

Abstract
Background and Purpose
PGE2 inhibits cytokine generation from human lung macrophages. However, the EP receptor that mediates this beneficial anti‐inflammatory effect of PGE2 has not been defined. The aim of this study was to identify the EP receptor by which PGE2 inhibits cytokine generation from human lung macrophages. This was determined by using recently developed EP receptor ligands.
Experimental Approach
The effects of PGE2 and EP‐selective agonists on LPS‐induced generation of TNF‐α and IL‐6 from macrophages were evaluated. The effects of EP2‐selective (PF‐04852946, PF‐04418948) and EP4‐selective (L‐161,982, CJ‐042794) receptor antagonists on PGE2 responses were studied. The expression of EP receptor subtypes by human lung macrophages was determined by RT‐PCR.
Key Results
PGE2 inhibited LPS‐induced and Streptococcus pneumoniae‐induced cytokine generation from human lung macrophages. Analysis of mRNA levels indicated that macrophages expressed EP2 and EP4 receptors. L‐902,688 (EP4 receptor‐selective agonist) was considerably more potent than butaprost (EP2 receptor‐selective agonist) as an inhibitor of TNF‐α generation from macrophages. EP2 receptor‐selective antagonists had marginal effects on the PGE2 inhibition of TNF‐α generation, whereas EP4 receptor‐selective antagonists caused rightward shifts in the PGE2 concentration–response curves.
Conclusions and Implications
These studies demonstrate that the EP4 receptor is the principal receptor that mediates the anti‐inflammatory effects of PGE2 on human lung macrophages. This suggests that EP4 receptor agonists could be effective anti‐inflammatory agents in human lung disease.
Abbreviation
- FCS
fetal calf serum
Tables of Links
| TARGETS |
|---|
| GPCRs |
| EP2 receptor |
| EP4 receptor |
| LIGANDS | |
|---|---|
| Butaprost | PF‐04418948 |
| CJ‐042794 | PF‐04852946 |
| L‐161,982 | PGE2 |
| L‐902,688 | Roflumilast |
| Misoprostol | Salbutamol |
| ONO‐AE1‐259 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
PGE2 is known to have wide‐ranging effects on a variety of tissues. These effects of PGE2 are mediated through specific EP receptors of which four have been identified (Coleman et al., 1994; Breyer et al., 2001; Woodward et al., 2011). In the lung, PGE2 can act on airway smooth muscle to mediate bronchodilation. This has led to suggestions that targeting EP receptors may be of benefit in the treatment of respiratory diseases (Kawakami et al., 1973; Melillo et al., 1994; Gauvreau et al., 1999). An undesirable effect of PGE2, however, is that it also induces cough (Maher et al., 2011). Nonetheless, cough and bronchodilation appear to be mediated by different receptors, suggesting that selective targeting of the beneficial receptor might be possible. The EP3 receptor has been linked to cough (Maher et al., 2011), whereas bronchodilation appears to be mediated by EP4 receptors (Buckley et al., 2011; Benyahia et al., 2012). Identification of the relevant EP receptor that mediates the beneficial effects of PGE2 is likely to be valuable information from a clinical perspective.
The human lung macrophage plays an important role in host defence in the lung. However, aberrant activation of lung macrophages has been linked to respiratory diseases, chronic obstructive pulmonary disease in particular (Barnes, 2008). PGE2 has been shown to inhibit pro‐inflammatory cytokine release from lung macrophages (Rowe et al., 1997; Ratcliffe et al., 2007; Buenestado et al., 2012). This effect of PGE2 on human lung macrophages has been reported to be mediated by EP2 and EP4 receptors (Ratcliffe et al., 2007). However, this conclusion was drawn at a time when the availability of selective pharmacological ligands at EP2 and EP4 receptors was limited. The situation has now changed with the recent emergence of novel ligands such as PF‐04418948, the first potent and selective EP2 receptor antagonist reported (af Forselles et al., 2011). Use of these novel experimental tools has provided an opportunity to reappraise the mechanism by which PGE2 stabilizes macrophage responses. In this regard, use of these tools has shown that the EP4 receptor is the main receptor regulating functional responses in THP‐1 cells, a human monocytic cell line (Birrell et al., 2015).
The aim of the present study was to identify the EP receptor responsible for mediating the inhibitory effects of PGE2 on pro‐inflammatory cytokine release from human lung macrophages. This was determined by using a variety of pharmacological ligands, principally, a range of EP2 receptor‐selective and EP4 receptor‐selective antagonists. These studies demonstrate that the EP4 receptor is the principal receptor that mediates the anti‐inflammatory effects of PGE2 on human lung macrophages, suggesting that EP4 receptor agonists could be effective anti‐inflammatory agents in human lung disease.
Methods
Lung tissue
The use of human lung tissue in this study was approved by the National Research Ethics' Service (REC reference: 15/NW/0657). Informed written consent was obtained.Non‐lesional lung tissue was obtained from surgical resections. Most patients were undergoing surgery for carcinoma. Sixty‐two preparations were used in this study, and these were derived from 31 male and 31 female participants. Ages of participants ranged from 49 to 88 years with a median age of 71. Details of the patients’ smoking status are presented in the Supporting information (Figure S1).
Macrophage isolation
Lung tissue was chopped with scissors in RPMI‐1640 (50 mL per 5 g of lung tissue) and filtered through 100 μm nylon mesh (Incamesh, Warrington, UK) over a collection vessel. This cycle of chopping and washing was repeated. The filtrate (100–200 mL) was centrifuged (300 × g, 10 min) at room temperature; the supernatant aspirated and the pellets resuspended in 40–50 mL of RPMI‐1640 supplemented with 10% FCS, penicillin (25 U·mL−1), streptomycin (25 μg·mL−1), gentamicin (50 μg·mL−1) and amphotericin B (1 μg·mL−1). The cell suspensions were inverted several times and left to sediment at 4°C for 1 h according to a protocol modified from Liu et al. (1984). After sedimentation, the supernatant was aspirated, and the sedimented material was resuspended in supplemented RPMI‐1640. This sedimentation step at 4°C was repeated. The sedimented material was resuspended in 30 mL PIPES buffer and centrifuged (300 × g, 10 min, room temperature). The resulting pellet was resuspended in PIPES buffer, and the suspension was filtered through nylon mesh before being layered on to a discontinuous Percoll gradient.
One 20 mL Percoll gradient was used for cells harvested from every 5 g of lung tissue. Isotonic Percoll (nine‐part Percoll to one‐part 10× PIPES buffer) was diluted with PIPES buffer to produce an 80% Percoll gradient. The cell suspension (20 mL) was layered onto the gradient and centrifuged (400 × g, 20 min, room temperature) resulting in a flocculent layer containing macrophages. The interface was harvested, and two washes were performed with PIPES buffer (50 mL). Following centrifugation, (488 × g, 10 min at room temperature) the resulting cell pellet was resuspended in 10 mL of supplemented RPMI‐1640 (or for infection experiments, supplemented RPMI‐1640 without antibiotics). The cells were counted using a haemocytometer. Macrophages were seeded at 2 × 105 per well in a 24‐well cell culture plate with 1 mL of supplemented RPMI‐1640 (or for infection experiments, supplemented RPMI‐1640 without antibiotics) and incubated overnight (37°C, 5% CO2).
The purity of cell suspensions was determined by morphology using cytospins (Thermo Shandon Cytospin 3). Cytospins were stained with Quick‐Diff and processed according to the manufacturer's instructions. Cell viability was assessed by erythrosin‐B exclusion. In this study, macrophage purity was 85 ± 2%, and cell viability was 92 ± 1%.
Macrophage activation protocol
After incubation overnight, medium from the wells was removed and replaced with fresh supplemented RPMI‐1640 (1 mL) 2 h before the start of the experiment. Where pharmacological agents were used, the cells were pretreated with these (30 to 60 min at 37°C, 5% CO2) before addition of stimulus. When agonists were used, macrophages were first incubated with or without indomethacin for 30 min and then with or without agonist for a further 30 min before addition of LPS. When antagonists were used, cells were incubated first with indomethacin (30 min), then with antagonist (1 h) followed by agonist (30 min) before activation. The cells were incubated (37°C, 5% CO2) for 22 h with the stimulus. The cell culture supernatants were then harvested and centrifuged (488 × g, 4 min, room temperature). The resulting supernatants were stored at −80°C until analysis for cytokine content. TNF‐α and IL‐6 were analysed using commercially available elisa kits (Rsg kits; Ebioscience, Hatfield, UK). PGE2 was also analysed using a commercially available kit (Cayman Chemical Company, Ann Arbour, MI, USA).
Preparation of Streptococcus pneumoniae
Type 2 S. pneumoniae (Spn) strain D39 was grown and stored as previously described (Dockrell et al., 2001). Bacteria were opsonized by resuspending pellets in RPMI‐1640 with 10% anti‐pneumococcal immune serum and incubating at 37°C for 30 min on a rotating stand. Pellets were then washed three times in PBS and resuspended in RPMI‐1640 supplemented with 10% fetal calf serum (FCS) without antibiotics.
Macrophage infection protocol
After incubation overnight, medium from the wells was removed and replaced with fresh supplemented RPMI‐1640 without antibiotics (1 mL) 2 h before the start of the experiment. Opsonized Type 2 Spn strain D39 (see above) were added to the cells at a multiplicity of infection (MOI) of 1, or the cells were mock‐infected. The cells were incubated at 4°C for 1 h to maximize bacterial adherence followed by incubation at 37°C for 3 h for internalization. The wells were then washed with PBS, and the cell culture medium was replaced with the re‐addition of pharmacological agents as appropriate. The cells were incubated at 37°C until 22 h post‐infection. The cell culture supernatants were then harvested and stored at −80°C until analysis for cytokine content.
Assessment of total cell cAMP
Macrophages (2 × 105 cells) were incubated (30 min) with or without indomethacin (1 μM) and then with PGE2 (0.5 to 5 h) in supplemented RPMI‐1640 (1 mL). After incubation, the supernatants were removed and the cells solubilized by addition of ice‐cold acidified ethanol and snap frozen in liquid nitrogen. After thawing, the ethanol was recovered and centrifuged (13 000 × g, 2 min) to pellet any cellular debris. The supernatant was then evaporated off under vacuum using a rotary evaporator. The dried residue was reconstituted in assay buffer (250 μL) and stored at −80°C. Total cell cAMP content was determined using a commercially available kit (Cayman Chemical Company).
RT‐PCR
RNA was extracted from purified macrophages (1 to 5 × 106 cells) using Tri‐Reagent (1 mL). In order to generate cDNA, samples were processed essentially as described elsewhere (Kay et al., 2013). Amplification of cDNA was performed by PCR using conditions and primer pairs for human EP receptor subtypes (Schlötzer‐Schrehardt et al., 2002; Thorat et al., 2008). The house‐keeping gene, β‐actin, was also amplified. Primers were synthesized by Sigma (Poole, UK). PCR products were sequenced in‐house to ensure that correct amplification had taken place as described in more detail elsewhere (Kay et al., 2013).
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Antagonist affinity (pKB) was determined by using the Gaddum equation: pKB = log(dose ratio − 1) − log(antagonist concentration) (Kenakin, 1984). Maximal responses (Emax) and potencies (EC50) were determined by nonlinear regression analysis (GraphPad Prism, version 5.0d, La Jolla, CA, USA). Statistical significance was determined utilizing Student's paired t‐tests or repeated measures anova as appropriate. When analysing data by anova, post hoc tests were either Dunnett's test or Tukey's test. Comparisons were considered significant when P < 0.05.
Materials
The buffers used were: PBS (composition, mM): NaCl 137, Na2HPO4.12H2O 8, KCl 2.7 and KH2PO4 1.5. PIPES buffer contained (mM) the following: PIPES (22), NaCl (110) and KCl (5), and the pH was titrated to 7.4 with NaOH.
Stock solutions (10 mM) of PGE2, butaprost (free acid), L‐902,688, misoprostol (free acid) and indomethacin were prepared in ethanol and stored at −20°C. ONO‐AE1‐259 was made up in distilled water (10 mM stock) and stored at −20°C. All antagonists, PF‐04852946, PF‐04418948, CJ‐042794 and L‐161,982, formerly known as EP4A (Machwate et al., 2001), were prepared as stock solutions (10 mM) in dimethyl sulphoxide and stored at −20°C. Salbutamol was prepared as a stock solution (10 mM), dissolved in distilled water and stored at 4°C. Roflumilast was prepared as a stock solution (10 mM) in dimethyl sulphoxide and stored at −20°C. LPS from Escherichia coli serotype R515 (Re) was provided as a 1 mg·mL−1 stock solution and stored at 4°C.
The materials used were supplied as follows: indomethacin, PGE2, Percoll, salbutamol, Tri‐Reagent (all Sigma); gentamicin, penicillin/streptomycin, fungizone, RPMI 1640, (Invitrogen, Paisley, UK); butaprost, misoprostol, L‐902,688 (Cayman Chemical Company); L‐161,982 (Tocris Bioscience, Bristol, UK); roflumilast (Santa Cruz Biotechnology, Heidelberg, Germany); Quick‐Diff (Reagena, Toivala, Finland); FCS (Promocell, Heidelberg, Germany); and LPS (Enzo Life Sciences, Exeter, UK).
PF‐04418948, PF‐04852946 and CJ‐042794 were obtained from Pfizer Global Research and Development (Sandwich, UK). PF‐04418948 will be available commercially from Sigma‐Aldrich, Tocris and Toronto Research Chemicals Inc (North York, ON, Canada). ONO‐AE1‐259 was a kind gift from Ono Pharmaceutical Company Ltd (Osaka, Japan).
Results
PGE2 inhibits cytokine generation from macrophages
In keeping with previous studies, PGE2 inhibited LPS‐induced TNF‐α generation from human lung macrophages in a concentration‐dependent manner. This experiment was carried out in the absence (Figure 1A) and presence (Figure 1B) of the cyclo‐oxygenase (COX) inhibitor indomethacin (1 μM). PGE2 was a more potent (EC50; 3.2 ± 0.6 cf 10.8 ± 2.0 nM) and efficacious (Emax; 77 ± 1.8 cf 53.5 ± 2.0% inhibition) inhibitor of LPS‐induced TNF‐α generation in the presence of indomethacin (Figure 1C). Moreover, in the presence of indomethacin (1 μM), TNF‐α generation by LPS was significantly (P < 0.05) higher than in its absence (2657 ± 496 cf 1648 ± 213 pg·mL−1; n = 13).
Figure 1.
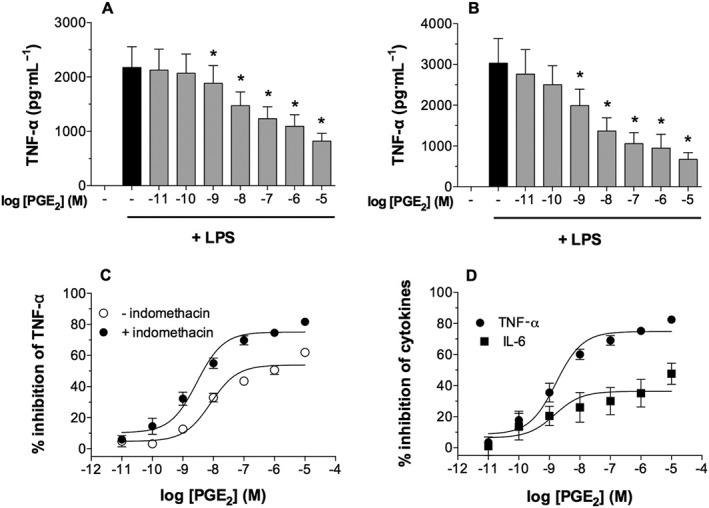
Effects of PGE2 on cytokine generation from macrophages. Macrophages were pre‐incubated without (A) or with (B) indomethacin (1 μM) for 30 min and then with or without PGE2 for 30 min before challenge with LPS (1 ng·mL−1) for 22 h after which supernatants were harvested and assayed for TNF‐α generation. The data in (A) and (B) were reworked as % inhibition of the control unblocked release of TNF‐α, and this is shown in (C). In further experiments, macrophages were pre‐incubated (30 min) with indomethacin (1 μM) and then with or without PGE2 for 30 min before challenge with LPS (1 ng·mL−1) for 22 h and both IL‐6 and TNF‐α measured in the supernatants (D). Values are expressed as the % inhibition of control cytokine releases, which were 2422 ± 510 pg·mL−1 of TNF‐α and 4992 ± 1980 pg·mL−1 of IL‐6. Data shown are means ± SEM, for nine (A, B and C) or six (D) experiments. * P < 0.05; significantly different from unblocked control levels.
These experiments suggested that macrophages produce PGE2 in response to LPS, which acts in a paracrine fashion to limit TNF‐α generation. Further experiments confirmed that macrophages generate a small amount of PGE2 spontaneously and larger quantities following challenge with LPS (data not shown). In order to eliminate the potentially confounding influence of endogenous PGE2 generation in the context of receptor characterizations, in all subsequent functional studies, indomethacin was also included.
In further studies, the effects of PGE2 on LPS‐induced IL‐6 as well as TNF‐α generation were determined (Figure 1D). PGE2 inhibited TNF‐α and IL‐6 generation with similar potency (EC50; ~1.6 nM), but PGE2 was less efficacious as an inhibitor of IL‐6 generation than TNF‐α.
Macrophages express EP2 and EP4 receptors
Expression of EP receptors by human lung macrophages was determined by RT‐PCR. The data indicate that human lung macrophages express message for EP2 and EP4 receptors but do not express message for EP1 or EP3 receptors (Figure 2).
Figure 2.
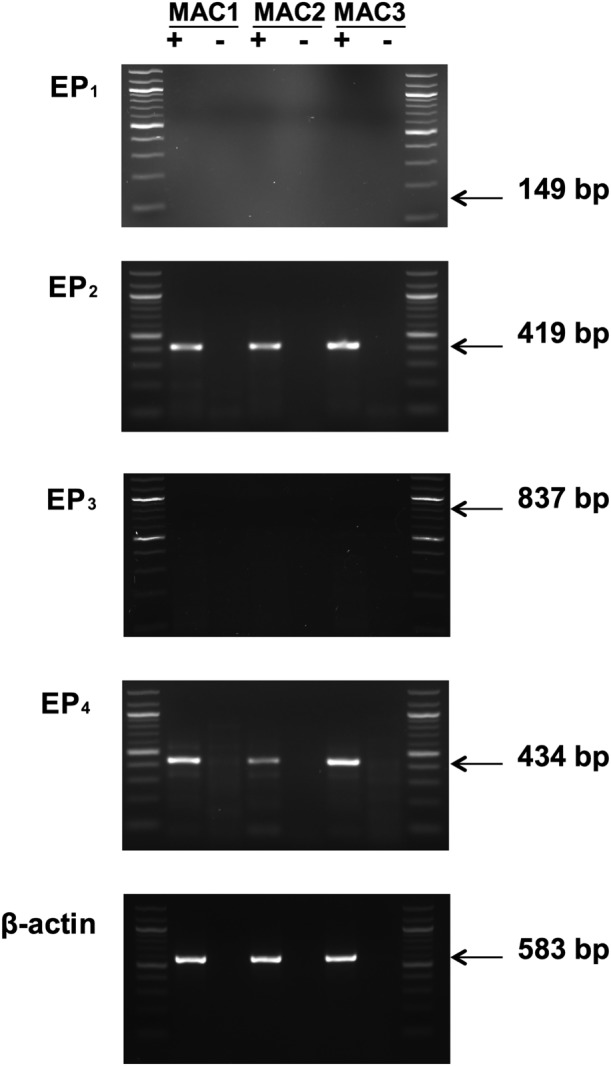
EP receptor expression in macrophages. Isolated RNA was converted to cDNA by reverse trancriptase (+), and as a control, this reaction step was also carried out in the absence of reverse transcriptase (−). Amplification of cDNA was performed using primers specific for each of the EP receptor subtypes and β‐actin. Expression profiles for three macrophage preparations (MAC1, MAC2 and MAC3) are shown. No mRNA for EP1 receptors was detected in macrophages but, in separate experiments, the presence of EP1 receptors could be readily demonstrated in several breast cancer cell lines, MDA‐MB‐468, MDA‐MB‐231 and ZR‐75‐1 (Kay et al., 2013). No mRNA for EP3 receptors was detected, but in separate experiments, EP3 receptors could be detected in the human mast cell line, LAD‐2 (Kay et al., 2013). These findings are representative of a total of five different macrophage preparations in excess of 95% purity. Lanes at either end of each gel represent a 100 bp ladder.
PGE2 increases macrophage cAMP levels
Since EP2 and EP4 receptors are G‐protein receptors coupled to adenylyl cyclase, we investigated whether exposure (30 min) of macrophages to PGE2 (1 μM) induced increases in total cell cAMP. Our data demonstrated that PGE2 induced statistically significant (P < 0.05) increases in total cell cAMP levels over basal (Figure 3). Further studies demonstrated that PGE2 maintained these increased cAMP levels in macrophages for up to 5 h (data not shown).
Figure 3.
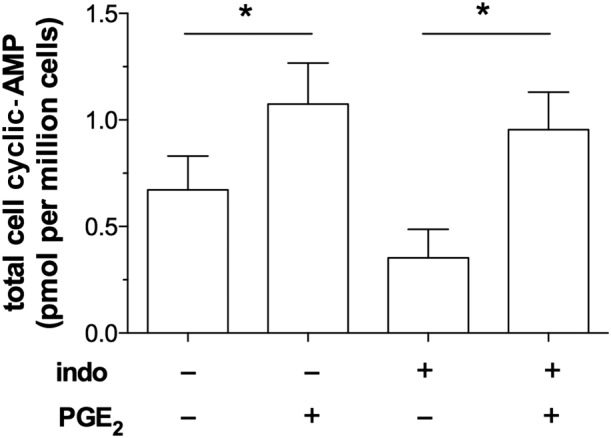
Effect of PGE2 on cAMP. Macrophages were pre‐incubated (30 min) with or without indomethacin (indo; 1 μM) and then with or without PGE2 (1 μM) for a further 30 min. After this treatment, the cells were solubilized and total cell cAMP levels measured. Data shown are means ± SEM for five experiments. * P < 0.05; significantly different from unstimulated control levels.
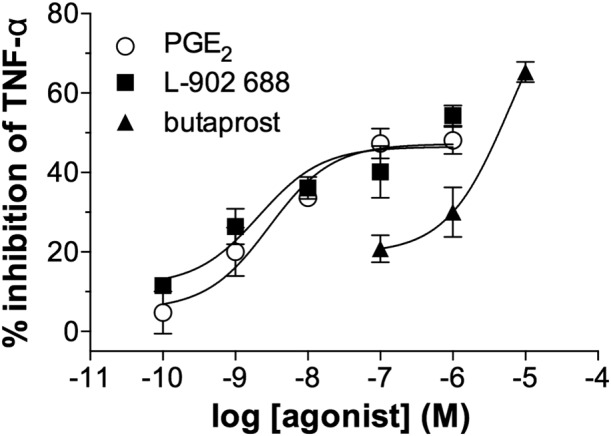
EP4 receptor agonists are far more potent inhibitors than EP2 receptor agonists
The effects of alternative EP agonists on macrophage function were explored. The effects of misoprostol (non‐selective), butaprost (EP2 receptor‐selective) and L‐902,688 (EP4 receptor‐selective) on LPS‐induced TNF‐α generation from macrophages were investigated. The data show that misoprostol (Figure 4A) was about 26‐fold less potent than PGE2 as an inhibitor of TNF‐α generation (Table 1). The EP4 receptor agonist, L‐902,688 (Figure 4B), was sevenfold more potent than PGE2 as an inhibitor of TNF‐α generation whereas, by contrast, the EP2 receptor‐selective agonist, butaprost (Figure 4C), was over 400‐fold less potent than PGE2 in this system (Table 1). In further studies, the effects of another EP2 receptor‐selective agonist, ONO‐AE1‐259, were determined and ONO‐AE1‐259 was about 40‐fold less potent than PGE2 (Table 1).
Figure 4.
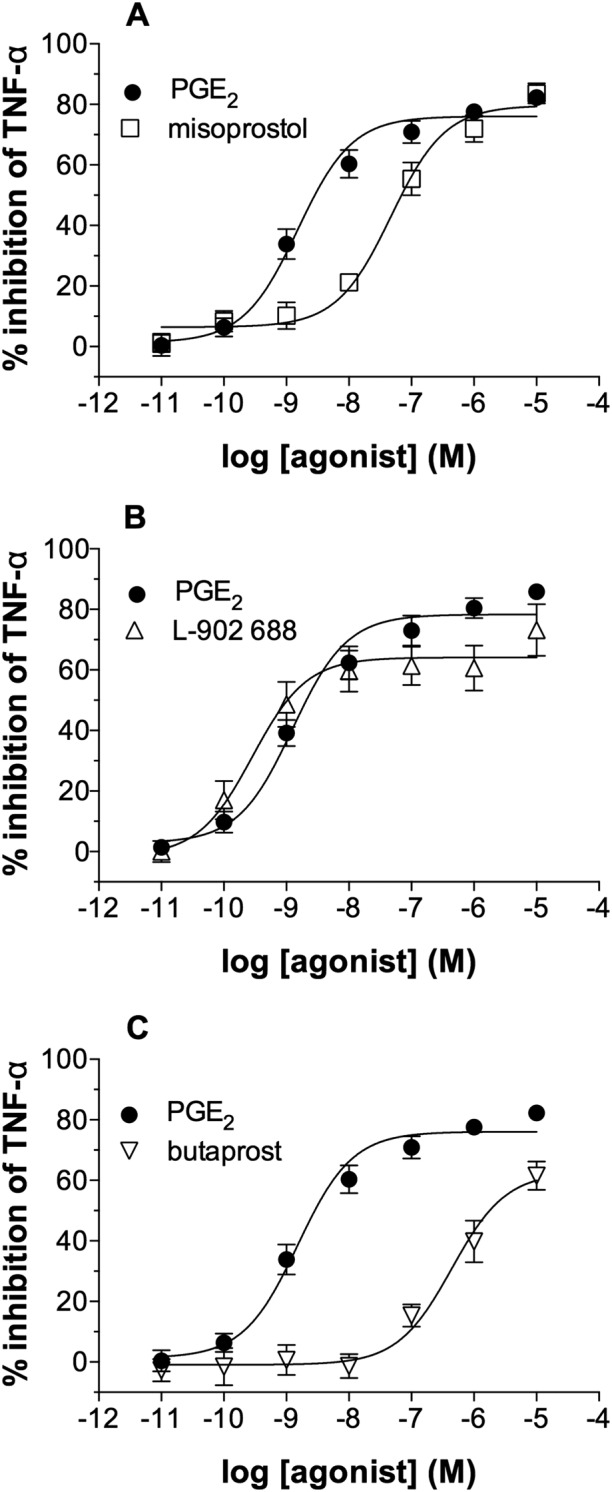
Effects of EP receptor agonists on macrophages. Macrophages were pre‐incubated (30 min) with indomethacin (1 μM) and then with or without either (A) misoprostol, (B) L‐902,688, (C) butaprost or PGE2 for 30 min before challenge with LPS (1 ng·mL−1) for 22 h after which TNF‐α was measured in the supernatants. Results are expressed as the % inhibition of control cytokine release, which was 1379 ± 431 pg·mL−1 of TNF‐α. Data shown are means ± SEM for five (A, B) or six (C) experiments.
Table 1.
EC50 and Emax values for the inhibition of TNF‐α generation by EP receptor agonists
| Agonist | EC50 (nM) | Emax (%) |
|---|---|---|
| PGE2 | 2.1 ± 0.6 | 77 ± 3 |
| Misoprostol | 54 ± 9.1 | 80 ± 4 |
| L‐902688 | 0.3 ± 0.1 | 63 ± 7 |
| Butaprost | 878 ± 340 | 67 ± 5 |
| ONO‐AE1‐259 | 82 ± 24 | 43 ± 4 |
Experimental details relevant to this Table can be found in the legend of Figure 4. Values are means ± SEM from five (misoprostol, l‐902,688, ONO‐AE1‐259), six (butaprost) and eight (PGE2) experiments.
EP4 receptor antagonists reverse the effects of PGE2
The effects of the antagonists PF‐04418948 (EP2 receptor‐selective) and CJ‐042794 (EP4 receptor‐selective) were investigated (Murase et al., 2008; af Forselles et al., 2011). Macrophages were incubated with either PF‐04418948 (300 nM) or CJ‐042794 (300 nM) before incubation with PGE2 and then challenged with LPS. CJ‐042 794 effectively antagonized the PGE2 inhibition of TNF‐α generation (Figure 5A). No antagonism of the PGE2 inhibition was seen with PF‐04418948 (Figure 5B).
Figure 5.
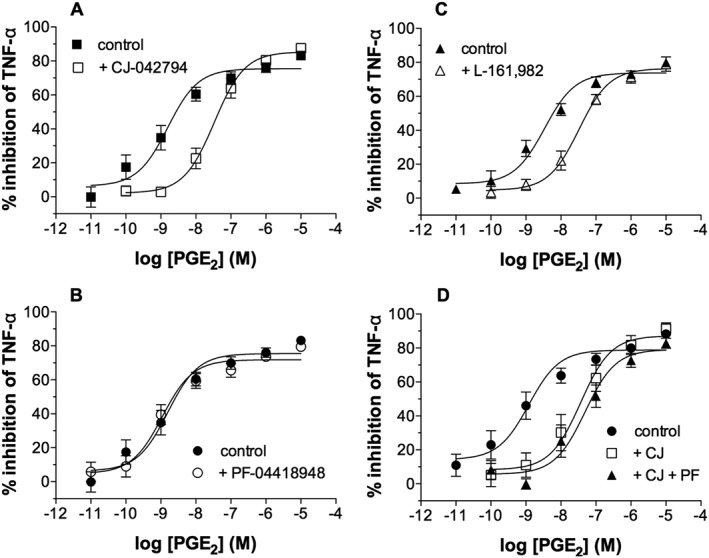
Effects of EP receptor antagonists on PGE2. Macrophages were pre‐incubated with indomethacin (1 μM) for 30 min and then without or with EP receptor‐selective antagonists (300 nM) for 1 h and then without or with PGE2 for 30 min before challenge with LPS (1 ng·mL−1) for 22 h after which TNF‐α was measured in the supernatants. The effects on PGE2 of (A) the EP4 receptor‐selective antagonist CJ‐042 794, (B) the EP2 receptor‐selective antagonist PF‐04 418 948, (C) the EP4 receptor‐selective antagonist L‐161,982 and (D) CJ‐042 794 with and without PF‐04 418 948 were evaluated. Results are expressed as the % inhibition of control TNF‐α releases which were, in the absence and presence of antagonist respectively, (A) 2646 ± 562 and 2582 ± 496 pg mL−1, (B) 2912 ± 532 and 2881 ± 507 pg·mL−1, (C) 2756 ± 882 and 2873 ± 862 pg·mL−1 and (D) 2672 ± 972 to 2212 ± 799 pg·mL−1. Data shown are means ± SEM for five (A, B and D) and six (C) experiments respectively.
Another EP4 receptor‐selective antagonist, L‐161,982 (Machwate et al., 2001), was also evaluated, and in agreement with data obtained with CJ‐042794, L‐161,982 (300 nM) was found to be effective as an antagonist (Figure 5C). Another EP2 receptor‐selective antagonist, PF‐04852946, was also studied. PF‐04852946 is structurally distinct from PF‐04418948 and is about 10‐fold more potent than PF‐04418948 at EP2 receptors (Kay et al., 2013). PF‐04852946 (30 nM) was found to be an ineffective antagonist of the PGE2 inhibition of TNF‐α generation (data not shown).
pKB estimates for the antagonism of PGE2 by CJ‐042794 and L‐161,982 were 8.77 ± 0.13 (KB, 1.7 nM) and 8.46 ± 0.12 (KB, 3.5 nM) respectively. These affinities are consistent with effects of these compounds at EP4 receptors (Jones et al., 2009).
In further studies to determine whether a contribution of the PGE2 effect on macrophages might be mediated by the EP2 receptor, the effect of a combination of EP2 and EP4 receptor‐selective antagonists on the PGE2 inhibition was investigated. The data demonstrate that combined use of PF‐04418948 (300 nM) and CJ‐042794 (300 nM) caused marginally greater antagonism of the PGE2 response than CJ‐042794 alone (Figure 5D). These data indicate that if the EP2 receptor does contribute to the PGE2 response in macrophages, then the contribution is, at best, minimal. These data further emphasize that EP4 receptors are the principal receptors mediating the anti‐inflammatory effects of PGE2 on macrophages.
PGE2 inhibits TNF‐α generation induced by S. pneumoniae
While LPS is an effective tool to activate macrophages, we also investigated whether the response of macrophages to a respiratory pathogen, S. pneumoniae (Spn), could be attenuated by PGE2 (Figure 6). Preliminary studies indicated that Spn‐induced TNF‐α generation from macrophages in a concentration‐dependent fashion with maximal levels of release at an MOI of 1 (data not shown). Further studies demonstrated that PGE2 concentration‐dependently inhibited TNF‐α generation induced by Spn (MOI of 1). The effects of alternative agonists, L‐902,688 and butaprost on Spn‐induced TNF‐α generation from macrophages were also investigated. The EP4 receptor agonist, L‐902,688 (EC50; ~2 nM), was slightly more potent than PGE2 (EC50; ~3 nM) as an inhibitor of TNF‐α generation, whereas by contrast, the EP2 receptor‐selective agonist, butaprost, was less potent than PGE2.
Figure 6.
Effects of PGE2 and other EP receptor agonists on Spn‐induced TNF‐α generation. Macrophages were pre‐incubated (30 min) with indomethacin (1 μM) and then with or without either PGE2, L‐902,688 or butaprost for 30 min before challenge with Spn (MOI of 1) for 22 h after which TNF‐α was measured in the supernatants. Results are expressed as the % inhibition of the control cytokine release, which was 1346 ± 669 pg·mL−1 of TNF‐α. Data shown are means ± SEM for four experiments.
PGE2 is more effective than either salbutamol or roflumilast
In further studies, we compared the effects of PGE2 with established drugs used in the treatment of respiratory diseases. PGE2 was found to be both more potent and efficacious than the β2‐adrenoceptor agonist salbutamol (Figure 7A) as an inhibitor of TNF‐α generation from macrophages driven by LPS. Similar studies with roflumilast, an inhibitor of the cAMP‐specific PDE (PDE4), demonstrated that roflumilast was a considerably weaker inhibitor than PGE2 (Figure 7B). Further studies were performed to determine whether roflumilast (30 nM) might enhance the effects of PGE2. The data show that, in the context of inhibiting LPS‐induced TNF‐α generation, the effect of roflumilast on the inhibition by PGE2 was at best additive (Figure 7C).
Figure 7.
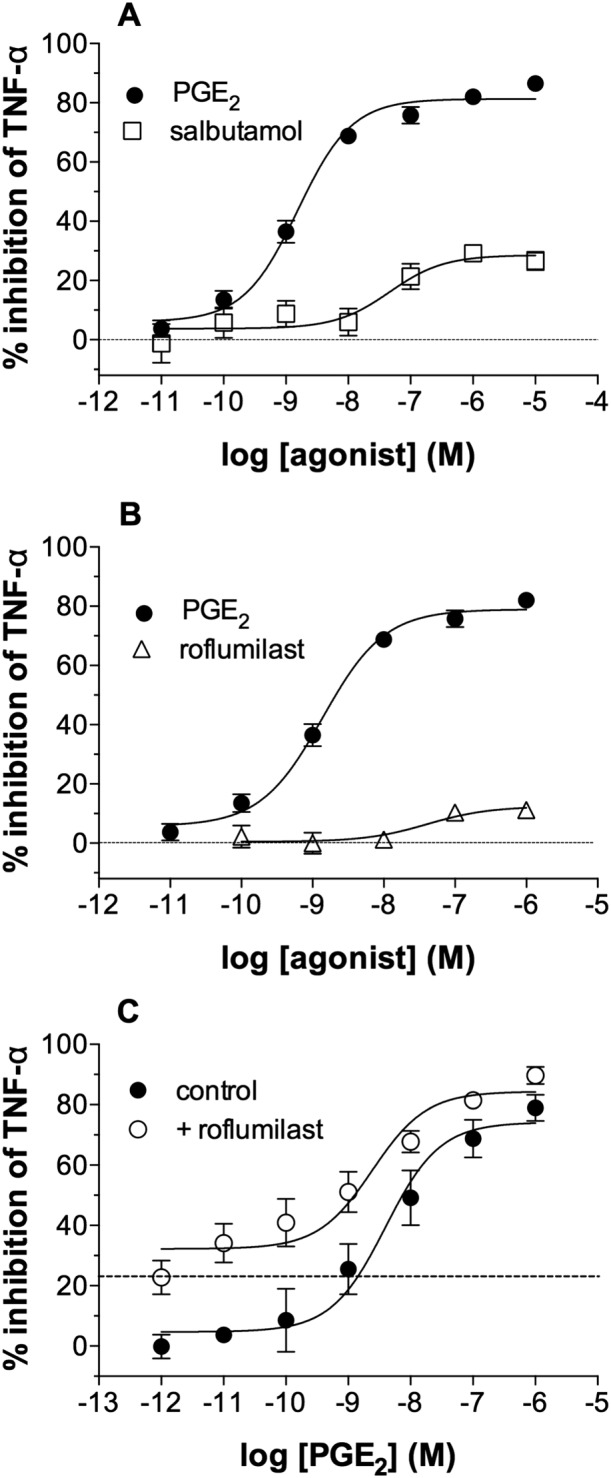
Effects of salbutamol and roflumilast on macrophages. Macrophages were pre‐incubated (30 min) with indomethacin (1 μM) and then with or without either (A) salbutamol, (B) roflumilast or (C) PGE2 in the absence (control) or presence of a single concentration of roflumilast (30 nM) for 30 min before challenge with LPS (1 ng·mL−1) for 22 h after which TNF‐α was measured in the supernatants. The horizontal grid line in (C) shows the inhibition seen with roflumilast alone (22 ± 5% inhibition). Results are expressed as the % inhibition of the unblocked control TNF‐α releases, which ranged from 2363 ± 835 to 2208 ± 969 pg·mL−1. Data shown are means ± SEM for five (A, B and C) experiments.
Discussion
In this study, we showed that PGE2 was an effective inhibitor of cytokine generation from activated macrophages. Furthermore, we showed that PGE2 acts principally through the EP4 receptor to stabilize the pro‐inflammatory responses of human lung macrophages. This suggests that, in lung diseases in which activated macrophages participate, EP4 agonists could be effective anti‐inflammatory agents.
In order to identify which EP receptors are expressed by macrophages, a number of approaches were adopted. Evaluation of mRNA expression by RT‐PCR demonstrated that lung macrophages express both EP2 and EP4 receptors but not EP1 or EP3 receptors. These data suggest that EP2 and/or EP4 receptors are involved in mediating the effects of PGE2 in human lung macrophages. This was further reinforced by the finding that PGE2 induced increases in total cell cAMP in macrophages. Because both EP2 and EP4 receptors are known to be coupled to adenylyl cyclase, increases in cAMP are consistent with the expression of EP2 and/or EP4 receptors in macrophages (Wilson et al., 2004).
In attempts to characterize EP receptors further, a range of EP receptor agonists were studied for effects on cytokine generation. The non‐selective agonist, misoprostol, was about 26‐fold less sensitive than PGE2 as an inhibitor of LPS‐induced TNF‐α generation. This potency ratio is consistent with an effect of misoprostol at EP4 receptors because misoprostol is about 29‐fold less potent than PGE2 at EP4 receptors, whereas at EP2 receptors misoprostol is about sevenfold less potent than PGE2 (Abramovitz et al., 2000). Other agonists were also studied, and it was of interest that the EP4 receptor agonist, L‐902,688, was about sevenfold more potent than PGE2. This finding provides preliminary evidence that the EP4 receptor is involved in mediating the effects of PGE2. Although EP2 receptor‐selective agonists were active in this system, the concentrations of both butaprost and ONO‐AE1–259 required for inhibition were higher than those usually associated with effects at EP2 receptors. In this system, butaprost was over 400‐fold less potent than PGE2, whereas at EP2 receptors, butaprost has been reported to be about 18‐fold less potent than PGE2 (Abramovitz et al., 2000). Also, it is noteworthy that butaprost is known to activate EP4 receptors when used at high enough concentrations (Tang et al., 2000; Clarke et al., 2004; Wilson et al., 2004; Benyahia et al., 2012). Overall, these data provide strong evidence that the EP4 receptor is responsible for mediating the effects of PGE2 but evidence for involvement of the EP2 receptor cannot be excluded.
In order to obtain a definitive characterization of EP receptors involved, the effects of EP2 and EP4 receptor‐selective antagonists on the PGE2 response in macrophages were evaluated. It is noteworthy that the EP2 receptor antagonists, PF‐04418948 and PF‐04852946, that were used in this study are highly selective ligands (af Forselles et al., 2011; Kay et al., 2013) and considerably superior to AH6809, which until now was the only EP2 receptor antagonist available. Indeed, AH6909 has been used in recent studies to invoke a role for EP2 receptors (O'Brien et al., 2014). However, AH6809 shows poor selectivity and potency such that data generated with this antagonist are unlikely to be reliable (Abramovitz et al., 2000; Jones et al., 2009). Neither of the two EP2 receptor antagonists used in this study had any effect on the PGE2 inhibition of TNF‐α generation. By contrast, two EP4 receptor antagonists, CJ‐042 794 (KB; 1.7 nM) and L‐161,982 (KB; 3.5 nM), effectively reversed the PGE2 inhibition of TNF‐α generation with affinities consistent with antagonism at EP4 receptors (Jones et al., 2009). Combining an EP2 receptor antagonist with an EP4 receptor antagonist did lead to a marginal rightward shift in the PGE2 concentration–response curve over that seen with an EP4 receptor antagonist alone. This could mean that a very small component of the PGE2 inhibition is driven by EP2 receptors. Overall, these data provide strong evidence that the principal receptor that mediates the anti‐inflammatory effects of PGE2 in human lung macrophages is the EP4 receptor.
The suggestion has been made that the EP4 receptor could be a target for respiratory diseases. This contention has been based largely on recent studies showing that PGE2 mediates bronchodilation via the EP4 receptor (Buckley et al., 2011; Benyahia et al., 2012). The present study has demonstrated that targeting the EP4 receptor may also provide desirable anti‐inflammatory effects by preventing cytokine generation from macrophages. In this regard, it is of interest that PGE2 attenuated the generation of both TNF‐α and IL‐6 in human lung macrophages, which differs from findings reported for mouse alveolar macrophages in which PGE2 inhibited TNF‐α but, by contrast, potentiated IL‐6 generation (Konya et al., 2015).
The potential therapeutic value of targeting EP receptors is reinforced by the finding that PGE2 was effective at attenuating cytokine generation from macrophages activated by not only LPS but also the respiratory pathogen, S.pneumoniae. Moreover, it is noteworthy that PGE2 was considerably more efficacious and potent than either salbutamol or roflumilast as an inhibitor of LPS‐induced TNF‐α generation from macrophages. Bronchodilators such as salbutamol are β2‐adrenoceptor agonists that may possess some anti‐inflammatory activity (Donnelly et al., 2010). The mechanism of action of the PDE4 inhibitor roflumilast is not entirely known although anti‐inflammatory effects have been suggested (Giembycz and Field, 2010). However, our data suggest that EP4 agonists are likely to show far greater anti‐inflammatory potential than either β2‐adrenoceptor agonists or PDE inhibitors.
In an allied context, it was notable that the PGE2 response was relatively consistent among macrophage preparations (see Supporting Information Fig. S1). This could be important from a therapeutic perspective, as it is possible that factors such as disease state, smoking status and age could influence macrophage functionality (Berenson et al., 2006; Hodge et al., 2007; Suzuki et al., 2008). While we were unable to stratify effectively our population according to disease state, we were able to stratify according to smoking status and age (see Supporting Information Fig. S1). There was clearly no difference in the inhibitory response to PGE2 among macrophages isolated from smokers, ex‐smokers or never smokers. Moreover, there was no influence of age on the inhibitory response to PGE2. This consistency in response could be an advantage when considering the potential of targeting the EP4 receptor therapeutically.
In conclusion, our studies demonstrated that the EP4 receptor was the principal receptor that mediated the anti‐inflammatory effects of PGE2 in human lung macrophages. This suggests that EP4 receptor agonists could be effective anti‐inflammatory agents in lung diseases that are associated with aberrant macrophage activation.
Author contributions
S.K.G., Y.Y., L.J.K. and M.A.B. performed the experimental work; H.M.M. and P.T.P. designed the study; P.T.P. and S.K.G. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Inter‐preparation responses to PGE2. Macrophages were pre‐incubated (30 min) with indomethacin (1 μM) and then with or without PGE2 for 30 min before challenge with LPS (1 ng mL‐1) for 22 h after which TNFα was measured in the supernatants. Values are expressed as the % inhibition of the unblocked control TNFα release. The effects of PGE2 in 32 macrophage preparations are shown (A). Data have been stratified according to age (B) or smoking status (C). Values are means ± SEM. (B) Values are for 5 people, 50‐60 years old; 7 people, 60‐70 years old, 17 people, 70‐80 years old and 3 people, 80‐90 years old. (C) Values are for 14 (smokers), 13 (ex‐smokers) and 5 (non‐smokers) preparations.
Supporting info item
Acknowledgements
The authors thank Mr J Edwards, Mr J Rao, Miss L Socci and Mr D Hopkinson (Cardiothoracic Surgery, Northern General Hospital) and Dr SK Suvarna, Dr P Kitsanta and Dr J Bury (Histopathology, Royal Hallamshire Hospital) for their invaluable help in providing lung tissue specimens. We thank Professor Stephen Renshaw for insightful suggestions. We thank Dr Nick Pullen (Pfizer, USA) for facilitating provision of PF‐04852946, PF‐04418948 and CJ‐042794. Sharonjit Gill is supported by a BBSRC‐Pfizer CASE studentship.
Gill, S. K. , Yao, Y. , Kay, L. J. , Bewley, M. A. , Marriott, H. M. , and Peachell, P. T. (2016) The anti‐inflammatory effects of PGE2 on human lung macrophages are mediated by the EP4 receptor. British Journal of Pharmacology, 173: 3099–3109. doi: 10.1111/bph.13565.
References
- Abramovitz M, Adam M, Boie Y, Carrière M‐C, Denis D, Godbout C et al. (2000). The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta 1483: 285–293. [DOI] [PubMed] [Google Scholar]
- af Forselles KJ, Root J, Clarke T, Davey D, Aughton K, Dack K et al. (2011). In vitro and in vivo characterisation of PF‐04418948, a novel, potent and selective prostaglandin E2 receptor‐2 (EP2) antagonist. Br J Pharmacol 164: 1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (2008). Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 8: 183–192. [DOI] [PubMed] [Google Scholar]
- Benyahia C, Gomez I, Kanyinda L, Boukais K, Danel C, Leséche D et al. (2012). PGE2 receptor (EP4) agonists: potent dilators of human bronchi and future asthma therapy. Pulm Pharmacol Ther 25: 115–118. [DOI] [PubMed] [Google Scholar]
- Berenson CS, Wrona CT, Grove LJ, Maloney J, Garlipp MA, Wallace PK et al. (2006). Impaired alveolar macrophage response to Haemophilus antigens in chronic obstructive lung disease. Am J Respir Crit Care Med 173: 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell MA, Maher SA, Dekkak B, Jones V, Wong S, Brook P et al. (2015). Anti‐inflammatory effects of PGE2 in the lung: role of the EP4 receptor. Thorax 70: 740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD (2001). Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 41: 661–690. [DOI] [PubMed] [Google Scholar]
- Buckley J, Birrell MA, Maher SA, Nials AT, Clarke DL, Belvisi MG (2011). EP4 receptor as a new target for bronchodilator therapy. Thorax 66: 1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenestado A, Grassin‐Delyle S, Guitard F, Naline E, Faisy C, Israël‐Biet D et al. (2012). Roflumilast inhibits the release of chemokines and TNF‐α from human lung macrophages stimulated with lipopolysaccharide. Br J Pharmacol 165: 1877–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DL, Belvisi MG, Catley MC, Yacoub MH, Newton R, Giembycz MA (2004). Identification in human airways smooth muscle cells of the prostanoid receptor and signalling pathway through which PGE2 inhibits the release of GM‐CSF. Br J Pharmacol 141: 1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S (1994). International union of pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 46: 205–229. [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockrell DH, Lee M, Lynch DH, Read RC (2001). Immune‐mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J Infect Dis 184: 713–722. [DOI] [PubMed] [Google Scholar]
- Donnelly LE, Tudhope SJ, Fenwick PS, Barnes PJ (2010). Effects of formoterol and salmeterol on cytokine release from monocyte‐derived macrophages. Eur Respir J 36: 178–186. [DOI] [PubMed] [Google Scholar]
- Gauvreau GM, Watson RM, O'Byrne PM (1999). Protective effects of inhaled PGE2 on allergen‐induced airway responses and airway inflammation. Am J Respir Crit Care Med 159: 31–36. [DOI] [PubMed] [Google Scholar]
- Giembycz MA, Field SA (2010). Roflumilast: first phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Des Devel Ther 4: 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN (2007). Smoking alters alveolar macrophage recognition and phagocytic ability. Am J Respir Cell Mol Biol 37: 748–755. [DOI] [PubMed] [Google Scholar]
- Jones RL, Giembycz MA, Woodward DF (2009). Prostanoid receptor antagonists: development strategies and therapeutic applications. Br J Pharmacol 158: 104–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Uchiyama K, Irie T, Murao M (1973). Evaluation of aerosols of prostaglandins E1 and E2 as bronchodilators. Eur J Clin Pharmacol 6: 127–132. [DOI] [PubMed] [Google Scholar]
- Kay LJ, Gilbert M, Pullen N, Skerratt S, Farrington J, Seward EP et al. (2013). Characterization of the EP receptor subtype that mediates the inhibitory effects of prostaglandin E2 on IgE‐dependent secretion from human lung mast cells. Clin Exp Allergy 43: 741–751. [DOI] [PubMed] [Google Scholar]
- Kenakin TP (1984). The classification of drugs and drug receptors in isolated tissues. Pharmacol Rev 36: 165–222. [PubMed] [Google Scholar]
- Konya V, Maric J, Jandl K, Luschnig P, Aringer I, Lanz I et al. (2015). Activation of EP4 receptors prevents endotoxin‐induced neutrophil infiltration into the airways and enhances microvascular barrier function. Br J Pharmacol 172: 4454–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MC, Proud D, Schleimer RP, Plaut M (1984). Human lung macrophages enhance and inhibit lymphocyte proliferation. J Immunol 132: 2895–2903. [PubMed] [Google Scholar]
- Machwate M, Harada S, Leu CT, Seedor G, Labelle M, Gallant M et al. (2001). Prostaglandin receptor EP4 mediates the bone anabolic effects of PGE2 . Mol Pharmacol 60: 36–41. [DOI] [PubMed] [Google Scholar]
- Maher SA, Dubuis ED, Belvisi MG (2011). G‐protein coupled receptors regulating cough. Curr Opin Pharmacol 11: 248–253. [DOI] [PubMed] [Google Scholar]
- Melillo E, Woolley KL, Manning PJ, Watson RM, O'Byrne PM (1994). Effect of inhaled PGE2 on exercise‐induced bronchoconstriction in asthmatic subjects. Am J Respir Crit Care Med 149: 1138–1141. [DOI] [PubMed] [Google Scholar]
- Murase A, Taniguchi Y, Tonai‐Kachi H, Nakao K, Takada J (2008). In vitro pharmacological characterization of CJ‐042794, a novel, potent, and selective prostaglandin EP(4) receptor antagonist. Life Sci 82: 226–232. [DOI] [PubMed] [Google Scholar]
- O'Brien AJ, Fullerton JN, Massey KA, Auld G, Sewell G, James S et al. (2014). Immunosuppression in acutely decompensated cirrhosis is mediated by prostaglandin E2 . Nat Med 20: 518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe MJ, Walding A, Shelton PA, Flaherty A, Dougall IG (2007). Activation of E‐prostanoid4 and E‐prostanoid2 receptors inhibits TNF‐α release from human alveolar macrophages. Eur Respir J 29: 986–994. [DOI] [PubMed] [Google Scholar]
- Rowe J, Finlay‐Jones JJ, Nicholas TE, Bowden J, Morton S, Hart PH (1997). Inability of histamine to regulate TNF‐α production by human alveolar macrophages. Am J Respir Cell Mol Biol 17: 218–226. [DOI] [PubMed] [Google Scholar]
- Schlötzer‐Schrehardt U, Zenkel M, Nüsing RM (2002). Expression and localization of FP and EP prostanoid receptor subtypes in human ocular tissues. Invest Opthalmol Vis Sci 43: 1475–1487. [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, Kaga K et al. (2008). Down‐regulated NF‐E2‐related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 39: 673–682. [DOI] [PubMed] [Google Scholar]
- Tang L, Loutzenhiser K, Loutzenhiser R (2000). Biphasic actions of prostaglandin E2 on the renal afferent arteriole, role of EP3 and EP4 receptors. Circ Res 86: 663–670. [DOI] [PubMed] [Google Scholar]
- Thorat MA, Morimiya A, Mehrotra S, Konger R, Badve SS (2008). Prostanoid receptor EP1 expression in breast cancer. Mod Pathol 21: 15–21. [DOI] [PubMed] [Google Scholar]
- Wilson RJ, Rhodes SA, Wood RL, Shield VJ, Noel LS, Gray DW (2004). Functional pharmacology of human prostanoid EP2 and EP4 receptors. Eur J Pharmacol 501: 49–58. [DOI] [PubMed] [Google Scholar]
- Woodward DF, Jones RL, Narumiya S (2011). International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev 63: 471–538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Inter‐preparation responses to PGE2. Macrophages were pre‐incubated (30 min) with indomethacin (1 μM) and then with or without PGE2 for 30 min before challenge with LPS (1 ng mL‐1) for 22 h after which TNFα was measured in the supernatants. Values are expressed as the % inhibition of the unblocked control TNFα release. The effects of PGE2 in 32 macrophage preparations are shown (A). Data have been stratified according to age (B) or smoking status (C). Values are means ± SEM. (B) Values are for 5 people, 50‐60 years old; 7 people, 60‐70 years old, 17 people, 70‐80 years old and 3 people, 80‐90 years old. (C) Values are for 14 (smokers), 13 (ex‐smokers) and 5 (non‐smokers) preparations.
Supporting info item