

Abstract
Targeted molecular therapy is an effective anticancer strategy. Anti‐EGFR monoclonal antibodies such as cetuximab (CTX) have been approved for the treatment of various malignancies, including colorectal cancer (CRC) with wild‐type KRAS. However, their efficacy in patients with KRAS mutations has not been established. Therefore, we investigated whether CTX treatment was effective as a single agent or in combination with zoledronic acid (ZOL) in human CRC cell lines with different KRAS status. CRC cell lines SW48 (wild‐type KRAS) and LS174T (mutant KRAS) were treated with ZOL, CTX and a combination of both drugs. Cytotoxicity was measured using the MTT assay. Changes in the levels of intracellular signaling proteins were evaluated using western blot analysis. Finally, we evaluated the efficacy of the combination treatment in an in vivo xenograft model. We observed that ZOL apparently inhibited growth in both cell lines, whereas CTX showed little effect. ZOL also increased the levels of unprenylated RAS. Combined ZOL and CTX treatment was synergistic in both cell lines and was associated with inhibition of the RAS‐MAPK and AKT‐mTOR signaling pathways. Furthermore, the combination treatment was more effective in suppressing the growth of xenografts derived from both SW48 and LS174T cells; this effect was associated with increased apoptosis. These results demonstrate that ZOL inhibits the growth of colon cancer cells regardless of KRAS status, and combination therapy using ZOL and CTX enhances this growth suppression. These findings suggest a novel strategy for the treatment of CRC independent of KRAS mutational status.
Keywords: colorectal cancer, k‐ras mutant, zoledronic acid, cetuximab
Short abstract
What's new?
A new combination therapy could be the one‐two punch that takes out treatment‐resistant colorectal cancer. The anti‐EGFR antibody cetuximab works well against colorectal cancer, but tumors with KRAS mutations can fend it off. Zoledronic acid, which can treat osteoporosis, also thwarts various types of cancer, and in this article the authors evaluated whether it could boost cetuximab's effectiveness. They showed that not only did zoledronic acid suppress colorectal tumor growth, even in KRAS mutants, but that the combination of both agents works better than either alone, both in cultured cell and in mice.
Abbreviations
- BRAF
B‐Raf proto‐oncogene, serine/threonine kinase
- DMSO
dimethyl sulfoxide
- EGFR
epidermal growth factor receptor
- FOLFILI
folinic acid, fuluorouracil, irinotecan
- FOLFOX
folinic acid, fuluorouracil, oxaliplatin
- GTP
guanosine‐5′‐triphosphate
- IGF‐1
insulin‐like growth factor‐1
- JAK
Janus kinase
- KRAS
v‐Ki‐ras2 kirsten rat sarcoma viral oncogene homolog
- MAPK
mitogen‐activated protein kinase
- mTOR
mammalian target of rapamycin
- PARP
poly(ADP‐ribose)polymerase
- PBS
phosphate‐buffered saline
- PI3K
phosphatidylinositol‐3 kinase
- PVDF
polyvinylidene difluoride
- RAP1A
RAS‐related protein‐1a
- RIPA
radio immunoprecipitation assay
- STAT
signal transducers and activators of transcription
- XELOX
xeloda, oxaliplatin
Colorectal cancer remains one of the major causes of cancer deaths worldwide.1, 2 Despite recent advances in the development of various diagnostic tools, in many patients, colorectal cancer is still diagnosed at an advanced stage, and recurrent tumors are often detected even after curative surgery. On the other hand, new chemotherapeutic regimens such as FOLFOX, FOLFIRI, and XELOX have been developed to exert a more potent activity.3, 4, 5, 6 More recently, molecular targeted therapies such as tyrosine kinase inhibitors and monoclonal antibodies have been shown to enhance tumor regression in combination with chemotherapy.7, 8
Epidermal growth factor receptor (EGFR) is a tyrosine kinase receptor that plays a key role in the development and progression of several human cancers. EGFR targeting has been successful in the treatment of several cancers.9 The EGFR family consists of at least four members, of which both EGFR and human epidermal growth factor 2 (HER2) are critical targets in cancers including breast and gastric malignancies.10, 11, 12, 13 Cetuximab (CTX), an anti‐EGFR monoclonal antibody, has been widely used particularly for treatment of colorectal and lung cancers.14, 15 However, some patients with colorectal cancer (CRC) are resistant to EGFR inhibitors because of the continuous activation of the RAS/mitogen‐activated protein kinase (MAPK) pathway by a mutation in codon 12 of the wild‐type v‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) gene.14, 15, 16, 17 Therefore, the use of CTX is currently restricted to patients with wild‐type KRAS. In human CRC, mutations in the KRAS gene have a frequency of around 30–40% and are linked to poor outcomes, whereas mutations of the B‐Raf proto‐oncogene, serine/threonine kinase (BRAF) gene, a downstream molecule of KRAS, occur in only 5–10% of patients with sporadic diseases. Mutations in the KRAS and BRAF genes are frequently found to be mutually exclusive in CRC.18
Zoledronic acid (ZOL) is a member of the bisphosphonate (BP) molecular class and is clinically used to treat osteoporosis and prevent skeletal events related to bone metastasis such as tumor‐induced osteolysis; these effects are mediated by suppression of osteoclast function.19 Clinical reports show that ZOL suppresses not only skeleton‐related events but also the incidence of invasive breast cancer.20 The results of previous studies have shown that ZOL has anticancer activity against several human neoplasms such as leukemia, breast, prostate, and pancreatic cancers in vivo . 21, 22, 23, 24 The mechanism underlying these anti‐proliferative effects is thought to involve the inhibition of RAS GTPase prenylation.25, 26
On the basis of these observations, we hypothesized that ZOL may be effective in the treatment of CRC with mutant KRAS. In this study, we demonstrate that ZOL has an anti‐proliferative effect against colon cancer, which is brought about via inhibition of RAS prenylation, and that it has synergistic effects when used in combination with CTX both in vitro and in vivo.
Material and Methods
Drugs
CTX was purchased from Merck (Darmstadt, Germany), and ZOL was purchased from Novartis Pharma (Basel, Switzerland).
Cell lines
We used eight colon cancer cell lines: SW48, CaCO2, LOVO, LS174T, SW1417, RKO, HCT116, and SW620; a gastric cancer cell line: MKN45; and a breast cancer cell line: MCF7 (all from ATCC, Manassas, VA). SW48 and CaCO2 cells carry the wild‐type KRAS gene, whereas LS174T (G12D), LOVO (G13D), HCT116 (G13D), and SW620 (G12V) cells exhibit KRAS mutations (indicated parenthetically); none of these cell lines carry BRAF mutations.27 In addition, SW1417 (V600E) and RKO (V600E) only exhibit BRAF mutations (Table 1). We focused on two of these cell lines (SW48 and LS174T) for much of our present study. SW48 and LS174T cells were cultured in RPMI 1640 medium (Wako, Osaka, Japan) supplemented with 10% fetal bovine serum (Sigma–Aldrich, St. Louis, MO), antibiotics (Sigma‐Aldrich), and HEPES (Sigma–Aldrich) in a humidified atmosphere of 5% CO2 at 37°C. SW1417 cells were cultured in Leibovitz's L‐15 Medium (Wako) supplemented with 10% fetal bovine serum (Sigma–Aldrich) and antibiotics (Sigma–Aldrich) in a humidified atmosphere of CO2 free at 37°C.
Table 1.
Status of KRAS and BRAF
| Cell line | KRAS | BRAF |
|---|---|---|
| SW48 | Wild‐type | Wild‐type |
| CaCO2 | Wild‐type | Wild‐type |
| LS174T | Mutation at exon2 (G12V) | Wild‐type |
| LOVO | Mutation at exon2 (G13D) | Wild‐type |
| HCT116 | Mutation at exon2 (G13D) | Wild‐type |
| SW620 | Mutation at exon2 (G12V) | Wild‐type |
| SW1417 | Wild‐type | Mutation at exon 15 (V600E) |
| RKO | Wild‐type | Mutation at exon 15 (V601E) |
Evaluation of the effects of CTX and/or ZOL on cell growth
Cell growth was assessed by a standard MTT assay, which detects dehydrogenase activity in viable cells. A total of 5 × 103 or 10 × 103 cells were seeded into each well of 96‐well culture plates. After 24 hrs, the cells were treated with various concentrations of the drugs. After another 72 hrs, the culture medium was removed and 100 μL 0.5 mg/mL MTT (Sigma–Aldrich) was added to each well. The plates were then incubated for 4 hrs at 37°C. The culture medium was replaced with 100 μL DMSO per well, and the absorbance at 540 nm was determined using an Envision 2104 Multilabel Reader (Perkin Elmer, Waltham, MA).
Clonogenic survival assay
A total of 1× 103 or 5 × 103 cells were seeded into 10‐cm dishes. After 24 hrs, the cells were treated with various concentrations of the drugs and incubated for 14–25 days until 1‐mm colonies were formed in control dishes for each cell line. Fresh media and drugs were added on the fifth day. After 14–25 days, media was removed from the dishes, and cells were washed three times with phosphate‐buffered saline (PBS). The colonies were fixed with 10% formalin for 10 min, washed three times with water, and stained with 2 mL 0.25% methylene blue for 10 min on a rocking platform. The dishes were rinsed three times with water and air‐dried, and the colonies were counted.28
Western blot analysis and antibodies
SW48 and LS174T cells (50% confluence) were grown for 24 hrs in medium. Then the cells were treated with ZOL (100 μM) for 24 hrs. Thereafter, the cells were treated with fibroblast growth factor (FGF: 20 ng/mL) and CTX (0, 10, 100 nM). After 30 min, the cells were harvested and lysed in RIPA buffer with phosphatase inhibitors (Sigma–Aldrich) for 30 min on ice. The protein concentration of the lysates was determined using a DC Protein Assay Kit (Bio‐Rad, Hercules, CA). Total cell protein extracts (20 μg/lane) were subjected to SDS‐PAGE analysis. The membranes were blocked with the PVDF blocking reagent (TOYOBO, Osaka, Japan) for 1 hr before incubation with primary antibodies (antibodies against β‐actin [rabbit], EGFR [rabbit], phospho (p)‐EGFR (Tyr1068) [rabbit], MAPK/Extracellular signal‐regulated kinase (MAPK/ERK) [rabbit], phosphor (p)‐ERK (Thr202/Tyr204 and Thr185/Tyr187) [rabbit], v‐akt murine thymoma viral oncogene homolog (AKT) [mouse] or phospho (p)‐AKT (Ser473) [rabbit]) (1:5,000) (Cell Signaling Technology, Danvers, MA), Ras (mouse, 1:5,000) (BD Biosciences, CA), a RAS‐related protein‐1a (RAP1A) antibody (goat, 1:1000) (Santa Cruz Biotechnologies, Santa Cruz, CA), and caspase‐3, cleaved PARP: poly (ADP‐ribose) polymerase (PERP) (mouse, 1:1,000) (Cell Signaling) overnight at 4°C. The primary antibodies were diluted with Can Get Signal Solution 1 (TOYOBO). The membranes were washed with the Dako Washing Buffer (Dako, Glostrup, Denmark) and incubated with the appropriate secondary antibodies (1:25,000) (Millipore, Billerica, MA). The secondary antibodies were diluted with Can Get Signal Solution 2 (TOYOBO). The immunoreactive proteins were visualized via chemiluminescence microscopy by using ImmunoStar LD reagents (Wako, Osaka, Japan).29 Images were captured using an LAS‐4000 camera system (FUJIFILM, Tokyo, Japan) and quantified using public ImageJ software from the NIH.
Nude mouse xenograft study
Five‐week‐old male athymic nude mice (BALB/c nu/nu) were obtained from SLC (Hamamatsu, Japan). All animals were bred in laminar‐flow cabinets under specific pathogen‐free conditions. Before implanting the SW48 and LS174T xenografts, the cells were briefly treated with trypsin‐EDTA and washed twice with serum‐free medium. The mice were anesthetized with ether and implanted subcutaneously with LS174T (2 × 106 cells) or SW48 (4 × 106 cells) cells (100 μL in serum‐free medium). Each mouse received subcutaneous injections in both flanks so that they would develop two tumors. When the tumors reached around 100 mm3, the mice (n = 6 mice per cell line per treatment) were assigned to one of four groups: CTX (10 mg/kg i.p. twice a week), ZOL (0.2 mg/kg i.p. once a week), combination of CTX and ZOL (CTX 10 mg/kg i.p. twice a week, ZOL 0.2 mg/kg i.p. once a week) or PBS as a control.
The tumor diameters were measured using calipers every 2–3 days, and the tumor volumes were estimated using the following formula: tumor volume = ab2/2, where “a” is the longest diameter of the tumor, and “b” is the shortest diameter. Mice were sacrificed, and the resected tumors were weighed.30 The study was independently repeated twice. The extracted tumors were minced, lysed in lysis buffer, and subjected to western blot to evaluate apoptosis‐related proteins. The animal experiments were performed in accordance with the legal and institutional guidelines.
Immunohistochemistry
A Dako LSAB Kit (Dako, Carpinteria, CA) was used for immunohistochemical analysis. In brief, sections were pretreated by microwave treatment in citrate buffer for 15 min to retrieve antigenicity. After peroxidase activity was blocked with 3% H2O2 methanol for 10 min, sections were incubated with normal goat serum (Dako) for 20 min to block nonspecific antibody binding sites. Thereafter, sections were incubated with the primary antibody against Ki67 (M7240, 1:1,000, Dakocytomation, Denmark) and p‐ERK (#4370, 1:800, Cell Signaling Technology) for 1 hr at 25°C, and p‐AKT (#4060, 1:25, Cell Signaling Technology) overnight at 4°C, followed by incubations with biotinylated anti‐mouse IgG and peroxidase‐labeled streptavidin for 10 min each. Staining was completed with the substrate‐chromogen solution followed by counterstaining with 0.1% hematoxylin. The Ki67 index was calculated by calculating the average number of Ki67‐positive cells/×200 field.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
The 3′‐OH groups of DNA fragments in apoptotic cells were labeled and stained using an apoptosis in situ TUNEL kit (Takara, Shiga, Japan) according to the manufacturer's recommended protocol using the provided positive controls. Fluorescent microscopy (Nikon Corporation, Tokyo, Japan) was used to image the FITC‐labeled TUNEL‐positive cells, which were then counted by experienced pathologists.
FACS (fluorescence activated cell sorting)
SW48 and LS174T cells were treated with 100 μM ZOL for 0, 12, 24, 48, 72, 96, and 120 hrs. Cells were then trypsinized, washed, collected, and fixed in 70% ethanol. Fixed samples were centrifuged, treated with RNase (0.2 mg/mL), and resuspended in propidium iodide (50 μg/mL). The stained cells were analyzed on a Becton‐Dickinson FACScan flow cytometer. The sub‐G1 fraction of cells was defined as the apoptotic portion, and the proportion of apoptotic to total cells was indicated as a percentage.
Statistical analysis
The mean tumor volume in each group was calculated as the total volume from all mice divided by the number of mice. The statistical significance of the differences between the tumor volumes and weights was calculated using Student's t test. All p values < 0.05 were considered statistically significant. All statistical tests were two‐sided.
Results
Expression levels of EGFR and RAS in six CRC cell lines
The expression levels of the EGFR, p‐EGFR, and RAS proteins were evaluated using western blot analysis. Among the cells with wild‐type KRAS, SW48 cells showed the highest EGFR and p‐EGFR expression levels. LS174T cells, which carry a KRAS mutation, showed relatively high levels of the EGFR and the highest level of KRAS among all the analyzed cells (Fig. 1a). Therefore, we investigated the effect of ZOL and/or CTX mainly in SW48 and LS174T cells.
Figure 1.
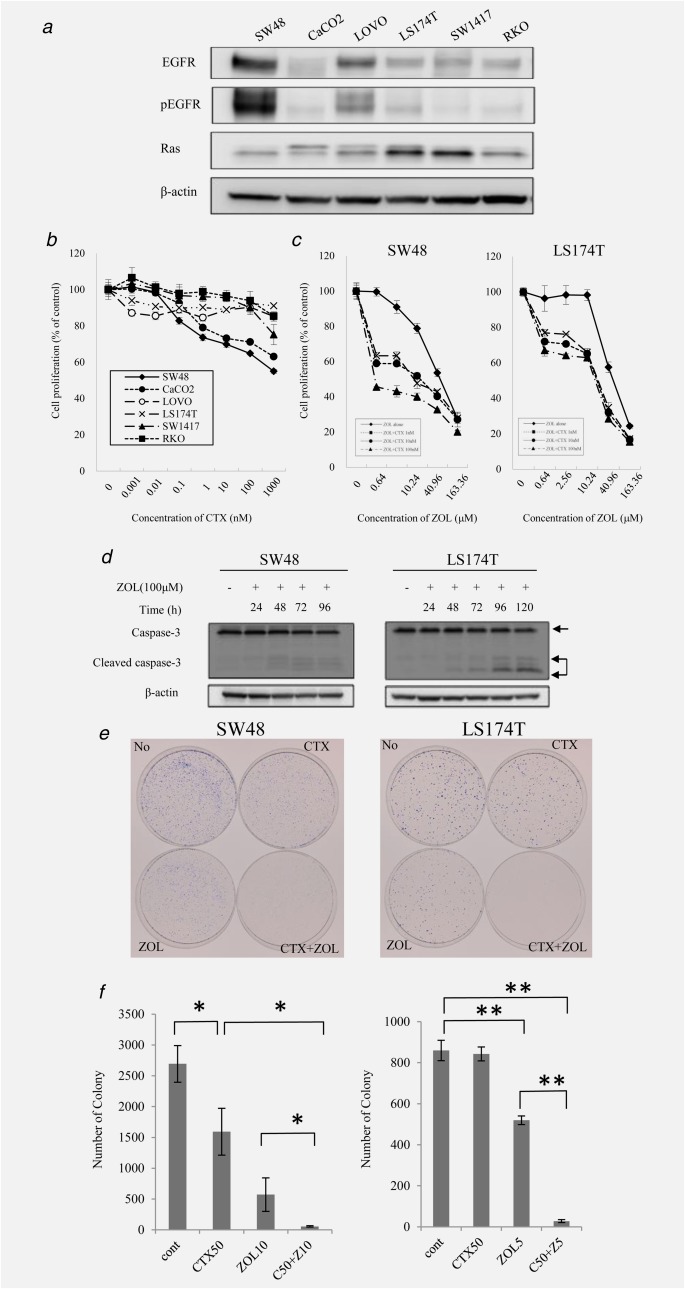
Effect of CTX and ZOL on colon cancer cell lines. (a) The expression levels of EGFR, p‐EGFR and RAS were determined via western blot analysis in six colorectal cancer cell lines. β‐actin was used as the loading control. (b) The in vitro sensitivity of six colorectal cancer cells to CTX was determined using the MTT assay. These cells were treated with 0.001–1000 nM CTX alone for 72 hr. (c) Synergistic effect of CTX and ZOL. SW48 and LS174T cells were treated with 1–100 nM CTX and/or 0–163.36 μM ZOL for 72 hr. (d) SW48 and LS174T cells were treated with ZOL (100 μM) for 96–120 hr. Cells were subjected to analysis of caspase‐3 by western blot. (e) The long‐term effects of combination treatment with CTX and ZOL. Representative plates are shown. Numbers of colonies are indicated. Error bars in (b), (c) and (e) indicate standard deviation (SD).
Inhibition of CRC cell growth by CTX and/or ZOL
We examined the growth inhibition of SW48 and LS174T cells by CTX, ZOL, or their combination. The cells were treated with CTX (0.01–1,000 nM) and/or ZOL (0.64–163.36 μM) for 72 hrs. Then, the MTT assay was performed to assess cell viability. Growth inhibition was observed in wild‐type KRAS cells but not in KRAS‐mutant cell lines (Fig. 1b). However, ZOL inhibited growth in a dose‐dependent manner in both SW48 and LS174T cell lines (Fig. 1c). We determined that the IC50 for ZOL was 65.3 μM in SW48 cells and 72.5 μM in LS174T cells. Next, we investigated the effect of combination treatment in these cell lines. Interestingly, although CTX was ineffective as a single agent, it significantly enhanced growth inhibition in combination with ZOL in LS174T cells. In SW48 cells with wild‐type KRAS, ZOL dramatically enhanced growth inhibition even in combination with a low concentration of CTX (0.16–2.56 μM). The IC50 values of ZOL when combined with 1, 10, and 100 nM CTX were 18.4, 17.1, and 5.28 µM, respectively. ZOL also synergized with LS174T cells (containing mutant KRAS) in a concentration‐dependent manner and showed IC50 values of 30.7, 25.1, and 11.68 µM when combined with 1, 10, and 100 nM CTX, respectively. Western blot analysis of the cells treated with combination therapy showed a time‐dependent increase in cleaved caspase‐3, particularly in LS174T cells. When we treated both cells by ZOL alone, cleaved PARP increased 48–72 hrs after treatment with ZOL (Fig. 1d, Supporting Information Fig. 2a). To further confirm that ZOL induced apoptosis, we performed FACS analysis. As we expected, the sub‐G1 fraction increased markedly in both cell types in a time‐dependent manner, indicating that ZOL induces apoptosis (Supporting Information Fig. 2b). The long‐term effects of combination treatment with CTX and ZOL were assessed by a clonogenic assay. Colony formation in SW48 cells was inhibited with CTX alone (Fig. 1b). Furthermore, colony formation in SW48 and LS174T cells treated with a combination of CTX and ZOL for 14 and 18 days was significantly and synergistically suppressed compared with monotherapy (Fig. 1e).
The effects of ZOL on the signaling pathway
We performed western blot analysis to investigate the signaling pathways downstream of EGFR in the presence of ZOL. SW48 and LS174T cells were treated with or without ZOL (100 μM) for 6, 12, 24 or 48 hrs, followed by treatment with EGF (20 ng/mL) for 30 min. After harvesting, the cells were subjected to western blot for ERK (downstream of EGFR), RAP1A (geranylgeranylated Ras, which is activated after geranylgeranylation), and AKT (upstream of mammalian target of rapamycin [mTOR]) because ZOL is reported to inhibit the AKT‐mTOR pathway.31 The RAP1A antibody we used detected only unfarnesylated RAP1A.32
In the presence of EGF, ZOL did not inhibit the phosphorylation of EGFR in these cells. However, the levels of p‐ERK and p‐AKT decreased after treatment with 100 μM ZOL for 48 hrs [SW48: 30% (p‐ERK) and 7% (p‐AKT) reduction, LS174T: 13% (p‐ERK) and 38% (p‐AKT) reduction] compared to the control. Particularly, levels of p‐AKT increased 6–12 hrs after EGF stimulation, then decreased between 12 and 24 hrs. On the other hand, in LS174T cells, Ras protein increased after 48 hrs [SW48: 26%, LS174T: 167%] compared to the control. Interestingly, unprenylated RAP1A also increased in these cells upon ZOL treatment (Fig. 2). These results indicate that ZOL inhibits both MAPK and AKT pathways in cells with either wild‐type or mutant KRAS.
Figure 2.
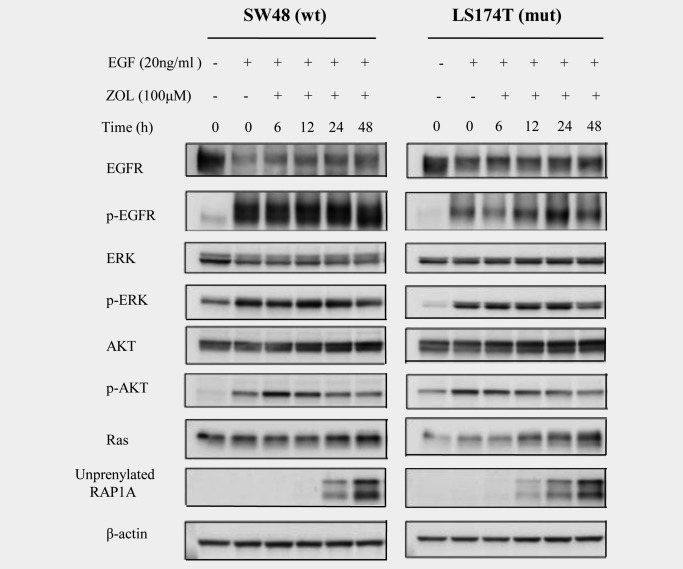
Signaling pathways affected by ZOL. SW48 and LS174T cells were treated with ZOL (100 μM) for 6, 12, 24 or 48 hrs, followed by treatment with EGF (20 ng/mL) before harvesting. The results of the western blot analysis for EGFR, p‐EGFR, ERK, p‐ERK, AKT, p‐AKT, RAS and unprenylated RAP1A are shown. β‐actin was used as the loading control. Quantification was done by public ImageJ software from the NIH.
The effects of CTX and ZOL combination treatment on signaling pathways
We investigated the signaling pathways associated with ZOL and CTX combination therapy. First, SW48 and LS174T cells were treated with/without ZOL (100 μM) for 24 hrs; then, the cells were treated with EGF (20 ng/mL) followed by CTX (0, 10 and 100 nM). The cells were harvested after 30 min of EGF and/or CTX treatment and subjected to western blot for phosphorylated intermediates of the MAPK and AKT pathways.
Upon EGF treatment, CTX inhibited phosphorylation of EGFR in both cells. As expected, the level of p‐ERK decreased in SW48 cells and showed only slight suppression in LS174T cells after treatment with 100 nM CTX alone (54 and 21% reduction). When 100 μM ZOL was added, p‐ERK and p‐AKT were substantially decreased in SW48 cells and slightly decreased in LS174T cells in a CTX‐dose‐dependent manner (78 and 93% reduction in SW48 cells; 57 and 77% reduction in LS174T cells). Unprenylated RAP1A noticeably increased after combination treatment with ZOL and CTX in all cells (Fig. 3). These results indicate that combination treatment with CTX and ZOL enhanced the growth inhibition of CRC cells, including those with KRAS mutations, through the RAS‐MAPK and/or AKT‐mTOR pathways.
Figure 3.
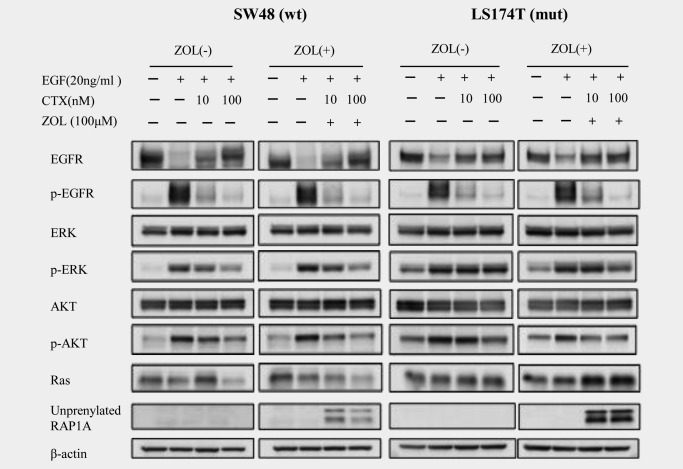
Signaling pathways modulated by CTX and ZOL. SW48 and LS174T cells were treated with/without ZOL (100 μM) for 24 hr, followed by treatment with EGF (20 ng/mL) and CTX (0, 10 and 100 nM). The results of the western blot analysis for EGFR, p‐EGFR, ERK, p‐ERK, AKT, p‐AKT, RAS and unprenylated RAP1A are shown. β‐Actin was used as the loading control.
The effects of ZOL and CTX on in vivo tumor growth
To determine whether ZOL enhanced the antitumor effect of CTX therapy in vivo, we assayed their effects on xenografted tumors in nude mice. Compared with the tumors in PBS‐treated controls, the xenografts from SW48 cells after treatment with CTX alone, ZOL alone, and the combination of CTX and ZOL were significantly reduced in size (CTX vs. control: p < 0.05, ZOL, CTX and ZOL vs. control: p < 0.01) and in weight (CTX, ZOL vs. control: p < 0.05; CTX and ZOL vs. control: p < 0.01) (Figs. 4a and 4b). There was no significant difference between treatment with ZOL alone and the combination of CTX and ZOL. In the xenografts from LS174T cells, CTX alone did not inhibit tumor growth compared to the control. However, tumor growth was significantly inhibited in the groups treated with ZOL and the combination of CTX and ZOL compared with the control group (p < 0.05 and p < 0.01, respectively). Furthermore, the sizes of xenografted tumors after combination therapy were significantly decreased relative to treatment with CTX or ZOL alone (p < 0.05).
Figure 4.
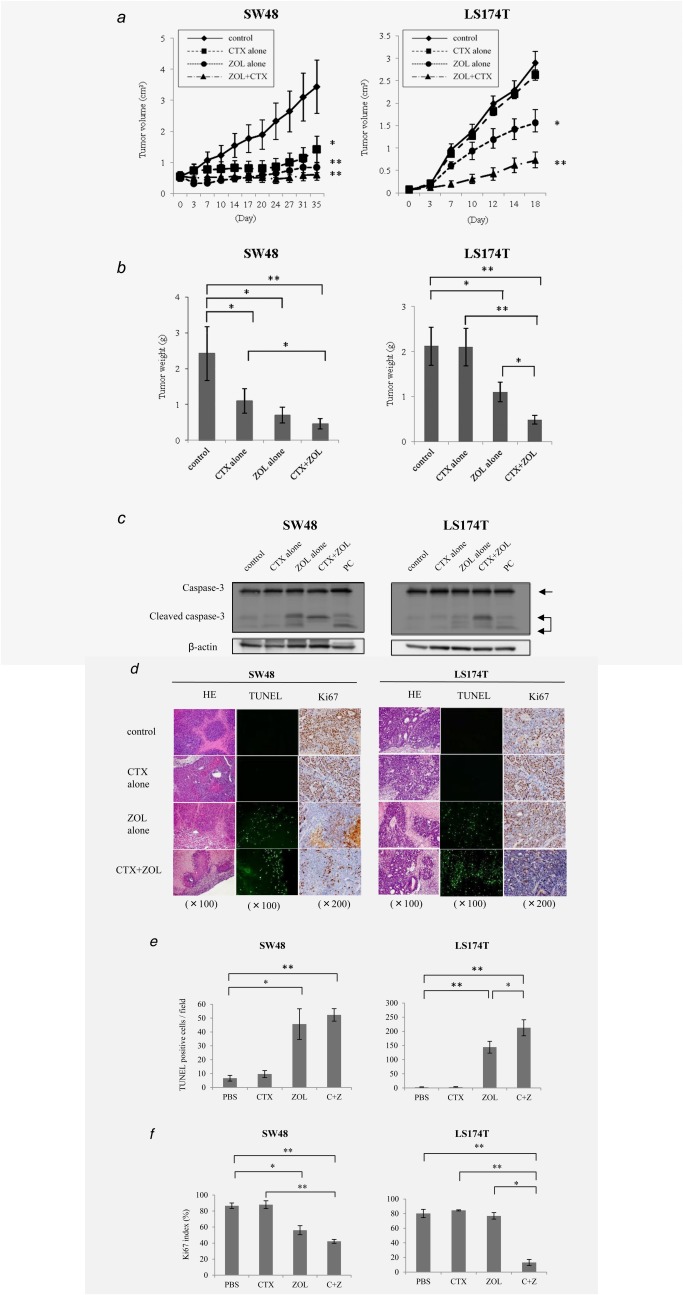
Nude mouse xenograft study. SW48 and LS174T cells were transplanted into nude mice, and the mice were treated with PBS as a control, CTX (1 mg/100 μL), ZOL (0.2 µM), or the combination of CTX and ZOL. (a) The volumes of implanted tumors compared to controls (left: SW48, right: LS174T). (b) The weights of the resected tumors. (c) Evaluation of xenografts. The results of western blot analysis for caspase‐3 and cleaved caspase‐3 are shown. (d) Histological evaluation of the xenografts. The expression of Ki67 and apoptosis indicated by TUNEL are shown. (e) Analysis of the TUNEL assay. Numbers of TUNEL‐positive cells per field are indicated. (f) Ki67 index is indicated. *p < 0.05; **p ≤ 0.01. Error bars in (a), (b), (e) and (f) show standard division (SD).
To elucidate the mechanism of tumor suppression in vivo, we examined the levels of apoptosis‐related effector. Xenografts were dissected, minced, lysed in lysis buffer, and then subjected to western blot. Interestingly, as indicated in Figure 1d, the level of cleaved caspase‐3 increased after combination treatment but not after treatment with CTX alone in both samples (Fig. 4c). Western blot analysis of minced xenografts revealed that p‐ERK was suppressed in both SW48‐ and LS174T‐derived tumors. The level of p‐AKT was also generally reduced in both samples (Supporting Information Fig. 3a). Immunohistochemistry showed that p‐ERK in SW48 cells and p‐AKT in LS174T cells were suppressed by combination therapy with CTX and ZOL (Supporting Information Fig. 3b).
TUNEL assays were performed to obtain more accurate and comprehensive information about the impact of ZOL on apoptosis. Tumors treated with ZOL showed an apparent increase in apoptosis, whereas the Ki67 index decreased relative to the control (86.6%) after ZOL or combination treatment with CTX and ZOL in SW48 cells (ZOL: 56.0%, p < 0.05; CTX and ZOL: 42.2%, p < 0.01) (Figs. 4d and 4e). Even in LS174T cells, Ki67 significantly decreased after combination treatment (13.1%, p < 0.01) compared with PBS (80.2%) but not after treatment with CTX (84.5%) or ZOL (76.9%) alone (Figs. 4d and 4f). These results suggest that combination therapy with CTX and ZOL suppressed tumor growth by inducing apoptosis.
Discussion
In this study, we showed that the combination of ZOL and CTX has a synergistic antitumor activity against CRC cells and tumors. Generally, CTX does not exhibit any significant antitumor effects in cancer cells with a KRAS mutation because of the continuous activation of signaling pathways downstream of RAS.33 However, we observed synergism between ZOL and CTX, and this was evident even in cancer cells harboring a KRAS mutation. Although we first focused predominantly on SW48 cells with wild‐type KRAS and LS174T cells with mutant KRAS, we also examined other CRC cell lines with mutant KRAS such as LOVO, HCT116, and SW620 under the same conditions. In these analyses, CTX and ZOL showed synergistic activity in LOVO and HCT116 cells; however, almost no synergy was observed in SW620 cells (Supporting Information Fig. 1a). We speculated that the reduced levels of EGFR (when compared with LS174T and LOVO) might underlie the effect (Supporting Information Fig. 1b). We also examined other cell lines with wild‐type KRAS such as CaCO2, SW1417 and RKO cells. CaCO2 cells showed responses similar to SW48 cells. However, neither CTX nor ZOL had a significant effect on cell growth in SW1417 and RKO cells; these cells contain mutant BRAF (V600E) despite their wild‐type KRAS status, suggesting that both EGFR and BRAF might be critical determinants of ZOL‐CTX combination efficacy. According to previous reports, ZOL exerted antitumor effects by inhibiting prenylation of RAS in bladder cancer and myeloma cells.25, 34 However, to our knowledge, no previous reports have described an antitumor effect of ZOL in CRC, suggesting that ZOL may work in a tissue‐ or tumor‐specific manner. We also examined the combination effect in gastric and breast cancer cells (MKN45 and MCF7 cells); however, in contrast to our results in CRC cells, combination treatment was ineffective (Supporting Information Fig. 1a).
Given the antitumor activity of combination therapy in KRAS mutant but not BRAF mutant CRC cells, ZOL may overcome CTX resistance by modulating ERK/AKT pathways and inducing apoptosis. In a mouse model, the growth of KRAS mutant xenografts was significantly inhibited when ZOL was added to CTX during treatment. To our knowledge, this is the first report to show the potential role of ZOL as an anticancer drug that may overcome CTX resistance in KRAS mutant CRC cells.
Why the increase in ZOL‐induced unprenylated RAS levels was higher in LS174T cells (mutant KRAS) compared to that in SW48 cells (wild‐type KRAS) remains unclear. However, based on these findings, we hypothesize that ZOL could preferentially inhibit mutated KRAS and that CTX could inhibit the residual wild‐type KRAS, resulting in a synergistic effect. There are various cell growth signals transmitted through the RAS protein, such as those mediated by the HER family members, insulin‐like growth factor‐1 (IGF‐1), and FGF. Furthermore, cancer cells might be able to grow without the wild‐type RAS protein because of the expression of survival signals, such as those occurring via the AKT‐phosphatidylinositol‐3 kinase (PI3K) and Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathways.35 ZOL is considered to have a strong antitumor effect because it inhibits not only the RAS protein but also mTOR‐mediated survival signals by inhibiting farnesylation of a RAS homolog enriched in the brain that activates mTOR.36
In the in vivo study in a mouse model, the addition of a low dose of ZOL to CTX therapy strongly inhibited the growth of xenograft tumors, especially in mice implanted with LS174T cells (KRAS mutant). Our data indicated that the expression of cleaved caspase‐3, TUNEL‐positive cells and cleaved PERP increased dramatically, suggesting that strong induction of apoptosis is one of the pivotal mechanisms for ZOL and CTX combination therapy. Another mechanism might also enhance this phenomenon in vivo because CTX may exert its antitumor effects via pathways unrelated to EGFR‐targeting, such as through antibody‐dependent cell cytotoxicity (ADCC).37 ZOL is also reported to increase the number of γδT cells, which are thought to activate ADCC.38 The evidence suggests that both reagents may synergistically enhance ADCC activity. Furthermore, ZOL is known to suppress visceral metastasis of breast cancer cells by inhibiting cell migration and invasion, thereby increasing apoptosis in metastatic lesions.36 These probable mechanisms of ZOL activity may inform development of novel strategies for anticancer therapy.
Recent clinical data indicate that BP suppresses bone‐related events and shows independent antitumor activity by inhibiting cellular proliferation or inducing apoptosis.21, 39, 40, 41 In most studies, a high concentration of ZOL (10–100 μM) was used to demonstrate antitumor effects in vitro. The dose of ZOL used to prevent skeleton‐related events associated with bone metastasis results in a serum concentration of 0.1–1 μM. In our current study, we therefore demonstrated the antitumor effect of ZOL using a clinically relevant concentration (0.1–10 μM) in combination with CTX. Furthermore, ZOL synergizes with chemotherapeutic agents such as docetaxel, paclitaxel, and cisplatin.34, 42 These findings suggest that ZOL might play an important role as an effector for cancer therapy. To our knowledge, this is the first report showing that ZOL overcomes resistance to CTX, even in tumors with a KRAS mutation, and that it enhances the antitumor activity of CTX. Further studies are needed to elucidate the mechanisms underlying the synergistic activity of ZOL and CTX.
In conclusion, our results demonstrate that ZOL inhibits growth of CRC cells with both wild‐type and mutant KRAS but not with mutant BRAF. Therefore, the effects of combination therapy with ZOL and CTX show great promise for the treatment of CRC, and further studies examining the clinical efficacy of this combination are warranted.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Acknowledgement
The authors thank Shizu K for technical assistance and Enya K, Iwata A, Takano K, Mori K, and Hirata M for administrative assistance.
References
- 1. Parkin DM, Bray F, Ferlay J, et al. Global cancer statistics, 2002. CA Cancer J Clin 2005;55:74–108. [DOI] [PubMed] [Google Scholar]
- 2. Center MM, Jemal A, Smith RA, et al. Worldwide variations in colorectal cancer. CA Cancer J Clin 2009;59:366–78. [DOI] [PubMed] [Google Scholar]
- 3. Ducreux M, Bennouna J, Hebbar M, et al. Capecitabine plus oxaliplatin (XELOX) versus 5‐fluorouracil/leucovorin plus oxaliplatin (FOLFOX‐6) as first‐line treatment for metastatic colorectal cancer. Int J Cancer 2011;128:682–90. [DOI] [PubMed] [Google Scholar]
- 4. Schmoll HJ, Cartwright T, Tabernero J, et al. Phase III trial of capecitabine plus oxaliplatin as adjuvant therapy for stage III colon cancer: a planned safety analysis in 1,864 patients. J Clin Oncol 2007;25:102–9. [DOI] [PubMed] [Google Scholar]
- 5. Alberts SR, Sargent DJ, Nair S, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ocvirk J, Brodowicz T, Wrba F, et al. Cetuximab plus FOLFOX6 or FOLFIRI in metastatic colorectal cancer: CECOG trial. World J Gastroenterol 2010;16:3133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saltz LB, Clarke S, Díaz‐Rubio E, et al. Bevacizumab in combination with oxaliplatin‐based chemotherapy as first‐line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9. [DOI] [PubMed] [Google Scholar]
- 8. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42. [DOI] [PubMed] [Google Scholar]
- 9. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008;358:1160–74. [DOI] [PubMed] [Google Scholar]
- 10. Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet 2010;376:687–97. [DOI] [PubMed] [Google Scholar]
- 11. Slamon DJ, Leyland‐Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783–92. [DOI] [PubMed] [Google Scholar]
- 12. Piccart‐Gebhart MJ, Procter M, Leyland‐Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2‐positive breast cancer. N Engl J Med 2005;353:1659–72. [DOI] [PubMed] [Google Scholar]
- 13. Terashima M, Kitada K, Ochiai A, et al. Impact of expression of human epidermal growth factor receptors EGFR and ERBB2 on survival in stage II/III gastric cancer. Clin Cancer Res 2012;18:5992–6000. [DOI] [PubMed] [Google Scholar]
- 14. Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009;360:1408–17. [DOI] [PubMed] [Google Scholar]
- 15. Bokemeyer C, Bondarenko I, Makhson A, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first‐line treatment of metastatic colorectal cancer. J Clin Oncol 2009;27:663–71. [DOI] [PubMed] [Google Scholar]
- 16. Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008;26:374–9. [DOI] [PubMed] [Google Scholar]
- 17. Karapetis CS, Khambata‐Ford S, Jonker DJ, et al. K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–65. [DOI] [PubMed] [Google Scholar]
- 18. Benvenuti S, Sartore‐Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti‐epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8. [DOI] [PubMed] [Google Scholar]
- 19. Hirsh V, Major PP, Lipton A, et al. Zoledronic acid and survival in patients with metastatic bone disease from lung cancer and elevated markers of osteoclast activity. J Thorac Oncol 2008;3:228–36. [DOI] [PubMed] [Google Scholar]
- 20. Chlebowski RT, Chen Z, Cauley JA, et al. Oral bisphosphonate use and breast cancer incidence in postmenopausal women. J Clin Oncol 2010;28:3582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuroda J, Kimura S, Segawa H, et al. The third‐generation bisphosphonate zoledronate synergistically augments the anti‐Ph+ leukemia activity of imatinib mesylate. Blood 2003;15:2229–35. [DOI] [PubMed] [Google Scholar]
- 22. Hiraga T, Williams PJ, Ueda A, et al. Zoledronic acid inhibits visceral metastases in the 4T1/luc mouse breast cancer model. Clin Cancer Res 2004;10:4559–67. [DOI] [PubMed] [Google Scholar]
- 23. Corey E, Brown LG, Quinn JE, et al. Zoledronic acid exhibits inhibitory effects on osteoblastic and osteolytic metastases of prostate cancer. Clin Cancer Res 2003;9:295–306. [PubMed] [Google Scholar]
- 24. Tassone P, Tagliaferri P, Viscomi C, et al. Zoledronic acid induces antiproliferative and apoptotic effects in human pancreatic cancer cells in vitro. Br J Cancer 2003;88:1971–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koizumi M, Nakaseko C, Ohwada C, et al. Zoledronate has an antitumor effect and induces actin rearrangement in dexamethasone‐resistant myeloma cells. Eur J Haematol 2007;79:382–91. [DOI] [PubMed] [Google Scholar]
- 26. Stathopoulos GT, Moschos C, Loutrari H, et al. Zoledronic acid is effective against experimental malignant pleural effusion. Am J Respir Crit Care Med 2008;178:50–9. [DOI] [PubMed] [Google Scholar]
- 27. Janakiraman M, Vakiani E, Zeng Z, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res 2010;70:5901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyamoto Y, Futamura M, Kitamura N, et al. Identification of UNC5A as a novel transcriptional target of tumor suppressor p53 and a regulator of apoptosis. Int J Oncol 2010;36:1253–60. [DOI] [PubMed] [Google Scholar]
- 29. Kanematsu M, Futamura M, Takata M, et al. Clinical significance of glycoprotein non‐metastatic B and its association with HER2 in breast cancer. Cancer Med 2015;4:1344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Futamura M, Kamino H, Miyamoto Y, et al. Possible role of semaphorin 3F, a candidate tumor suppressor gene at 3p21.3, in p53‐regulated tumor angiogenesis suppression. Cancer Res 2007;67:1451–60. [DOI] [PubMed] [Google Scholar]
- 31. Moriceau G, Ory B, Mitrofan L, et al. Zoledronic acid potentiates mTOR inhibition and abolishes the resistance of osteosarcoma cells to RAD001 (Everolimus): pivotal role of the prenylation process. Cancer Res 2010;70:10329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reszka AA, Halasy‐Nagy J, Rodan GA. Nitrogen‐bisphosphonates block retinoblastoma phosphorylation and cell growth by inhibiting the cholesterol biosynthetic pathway in a keratinocyte model for esophageal irritation. Mol Pharmacol 2001;59:193–202. [DOI] [PubMed] [Google Scholar]
- 33. De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy‐refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010;304:1812–20. [DOI] [PubMed] [Google Scholar]
- 34. Sato K, Yuasa T, Nogawa M, et al. A third‐generation bisphosphonate, minodronic acid (YM529), successfully prevented the growth of bladder cancer in vitro and in vivo. Br J Cancer 2006;95:1354–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Slattery ML, Lundgreen A, Kadlubar SA, et al. JAK/STAT/SOCS‐signaling pathway and colon and rectal cancer. Mol Carcinog 2013;52:155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Niessner H, Beck D, Sinnberg T, et al. The farnesyl transferase inhibitor lonafarnib inhibits mTOR signaling and enforces sorafenib‐induced apoptosis in melanoma cells. J Invest Dermatol 2011;131:468–79. [DOI] [PubMed] [Google Scholar]
- 37. Correal P, Marra M, Remondo C, et al. Cytotoxic drugs up‐regulate epidermal growth factor receptor (EGFR) expression in colon cancer cells and enhance their susceptibility to EGFR‐targeted antibody‐dependent cell‐mediated‐cytotoxicity (ADCC). Eur J Cancer 2010;46:1703–11. [DOI] [PubMed] [Google Scholar]
- 38. Maniar A, Zhang X, Lin W, et al. Human gammadelta T lymphocytes induce robust NK cell‐mediated antitumor cytotoxicity through CD137 engagement. Blood 2010;116:1726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hortobagyi GN, Theriault RL, Porter L, et al. Efficacy of pamidronate in reducing skeletal complications in patients with breast cancer and lytic bone metastases. Protocol 19 Aredia Breast Cancer Study Group. N Engl J Med 1996;335:1785–91. [DOI] [PubMed] [Google Scholar]
- 40. Kanis JA, Powles T, Paterson AH, et al. Clodronate decreases the frequency of skeletal metastases in woman with breast cancer. Bone 1996;19:663–7. [DOI] [PubMed] [Google Scholar]
- 41. Theriault RL, Lipton A, Hortobagyi GN, et al. Pamidronate reduces skeletal morbidity in woman with breast cancer and lytic bone lesions: a randomized, placebo‐controlled trial. Protocol 18 Aredia Breast Cancer Study Group. J Clin Oncol 1999;17:846–54. [DOI] [PubMed] [Google Scholar]
- 42. Inoue K, Karashima T, Fukata S, et al. Effect of combination therapy with a novel bisphosphonate, minodronate (YM529), and docetaxel on a model of bone metastasis by human transitional cell carcinoma. Clin Cancer Res 2005;11:6669–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.