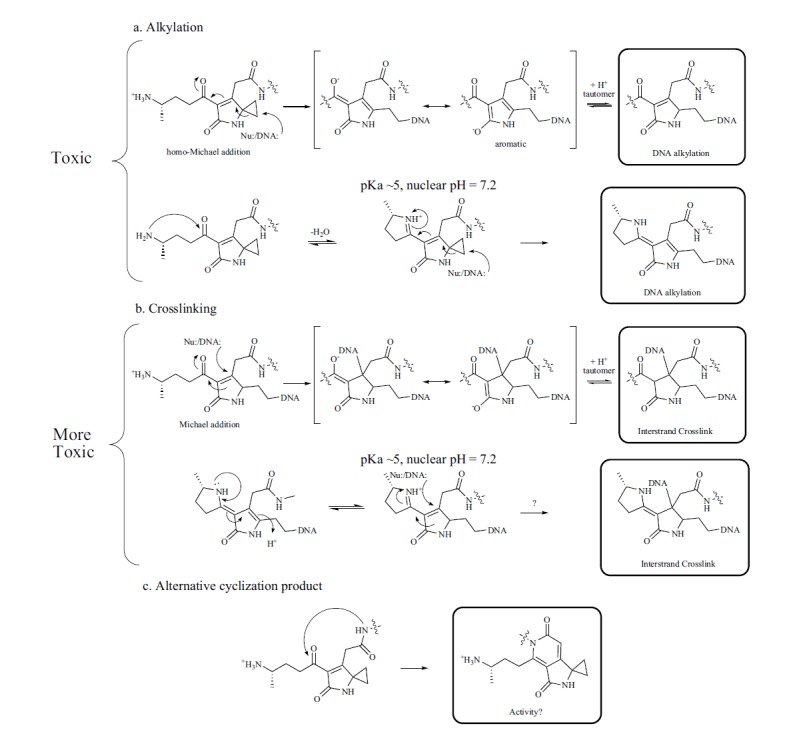
Possible modes of action for the colibactin warhead.
In considering the above possible modes of action, several structural and biosynthetic features need to be considered. As discussed earlier, during export out of the cell, precolibactins are cleaved releasing a primary amine. The amine would largely be protonated under physiological conditions, and may help colibactin bind DNA. Alternatively, the amine could undergo an intramolecular cyclization, forming a reversible cyclic imine (Fig. 4a and b, bottom). The calculated pKa for its conjugate acid iminium species is predicted to be approximately 5. With a nuclear pH of 7.2, less than one percent of the reactive iminium would be expected in the nucleus. While its neutrality may reduce inter-
The colibactin warhead contains a ring-strained cyclopropane. Nucleophilic attack by DNA could lead to opening of the ring and DNA-alkylation (a). Cleavage of precolibactin liberates a primary amino group, which may be involved in DNA binding or modulating the warhead activity by forming a cyclic imine (bottom). A Michael addition (or analogous iminium addition) into the alkylated warhead could result in a DNA-interstrand crosslink (b). Formation of alternative cyclization products (pyridones) could compete with the five-membered cyclic imine cyclization route. No biological activities have yet to be reported for the stable pyridone-containing molecules (c). action with the DNA phosphate backbone, it may alternatively participate in DNA binding, and transient protonation (or DNA binding-induced protonation, pKa perturbation) of the imine may serve to activate the cyclized warhead and promote attack by DNA. Additionally, a cyclopropane-containing metabolite arising from an alternative cyclization mode featuring a pyridone scaffold was recently proposed from the colibactin pathway by MS (Fig. 2 and Fig. 4c) [61]. This compound was isolated in low yields (0.1 mg/ 200 L) from a ΔclbP strain overexpressing the colibactin pathway. In a wildtype strain, protease ClbP would cleave the N-acyl-D-Asn moiety, and consequently, two competing cyclization routes can explain the structural differences (Fig. 4). As no functional data was reported for the pyridone-containing colibactin metabolites, it is currently unclear if these molecules are stable shunt metabolites or advanced biosynthetic products (i.e., precolibactin B?).
Precolibactin A has a predicted thiazolinyl- and thiazole containing moiety that presumably participates in DNA binding and poises the ClbP-cleaved warhead (open- or closed-chain) for electrophilic attack [57]. A bithiazole-containing structure from the colibactin pathway has also been proposed (Fig. 4) [61]. Bleomycin, an antitumor compound produced by Streptomyces, contains a C-terminal bithiazole and is capped with a C-terminal cationic group, whereas phleomycin contains a thiazolinyl-thiazole moiety similarly capped with a cationic group [116]. These structural motifs are required for the action of bleomycin and phleomycin, presumably by mediating DNA binding [117]. While the diverse colibactin products proposed to date may have distinct activities, further mechanistic studies are required to assess the dominant molecular route(s) for colibactin genotoxicty in mammalian cells.
CONCLUSION
The continued expansion of microbial genome sequence information has provided enormous promise of much more to come in novel biosynthesis and secondary metabolite discovery. Based on an overabundance of historical precedence, novel structural scaffolds to come will continue to serve as leads for the development of new-in-class (ant)agonists, molecular probes, and pharmaceuticals. Discovery and characterization of novel biocatalysts will continue to expand our arsenal of biocatalytic reactions while assigning functions to the unannotated majority, hypothetical proteins. Multidisciplinary and systematic genes-to-molecules approaches have been effective at aiding in these overall efforts. In particular, molecular networking has provided a route to begin to overcome current metabolite database inadequacies, especially for groups focused on novel secondary metabolite discovery from functional or activated pathways. Pathway-targeted molecular networking, as highlighted for the genotoxic colibactin pathway, enables a detailed systems biosynthesis-level view of a targeted secondary metabolic pathway within a complex metabolome, and ties into existing molecular networking workflows. The colibactin pathway provides a nice example of the sequence first, structures to follow paradigm in the human microbiome. Deciphering the unexpected structural and functional outcomes for the colibactin pathway and determining its mode of action in vivo remain subjects of rigorous inquiry.
ACKNOWLEDGEMENTS
Our work on the discovery of secondary metabolites in host-bacteria interactions was supported by the National Institutes of Health (National Cancer Institute grant 1DP2-CA186575 and National Institute of General Medical Sciences grant R00-GM097096), the Searle Scholars Program (grant 13-SSP-210), and the Damon Runyon Cancer Research Foundation (grant DFS:05-12).
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.Newman D.J., Cragg G.M. Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012;75(3):311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler M.S., Robertson A.A., Cooper M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014;31(11):1612–1661. doi: 10.1039/c4np00064a. [DOI] [PubMed] [Google Scholar]
- 3.Li J.W, Vederas J.C. Drug Discovery and Natural Products, End of an Era or an Endless Frontier? Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 4.Demain A.L. Importance of microbial natural products and the need to revitalize their discovery. J. Ind. Microbiol. Biotechnol. 2014;41(2):185–201. doi: 10.1007/s10295-013-1325-z. [DOI] [PubMed] [Google Scholar]
- 5.Challis G.L. Mining microbial genomes for new natural products and biosynthetic pathways. Microbiology. 2008;154(Pt 6):1555–1569. doi: 10.1099/mic.0.2008/018523-0. [DOI] [PubMed] [Google Scholar]
- 6.Nett M., Ikeda H., Moore B.S. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 2009;26(11):1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brady S.F., Simmons L., Kim J.H., Schmidt E.W. Metagenomic approaches to natural products from free-living and symbiotic organisms. Nat. Prod. Rep. 2009;26(11):1488–1503. doi: 10.1039/b817078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winter J.M., Behnken S., Hertweck C. Genomics-inspired discovery of natural products. Curr. Opin. Chem. Biol. 2011;15(1):22–31. doi: 10.1016/j.cbpa.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 9.Zotchev S.B., Sekurova O.N., Katz L. Genome-based bioprospecting of microbes for new therapeutics. Curr. Opin. Biotechnol. 2012;23(6):941–947. doi: 10.1016/j.copbio.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Doroghazi J.R., Albright J.C., Goering A.W., Ju K.S., Haines R.R., Tchalukov K.A., Labeda D.P., Kelleher N.L., Metcalf W.W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014;10(11):963–968. doi: 10.1038/nchembio.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bachmann B.O., Van Lanen S.G., Baltz R.H. Microbial genome mining for accelerated natural products discovery, is a renaissance in the making? J. Ind. Microbiol. Biotechnol. 2014;41(2):175–184. doi: 10.1007/s10295-013-1389-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Staley J.T., Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu. Rev. Microbiol. 1985;39:321–346. doi: 10.1146/annurev.mi.39.100185.001541. [DOI] [PubMed] [Google Scholar]
- 13.Kaeberlein T., Lewis K., Epstein S.S. Isolating Uncultivable. microorganisms in pure culture in a simulated natural environment. Science. 2002;296(5570):1127–1129. doi: 10.1126/science.1070633. [DOI] [PubMed] [Google Scholar]
- 14.Nichols D., Cahoon N., Trakhtenberg E.M., Pham L., Mehta A., Belanger A., Kanigan T., Lewis K., Epstein S.S. Use of Ichip for High-Throughput In Situ Cultivation of Uncultivable. Microbial Species. Appl. Environ. Microbiol. 2010;76(8):2445–2450. doi: 10.1128/AEM.01754-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Onofrio A., Crawford J.M., Stewart E.J., Witt K., Gavrish E., Epstein S., Clardy J., Lewis K. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem. Biol. 2010;17(3):254–264. doi: 10.1016/j.chembiol.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis K., Epstein S., D'Onofrio A., Ling L.L. Uncultured microorganisms as a source of secondary metabolites. J. Antibiot. (Tokyo) 2010;63(8):468–476. doi: 10.1038/ja.2010.87. [DOI] [PubMed] [Google Scholar]
- 17.Ling L.L., Schneider T., Peoples A.J., Spoering A.L., Engels I., Conlon B.P., Mueller A., Schaberle T.F., Hughes D.E., Epstein S., Jones M., Lazarides L., Steadman V.A., Cohen D.R., Felix C.R., Fetterman K.A., Millett W.P., Nitti A.G., Zullo A.M., Chen C., Lewis K. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517(7535):455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scherlach K., Hertweck C. Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 2009;7(9):1753–1760. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 19.Williams R.B., Henrikson J.C., Hoover A.R., Lee A.E., Cichewicz R.H. Epigenetic remodeling of the fungal secondary metabolome. Org. Biomol. Chem. 2008;6(11):1895–1897. doi: 10.1039/b804701d. [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Escribano J.P., Bibb M.J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011;4(2):207–215. doi: 10.1111/j.1751-7915.2010.00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen P.R., Chavarria K.L., Fenical W., Moore B.S., Ziemert N. Challenges and triumphs to genomics-based natural product discovery. J. Ind. Microbiol. Biotechnol. 2014;41(2):203–209. doi: 10.1007/s10295-013-1353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J.Y., Sanchez L.M., Rath C.M., Liu X., Boudreau P.D., Bruns N., Glukhov E., Wodtke A., de Felicio R., Fenner A., Wong W.R., Linington R.G., Zhang L., Debonsi H.M., Gerwick W.H., Dorrestein P.C. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013;76(9):1686–1699. doi: 10.1021/np400413s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crawford J.M., Clardy J. Bacterial symbionts and natural products. Chem. Commun. (Camb.) 2011;47(27):7559–7566. doi: 10.1039/c1cc11574j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt E.W., Donia M.S., McIntosh J.A., Fricke W.F., Ravel J. Origin and variation of tunicate secondary metabolites. J. Nat. Prod. 2012;75(2):295–304. doi: 10.1021/np200665k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hentschel U., Piel J., Degnan S.M., Taylor M.W. Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 2012;10(9):641–654. doi: 10.1038/nrmicro2839. [DOI] [PubMed] [Google Scholar]
- 26.Rath C.M., Dorrestein P.C. The bacterial chemical repertoire mediates metabolic exchange within gut microbiomes. Curr. Opin. Microbiol. 2012;15(2):147–154. doi: 10.1016/j.mib.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brachmann A.O., Bode H.B. Identification and Bioanalysis of Natural Products from Insect Symbionts and Pathogens. Adv. Biochem. Eng. Biotechnol. 2013;135:123–155. doi: 10.1007/10_2013_192. [DOI] [PubMed] [Google Scholar]
- 28.Vizcaino M.I., Guo X., Crawford J.M. Merging chemical ecology with bacterial genome mining for secondary metabolite discovery. J. Ind. Microbiol. Biotechnol. 2014;41(2):285–299. doi: 10.1007/s10295-013-1356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lederberg, J. Infectious history. Science. 2000;288(5464):287–293. doi: 10.1126/science.288.5464.287. [DOI] [PubMed] [Google Scholar]
- 30.Donia M.S., Cimermancic P., Schulze C.J., Wieland Brown L.C., Martin J., Mitreva M., Clardy J., Linington R.G., Fischbach M.A. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell. 2014;158(6):1402–1414. doi: 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharon G., Garg N., Debelius J., Knight R., Dorrestein P.C., Mazmanian S.K. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014;20(5):719–730. doi: 10.1016/j.cmet.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donia M.S., Fischbach M.A. Small molecules from the human microbiota. Science. 2015;349(6246):1254766. doi: 10.1126/science.1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hooper L.V., Gordon J.I. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 34.Round J.L., Mazmanian S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009;9(5):313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cani P.D., Delzenne N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009;15:1546–1558. doi: 10.2174/138161209788168164. [DOI] [PubMed] [Google Scholar]
- 36.Ley R.E. Obesity and the human microbiome. Curr. Opin. Gastroenterol. 2010;26(1):5–11. doi: 10.1097/MOG.0b013e328333d751. [DOI] [PubMed] [Google Scholar]
- 37.DiBaise J.K., Frank D.N., Mathur R. Impact of the Gut Microbiota on the Development of Obesity, Current Concepts. Am. J. Gastroenterol. Suppl. 2012;1(1):22–27. [Google Scholar]
- 38.Arthur J.C., Perez-Chanona E., Mühlbauer M., Tomkovich S., Uronis J.M., Fan T-J., Campbell B.J., Abujamel T., Dogan B., Rogers A.B., Rhodes J.M., Stintzi A., Simpson K.W., Hansen J.J., Keku T.O., Fodor A.A., Jobin C. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science. 2012;338(6103):120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arthur J.C., Jobin C. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes. 2013;4(3):253–258. doi: 10.4161/gmic.24220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Vos W.M., de Vos E.A. Role of the intestinal microbiome in health and disease, from correlation to causation. Nutr. Rev. 2012;70(Suppl. 1):S45–S56. doi: 10.1111/j.1753-4887.2012.00505.x. [DOI] [PubMed] [Google Scholar]
- 41.Human Microbiome Project C. A framework for human microbiome research. Nature. 2012;486(7402):215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nougayrede J.P., Homburg S., Taieb F., Boury M., Brzuszkiewicz E., Gottschalk G., Buchrieser C., Hacker J., Dobrindt U., Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313(5788):848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 44.Putze J., Hennequin C., Nougayrede J.P., Zhang W., Homburg S., Karch H., Bringer M.A., Fayolle C., Carniel E., Rabsch W., Oelschlaeger T.A., Oswald E., Forestier C., Hacker J., Dobrindt U. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect. Immun. 2009;77(11):4696–4703. doi: 10.1128/IAI.00522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bondarev V., Richter M., Romano S., Piel J., Schwedt A., Schulz-Vogt H.N. The genus Pseudovibrio contains metabolically versatile bacteria adapted for symbiosis. Environ. Microbiol. 2013;15(7):2095–2113. doi: 10.1111/1462-2920.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engel P., Vizcaino M.I., Crawford J.M. Gut symbionts from distinct hosts exhibit genotoxic activity via divergent colibactin biosynthetic pathways. Appl. Environ. Microbiol. 2014;81(4):1502–1512. doi: 10.1128/AEM.03283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engel P., Bartlett K.D., Moran N.A. The Bacterium Frischella perrara Causes Scab Formation in the Gut of its Honeybee Host. MBio. 2015;6(3) doi: 10.1128/mBio.00193-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nougayrede J.P., Homburg S., Taieb F., Boury M., Brzuszkiewicz E., Gottschalk G., Buchrieser C., Hacker J., Dobrindt U., Oswald E. Escherichia coli induces DNA double-stranded breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 49.Cuevas-Ramos G., Petit C., Marcq I., Boury M., Oswald E., Nougayrede J.P., Isberg R. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA. 2010;107(25):11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nowrouzian F.L., Oswald E. Escherichia coli strains with the capacity for long-term persistence in the bowel microbiota carry the potentially genotoxic pks island. Microb. Pathog. 2012;53(3-4):180–182. doi: 10.1016/j.micpath.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 51.Arthur J.C., Perez-Chanona E., Muhlbauer M., Tomkovich S., Uronis J.M., Fan T.J., Campbell B.J., Abujamel T., Dogan B., Rogers A.B., Rhodes J.M., Stintzi A., Simpson K.W., Hansen J.J., Keku T.O., Fodor A.A., Jobin C. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science. 2012;338(6103):120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dalmasso G., Cougnoux A., Delmas J., Darfeuille-Michaud A., Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. 2014;5(5):675–680. doi: 10.4161/19490976.2014.969989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olier M., Marcq I., Salvador-Cartier C., Secher T., Dobrindt U., Boury M., Bacquie V., Penary M., Gaultier E., Nougayrede J.P., Fioramonti J., Oswald E. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes. 2012;3(6):501–509. doi: 10.4161/gmic.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brotherton C.A., Balskus E.P. A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J. Am. Chem. Soc. 2013;135(9):3359–3362. doi: 10.1021/ja312154m. [DOI] [PubMed] [Google Scholar]
- 55.Bian X., Fu J., Plaza A., Herrmann J., Pistorius D., Stewart A.F., Zhang Y., Muller R. In vivo evidence for a prodrug activation mechanism during colibactin maturation. ChemBioChem. 2013;14(10):1194–1197. doi: 10.1002/cbic.201300208. [DOI] [PubMed] [Google Scholar]
- 56.Vizcaino M.I., Engel P., Trautman E., Crawford J.M. Comparative metabolomics and structural characterizations illuminate colibactin pathway-dependent small molecules. J. Am. Chem. Soc. 2014;136(26):9244–9247. doi: 10.1021/ja503450q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vizcaino M.I., Crawford J.M. The colibactin warhead crosslinks DNA. Nat. Chem. 2015;7(5):411–417. doi: 10.1038/nchem.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bian X.Y., Plaza A., Zhang Y.M., Muller R. Two more pieces of the colibactin genotoxin puzzle from Escherichia coli show incorporation of an unusual 1-aminocyclopropanecarboxylic acid moiety. Chem. Sci. (Camb.) 2015;6(5):3154–3160. doi: 10.1039/c5sc00101c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brotherton C.A., Wilson M., Byrd G., Balskus E.P. Isolation of a metabolite from the pks island provides insights into colibactin biosynthesis and activity. Org. Lett. 2015;17(6):1545–1548. doi: 10.1021/acs.orglett.5b00432. [DOI] [PubMed] [Google Scholar]
- 60.Brotherton C.A., Wilson M., Byrd G., Balskus E.P. Correction to Isolation of a Metabolite from the pks Island Provides Insights into Colibactin Biosynthesis and Activity. Org. Lett. 2015;17(9):2294. doi: 10.1021/acs.orglett.5b01056. [DOI] [PubMed] [Google Scholar]
- 61.Li Z.R., Li Y., Lai J.Y., Tang J., Wang B., Lu L., Zhu G., Wu X., Xu Y., Qian P.Y. Critical intermediates reveal novel biosynthetic events in the enigmatic colibactin pathway. ChemBioChem. 2015;16(12):1715–1719. doi: 10.1002/cbic.201500239. [DOI] [PubMed] [Google Scholar]
- 62.Vizcaino M.I., Crawford J.M. Secondary metabolic pathway-targeted metabolomics. Methods Mol. Biol. doi: 10.1007/978-1-4939-3375-4_12. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khosla C. Quo vadis, enzymology? Nat. Chem. Biol. 2015;11(7):438–441. doi: 10.1038/nchembio.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fischbach M.A., Walsh C.T. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics, logic, machinery, and mechanisms. Chem. Rev. 2006;106(8):3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 65.Strieker M., Tanovic A., Marahiel M.A. Nonribosomal peptide synthetases, structures and dynamics. Curr. Opin. Struct. Biol. 2010;20(2):234–240. doi: 10.1016/j.sbi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 66.Walsh C.T., O'Brien R.V., Khosla C. Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem. Int. Ed. Engl. 2013;52(28):7098–7124. doi: 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Medema M. H., Blin K., Cimermancic P., de Jager V., Zakrzewski P., Fischbach M. A., Weber T., Takano E., Breitling R. antiSMASH, rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. . Nucleic Acids Res. 2011. [DOI] [PMC free article] [PubMed]
- 68.Blin K., Medema M.H., Kazempour D., Fischbach M.A., Breitling R., Takano E., Weber T. antiSMASH 2.0--a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41(W1):W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blin K., Kazempour D., Wohlleben W., Weber T. Improved lanthipeptide detection and prediction for antiSMASH. PLoS One. 2014;9(2):e89420. doi: 10.1371/journal.pone.0089420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weber T., Blin K., Duddela S., Krug D., Kim H.U., Bruccoleri R., Lee S.Y., Fischbach M.A., Muller R., Wohlleben W., Breitling R., Takano E., Medema M.H. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43(W1):W237-43. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cimermancic P., Medema M.H., Claesen J., Kurita K., Wieland Brown L.C., Mavrommatis K., Pati A., Godfrey P.A., Koehrsen M., Clardy J., Birren B.W., Takano E., Sali A., Linington R.G., Fischbach M.A. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014;158(2):412–421. doi: 10.1016/j.cell.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dantas G., Sommer M.O., Degnan P.H., Goodman A.L. Experimental approaches for defining functional roles of microbes in the human gut. Annu. Rev. Microbiol. 2013;67:459–475. doi: 10.1146/annurev-micro-092412-155642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fang J., Dorrestein P.C. Emerging mass spectrometry techniques for the direct analysis of microbial colonies. Curr. Opin. Microbiol. 2014;19:120–129. doi: 10.1016/j.mib.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watrous J., Roach P., Alexandrov T., Heath B.S., Yang J.Y., Kersten R.D., van der Voort M., Pogliano K., Gross H., Raaijmakers J.M., Moore B.S., Laskin J., Bandeira N., Dorrestein P.C. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA. 2012;109(26):E1743–E1752. doi: 10.1073/pnas.1203689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moree W.J., Phelan V.V., Wu C.H., Bandeira N., Cornett D.S., Duggan B.M., Dorrestein P.C. Interkingdom metabolic transformations captured by microbial imaging mass spectrometry. Proc. Natl. Acad. Sci. USA. 2012;109(34):13811–13816. doi: 10.1073/pnas.1206855109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Traxler M.F., Watrous J.D., Alexandrov T., Dorrestein P.C., Kolter R. Interspecies interactions stimulate diversification of the Streptomyces coelicolor secreted metabolome. MBio. 2013;4(4) doi: 10.1128/mBio.00459-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nguyen D.D., Wu C.H., Moree W.J., Lamsa A., Medema M.H., Zhao X., Gavilan R.G., Aparicio M., Atencio L., Jackson C., Ballesteros J., Sanchez J., Watrous J.D., Phelan V.V., van de Wiel C., Kersten R.D., Mehnaz S., De Mot R., Shank E.A., Charusanti P., Nagarajan H., Duggan B.M., Moore B.S., Bandeira N., Palsson B.O., Pogliano K., Gutierrez M., Dorrestein P.C. MS/MS networking guided analysis of molecule and gene cluster families. Proc. Natl. Acad. Sci. USA. 2013;110(28):E2611–E2620. doi: 10.1073/pnas.1303471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reimer D., Bode H.B. A natural prodrug activation mechanism in the biosynthesis of nonribosomal peptides. Nat. Prod. Rep. 2014;31(2):154–159. doi: 10.1039/c3np70081j. [DOI] [PubMed] [Google Scholar]
- 79.Arnison P.G., Bibb M.J., Bierbaum G., Bowers A.A., Bugni T.S., Bulaj G., Camarero J.A., Campopiano D.J., Challis G.L., Clardy J., Cotter P.D., Craik D.J., Dawson M., Dittmann E., Donadio S., Dorrestein P.C., Entian K.D., Fischbach M.A., Garavelli J.S., Goransson U., Gruber C.W., Haft D.H., Hemscheidt T.K., Hertweck C., Hill C., Horswill A.R., Jaspars M., Kelly W.L., Klinman J.P., Kuipers O.P., Link A.J., Liu W., Marahiel M.A., Mitchell D.A., Moll G.N., Moore B.S., Muller R., Nair S.K., Nes I.F., Norris G.E., Olivera B.M., Onaka H., Patchett M.L., Piel J., Reaney M.J., Rebuffat S., Ross R.P., Sahl H.G., Schmidt E.W., Selsted M.E., Severinov K., Shen B., Sivonen K., Smith L., Stein T., Sussmuth R.D., Tagg J.R., Tang G.L., Truman A.W., Vederas J.C., Walsh C.T., Walton J.D., Wenzel S.C., Willey J.M., van der Donk W.A. Ribosomally synthesized and post-translationally modified peptide natural products, overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013;30(1):108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hashimoto M., Komori T., Kamiya T., Nocardicin A. A New Monocyclic 8-Lactam Antibiotic II. Structure Determination Of Nocardicins A and B. J. Antibiot. 1976;29:890–901. doi: 10.7164/antibiotics.29.890. [DOI] [PubMed] [Google Scholar]
- 81.Gunsior M., Breazeale S.D., Lind A.J., Ravel J., Janc J.W., Townsend C.A. The biosynthetic gene cluster for a monocyclic beta-lactam antibiotic, nocardicin A. Chem. Biol. 2004;11(7):927–938. doi: 10.1016/j.chembiol.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 82.Davidsen J.M., Bartley D.M., Townsend C.A. Non-ribosomal propeptide precursor in nocardicin A biosynthesis predicted from adenylation domain specificity dependent on the MbtH family protein NocI. J. Am. Chem. Soc. 2013;135(5):1749–1759. doi: 10.1021/ja307710d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gaudelli N.M., Townsend C.A. Epimerization and substrate gating by a TE domain in beta-lactam antibiotic biosynthesis. Nat. Chem. Biol. 2014;10(4):251–258. doi: 10.1038/nchembio.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gaudelli N.M., Long D.H., Townsend C.A. beta-Lactam formation by a non-ribosomal peptide synthetase during antibiotic biosynthesis. Nature. 2015;520(7547):383–387. doi: 10.1038/nature14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kevany B.M., Rasko D.A., Thomas M.G. Characterization of the complete zwittermicin A biosynthesis gene cluster from Bacillus cereus. Appl. Environ. Microbiol. 2009;75(4):1144–1155. doi: 10.1128/AEM.02518-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Silo-Suh L.A., Lethbridge B.J., Raffel S.J., He H., Clardy J., Handelsman J. Biological Activities of Two Fungistatic Antibiotics Produced by Bacillus cereus UW85. Appl. Environ. Microbiol. 1994;60:2023–2030. doi: 10.1128/aem.60.6.2023-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reimer D., Pos K.M., Thines M., Grun P., Bode H.B. A natural prodrug activation mechanism in nonribosomal peptide synthesis. Nat. Chem. Biol. 2011;7(12):888–890. doi: 10.1038/nchembio.688. [DOI] [PubMed] [Google Scholar]
- 88.Dubois D., Baron O., Cougnoux A., Delmas J., Pradel N., Boury M., Bouchon B., Bringer M.A., Nougayrede J.P., Oswald E., Bonnet R. ClbP is a prototype of a peptidase subgroup involved in biosynthesis of nonribosomal peptides. J. Biol. Chem. 2011;286(41):35562–35570. doi: 10.1074/jbc.M111.221960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cougnoux A., Gibold L., Robin F., Dubois D., Pradel N., Darfeuille-Michaud A., Dalmasso G., Delmas J., Bonnet R. Analysis of structure-function relationships in the colibactin-maturating enzyme ClbP. J. Mol. Biol. 2012;424(3-4):203–214. doi: 10.1016/j.jmb.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 90.Xu Y., Kersten R.D., Nam S.J., Lu L., Al-Suwailem A.M., Zheng H., Fenical W., Dorrestein P.C., Moore B.S., Qian P.Y. Bacterial biosynthesis and maturation of the didemnin anti-cancer agents. J. Am. Chem. Soc. 2012;134(20):8625–8632. doi: 10.1021/ja301735a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Masschelein J., Mattheus W., Gao L.J., Moons P., Van Houdt R., Uytterhoeven B., Lamberigts C., Lescrinier E., Rozenski J., Herdewijn P., Aertsen A., Michiels C., Lavigne R.A. PKS/NRPS/FAS hybrid gene cluster from Serratia plymuthica RVH1 encoding the biosynthesis of three broad spectrum, zeamine-related antibiotics. PLoS One. 2013;8(1):e54143. doi: 10.1371/journal.pone.0054143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wendenbaum S., Demange P., Dell A., Meyer J.M., Abdallah M.A. The Structure of Pyoverdine Pa, the Siderophore of Pseudomonas-Aeruginosa. Tetrahedron Lett. 1983;24(44):4877–4880. [Google Scholar]
- 93.Schalk I.J. Metal trafficking via siderophores in Gram-negative bacteria, specificities and characteristics of the pyoverdine pathway. J. Inorg. Biochem. 2008;102(5-6):1159–1169. doi: 10.1016/j.jinorgbio.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 94.Yeterian E., Martin L.W., Guillon L., Journet L., Lamont I.L., Schalk I.J. Synthesis of the siderophore pyoverdine in Pseudomonas aeruginosa involves a periplasmic maturation. Amino Acids. 2010;38(5):1447–1459. doi: 10.1007/s00726-009-0358-0. [DOI] [PubMed] [Google Scholar]
- 95.Drake E.J., Gulick A.M. Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in pyoverdine synthesis. ACS Chem. Biol. 2011;6(11):1277–1286. doi: 10.1021/cb2002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koketsu K., Watanabe K., Suda H., Oguri H., Oikawa H. Reconstruction of the saframycin core scaffold defines dual Pictet-Spengler mechanisms. Nat. Chem. Biol. 2010;6(6):408–410. doi: 10.1038/nchembio.365. [DOI] [PubMed] [Google Scholar]
- 97.Itoh J., Omoto S., Shomura T., Nishizawa N., Miyado S., Yuda Y., Shibata U., Inouye S. Amicoumacin-A, A new antibiotic with strong antiinflammatory and antiulcer activity. J. Antibiot. 1981;34:611–613. doi: 10.7164/antibiotics.34.611. [DOI] [PubMed] [Google Scholar]
- 98.Li Y., Xu Y., Liu L., Han Z., Lai P.Y., Guo X., Zhang X., Lin W., Qian P.Y. Five new amicoumacins isolated from a marine-derived bacterium Bacillus subtilis. Mar. Drugs. 2012;10(2):319–328. doi: 10.3390/md10020319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li Y., Li Z., Yamanaka K., Xu Y., Zhang W., Vlamakis H., Kolter R., Moore B.S., Qian P.Y. Directed natural product biosynthesis gene cluster capture and expression in the model bacterium Bacillus subtilis. Sci. Rep. 2015;5:9383. doi: 10.1038/srep09383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Freeman M.F., Moshos K.A., Bodner M.J., Li R., Townsend C.A. Four enzymes define the incorporation of coenzyme A in thienamycin biosynthesis. Proc. Natl. Acad. Sci. USA. 2008;105(32):11128–11133. doi: 10.1073/pnas.0804500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bell M.R., Johnson J.R., Wildi B.S., Woodward R.B. The structure of gliotoxin. J. Am. Chem. Soc. 1958;80(4):1001–1001. [Google Scholar]
- 102.Scharf D.H., Heinekamp T., Remme N., Hortschansky P., Brakhage A.A., Hertweck C. Biosynthesis and function of gliotoxin in Aspergillus fumigatus. Appl. Microbiol. Biotechnol. 2012;93(2):467–472. doi: 10.1007/s00253-011-3689-1. [DOI] [PubMed] [Google Scholar]
- 103.Scharf D.H., Chankhamjon P., Scherlach K., Heinekamp T., Willing K., Brakhage A.A., Hertweck C. Epidithiodiketopiperazine Biosynthesis, A Four-Enzyme Cascade Converts Glutathione Conjugates into Transannular Disulfide Bridges. Angew. Chem. Int. Ed. 2013;52(42):11092–11095. doi: 10.1002/anie.201305059. [DOI] [PubMed] [Google Scholar]
- 104.Zhao Q., Wang M., Xu D., Zhang Q., Liu W. Metabolic coupling of two small-molecule thiols programs the biosynthesis of lincomycin A. Nature. 2015;518(7537):115–119. doi: 10.1038/nature14137. [DOI] [PubMed] [Google Scholar]
- 105.Dobrindt U., Hochhut B., Hentschel U., Hacker J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004;2(5):414–424. doi: 10.1038/nrmicro884. [DOI] [PubMed] [Google Scholar]
- 106.Guo X., Crawford J.M. An atypical orphan carbohydrate-NRPS genomic island encodes a novel lytic transglycosylase. Chem. Biol. 2014;21(10):1271–1277. doi: 10.1016/j.chembiol.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Adams D.O., Yang S.F. Ethylene Biosynthesis - Identification of 1-Aminocyclopropane-1-Carboxylic Acid as an Intermediate in the Conversion of Methionine to Ethylene. Proc. Natl. Acad. Sci. USA. 1979;76(1):170–174. doi: 10.1073/pnas.76.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thibodeaux C.J., Chang W.C., Liu H.W. Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem. Rev. 2012;112(3):1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vaillancourt F.H., Yeh E., Vosburg D.A., O'Connor S.E., Walsh C.T. Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature. 2005;436(7054):1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 110.Zhang H., Kakeya H., Osada H. Biosynthesis of 1-aminoeyclopropane-l-carboxylic acid moiety on cytotrienin A in Streptomyces sp. Tetrahedron Lett. 1998;39:6947–6948. [Google Scholar]
- 111.Brachmann A.O., Garcie C., Wu V., Martin P., Ueoka R., Oswald E., Piel J. Colibactin biosynthesis and biological activity depend on the rare aminomalonyl polyketide precursor. Chem. Commun. (Camb.) 2015;51(66):13138–13141. doi: 10.1039/c5cc02718g. [DOI] [PubMed] [Google Scholar]
- 112.Chan Y.A., Boyne M.T., II, Podevels A.M., Klimowicz A.K., Handelsman J., Kelleher N.L., Thomas M.G. Hydroxymalonyl-acyl carrier protein (ACP) and aminomalonyl-ACP are two additional type I polyketide synthase extender units. Proc. Natl. Acad. Sci. USA. 2006;103(39):14349–14354. doi: 10.1073/pnas.0603748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Holmes T.C., May A.E., Zaleta-Rivera K., Ruby J.G., Skewes-Cox P., Fischbach M.A., DeRisi J.L., Iwatsuki M., Omura S., Khosla C. Molecular insights into the biosynthesis of guadinomine, a type III secretion system inhibitor. J. Am. Chem. Soc. 2012;134(42):17797–17806. doi: 10.1021/ja308622d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park H.B., Crawford J.M. Lumiquinone A, an alpha-Aminomalonate-Derived Aminobenzoquinone from Photorhabdus luminescens. J. Nat. Prod. 2015;78(6):1437–1441. doi: 10.1021/np500974f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fu D., Calvo J.A., Samson L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer. 2012;12(2):104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Takita T., Muraoka Y., Yoshioka T., Fujii A., Maeda K., Umezawa H. Chemistry of Bleomycin. 9. Structures of Bleomycin and Phleomycin. J. Antibiot. 1972;25(12):755–758. doi: 10.7164/antibiotics.25.755. [DOI] [PubMed] [Google Scholar]
- 117.Stubbe J., Kozarich J.W. Mechanisms of bleomycin-induced DNA degradation. Chem. Rev. 1987;87:1107–1136. [Google Scholar]