

Abstract
Many Gram-negative bacterial pathogens express contact-dependent growth inhibition (CDI) systems that promote cell-cell interaction. CDI+ bacteria express surface CdiA effector proteins, which transfer their C-terminal toxin domains into susceptible target cells upon binding to specific receptors. CDI+ cells also produce immunity proteins that neutralize the toxin domains delivered from neighboring siblings. Here, we show that CdiAEC536 from uropathogenic Escherichia coli 536 (EC536) uses OmpC and OmpF as receptors to recognize target bacteria. E. coli mutants lacking either ompF or ompC are resistant to CDIEC536-mediated growth inhibition, and both porins are required for target-cell adhesion to inhibitors that express CdiAEC536. Experiments with single-chain OmpF fusions indicate that the CdiAEC536 receptor is heterotrimeric OmpC-OmpF. Because the OmpC and OmpF porins are under selective pressure from bacteriophages and host immune systems, their surface-exposed loops vary between E. coli isolates. OmpC polymorphism has a significant impact on CDIEC536 mediated competition, with many E. coli isolates expressing alleles that are not recognized by CdiAEC536. Analyses of recombinant OmpC chimeras suggest that extracellular loops L4 and L5 are important recognition epitopes for CdiAEC536. Loops L4 and L5 also account for much of the sequence variability between E. coli OmpC proteins, raising the possibility that CDI contributes to the selective pressure driving OmpC diversification. We find that the most efficient CdiAEC536 receptors are encoded by isolates that carry the same cdi gene cluster as E. coli 536. Thus, it appears that CdiA effectors often bind preferentially to "self" receptors, thereby promoting interactions between sibling cells. As a consequence, these effector proteins cannot recognize nor suppress the growth of many potential competitors. These findings suggest that self-recognition and kin selection are important functions of CDI.
Author Summary
Bacterial pathogens often live in crowded communities where cells reside in close contact with one another. Many of these bacteria possess contact-dependent growth inhibition (CDI) systems, which allow cells to touch and inhibit each other using toxic CdiA proteins. CDI+ bacteria also produce immunity proteins that specifically protect the cell from the CdiA toxins of neighboring sibling cells. The CDI system from Escherichia coli EC93 was the first to be characterized and its CdiA toxin recognizes a receptor (BamA) that is identical in virtually all E. coli isolates. Here, we describe a different CDI system from uropathogenic E. coli 536, which causes urinary tract infections. In contrast to E. coli EC93, CdiA from E. coli 536 binds to receptor proteins (OmpC/OmpF) that vary widely between different E. coli isolates. Thus, uropathogenic E. coli preferentially bind and deliver toxins into sibling cells and other closely related E. coli strains. These results suggest that CDI systems distinguish between "self" and "non-self" cells. Moreover, because sibling cells are immune to CdiA-mediated growth inhibition, these findings raise the possibility that toxin exchange may be used for communication and cooperative behavior between genetically identical bacteria.
Introduction
Contact-dependent growth inhibition (CDI) systems mediate the transfer of protein toxins between Gram-negative bacteria. CDI was first discovered and characterized in Escherichia coli EC93, which uses CdiBEC93/CdiAEC93 two-partner secretion proteins to inhibit the growth of other E. coli isolates [1]. CdiBEC93 is an Omp85 family β-barrel protein that exports and presents CdiAEC93 on the cell surface. CdiAEC93 carries toxic effector activity and is predicted to form a β-helical filament that projects several hundred angstroms from the inhibitor cell [2, 3]. CdiAEC93 binds to BamA on the surface of neighboring E. coli cells, then delivers its C-terminal toxin domain (CdiA-CTEC93) into the target cell to inhibit growth [4]. The proton gradient is rapidly dissipated in intoxicated cells [5, 6], suggesting that the CdiA-CTEC93 toxin forms pores in the cytoplasmic membranes of target bacteria. Because E. coli EC93 cells also express BamA receptors, they receive toxin domains from neighboring siblings. However, inter-sibling toxin exchange does not induce growth arrest because the cdi locus encodes the CdiIEC93 immunity protein, which neutralizes CdiA-CTEC93 toxicity [1, 7]. Thus, E. coli EC93 deploys CDI to inhibit competing bacteria, but sibling cells are immune to growth inhibition. Since their discovery in E. coli EC93, cdi genes have been identified and characterized in several other proteobacteria [8–13]. CDI systems are typically encoded on plasmids and genomic islands and therefore are not necessarily found in all isolates of given species. This is exemplified by E. coli, for which ~25% of sequenced isolates carry cdi gene clusters. By contrast, every sequenced strain of Neisseria meningitidis and Burkholderia pseudomallei encodes at least one CDI system. CDI is also characterized by toxin diversity [3, 14]. CdiA-CT sequences vary widely between bacteria, with toxins exhibiting several distinct activities [3, 15]. CdiI immunity proteins are also diverse and specifically protect against cognate CdiA-CT toxins. Thus, cdi genes collectively encode a network of toxin/immunity protein pairs that appear to be rapidly diversifying. Toxin diversity and the specificity of immunity protection suggest that CDI systems are deployed to compete for environmental resources and growth niches.
The molecular mechanisms of CDI toxin delivery have been explored largely using the E. coli EC93 system as a model. Early work showed that CDIEC93 expression in E. coli K-12 strains promotes auto-adhesion [1] and that CdiA-CTEC93 delivery is receptor-dependent [4]. The CdiAEC93 receptor, BamA, was identified from selections for CDI-resistant (CDIR) target cells. E. coli bamA101 mutants carry a transposon insertion that reduces BamA expression five-fold and confers partial resistance to CDIEC93-mediated growth inhibition [4]. BamA is an essential outer-membrane protein that forms the core of the β-barrel assembly machine (BAM) complex in Gram-negative bacteria [16–18]. The BAM complex inserts β-barrel proteins into the outer membrane, and BamA orthologues are found in all Gram-negative bacteria and eukaryotic plastids. Though BamA is conserved, its extracellular loops vary considerably between closely related enterobacterial species [19]. For example, E. coli BamAEco shares ~94% overall sequence identity with BamAECL from Enterobacter cloacae ATCC 13047, but the surface-exposed portions of extracellular loops L4, L6 and L7 are unrelated in sequence [20]. Consequently, E. coli EC93 does not recognize E. cloacae cells as targets and cannot inhibit the growth of this species. Similarly, replacement of bamA Eco with bamA genes from other enterobacterial species confers CDI-resistance to E. coli cells [20]. Thus, receptor polymorphism restricts the CDIEC93 target-cell range to E. coli and Shigella isolates, which share identical BamA proteins. Recognition of "self" receptors appears to be a general feature of CDI, because CdiA effectors from Dickeya dadantii 3937, Burkholderia thailandensis E264, Neisseria meningitidis B16B6 and Pseudomonas aeruginosa PAO1 all deliver toxins to sibling cells [8–13]. This exchange of toxins provides no apparent competitive advantage because siblings are immune. However, the accompanying cell-cell adhesion likely imparts significant benefits by promoting auto-aggregation and biofilm formation [7, 11, 13, 21–25]. Thus, in addition to suppressing the growth of competing bacteria, CDI also contributes to bacterial fitness by mediating cooperative behaviors between sibling cells.
Bacterial surfaces interact directly with the environment and are therefore under strong selective pressure. Bacteriophages and adaptive immune systems exploit surface epitopes to identify bacteria for infection and elimination, respectively. In response, bacteria have evolved mechanisms to rapidly alter their repertoire of surface molecules such that different isolates of the same species commonly express unique extracellular antigens [26–29]. Therefore, CdiA effectors from different bacterial species must presumably recognize distinct receptors. Here, we begin to explore CDI receptor diversity using the CdiAEC536 effector from uropathogenic E. coli 536 (EC536). We find that E. coli ompC and ompF mutants are resistant to CDIEC536-mediated growth inhibition, suggesting that the OmpC and OmpF osmoporins function as receptors for CdiAEC536. CDIEC536 inhibitor cells only bind to target bacteria that express OmpC and OmpF, indicating that both porins are required for receptor function. Using single-chain OmpF dimers, we provide evidence that heterotrimeric OmpC-OmpF constitutes the receptor for CdiAEC536. Because OmpF and OmpC exhibit considerable sequence variability in their extracellular loops [30, 31], we examined the effects of these polymorphisms on receptor function. We find that several OmpC proteins from uropathogenic E. coli isolates are not recognized by CdiAEC536, suggesting that many potential competitors are resistant to CDIEC536. By contrast, OmpC proteins from E. coli UTI89 and F11 are effective receptors for CdiAEC536. Notably, E. coli UTI89 and F11 also encode CdiA proteins that are >98% identical to CdiAEC536, indicating that their effectors also recognize OmpC-OmpF receptors and mediate self-recognition. In these instances, CdiA effector proteins preferentially recognize "self" receptors, suggesting that auto-adhesion and kin selection are important functions of CDI.
Results
Genetic analysis of the CDIEC536 system
We previously reported that E. coli bamA101 mutants are resistant to CDIEC536 [32], though the level of protection is less than that observed against CDIEC93 inhibitors [4]. Because the bamA101 mutation confers incomplete resistance, we revisited these experiments using E. coli target strains that express E. cloacae bamA (bamA ECL), which provides full resistance to CDIEC93 [20]. We first confirmed that E. coli bamA ECL target cells are resistant to CDIEC93 using E. coli EPI100 inhibitor cells that express the cdiBAI EC93 gene cluster from a cosmid vector. As expected, E. coli bamA ECL targets grew to the same level in co-culture with either CDIEC93 inhibitors or CDI−cells that carry an empty cosmid vector (Fig 1). By contrast, bamA Eco target-cell viability decreased ~600-fold during co-culture with CDIEC93 inhibitors (Fig 1). We then tested bamA Eco and bamA ECL cells in competitions against E. coli EPI100 that express the E. coli 536 cdiBAI EC536 genes from a cosmid and found that both target strains were inhibited to the same extent (Fig 1). These results indicate that BamAECL does not protect against CDIEC536, suggesting that the originally observed bamA101 resistance may be due to decreased expression of an unidentified outer-membrane receptor that requires BamA for assembly.
Fig 1. Heterologous BamA does not protect against CDIEC536.

Target bacteria that express bamA Eco (CH10226) or bamA ECL (CH10227) were cultured at a 1:10 ratio with E. coli EPI100 inhibitor cells that carry cosmids pDAL660Δ1–39 (CDIEC93), pDAL866 (CDIEC536) or pWEB::TNC (CDI–). Viable target bacteria are reported as colony-forming units per milliliter (cfu/mL) ± SEM for two independent experiments.
We used a genetic approach to identify the CdiAEC536 receptor, reasoning that its disruption should confer CDI resistance (CDIR). We subjected CDI-sensitive E. coli target cells to transposon mutagenesis, then selected CDIR mutants from co-cultures with E. coli 536 cells that express the cdi locus under control of an arabinose-inducible promoter. Additionally, because CysK (O-acetylserine sulfhydrylase A) is required to activate the CdiA-CTEC536 toxin domain [33, 34], we provided target cells with plasmid-borne cysK to avoid isolating CDIR cysK null mutants. CDIR populations were enriched after three rounds of selection, and randomly isolated clones exhibited complete resistance to growth inhibition. Transposon insertions from CDIR clones were transduced into CDI-sensitive cells and genetic linkage between insertions and CDIR phenotypes was established for each transductant. The disrupted genes were then identified from ten randomly selected CDIR mutants, revealing five independent insertions each in ompC and ompF (Fig 2A). The ompC and ompF genes encode closely related osmoporins, which are trimeric β-barrel proteins that facilitate the diffusion of small hydrophilic molecules across the outer membranes of Gram-negative bacteria [35]. To confirm the roles of OmpC and OmpF in CDIEC536, we generated an E. coli ΔompC ΔompF double mutant and performed complementation analysis. As expected, the growth of ΔompC ΔompF cells was not inhibited in co-culture with E. coli 536 (Fig 2B). By contrast, wild-type ompC + ompF + target cells showed a 50-fold loss in viability under the same co-culture conditions (Fig 2B). This latter growth inhibition is due to CDIEC536, because the growth of wild-type E. coli was not affected by E. coli 536 mock inhibitors that carry a deletion of the cdiA-CT coding region (Fig 2C). Complementation of the ΔompC ΔompF mutant with a plasmid expressing either ompC or ompF had no effect on the resistance phenotype, but the strain became CDI-sensitive when it harbored plasmids expressing both osmoporins (Fig 2B). Together, these findings demonstrate that OmpC and OmpF expression is required to render bacteria sensitive to the CDIEC536 system.
Fig 2. E. coli ΔompC and ΔompF mutants are resistant to CDIEC536.
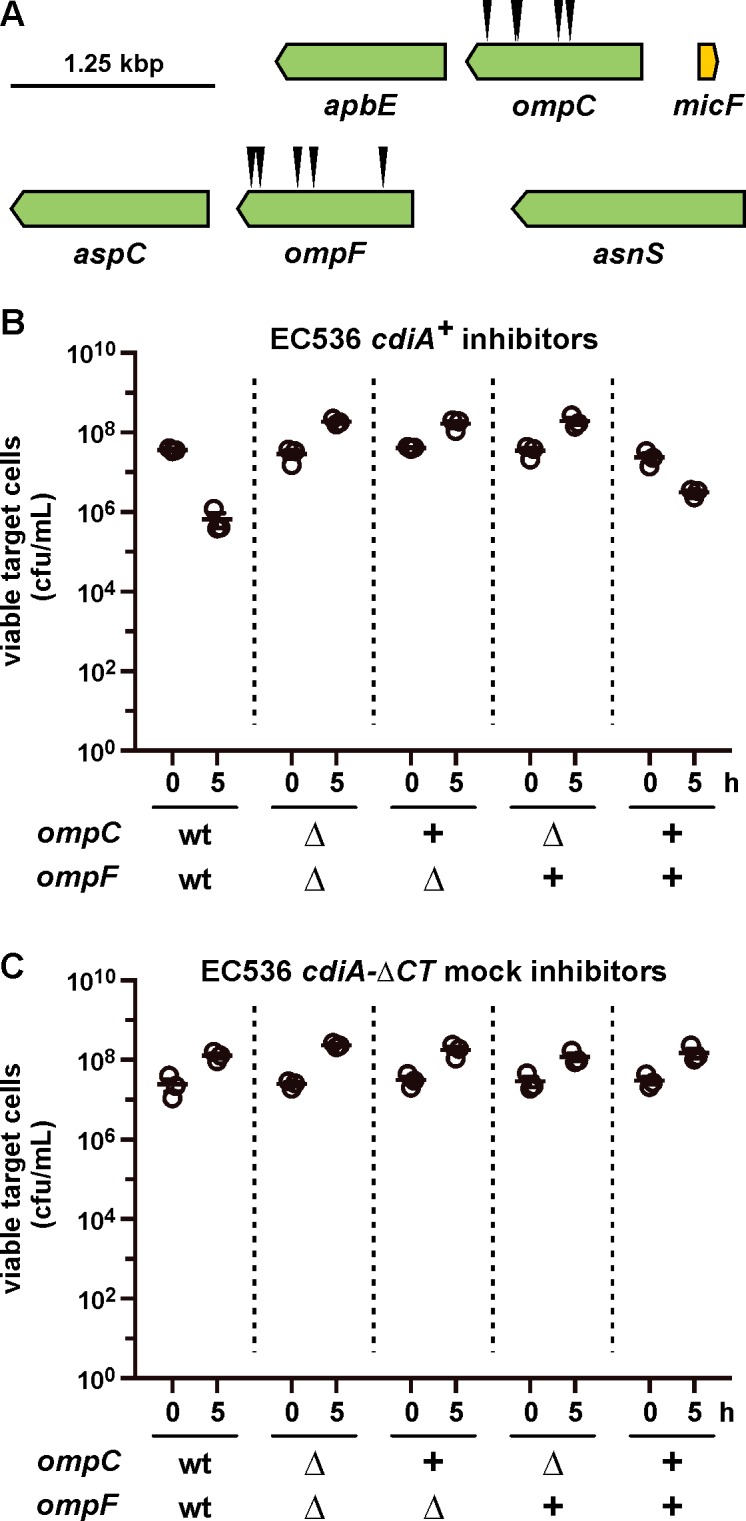
(A) Positions of mariner transposon insertion sites from CDIR mutants. (B) Competition co-cultures with CDI+ E. coli 536 inhibitors. E. coli CH10013 (ompC + ompF +) and CH10136 (ΔompC ΔompF) target cells were co-cultured at a 1:10 ratio with CDI-inducible E. coli 536 cells. The plus (+) symbols indicate complementation of ΔompC ΔompF target cells with plasmid-borne ompC K-12 (pCH10138) and ompF K-12 (pCH10202). Viable target bacteria are reported as colony-forming units per milliliter (cfu/mL) ± SEM for three independent experiments. (C) Competition co-cultures with CDI− E. coli 536. Co-cultures were performed as in panel B, except E. coli 536 cells deleted for the cdiA-CT region were used as mock inhibitors.
OmpC and OmpF are required for CdiAEC536-mediated cell-cell adhesion
CdiA-mediated adhesion between inhibitor and target cells is a hallmark of CDI. To determine whether OmpC and OmpF are required for CDI-dependent adhesion, we monitored cell-cell binding using a flow-cytometry approach. GFP-labeled inhibitor cells were mixed with DsRed-labeled target bacteria at a 5:1 ratio, and the suspensions analyzed by flow cytometry. Flow events registering both green and red fluorescence correspond to aggregates containing inhibitor and target bacteria. The flow cytometric assay for CDI-mediated adhesion generally requires CdiA over-expression [4, 36], therefore we used inhibitor cells that express cdi genes from cosmids for these experiments. Under these conditions, CDIEC93 inhibitor cells bind to 60–80% of target bacteria (Fig 3A). This cell-cell adhesion is dependent on CDI, with only about 5% of target cells associated with CDI−mock inhibitors (Fig 3A). We then tested whether CDIEC536 expression also promotes stable adhesion to target cells. As expected, we did not detect CDIEC536-dependent adhesion to ΔompC target bacteria; but surprisingly there was little increase in adhesion upon complementation with ompC K-12 from E. coli K-12 (Fig 3A). Despite the lack of detectable cell-cell adhesion, ompC K-12-complemented cells were inhibited during co-culture with E. coli 536 cells (Fig 3B) and immunoblotting confirmed that they produced OmpCK-12 (Fig 3C). OmpC porins vary significantly between E. coli isolates [30], and the E. coli K-12 and 536 proteins carry different surface-exposed loops (Fig 3D). Because extracellular loop polymorphism could affect receptor function, we tested whether ompC EC536 better supports CDIEC536-dependent cell adhesion. Strikingly, over 40% of ompC EC536 complemented bacteria adhered to CDIEC536 inhibitors, and this adhesion was specific because these target cells did not associate with CDI−mock inhibitors (Fig 3A). These results suggest that CdiAEC536 binds to OmpCEC536 with higher avidity than OmpCK-12. We also found that ompC EC536 target cells were inhibited to a greater extent than ompC K-12 targets in co-cultures with E. coli 536 (Fig 3B). We next tested OmpC proteins from Salmonella Typhimurium LT2 and E. cloacae ATCC 13047 (Fig 3D) to determine whether other species are potential targets for the CDIEC536 system. E. coli ΔompC cells complemented with S. Typhimurium ompC LT2 did not adhere to CDIEC536 inhibitors (Fig 3A) and were not inhibited in competition co-culture (Fig 3B), though they produced substantial OmpCLT2 protein (Fig 3C). By contrast, CDIEC536 inhibitors bound to ompC ECL complemented cells and inhibited their growth (Fig 3A and 3B). Together, these results support a role for OmpC as the CdiAEC536 receptor and indicate that OmpC polymorphism determines whether bacteria are susceptible to CDIEC536-mediated growth inhibition.
Fig 3. OmpC is required for CDIEC536 dependent cell-cell adhesion.

(A) Analysis of cell-cell adhesion by flow cytometry. E. coli DL4905 (GFP+) inhibitors carrying cosmids pDAL660Δ1–39 (CDIEC93), pDAL866 (CDIEC536) or pWEB::TNC (CDI–) were mixed at a 5:1 ratio with DsRed-labeled CH10099 (ΔompC) target cells. Where indicated, the target cells were complemented with plasmid-borne ompC alleles from E. coli K-12, E. coli 536 (EC536), S. Typhimurium LT2 or E. cloacae (ECL). The suspensions were analyzed by flow cytometry using FL1 (533/30nm, GFP) and FL2 (585/40nm, DsRed) fluorophore filters. The percentage of bound target cells was calculated as the number of dual green/red fluorescent events divided by the total number of red fluorescent events. The average ± SEM is presented for two independent experiments. (B) Competition co-cultures with CDI+ E. coli 536. E. coli CH10099 (ΔompC) target cells were co-cultured at a 1:10 ratio with CDI-inducible E. coli 536 cells. Where indicated, the target cells were complemented with plasmid-borne ompC alleles from E. coli K-12, E. coli 536 (EC536), S. Typhimurium LT2 or E. cloacae (ECL). Viable target bacteria are reported as colony-forming units per milliliter (cfu/mL) ± SEM for two independent experiments. (C) Protein from the target bacteria in panel A was subjected to immunoblot analysis using polyclonal antisera against OmpC/F from E. coli K-12. (D) Sequence alignment of OmpC proteins from E. coli K-12, E. coli 536, S. Typhimurium LT2 and E. cloacae ATCC 13047 (ECL). Extracellular loop sequences are indicated above the alignment.
We next examined the contribution of OmpF to CDIEC536-dependent adhesion by testing whether porins from different bacteria influence receptor activity (Fig 4A). Because OmpCK-12 does not support detectable adhesion to CDIEC536 inhibitor cells, we replaced the native ompC K-12 locus with the ompC EC536 allele to generate a target-cell strain for cell-cell binding studies. The resulting ΔompF ompC EC536 cells did not form aggregates with CDIEC536 inhibitors, but adhered normally to CDIEC93 inhibitor cells (Fig 4B). We then expressed plasmid-borne ompF from E. coli K-12, E. coli 536, S. Typhimurium and E. cloacae and found that each allele supported adhesion to CDIEC536 inhibitors (Fig 4B and 4C). These results demonstrate that both OmpC and OmpF are required for CDIEC536-dependent cell adhesion, indicating that each porin contributes to receptor function.
Fig 4. OmpF is required for CDIEC536 dependent cell-cell adhesion.

(A) Sequence alignment of OmpF proteins from E. coli K-12, E. coli 536, S. Typhimurium LT2 and E. cloacae ATCC 13047 (ECL). Extracellular loop sequences are indicated above the alignment. (B) Analysis of cell-cell adhesion by flow cytometry. E. coli DL4905 (GFP+) inhibitors carrying cosmids pDAL660Δ1–39 (CDIEC93), pDAL866 (CDIEC536) or pWEB::TNC (CDI–) were mixed at a 5:1 ratio with DsRed-labeled CH12134 (ΔompF ompC EC536) target cells. Where indicated, target cells were complemented with plasmid-borne ompF from E. coli K-12, E. coli 536 (EC536), S. Typhimurium LT2 or E. cloacae (ECL). Cells suspensions were analyzed by flow cytometry using FL1 (533/30nm, GFP) and FL2 (585/40nm, DsRed) fluorophore filters. The percentage of bound target cells was calculated as the number of dual green/red fluorescent events divided by the total number of red fluorescent events. The average ± SEM is presented for two independent experiments. (C) Protein from the target bacteria in panel B was subjected to immunoblot analysis using polyclonal antisera against OmpC/F from E. coli K-12.
CdiAEC536 recognizes OmpC-OmpF heterotrimers
At least two models can explain the roles of OmpC and OmpF in CDI. CdiAEC536 could interact simultaneously with adjacent OmpC and OmpF homotrimers on the target-cell surface. Alternatively, the effector could recognize OmpC and OmpF displayed as heterotrimers, which have been reported previously [37]. To differentiate between these models, we generated and tested the function of covalently constrained OmpC-OmpF heterotrimers. Because osmoporins are obligate trimers, we reasoned that porin dimers produced from a gene fusion should require an additional monomer to assemble properly in the outer membrane. We tested this prediction using dimeric OmpF proteins tethered by 8- and 16-residue linker peptides. OmpF assembly and function was assessed by monitoring sensitivity to colicin E5, which requires OmpF to translocate its C-terminal toxin domain into E. coli cells [38–41]. E. coli ΔompF ΔompC mutants are completely resistant to colicin E5, but become susceptible when complemented with plasmid-borne ompF EC536 (Fig 5A). By contrast, neither of the dimeric ompF constructs restored colicin sensitivity (Fig 5A), even though immunoblot analysis confirmed the presence of OmpF dimers in these cells (Fig 5B). We then expressed ompF-ompF fusions in the ΔompF ompC EC536 background to test whether monomeric OmpC supports heterotrimer formation. E. coli ΔompF ompC EC536 cells are somewhat sensitive to colicin E5 (Fig 5C), consistent with a prior report showing that OmpC can support colicin entry in the absence of OmpF [42]. However, colicin E5 had a greater inhibitory effect on ΔompF ompC EC536 cells that were complemented with plasmids that produce either monomeric or dimeric OmpF (Fig 5C). Together, these results indicate that OmpF2-OmpC heterotrimers assemble in the outer membrane and are active in colicin translocation. Having established a strategy to generate heterotrimeric porin complexes, we tested whether OmpF2-OmpC is a receptor for CdiAEC536. Flow cytometry showed that each OmpF dimer supported CDIEC536-dependent cell-cell adhesion to the same extent as monomeric OmpF (Fig 6A). Moreover, we also found that ΔompF ompC EC536 cells expressing dimeric OmpF were inhibited 10- to 100-fold by E. coli 536 cells in competition co-cultures (Fig 6B). Taken together, these results indicate that heterotrimeric OmpF-OmpC is the receptor for CdiAEC536.
Fig 5. Characterization of functional OmpF-OmpC heterotrimers.

(A) E. coli CH11346 (ΔompF ΔompC) cells were complemented with ompF (pCH11850), ompF-ompF with 8-residue linker (ompF 2, pCH10796), or ompF-ompF with 16-residue linker (ompF 2 *, pCH12054) and the resulting strains cultured in LB media without or with colicin E5 (added at 30 min as indicated by the arrow). Cell growth was monitored by measuring the optical density at 600 nm (OD600) of the cultures. Data are from two independent experiments. (B) Protein from the strains in panel A was subjected to immunoblot analysis using polyclonal antisera against OmpC/F and BamA (as a loading control) from E. coli K-12. (C) E. coli CH12180 (ΔompF ompC EC536) cells were complemented with plasmid-borne ompF, ompF 2 or ompF 2* and cultured as described in panel A. (D) Protein from the strains in panel C was subjected to immunoblot analysis using polyclonal antisera against OmpC/F from E. coli K-12.
Fig 6. Heterotrimeric OmpF-OmpC supports CDIEC536-mediated growth inhibition.

(A) E. coli DL4905 (GFP+) inhibitors carrying pDAL866 (CDIEC536) or pWEB::TNC (CDI–) were mixed at a 5:1 ratio with DsRed-labeled E. coli CH12180 (ΔompF ompC EC536) target cells complemented with ompF (pCH11850), ompF-ompF with 8-residue linker (ompF 2, pCH10796), or ompF-ompF with 16-residue linker (ompF 2 *, pCH12054). Cell suspensions were analyzed by flow cytometry using FL1 (533/30nm, GFP) and FL2 (585/40nm, DsRed) fluorophore filters. The fraction of bound target cells was calculated as the number of dual green/red fluorescent events divided by the total number of red fluorescent events. The average ± SEM is presented for two independent experiments. (B) E. coli CH12180 (ΔompF ompC EC536) target cells were complemented were complemented with plasmid-borne ompF, ompF 2 or ompF 2* and the resulting strains co-cultured at a 1:10 ratio with CDI+ (filled symbols) or CDI−(open symbols) E. coli 536 cells. Viable target bacteria are reported as colony-forming units per milliliter (cfu/mL) ± SEM for three independent experiments.
OmpC loops L4 and L5 are recognition determinants for CdiAEC536
We next used natural variations between E. coli OmpC porins to identify potential CdiAEC536 recognition epitopes. We surveyed predicted OmpC proteins from E. coli isolates and identified over 220 distinct sequences (S1 Table). Though some E. coli OmpC variants differ by single amino-acid residue substitutions, much of the sequence variation is localized to extracellular loops L4 (33 sequences), L5 (30 sequences) and L7 (13 sequences) (S2 Table). Moreover, these loops are found in many different combinations (S1 Table). To examine the influence of OmpC polymorphism on CDIEC536, we tested a subset of E. coli OmpC proteins with unique extracellular loop combinations (Figs 7A and S1). E. coli ΔompC target cells were provided with plasmid-borne ompC alleles and co-cultured with E. coli 536 inhibitor cells. The ompC alleles from E. coli UTI89, F11 and A33H rendered ΔompC cells sensitive to growth inhibition to roughly the same extent as ompC EC536 (Fig 7B). Targets that express ompC EC93 from E. coli EC93 were also inhibited in co-culture with E. coli 536, but ompC alleles from uropathogenic isolates CFT073, 6104H, A54H, A42H and A35H were all associated with resistance (Fig 7B and 7C). Immunoblot analysis confirmed that OmpC was produced in all of the complemented strains (Fig 7D), indicating that resistance is not due to a lack of ompC expression. Thus, E. coli ompC allele polymorphism appears to dictate cell susceptibility to CDIEC536-mediated intoxication.
Fig 7. E. coli OmpC polymorphism restricts the target-cell range of CDIEC536.

(A) Extracellular loop L4, L5 and L7 sequences from selected E. coli isolates. S1 Fig shows the complete sequence alignment, and the alphanumeric classification scheme for L4, L5 and L7 sequences is presented in S2 Table. (B) E. coli CH10099 (ΔompC) target cells were co-cultured at a 1:10 ratio with CDI+ E. coli 536 cells. Target cells were complemented with plasmid-borne ompC alleles from the indicated E. coli isolates. Viable target bacteria are reported as colony-forming units per milliliter (cfu/mL) ± SEM for three independent experiments. (C) The same target-cell strains from panel B were co-cultured at a 1:10 ratio with CDI− E. coli 536 and viable cells quantified in the same manner. (D) Protein from ompC complemented strains was subjected to immunoblot analysis using polyclonal antisera against OmpC/F from E. coli K-12.
To determine which OmpC sequences are critical for recognition by CdiAEC536, we generated chimeric porins using OmpCCFT073 as a framework upon which to graft heterologous OmpC loops. OmpCCFT073 and OmpCEC536 share loop L4, but differ in their L5, L7 and L8 sequences (Figs 7A and S1). OmpCCFT073 chimeras carrying L7 or L8 from OmpCEC536 still provided resistance to CDIEC536 (Fig 8A), indicating that these loops probably have no role in binding CdiAEC536. However, replacement of loop L5 converted OmpCCFT073 into a functional receptor that supports the same level of inhibition as OmpCEC536 (Fig 8A). Similarly, OmpCCFT073 chimeras carrying OmpCEC536 L7 or L8 were functional receptors, provided that they also carried loop L5 from OmpCEC536 (Fig 8A). We tested the role of loop L4 by grafting the sequence from OmpCF11 (which is a receptor for CdiAEC536) onto OmpCCFT073. Targets that express the resulting chimera were inhibited by E. coli 536 to about the same extent as targets that express OmpCF11 (Fig 8A). This latter observation shows that OmpCCFT073 loop L5 is not an anti-determinant, because it supports CdiAEC536 recognition in other contexts. Collectively, these results indicate that OmpC loops L4 and L5 contribute to target-cell selection, suggesting that the loops may be recognized directly by CdiAEC536.
Fig 8. OmpC extracellular loops L4 and L5 are important recognition determinants for CdiAEC536.

(A) E. coli CH10099 (ΔompC) target cells were co-cultured at a 1:10 ratio with CDI+ E. coli 536 cells. Target cells were complemented with the indicated chimeric ompC alleles. Viable target bacteria are reported as colony-forming units per milliliter (cfu/mL) ± SEM for three independent experiments. (B) Protein from the strains in panel A was subjected to immunoblot analysis using polyclonal antisera against OmpC/F from E. coli K-12.
Discussion
The results presented here establish a critical role for OmpC and OmpF in the CDIEC536 pathway. CDIEC536 inhibitor cells only adhere to target bacteria that express OmpC and OmpF, indicating that both porins contribute to receptor function. Though osmoporins are generally assumed to form homotrimers, we show that heterotrimeric OmpC-OmpF constitutes the CdiAEC536 receptor. Heterotrimeric porins were first reported by Gehring and Nikaido, who found that OmpC, OmpF and PhoE can form mixed trimers in E. coli K-12 [37]. OmpCK-12 and OmpFK-12 share 58.5% sequence identity (S2 Fig), and their β-barrels superimpose with root-square mean deviation of 0.78 Å [43]. The trimer interaction surface is dominated by hydrophobic contacts between β-barrel strands β1, β2, β3, β4, β5 and β1´, and these regions are highly conserved between OmpCK-12 and OmpFK-12 (S2 and S3 Figs, S3 Table). The porins also share an inter-protomer ion-pair between a conserved Glu residue (Glu66 in OmpCK-12 and Glu71 in OmpFK-12) at the tip of loop L2 and an Arg residue within L3 of the adjacent β-barrel (S3 Fig and S3 Table). Thus, the structures of OmpF and OmpC are compatible with heterotrimer formation. The targeting of heterotrimeric OmpF-OmpC receptors has a number of implications. Firstly, residues from both porins should be directly recognized by CdiAEC536. The results presented here suggest that OmpC loops L4 and L5 are important CdiAEC536-binding determinants, but the contribution of OmpF is less clear. OmpF porins from E. cloacae and S. Typhimurium are effective co-receptors in conjunction with OmpCEC536, so the CdiA-binding epitope is presumably shared by all of the tested OmpF variants. Additionally, because OmpC and OmpF are regulated reciprocally, it is possible that receptor availability fluctuates in response to environmental conditions. OmpF is expressed preferentially in response to low osmolarity, whereas OmpC dominates under hyperosmotic conditions [35]. We find that target cells are effectively inhibited when they over-express ompC alleles, suggesting that OmpF-OmpC2 heterotrimers are recognized by CdiAEC536. We have also shown that covalently constrained OmpF2-OmpC heterotrimers are functional receptors. Because both heterotrimeric forms support CDIEC536, we conclude that CdiAEC536 effectors probably make contact with one OmpC and one OmpF protomer in the receptor complex.
The mechanism(s) by which CDI toxins are translocated across the target-cell outer membrane is unknown, but in principle, receptors could mediate this transport. Group A colicins appear to exploit the central pore of OmpF to cross the outer membrane, and this translocation is powered by the proton-motive force via the Tol protein complex [40, 41, 44, 45]. However, CDI toxin translocation is not Tol-dependent, nor does it require the Ton-Exb system, which energizes the import of group B colicins [6, 44]. Further, CDI toxin domains are modular and can be exchanged between CdiA proteins for delivery through different receptor pathways [8, 10, 46]. Therefore, if CdiA receptors mediate outer-membrane transport, then BamA must also be capable of CDI toxin translocation. Like OmpF and OmpC, BamA is a 16-strand β-barrel protein, but its lumen is not exposed to the extracellular milieu. Instead, extracellular loops L3, L4, L6, L7 and L8 form a dome that shields the BamA lumen from the environment [47, 48]. It is possible that the BamA barrel opens upon binding to CdiAEC93, consistent with work showing that extracellular loop L6 is dynamic during the OMP assembly duty cycle [49, 50]. Buchanan and colleagues have also suggested that the BamA β-barrel opens a lateral gate during OMP biogenesis [51]. These conformational changes could be exploited to translocate toxin across the outer membrane, but we note that inactive BamA lacking the POTRA-3 (polypeptide-transport associated) domain is able to complement the bamA101 mutation and restore sensitivity to CDIEC93 [4]. Therefore, OMP biogenesis activity is not required for CDI, and thus it remains unclear whether BamA dynamics contribute to toxin translocation.
Putative CdiA-binding epitopes have been localized on BamA and OmpC, but the corresponding receptor-binding regions within CdiAEC93 and CdiAEC536 have not yet been identified. Given that BamA and OmpF/OmpC have distinct structures, the two CdiA effectors should contain unique receptor-binding domains. Alignment of CdiAEC93 and CdiAEC536 shows several regions of sequence divergence. Prominent among these are the C-terminal toxin region and the sequence corresponding to residues Ser1379 –Tyr1636 of CdiAEC93 (S4 Fig). Additionally, the CdiAEC536 effector contains a 69-residue insertion (Ser1988 –Ser2056) relative to CdiAEC93 (S4 Fig). We recently reported that truncated CdiAEC93 proteins lacking up to ~1,000 C-terminal residues retain BamA-binding function [7]. Furthermore, CdiAEC93 lacking internal residues Ala722 –Gly1624 is still secreted and assembled onto the cell surface, but does not bind to BamA [7]. Together, these observations suggest that the receptor-binding domain lies between residues Ser1300 –Asn1600 of CdiAEC93. There are four distinct classes of E. coli CdiA proteins based on sequence comparisons of the 1300–1600 region, suggesting that there may be additional CDI receptors in E. coli. Alternatively, the other uncharacterized E. coli CdiA effectors could bind to BamA or OmpC/OmpF using epitopes that are distinct from those recognized by CdiAEC93 and CdiAEC536 (respectively). Intriguingly, the putative receptor-binding domain is located in the middle of the CdiA primary sequence, between the FHA-1 and FHA-2 peptide repeat regions (S4 Fig). This position is unexpected based on current models of CdiA structure and cell-surface presentation. According to the β-helix structural model [2], the receptor-binding region would be positioned near the center of the CdiA filament rather than the distal tip. It is unclear how a central location is compatible with receptor binding, because the distal end of filament would presumably have to perforate the target-cell outer membrane. Penetration of target bacteria could facilitate toxin delivery, but this model raises the question of why specific receptors should be required for CDI.
Bacterial surface antigens are under intense pressure to diversify, yet E. coli BamA and OmpC proteins show remarkably different responses to this selective pressure. BamA is invariant in over 2,500 E. coli isolates for which sequence information is available. By contrast, E. coli strains encode at least 220 distinct OmpC proteins, many of which are distinguished by unique combinations of extracellular loops L4, L5 and L7. Crystal structures are available for the OmpCK-12, OmpCCFT073 and OmpC6104H porins studied here, and each exhibits a distinct surface topology and electrostatic potential (S5 Fig) [43, 52]. The large number of extracellular loop combinations seen in E. coli OmpC proteins suggests that these elements are modular and can be rearranged independently to generate novel porins. However, OmpC loops L4, L5, L6, L7 and L8 engage in a series of intra-protomer interactions (S4 Table), and therefore some loop combinations may be incompatible with a properly folded porin structure. Most intra-protomer loop interactions (e.g. L5-L7 and L7-L8) are mediated by invariant residues, though the contacts between L4 and L5 in OmpCCFT073 and OmpC6104H involve non-conserved residues (S4 Table). We note that chimeric OmpCCFT073 carrying L4 from OmpCF11 represents a L4-L5 combination that has yet to be found in any E. coli isolate. Because this OmpC chimera is functional as a CdiAEC536 receptor, it must be assembled properly in the outer membrane. Therefore, in this instance, loop L4 is clearly modular. This combinatorial complexity has implications for CDI, and almost certainly governs bacterial susceptibility to bacteriophages and immune recognition [53, 54].
CDI receptor polymorphism restricts the number of susceptible target bacteria, yet CdiAEC536 recognizes multiple OmpC variants. OmpC proteins from E. coli EC93, 536, F11, UTI89 and A33H are all recognized efficiently by CdiAEC536 though each receptor carries a distinct combination of extracellular loops. E. coli F11 and UTI89 encode CdiA proteins that share >98% identity with CdiAEC536. Taken together, these observations indicate that the CdiAF11 and CdiAUTI89 effectors also bind to OmpF-OmpC and preferentially recognize self-receptors. These findings are consistent with numerous reports that CDI+ bacteria tend to auto-aggregate [1, 7, 11, 13, 21–23]. Intriguingly, E. coli CFT073 also encodes a CdiA effector that shares 89.7% identity with CdiAEC536 over 3,037 N-terminal residues, yet OmpCCFT073 does not function as a CdiAEC536 receptor. Inspection of the cdiB gene from E. coli CFT073 reveals that it contains an in-frame stop codon [55], and therefore this strain presumably cannot secrete its CdiA effector. It is tempting to speculate that the loss of CdiACFT073 surface expression has allowed ompC CFT073 to diverge in response to other selective pressures. However, there are other E. coli isolates with intact cdi loci that also share ompC and ompF alleles with CFT073. Based on the data presented here, it appears that the CdiA proteins from these latter isolates would not preferentially recognize self and therefore may function primarily in competition. Though auto-adhesion is beneficial for bacteria, these phenomena can also be viewed from the perspective of cdi genes as selfish elements. CDI systems are encoded on mobile genetic elements and their toxin/immunity activities act as "gene-addiction" modules [56]. Cells that lose CDI-encoding plasmids or genomic islands are suddenly susceptible to inhibition and risk elimination by neighboring CDI+ siblings. Thus, self-recognition would ensure continual selective pressure to retain cdi genes in a population. We speculate that CdiA effectors may have first evolved as adhesins used exclusively for cooperation. Toxin-delivery capabilities could then be grafted onto the ancestral CdiA protein, providing a strong positive selection for cdi retention. Thus, the antagonistic activity of CDI systems may have arisen from evolutionary pressure to retain cdi genes in fluid bacterial genomes.
Materials and Methods
Bacterial strains
Bacterial strains are listed in Table 1. Bacteria were grown in lysogeny broth (LB) or on LB agar unless otherwise noted. Where indicated, media were supplemented with antibiotics at the following concentrations: ampicillin, 150 μg mL−1; kanamycin, 50 μg mL−1; chloramphenicol, 66 μg mL−1; spectinomycin, 50 μg mL−1; rifampicin, 200 μg mL−1; streptomycin, 100 μg mL−1. The bamA::cat allele was introduced into MC4100 pZS21-bamA + cells by bacteriophage P1-mediated transduction [20]. The ΔompC::kan, ΔompF::kan, Δwzb::kan and ΔrecA::kan disruptions were obtained from the Keio collection [57] and transduced into E. coli strain CH10013. Kanamycin-resistance cassettes were subsequently removed with FLP recombinase expressed from plasmid pCP20 [58]. The ompC K-12 gene of E. coli CH10013 was replaced with ompC EC536 using Red-mediated recombination. ompC EC536 was amplified by PCR with oligonucleotides 3322 and 3323, and a fragment upstream of ompC K-12 was amplified using oligonucleotide pair 3320/3321 (all oligonucleotide primers are listed in S5 Table). The two DNA fragments were combined into one fragment by overlap-extension PCR (OE-PCR) using primers 3320/3323 and ligated to pKAN [59] using SacI/BamHI sites to generate plasmid pCH11341. A fragment downstream of ompC K-12 was then amplified with primers 3324/3325 and ligated to pKAN using EcoRI/KpnI sites to generate plasmid pCH11342. Fragments from pCH11341 and pCH11342 were amplified with primer pairs 3320/3505 and 3504/3325 (respectively) and the resulting products combined into one fragment by OE-PCR using primers 3320/3325. The final product was introduced into E. coli CH10013 that express phage λ Red proteins from plasmid pSIM6 [60] and recombinant bacteria selected on LB agar supplemented with kanamycin.
Table 1. Bacterial strains.
| Bacterial strain | Description a | Reference |
|---|---|---|
| EC536 | uropathogenic isolate of E. coli | [66] |
| CFT073 | uropathogenic isolate of E. coli | [67] |
| F11 | uropathogenic isolate of E. coli | [68] |
| UTI89 | uropathogenic isolate of E. coli | [69] |
| 6104H | uropathogenic isolate of E. coli | [70] |
| A54H | uropathogenic isolate of E. coli | [70] |
| A42H | uropathogenic isolate of E. coli | [70] |
| A35H | uropathogenic isolate of E. coli | [70] |
| A33H | uropathogenic isolate of E. coli | [70] |
| EC93 | E. coli isolate from rat feces | [1] |
| EPI100 | F− mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacXcZΔM15 ΔlacX recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ−rpsL nupG | Epicentre |
| MFDpir | RP4-2 Tc::[ΔMu1::aac(3)IV-ΔaphA-Δnic35-ΔMu2::zeo] ΔdapA::(erm-pir) ΔrecA | [62] |
| DL4905 | MC4100 λ640–13 PpapBA-gfp-mut3, KanR | [71] |
| DL6536 | E. coli 536 Δkps15::cat ΔaraBAD spc-araC-Para::cdiBAI, CmR StrR SpcR | [8] |
| DL6381 | E. coli 536 Δkps15::cat ΔaraBAD spec-araC-Para::cdiBA(ΔCT)I, CmR StrR SpcR | [8] |
| CH2016 | X90 (DE3) Δrna ΔslyD::kan, KanR | [72] |
| CH10013 | spontaneous rifampicin-resistant isolate of JCM158, RifR | [73] |
| CH10099 | CH10013 ΔompC | This study |
| CH10100 | CH10013 ΔompF | This study |
| CH10136 | CH10013 ΔompC ΔompF | This study |
| CH10226 | MC4100 Δbam::cat pZS21amp-bamA Eco, AmpR | This study |
| CH10227 | MC4100 Δbam::cat pZS21amp-bamA ECL, AmpR | This study |
| CH11346 | CH10013 Δwzb ΔompF ΔompC ΔrecA, RifR | This study |
| CH12133 | CH10013 ompC EC536 -kan, KanR | This study |
| CH12134 | CH10013 ΔompF ompC EC536 -kan, KanR | This study |
| CH12180 | CH10013 Δwzb ΔompF ompC EC536 ΔrecA::kan, RifR KanR | This study |
| CH12345 | JCM158 Δwzb ΔompF ΔompC ΔrecA::kan, KanR | This study |
aAbbreviations: AmpR, ampicillin resistant; CmR, chloramphenicol resistant; KanR, kanamycin resistant; RifR, rifampicin resistant; SpcR, spectinomycin resistant; StrR, streptomycin resistant
Plasmid constructions
Plasmids constructs used in this study are listed in Table 2. The E. coli cysK gene was amplified with primer pair 2111/2110 and ligated to pBR322 using EcoRV and BamHI restriction sites to generate plasmid pCH9763. The ompF K-12 allele was amplified from E. coli JCM158 with primer pair 2385/2386, and the fragment ligated to pZS21 using BamHI and XbaI restriction sites to generate plasmid pCH10202. Additionally, ompF K-12 and ompF EC536 were amplified with primers 2729/2734, and the fragments ligated to pTrcKX using KpnI and XhoI restriction sites to generate plasmids pCH11850 and pCH10780. The ompF genes from S. Typhimurium LT2 and E. cloacae ATCC 13047 were amplified with primer pairs 3468/3469 and 2729/3470 (respectively) and ligated to pTrcKX as described for the E. coli genes. The ompC genes from S. Typhimurium LT2 and E. cloacae were amplified with primers 3326/2388, digested with NcoI/XhoI and ligated to plasmid pTrc99a to generate pCH11852 and pCH11853, respectively. All E. coli ompC alleles were amplified with primer pair 2387/2388, followed by ligation to plasmid pTrcKX using KpnI and XhoI restriction sites. The coding sequences for OmpCCFT073 loops L4, L5, L7 and L8 of OmpCCFT073 were replaced using megaprimer PCR [61]. Plasmid pCH10778 template DNA was amplified with reverse primer 2388 in conjunction with primers 2935 (L5EC536), 2936 (L7EC536) and 2937 (L8EC536). The resulting products were used as megaprimers to amplify ompC CFT073 in conjunction with forward primer 2387. The chimeric products were digested with KpnI/XhoI and ligated to pTrcKX to generate plasmids pCH11334, pCH11765 and pCH11335. The same procedure and primers were used to generate ompC CFT073 constructs encoding two loops from ompC EC536. The loop L4 exchange megaprimer was generated with primers 2387/2807 and used with 2388 to generate the final product. All PCR products were digested with KpnI and XhoI and ligated to plasmid pTrcKX.
Table 2. Plasmid constructs.
| Plasmids | Description a | Reference |
|---|---|---|
| pTrc99a | IPTG-inducible expression vector, AmpR | Amersham |
| pTrcKX | Derivative of pTrc99a with additional KpnI and XhoI restriction sites, AmpR | [74] |
| pCP20 | temperature-inducible expression of FLP recombinase, CmR AmpR | [58] |
| pSC189kan | Mobilizable plasmid with R6Kγ replication origin. Carries the mariner transposon containing a kanamycin-resistance cassette, AmpR KanR | [63] |
| pWEB::TNC | pWEB::TNC (CDI–), AmpR, CmR | Epicentre |
| pDAL660Δ1–39 | Constitutively expresses the E. coli EC93 cdiBAI gene cluster, AmpR | [1] |
| pDAL866 | Arabinose-inducible expression of the E. coli 536 cdiBAI gene cluster, AmpR CmR | [32] |
| pZS21 | pSC101-derived plasmid vector, KanR | [75] |
| pZS21amp | pZS21 derivative with ampicillin-resistance cassette, AmpR | [4] |
| pZS21amp-bamA Eco | Constitutive expression of E. coli bamA, AmpR | [4] |
| pZS21amp-bamA ECL | Constitutive expression of E. cloacae bamA, AmpR | [20] |
| pET21::colE5-immE5 | Over-produces colicin E5 and ImmE5-His6, AmpR | [74] |
| pCH450 | pACY184 derivative that carries E. coli araC and the L-arabinose-inducible PBAD promoter. TetR | [59] |
| pCH9181 | pTrcKX::cdiA-CT/cdiI Dd3937-his 6, AmpR | [76] |
| pCH9763 | pBR322::cysK, AmpR | This study |
| pCH10138 | pTrcKX::ompC K-12, AmpR | This study |
| pCH10201 | pTrcKX::ompC F11, AmpR | This study |
| pCH10202 | pZS21-ompF K-12, KanR | This study |
| pCH11338 | pTrcKX::ompC EC536, AmpR | This study |
| pCH10458 | pTrcKX::ompC EC869, AmpR | This study |
| pCH10460 | pTrcKX::ompC EC93, AmpR | This study |
| pCH10778 | pTrcKX::ompC CFT073, AmpR | This study |
| pCH10779 | pTrcKX::ompC UTI89, AmpR | This study |
| pCH10780 | pTrcKX::ompF EC536, AmpR | This study |
| pCH10796 | pTrcKX::link8-ompF, AmpR | This study |
| pCH10797 | pTrc99a::ompF 2, AmpR | This study |
| pCH10959 | pTrcKX::ompC(L4F11)CFT073, AmpR | This study |
| pCH11330 | pTrcKX::ompC 6104H, AmpR | This study |
| pCH11331 | pTrcKX::ompC A54H, AmpR | This study |
| pCH11332 | pTrcKX::ompC A42H, AmpR | This study |
| pCH11333 | pTrcKX::ompC A35H, AmpR | This study |
| pCH11341 | pKAN-micF-ompC EC536, AmpR KanR | This study |
| pCH11342 | pKAN-abpE´, AmpR KanR | This study |
| pCH11496 | pCH450::DsRed; arabinose-inducible expression of DsRed, TetR | This study |
| pCH11627 | pTrcKX::ompC A33H, AmpR | This study |
| pCH11334 | pTrcKX::ompC(L5536)CFT073, AmpR | This study |
| pCH11335 | pTrcKX::ompC(L8536)CFT073, AmpR | This study |
| pCH11336 | pTrcKX::ompC(L5/L7536)CFT073, AmpR | This study |
| pCH11337 | pTrcKX::ompC(L7/L8536)CFT073, AmpR | This study |
| pCH11765 | pTrcKX::ompC(L7536)CFT073, AmpR | This study |
| pCH11767 | pTrcKX::ompC(L5/L8536)CFT073, AmpR | This study |
| pCH11850 | pTrcKX::ompF K-12, AmpR | This study |
| pCH11851 | pTrcKX::ompF ECL, AmpR | This study |
| pCH11852 | pTrcKX::ompF LT2, AmpR | This study |
| pCH11853 | pTrcKX::ompC ECL, AmpR | This study |
| pCH11854 | pTrcKX::ompC LT2, AmpR | This study |
| pCH12054 | pTrc99a::ompF 2*, AmpR | This study |
aAbbreviations: AmpR, ampicillin resistant; CmR, chloramphenicol resistant; KanR, kanamycin resistant; TetR, tetracycline resistant
Dimeric OmpF expression plasmids were constructed through sequential ligation of ompF coding sequences. The ompF EC536 allele was amplified with primers 2733/2734 and ligated to pCH9181 using SpeI/XhoI restriction sites to generate intermediate construct pCH10796. A second ompF fragment was amplified with primers 2732/2729 and ligated to pCH10796 using KpnI/SpeI restriction sites. The resulting pCH10797 (ompF 2) construct produces OmpFEC536 dimers linked by a Ser-Gly-Thr-Thr-Ser-Thr-Gly-Gly peptide. To introduce a longer linker sequence, ompF was amplified with primers 2729/2765 and ligated to pCH9181 using KpnI/SpeI sites to generate plasmid pCH11893. The second ompF module was amplified with primers 3008/2734 and ligated to pCH11893 using BamHI/XhoI restriction sites. The resulting pCH12054 (ompF 2*) construct produces OmpFEC536 dimers linked by a Ser-Gly-Thr-Gly-Ser-Asp-Thr-Ser-Gly-Gly-Thr-Asp-Gly-Thr-Gly-Gly peptide.
Competition co-cultures
Inhibitor and target bacteria were grown to mid-log phase in LB media supplemented with the appropriate antibiotics, then mixed at a 10:1 ratio in fresh LB medium without antibiotics and incubated for 4–5 h at 37°C with vigorous shaking in baffled flasks. Viable target-cell counts were enumerated on selective LB-agar as colony forming units (cfu) per mL. E. coli EPI100 carrying pDAL660Δ1–39 [1] or pDAL866 [33] were used as inhibitor cells for the comparison of CDIEC93 and CDIEC536 systems. The mock CDI−inhibitors were E. coli EPI100 carrying the pWEB::TNC vector. All other competitions used the E. coli 536 derivative DL6536, which expresses cdiBAI EC536 under the control of an arabinose-inducible promoter [8]. The mock inhibitor strain DL6381 is a derivative of DL6536 that carries a deletion of the cdiA-CT coding sequence [8]. DL6536 and DL6381 were cultured in 0.2% arabinose to induce cdiBAI expression and competition co-cultures were supplemented with 0.2% arabinose.
Transposon library construction and selection for CDIR mutants
Plasmid pSC189 (encoding the mariner transposon) was introduced into E. coli CH10013 cells carrying pCH9763 (pBR322::cysK) through conjugation with E. coli MFDpir donors [62, 63]. Donor and recipient cells were grown to mid-log phase in LB media supplemented with ampicillin and 30 μM diaminopimelic acid (for donors). Donors (~6.0 ×108 colony forming units, cfu) and recipients (~3 ×108 cfu) were mixed and collected by centrifugation for 2 min at 9,000 rpm in a microcentrifuge. The supernatant was removed by aspiration and the cell pellet resuspended in 100 μL of 1× M9 salts. Cell mixtures were spotted onto 0.45 μm nitrocellulose membranes and incubated on LB-agar without inversion for 4 h at 37°C. Cells were then harvested into 2 mL of 1× M9 salts and transposon-insertion mutants selected on LB-agar supplemented with rifampicin and kanamycin. Over 50,000 colonies from each mating were harvested into 1× M9 salts, and inoculated into 50 mL of LB medium in a 250 mL baffled flask. Inhibitor cells (E. coli 536 derivative DL6536) were grown in parallel in LB medium supplemented with L-arabinose until mid-log phase. Inhibitors and targets were mixed at a 10:1 ratio in fresh LB medium supplemented with L-arabinose and cultured for 4 h with shaking at 37°C. Viable target cells were enumerated as cfu/mL on LB agar supplemented with rifampicin and kanamycin. Survivors from the first round of CDIEC536 selection were harvested into 1× M9 salts and inoculated into 50 mL of LB medium supplemented with L-arabinose for a second round of selection. After the third round of selection, target-cell populations were completely resistant to the CDI inhibitor cells. Individual colonies were screened for CysK activity in MOPS minimal media containing 3 mM 1,2,4-triazole, which inhibits the growth of cysK + bacteria [64]. CDIR phenotypes for all cysK + clones were confirmed in competition co-cultures with E. coli 536 inhibitors. Transposon mutations were transduced into CDI-sensitive cells, and the resulting transductants tested in competition co-cultures for CDIR phenotypes.
Flow cytometry analysis of cell-cell adhesion
Overnight cultures of GFP-labeled E. coli DL4905 cells carrying pDAL660Δ1–39 (CDIEC93), pDAL866 (CDIEC536) or pWEB::TNC (CDI−) were diluted into fresh tryptone broth (TB) and grown to mid-log phase at 30°C. Inhibitor cells were then mixed at a 5:1 ratio with DsRed-labeled E. coli CH10099 (ΔompC) or CH10100 (ΔompF) cells complemented with the indicated ompC and ompF expressing plasmids. Cell suspensions were incubated with aeration for 15 min at 30°C, diluted 1:50 into filtered 1× phosphate-buffered saline (PBS), and then analyzed on an Accuri C6 flow cytometer using FL1 (533/30 nm, GFP) and FL2 (585/40 nm, DS-Red) fluorophore filters (Becton Dickinson) as described [36].
Immunoblot analysis
Bacteria were grown to mid-log phase in LB media supplemented with ampicillin. Cells were collected by centrifugation at 6,000 ×g. Cell were frozen at –80°C, then broken by freeze-thaw cycles in 8 M urea, 150 mM NaCl, 50 mM Tris-HCl (pH 8.0). Extracts were quantified by Bradford assay and equal amounts of protein were resolved on 6 M urea/10% SDS polyacrylamide gels for 4 h at 100 V. Gels were electroblotted onto nitrocellulose membranes and OMPs detected using polyclonal antisera raised against E. coli OmpC/OmpF and BamA. Immunoblots were visualized using IRDye 680 (LI-COR) labeled anti-rabbit secondary antibodies and an Odyssey infrared imager.
Colicin E5 purification and activity assays
Colicin E5 was purified in complex with its His6-tagged immunity protein as described previously [6, 65]. E. coli strains CH12180 and CH12345 carrying pCH11850 (ompF), pCH10796 (ompF-ompF, 8-residue linker), or pCH12054 (ompF-ompF, 16-residue linker) were grown to mid-log phase in LB media supplemented with ampicillin, then collected by centrifugation and re-suspended in fresh pre-warmed media at OD600 = 0.05. After 30 min of incubation with shaking, purified colicin E5 was added to a final concentration of 1 μM and cell growth monitored by measuring the OD600 every 60 min.
Supporting Information
The sequences of mature OmpC proteins were aligned using Clustal-Omega and identical residues indicated with asterisks (*). Extracellular loop sequences are shown in red and β-strands in boldface.
(PDF)
The sequences of mature OmpCK-12 and OmpFK-12 were aligned using Clustal-Omega and identical residues indicated with asterisks (*). Extracellular loops are shown in red font and β-strands are underlined. Buried interfacial residues are shown in blue, and residues involved in direct inter-protomer H-bonds and salt-bridges are in orange. Contacts were determined with PDBePISA using PDB:2J1N (OmpCK-12) and PDB:3POX (OmpFK-12).
(PDF)
Individual protomers of (A) OmpCK-12 (PDB:2J1N) and (B) OmpFK-12 (PDB:3POX) are viewed from the inter-protomer interface (left) and from the extracellular milieu (right). Extracellular loops L2 and L4 from adjacent protomers are shown as yellow sticks and β-strands involved inter-subunit contacts are labeled in the left panels. Buried interfacial residues are shown in blue, and residues involved in direct inter-protomer H-bonds and salt-bridges are in orange. Contacts were determined with PDBePISA. The conserved inter-subunit ion-pairs are shown in the right panels with residues labeled.
(TIF)
CdiAEC93 and CdiAEC536 sequences were aligned using Clustal-Omega and identical residues are highlighted in blue. Predicted domains and peptide motifs are as described by the InterPro web server. CdiAEC93 annotations are available at http://www.ebi.ac.uk/interpro/protein/Q3YL96, and CdiAEC536 at http://www.ebi.ac.uk/interpro/protein/Q0T963. The signal sequence and TPS transport domain are required for CdiA export. FHA-1 (Pfam: PF05594) and FHA-2 (PF13332) peptide repeats are predicted to form a β-helix. The pretoxin-VENN domain (PT-VENN; PF04829) demarcates the variable C-terminal toxin region.
(TIF)
Individual OmpC protomers from E. coli K-12 (PDB:2J1N), CFT073 (PDB:2XE1) and 6104H (PDB:2XE2) are shown in surface representation as viewed from the extracellular milieu. Extracellular loops L4 (maroon), L5 (green) and L7 (orange) are labeled in the top row of images. The bottom row shows the OmpC protomers in the same orientation, but with the surface rendered as an electrostatic potential map. Red areas are electronegative, blue are electropositive and white are neutral.
(TIF)
OmpC from E. coli str. K-12 substr. MG1655 was used to query all E. coli isolates using BLASTP 2.2.32+ at https://blast.ncbi.nlm.nih.gov/Blast.cgi. Proteins with identical amino acid sequences are grouped with colored backgrounds. Extracellular loop L4, L5 and L7 variations are indicated according the scheme outlined in S2 Table. Asterisks (*) indicate OmpC variants tested in this study.
(XLSX)
Loop sequences were determined for 2707 predicted E. coli OmpC proteins. Sequences were grouped and numbered according to sequence identity. Red residues are shared with OmpC from E. coli K-12.
(XLSX)
Predicted hydrogen-bonds (H-bonds) and salt-bridges between individual protomers of E. coli K-12 OmpC (PDB: 2J1N) and OmpF (PDB: 3POX) were determined using PDBePISA (http://www.ebi.ac.uk/pdbe/pisa/). Solvent accessible surface areas (ASA) and buried surface areas (BSA) were determined using PDBePISA (http://www.ebi.ac.uk/pdbe/pisa/). Residues highlighted in orange are buried at interprotomer interfaces. Predicted hydrogen bonds are indicated by H and salt-bridges by SH.
(XLSX)
Extracellular loop residues involved in intraprotomer loop-loop interactions are presented for three E. coli OmpC proteins. Loop L4, L5 and L7 sequences are labeled according to the scheme outlined in S2 Table.
(XLSX)
(DOCX)
Acknowledgments
We thank Xiangwu Nou for providing E. coli EC869 genomic DNA and Thomas Silhavy for providing antisera to E. coli OmpC/OmpF and BamA.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grant R01 GM117930 (CSH) from the National Institutes of Health. CMB was supported in part by Dean's and Chang Fellowships from the University of California, Santa Barbara, and JLEW was supported by National Science Foundation Graduate Research Fellowship DGE-1144085. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. Contact-dependent inhibition of growth in Escherichia coli . Science. 2005;309(5738):1245–8. 10.1126/science.1115109 . [DOI] [PubMed] [Google Scholar]
- 2. Kajava AV, Cheng N, Cleaver R, Kessel M, Simon MN, Willery E, et al. Beta-helix model for the filamentous haemagglutinin adhesin of Bordetella pertussis and related bacterial secretory proteins. Mol Microbiol. 2001;42(2):279–92. Epub 2001/11/13. doi: 2598 [pii]. 10.1046/j.1365-2958.2001.02598.x . [DOI] [PubMed] [Google Scholar]
- 3. Willett JL, Ruhe ZC, Goulding CW, Low DA, Hayes CS. Contact-Dependent Growth Inhibition (CDI) and CdiB/CdiA Two-Partner Secretion Proteins. J Mol Biol. 2015;427(23):3754–65. Epub 2015/09/22. S0022-2836(15)00521-5 [pii] 10.1016/j.jmb.2015.09.010 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aoki SK, Malinverni JC, Jacoby K, Thomas B, Pamma R, Trinh BN, et al. Contact-dependent growth inhibition requires the essential outer membrane protein BamA (YaeT) as the receptor and the inner membrane transport protein AcrB. Mol Microbiol. 2008;70(2):323–40. 10.1111/j.1365-2958.2008.06404.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aoki SK, Webb JS, Braaten BA, Low DA. Contact-dependent growth inhibition causes reversible metabolic downregulation in Escherichia coli . J Bacteriol. 2009;191(6):1777–86. 10.1128/JB.01437-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruhe ZC, Nguyen JY, Beck CM, Low DA, Hayes CS. The proton-motive force is required for translocation of CDI toxins across the inner membrane of target bacteria. Mol Microbiol. 2014;94(2):466–81. Epub 2014/09/02. 10.1111/mmi.12779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruhe ZC, Townsley L, Wallace AB, King A, Van der Woude MW, Low DA, et al. CdiA promotes receptor-independent intercellular adhesion. Mol Microbiol. 2015;98(1):175–92. 10.1111/mmi.13114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, et al. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature. 2010;468(7322):439–42. Epub 2010/11/19. nature09490 [pii] 10.1038/nature09490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koskiniemi S, Garza-Sanchez F, Edman N, Chaudhuri S, Poole SJ, Manoil C, et al. Genetic analysis of the CDI pathway from Burkholderia pseudomallei 1026b. PLoS One. 2015;10(3):e0120265 Epub 2015/03/19. 10.1371/journal.pone.0120265 PONE-D-14-52801 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nikolakakis K, Amber S, Wilbur JS, Diner EJ, Aoki SK, Poole SJ, et al. The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol Microbiol. 2012;84(3):516–29. Epub 2012/03/23. 10.1111/j.1365-2958.2012.08039.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderson MS, Garcia EC, Cotter PA. The Burkholderia bcpAIOB genes define unique classes of two-partner secretion and contact dependent growth inhibition systems. PLoS Genet. 2012;8(8):e1002877 Epub 2012/08/23. 10.1371/journal.pgen.1002877 PGENETICS-D-12-00079 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arenas J, Schipper K, van Ulsen P, van der Ende A, Tommassen J. Domain exchange at the 3´ end of the gene encoding the fratricide meningococcal two-partner secretion protein A. BMC Genomics. 2013;14:622 Epub 2013/09/17. 1471-2164-14-622 [pii] 10.1186/1471-2164-14-622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mercy C, Ize B, Salcedo SP, de Bentzmann S, Bigot S. Functional Characterization of Pseudomonas Contact Dependent Growth Inhibition (CDI) Systems. PLoS One. 2016;11(1):e0147435 10.1371/journal.pone.0147435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ruhe ZC, Low DA, Hayes CS. Bacterial contact-dependent growth inhibition. Trends Microbiol. 2013;21(5):230–7. Epub 2013/03/12. S0966-842X(13)00023-1 [pii] 10.1016/j.tim.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct. 2012;7:18 Epub 2012/06/27. 1745-6150-7-18 [pii] 10.1186/1745-6150-7-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science. 2003;299(5604):262–5. Epub 2003/01/11. 10.1126/science.1078973 299/5604/262 [pii]. . [DOI] [PubMed] [Google Scholar]
- 17. Gentle I, Gabriel K, Beech P, Waller R, Lithgow T. The Omp85 family of proteins is essential for outer membrane biogenesis in mitochondria and bacteria. J Cell Biol. 2004;164(1):19–24. Epub 2003/12/31. 10.1083/jcb.200310092 jcb.200310092 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli . Cell. 2005;121(2):235–45. Epub 2005/04/27. S0092-8674(05)00160-1 [pii] 10.1016/j.cell.2005.02.015 . [DOI] [PubMed] [Google Scholar]
- 19. Smith DL, James CE, Sergeant MJ, Yaxian Y, Saunders JR, McCarthy AJ, et al. Short-tailed stx phages exploit the conserved YaeT protein to disseminate Shiga toxin genes among enterobacteria. J Bacteriol. 2007;189(20):7223–33. Epub 2007/08/19. 10.1128/JB.00824-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ruhe ZC, Wallace AB, Low DA, Hayes CS. Receptor polymorphism restricts contact-dependent growth inhibition to members of the same species. MBio. 2013;4(4). Epub 2013/07/25. mBio.00480-13 [pii] 10.1128/mBio.00480-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rojas CM, Ham JH, Deng WL, Doyle JJ, Collmer A. HecA, a member of a class of adhesins produced by diverse pathogenic bacteria, contributes to the attachment, aggregation, epidermal cell killing, and virulence phenotypes of Erwinia chrysanthemi EC16 on Nicotiana clevelandii seedlings. Proc Natl Acad Sci U S A. 2002;99(20):13142–7. Epub 2002/09/25. 10.1073/pnas.202358699 202358699 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guilhabert MR, Kirkpatrick BC. Identification of Xylella fastidiosa antivirulence genes: hemagglutinin adhesins contribute X. fastidiosa biofilm maturation and colonization and attenuate virulence. Mol Plant Microbe Interact. 2005;18(8):856–68. Epub 2005/09/02. 10.1094/MPMI-18-0856 . [DOI] [PubMed] [Google Scholar]
- 23. Neil RB, Apicella MA. Role of HrpA in biofilm formation of Neisseria meningitidis and regulation of the hrpBAS transcripts. Infect Immun. 2009;77(6):2285–93. Epub 2009/03/18. IAI.01502-08 [pii] 10.1128/IAI.01502-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gottig N, Garavaglia BS, Garofalo CG, Orellano EG, Ottado J. A filamentous hemagglutinin-like protein of Xanthomonas axonopodis pv. citri, the phytopathogen responsible for citrus canker, is involved in bacterial virulence. PLoS One. 2009;4(2):e4358 Epub 2009/02/06. 10.1371/journal.pone.0004358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia EC, Anderson MS, Hagar JA, Cotter PA. Burkholderia BcpA mediates biofilm formation independently of interbacterial contact-dependent growth inhibition. Mol Microbiol. 2013;89(6):1213–25. Epub 2013/07/25. 10.1111/mmi.12339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lerouge I, Vanderleyden J. O-antigen structural variation: mechanisms and possible roles in animal/plant-microbe interactions. FEMS Microbiol Rev. 2002;26(1):17–47. 10.1111/j.1574-6976.2002.tb00597.x . [DOI] [PubMed] [Google Scholar]
- 27. Deitsch KW, Lukehart SA, Stringer JR. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat Rev Microbiol. 2009;7(7):493–503. 10.1038/nrmicro2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Foley J. Mini-review: Strategies for Variation and Evolution of Bacterial Antigens. Comput Struct Biotechnol J. 2015;13:407–16. 10.1016/j.csbj.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Obergfell KP, Seifert HS. Mobile DNA in the Pathogenic Neisseria . Microbiol Spectr. 2015;3(1):MDNA3-0015-2014. 10.1128/microbiolspec.MDNA3-0015-2014 . [DOI] [PubMed] [Google Scholar]
- 30. Chen SL, Hung CS, Xu J, Reigstad CS, Magrini V, Sabo A, et al. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc Natl Acad Sci U S A. 2006;103(15):5977–82. Epub 2006/04/06. 0600938103 [pii] 10.1073/pnas.0600938103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petersen L, Bollback JP, Dimmic M, Hubisz M, Nielsen R. Genes under positive selection in Escherichia coli . Genome Res. 2007;17(9):1336–43. Epub 2007/08/07. gr.6254707 [pii] 10.1101/gr.6254707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Webb JS, Nikolakakis KC, Willett JLE, Aoki SK, Hayes CS, Low DA. Delivery of CdiA nuclease toxins into target cells during contact-dependent growth inhibition. PLoS ONE. 2013;8(2):e57609 10.1371/journal.pone.0057609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diner EJ, Beck CM, Webb JS, Low DA, Hayes CS. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev. 2012;26(5):515–25. Epub 2012/02/16. gad.182345.111 [pii] 10.1101/gad.182345.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnson PM, Beck CM, Morse RP, Garza-Sanchez F, Low DA, Hayes CS, et al. Unraveling the essential role of CysK in CDI toxin activation. Proc Natl Acad Sci U S A. 2016;113(35):9792–7. 10.1073/pnas.1607112113 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pratt LA, Hsing W, Gibson KE, Silhavy TJ. From acids to osmZ: multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli . Mol Microbiol. 1996;20(5):911–7. Epub 1996/06/01. 10.1111/j.1365-2958.1996.tb02532.x . [DOI] [PubMed] [Google Scholar]
- 36. Ruhe ZC, Hayes CS, Low DA. Measuring Cell-Cell Binding Using Flow-Cytometry. Methods Mol Biol. 2015;1329:127–36. Epub 2015/10/03. 10.1007/978-1-4939-2871-2_9 . [DOI] [PubMed] [Google Scholar]
- 37. Gehring KB, Nikaido H. Existence and purification of porin heterotrimers of Escherichia coli K12 OmpC, OmpF, and PhoE proteins. J Biol Chem. 1989;264(5):2810–5. Epub 1989/02/15. . [PubMed] [Google Scholar]
- 38. Kurisu G, Zakharov SD, Zhalnina MV, Bano S, Eroukova VY, Rokitskaya TI, et al. The structure of BtuB with bound colicin E3 R-domain implies a translocon. Nat Struct Biol. 2003;10(11):948–54. 10.1038/nsb997 . [DOI] [PubMed] [Google Scholar]
- 39. Yamashita E, Zhalnina MV, Zakharov SD, Sharma O, Cramer WA. Crystal structures of the OmpF porin: function in a colicin translocon. EMBO J. 2008;27(15):2171–80. Epub 2008/07/19. emboj2008137 [pii] 10.1038/emboj.2008.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Housden NG, Wojdyla JA, Korczynska J, Grishkovskaya I, Kirkpatrick N, Brzozowski AM, et al. Directed epitope delivery across the Escherichia coli outer membrane through the porin OmpF. Proc Natl Acad Sci U S A. 2010;107(50):21412–7. Epub 2010/11/26. 1010780107 [pii] 10.1073/pnas.1010780107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Housden NG, Hopper JT, Lukoyanova N, Rodriguez-Larrea D, Wojdyla JA, Klein A, et al. Intrinsically disordered protein threads through the bacterial outer-membrane porin OmpF. Science. 2013;340(6140):1570–4. 10.1126/science.1237864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mock M, Pugsley AP. The BtuB group col plasmids and homology between the colicins they encode. J Bacteriol. 1982;150(3):1069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Basle A, Rummel G, Storici P, Rosenbusch JP, Schirmer T. Crystal structure of osmoporin OmpC from E. coli at 2.0 A. J Mol Biol. 2006;362(5):933–42. Epub 2006/09/05. S0022-2836(06)00985-5 [pii] 10.1016/j.jmb.2006.08.002 . [DOI] [PubMed] [Google Scholar]
- 44. Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, Postle K, et al. Colicin biology. Microbiol Mol Biol Rev. 2007;71(1):158–229. Epub 2007/03/10. 71/1/158 [pii] 10.1128/MMBR.00036-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Housden NG, Kleanthous C. Colicin translocation across the Escherichia coli outer membrane. Biochem Soc Trans. 2012;40(6):1475–9. 10.1042/BST20120255 . [DOI] [PubMed] [Google Scholar]
- 46. Willett JL, Gucinski GC, Fatherree JP, Low DA, Hayes CS. Contact-dependent growth inhibition toxins exploit multiple independent cell-entry pathways. Proc Natl Acad Sci U S A. 2015;112(36):11341–6. Epub 2015/08/26. 1512124112 [pii] 10.1073/pnas.1512124112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, et al. Structural insight into the biogenesis of beta-barrel membrane proteins. Nature. 2013;501(7467):385–90. Epub 2013/09/03. nature12521 [pii] 10.1038/nature12521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ni D, Wang Y, Yang X, Zhou H, Hou X, Cao B, et al. Structural and functional analysis of the beta-barrel domain of BamA from Escherichia coli . FASEB J. 2014;28(6):2677–85. Epub 2014/03/13. fj.13-248450 [pii] 10.1096/fj.13-248450 . [DOI] [PubMed] [Google Scholar]
- 49. Leonard-Rivera M, Misra R. Conserved residues of the putative L6 loop of Escherichia coli BamA play a critical role in the assembly of beta-barrel outer membrane proteins, including that of BamA itself. J Bacteriol. 2012;194(17):4662–8. Epub 2012/07/04. JB.00825-12 [pii] 10.1128/JB.00825-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rigel NW, Ricci DP, Silhavy TJ. Conformation-specific labeling of BamA and suppressor analysis suggest a cyclic mechanism for beta-barrel assembly in Escherichia coli . Proc Natl Acad Sci U S A. 2013;110(13):5151–6. Epub 2013/03/13. 1302662110 [pii] 10.1073/pnas.1302662110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Noinaj N, Kuszak AJ, Balusek C, Gumbart JC, Buchanan SK. Lateral opening and exit pore formation are required for BamA function. Structure. 2014;22(7):1055–62. 10.1016/j.str.2014.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lou H, Chen M, Black SS, Bushell SR, Ceccarelli M, Mach T, et al. Altered antibiotic transport in OmpC mutants isolated from a series of clinical strains of multi-drug resistant E. coli. PLoS One. 2011;6(10):e25825 Epub 2011/11/05. 10.1371/journal.pone.0025825 PONE-D-11-12617 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nart P, Holden N, McAteer SP, Wang D, Flockhart AF, Naylor SW, et al. Mucosal antibody responses of colonized cattle to Escherichia coli O157-secreted proteins, flagellin, outer membrane proteins and lipopolysaccharide. FEMS Immunol Med Microbiol. 2008;52(1):59–68. 10.1111/j.1574-695X.2007.00341.x . [DOI] [PubMed] [Google Scholar]
- 54. Yu SL, Ko KL, Chen CS, Chang YC, Syu WJ. Characterization of the distal tail fiber locus and determination of the receptor for phage AR1, which specifically infects Escherichia coli O157:H7. J Bacteriol. 2000;182(21):5962–8. 10.1128/jb.182.21.5962-5968.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Welch RA, Burland V, Plunkett G 3rd, Redford P, Roesch P, Rasko D, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli . Proc Natl Acad Sci U S A. 2002;99(26):17020–4. 10.1073/pnas.252529799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ruhe ZC, Nguyen JY, Chen AJ, Leung NY, Hayes CS, Low DA. CDI Systems Are Stably Maintained by a Cell-Contact Mediated Surveillance Mechanism. PLoS Genet. 2016;12(6):e1006145 10.1371/journal.pgen.1006145 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006 0008. 10.1038/msb4100050 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158(1):9–14. Epub 1995/05/26. 037811199500193A [pii]. 10.1016/0378-1119(95)00193-a . [DOI] [PubMed] [Google Scholar]
- 59. Hayes CS, Sauer RT. Cleavage of the A site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol Cell. 2003;12(4):903–11. Epub 2003/10/29. S109727650300385X [pii]. 10.1016/s1097-2765(03)00385-x . [DOI] [PubMed] [Google Scholar]
- 60. Datta S, Costantino N, Court DL. A set of recombineering plasmids for gram-negative bacteria. Gene. 2006;379:109–15. Epub 2006/06/06. 10.1016/j.gene.2006.04.018 . [DOI] [PubMed] [Google Scholar]
- 61. Aiyar A, Leis J. Modification of the megaprimer method of PCR mutagenesis: improved amplification of the final product. Biotechniques. 1993;14(3):366–9. Epub 1993/03/01. . [PubMed] [Google Scholar]
- 62. Ferrieres L, Hemery G, Nham T, Guerout AM, Mazel D, Beloin C, et al. Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J Bacteriol. 2010;192(24):6418–27. Epub 2010/10/12. JB.00621-10 [pii] 10.1128/JB.00621-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chiang SL, Rubin EJ. Construction of a mariner-based transposon for epitope-tagging and genomic targeting. Gene. 2002;296(1–2):179–85. Epub 2002/10/18. S0378111902008569 [pii]. 10.1016/s0378-1119(02)00856-9 . [DOI] [PubMed] [Google Scholar]
- 64. Kredich NM, Foote LJ, Hulanicka MD. Studies on the mechanism of inhibition of Salmonella typhimurium by 1,2,4-triazole. J Biol Chem. 1975;250(18):7324–31. . [PubMed] [Google Scholar]
- 65. Garza-Sánchez F, Gin JG, Hayes CS. Amino acid starvation and colicin D treatment induce A-site mRNA cleavage in Escherichia coli. J Mol Biol. 2008;378(3):505–19. Epub 2008/04/02. S0022-2836(08)00275-1 [pii] 10.1016/j.jmb.2008.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Berger H, Hacker J, Juarez A, Hughes C, Goebel W. Cloning of the chromosomal determinants encoding hemolysin production and mannose-resistant hemagglutination in Escherichia coli . J Bacteriol. 1982;152(3):1241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, et al. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun. 1990;58(5):1281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stapleton A, Moseley S, Stamm WE. Urovirulence determinants in Escherichia coli isolates causing first-episode and recurrent cystitis in women. J Infect Dis. 1991;163(4):773–9. 10.1093/infdis/163.4.773 . [DOI] [PubMed] [Google Scholar]
- 69. Mulvey MA, Schilling JD, Hultgren SJ. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun. 2001;69(7):4572–9. 10.1128/IAI.69.7.4572-4579.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stamey TA, Timothy M, Millar M, Mihara G. Recurrent urinary infections in adult women. The role of introital enterobacteria. Calif Med. 1971;115(1):1–19. [PMC free article] [PubMed] [Google Scholar]
- 71. Morse RP, Nikolakakis KC, Willett JL, Gerrick E, Low DA, Hayes CS, et al. Structural basis of toxicity and immunity in contact-dependent growth inhibition (CDI) systems. Proc Natl Acad Sci U S A. 2012;109:21480–5. Epub 2012/12/14. 1216238110 [pii] 10.1073/pnas.1216238110 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Garza-Sánchez F, Janssen BD, Hayes CS. Prolyl-tRNA(Pro) in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J Biol Chem. 2006;281(45):34258–68. Epub 2006/09/14. M608052200 [pii] 10.1074/jbc.M608052200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Beck CM, Morse RP, Cunningham DA, Iniguez A, Low DA, Goulding CW, et al. CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure. 2014;22:707–18. Epub 2014/03/25. S0969-2126(14)00052-5 [pii] 10.1016/j.str.2014.02.012 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Beck CM, Diner EJ, Kim JJ, Low DA, Hayes CS. The F pilus mediates a novel pathway of CDI toxin import. Mol Microbiol. 2014;93(2):276–90. Epub 2014/06/04. 10.1111/mmi.12658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25(6):1203–10. Epub 1997/03/15. 10.1093/nar/25.6.1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Poole SJ, Diner EJ, Aoki SK, Braaten BA, T'Kint de Roodenbeke C, Low DA, et al. Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet. 2011;7(8):e1002217 Epub 2011/08/11. 10.1371/journal.pgen.1002217 PGENETICS-D-11-00234 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The sequences of mature OmpC proteins were aligned using Clustal-Omega and identical residues indicated with asterisks (*). Extracellular loop sequences are shown in red and β-strands in boldface.
(PDF)
The sequences of mature OmpCK-12 and OmpFK-12 were aligned using Clustal-Omega and identical residues indicated with asterisks (*). Extracellular loops are shown in red font and β-strands are underlined. Buried interfacial residues are shown in blue, and residues involved in direct inter-protomer H-bonds and salt-bridges are in orange. Contacts were determined with PDBePISA using PDB:2J1N (OmpCK-12) and PDB:3POX (OmpFK-12).
(PDF)
Individual protomers of (A) OmpCK-12 (PDB:2J1N) and (B) OmpFK-12 (PDB:3POX) are viewed from the inter-protomer interface (left) and from the extracellular milieu (right). Extracellular loops L2 and L4 from adjacent protomers are shown as yellow sticks and β-strands involved inter-subunit contacts are labeled in the left panels. Buried interfacial residues are shown in blue, and residues involved in direct inter-protomer H-bonds and salt-bridges are in orange. Contacts were determined with PDBePISA. The conserved inter-subunit ion-pairs are shown in the right panels with residues labeled.
(TIF)
CdiAEC93 and CdiAEC536 sequences were aligned using Clustal-Omega and identical residues are highlighted in blue. Predicted domains and peptide motifs are as described by the InterPro web server. CdiAEC93 annotations are available at http://www.ebi.ac.uk/interpro/protein/Q3YL96, and CdiAEC536 at http://www.ebi.ac.uk/interpro/protein/Q0T963. The signal sequence and TPS transport domain are required for CdiA export. FHA-1 (Pfam: PF05594) and FHA-2 (PF13332) peptide repeats are predicted to form a β-helix. The pretoxin-VENN domain (PT-VENN; PF04829) demarcates the variable C-terminal toxin region.
(TIF)
Individual OmpC protomers from E. coli K-12 (PDB:2J1N), CFT073 (PDB:2XE1) and 6104H (PDB:2XE2) are shown in surface representation as viewed from the extracellular milieu. Extracellular loops L4 (maroon), L5 (green) and L7 (orange) are labeled in the top row of images. The bottom row shows the OmpC protomers in the same orientation, but with the surface rendered as an electrostatic potential map. Red areas are electronegative, blue are electropositive and white are neutral.
(TIF)
OmpC from E. coli str. K-12 substr. MG1655 was used to query all E. coli isolates using BLASTP 2.2.32+ at https://blast.ncbi.nlm.nih.gov/Blast.cgi. Proteins with identical amino acid sequences are grouped with colored backgrounds. Extracellular loop L4, L5 and L7 variations are indicated according the scheme outlined in S2 Table. Asterisks (*) indicate OmpC variants tested in this study.
(XLSX)
Loop sequences were determined for 2707 predicted E. coli OmpC proteins. Sequences were grouped and numbered according to sequence identity. Red residues are shared with OmpC from E. coli K-12.
(XLSX)
Predicted hydrogen-bonds (H-bonds) and salt-bridges between individual protomers of E. coli K-12 OmpC (PDB: 2J1N) and OmpF (PDB: 3POX) were determined using PDBePISA (http://www.ebi.ac.uk/pdbe/pisa/). Solvent accessible surface areas (ASA) and buried surface areas (BSA) were determined using PDBePISA (http://www.ebi.ac.uk/pdbe/pisa/). Residues highlighted in orange are buried at interprotomer interfaces. Predicted hydrogen bonds are indicated by H and salt-bridges by SH.
(XLSX)
Extracellular loop residues involved in intraprotomer loop-loop interactions are presented for three E. coli OmpC proteins. Loop L4, L5 and L7 sequences are labeled according to the scheme outlined in S2 Table.
(XLSX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.