

Abstract
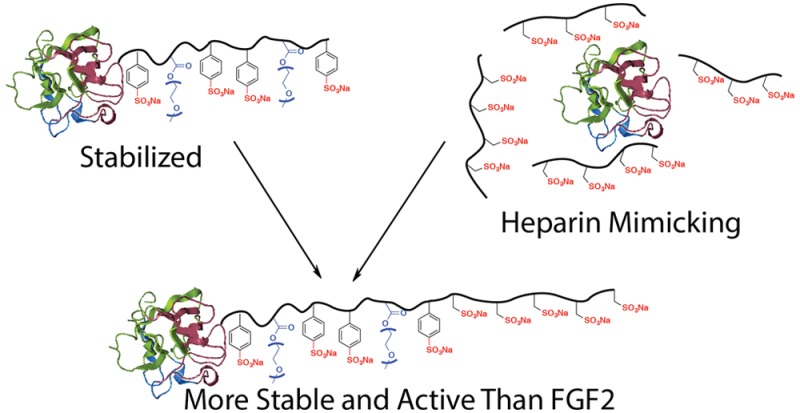
Fibroblast growth factor 2 (FGF2) is a protein involved in cellular functions in applications such as wound healing and tissue regeneration. Stabilization of this protein is important for its use as a therapeutic since the native protein is unstable during storage and delivery. Additionally, the ability to increase the activity of FGF2 is important for its application, particularly in chronic wound healing and the treatment of various ischemic conditions. Here we report a heparin mimicking block copolymer, poly(styrenesulfonate-co-poly(ethylene glycol) methyl ether methacrylate)-b-vinyl sulfonate) (p(SS-co-PEGMA)-b-VS, that contains a segment that enhances the stability of FGF2 and one that binds to the FGF2 receptor. The FGF2 conjugate retained activity after exposure to refrigeration (4 °C) and room temperature (23 °C) for 7 days, while unmodified FGF2 was inactive after these standard storage conditions. A cell study performed with a cell line lacking native heparan sulfate proteoglycans indicated that the conjugated block copolymer facilitated binding of FGF2 to its receptor similar to the addition of heparin to FGF2. A receptor-based enzyme-linked immunosorbant assay (ELISA) confirmed the results. The conjugate also increased the migration of endothelial cells by 80% compared to FGF2 alone. Additionally, the FGF2-p(SS-co-PEGMA)-b-VS stimulated endothelial cell sprouting 250% better than FGF2 at low concentration. These data verify that this rationally designed protein-block copolymer conjugate enhances receptor binding, cellular processes such as migration and tube-like formation, and stability, and suggest that it may be useful for applications in biomaterials, tissue regeneration, and wound healing.
Introduction
There is growing interest in biomaterials capable of treating ischemic conditions and promoting healing in burned and wounded tissues. Approximately 0.15% of Americans suffer from limb ischemia each year (2011),1 1–2% from chronic wounds (2004)2,3 and 450 000 from acute burn injuries (2014).4 Together, treatments for chronic wounds cost over 35 billion dollars annually as of 2007 in the United States alone.2,5 One important factor to consider in the successful development of biomaterials for treatment of ischemia and wound repair is the promotion of vascularization. Cellular proliferation, migration and angiogenesis are crucial for formation of new vasculature and are key processes in tissue repair. In normal healing processes, growth factors are typically produced in wounds and act as signaling molecules to stimulate growth and new tissue formation.6 In ischemic, burned and chronically wounded tissues, these processes are impaired.7 Indeed, many biomaterials have been developed to promote angiogenesis. These include hydrogels, topical creams, growth factors, small molecules and polymers.8,9 Yet, there is still interest in developing new options for successful treatment of ischemic and chronic wounds.
Fibroblast growth factor 2, or FGF2, is a 17 kDa heparin binding protein that promotes a variety of cellular processes including cell proliferation, migration, vasculogenesis, cell differentiation and stem cell self-renewal.10,11 Additionally, FGF2 has been shown to play a crucial role in tissue repair, angiogenesis, bone growth, and neuroregeneration.12 Decreased concentrations of growth factors including FGF2 in chronic wounds and ischemic conditions are known to inhibit these cellular processes, thereby preventing healing and angiogenesis.7,13 This decrease in FGF2 combined with the advantageous effects of FGF2 in tissue regeneration have led to new biomaterials and topical applications of FGF2 and other growth factors for treatment of chronic wounds.14,15 For example, it has been shown that delivery of various growth factors such as FGF and VEGF increases the amount of angiogenesis, and thus pro-angiogenic treatments involving these proteins have been widely studied.16 However, while these treatments have shown improved angiogenesis and tissue healing in vitro, their efficacy in clinical trials has been limited.17 To overcome this obstacle, superagonists of FGF2 and other growth factors have been studied to increase the mitogenic response in chronic wounds. These agonists have shown improved cellular response when compared to FGF2 alone and therefore, can be used to make up for the lower receptor count and lower growth factor concentrations due to growth factor degradation in diabetic patients.18 There are several known superagonists of FGF2 including protein mutants,19−21 protein dimers,22,23 FGF2 oligomers,24 peptide sequences,25,26 and protein conjugates.27 Additionally, growth factor combination therapies have been employed to improve the activity of exogenously applied growth factors.28,29
Heparinoid complexes and heparin have also been employed to increase the activity of FGF2.30 Heparin, a highly sulfated glycosaminoglycan, stabilizes FGF231 and promotes protein dimerization resulting in receptor dimerization and triggering of phosphorylation and eventual cell growth, migration and angiogenesis.32 Heparinoids are derivatives of heparin and typically are sulfated oligoheparin fragments, often well-defined in length. Due to the important role of heparin in FGF2 activity, both heparin and heparinoids have been used to stabilize33 or alter the activity of FGF2.34 In addition, heparin has been employed in many other Federal Drug Administration (FDA) approved therapeutics including treatment of angina,35 thrombosis36 and myocardial infarction.37 Heparin has also been shown to be efficacious in the treatment of chronic wounds and burns,38,39 prevention of metastatic cancer,40 reduction of inflammation41 and is FDA approved as an antithrombotic and anticoagulant.42
Growth factor superagonists are helpful in increasing cellular response for new therapeutics; however, another obstacle in the successful use of FGF2 as a therapeutic protein drug is its instability.43,44 FGF2 is quickly degraded during storage and upon delivery in vivo. Covalent protein–polymer conjugates have been used as a means of protein stabilization in the past45−48 and many conjugation chemistries have been explored.49 The covalent conjugation of poly(ethylene glycol) (PEG) to proteins (PEGylation) has become a popular means to stabilize proteins, with 10 FDA approved PEGylated proteins on the market.50 While PEGylation provides increased stability, decreased immunogenicity and increased blood half-life, moving toward biomimetic polymers for use in protein conjugates could improve biological function and provide better stability. Additional improvements to protein polymer conjugates include use of site specific conjugations51 and stimuli responsive polymer conjugates.52
In vivo, FGF2 is stabilized by heparin, allowing the protein to reach its target. While heparin and heparinoids provide many desirable therapeutic effects, they are susceptible to in vivo degradation and desulfation by heparinases.53 Additionally, heparin is isolated from animal tissues and is susceptible to high batch-to-batch variability.54 Fractionating the biomolecule has circumvented some negative effects of heparin, but this process is often costly.55 Because of the downsides of heparin, there have been many reports of heparin mimics designed to provide the desired effects of heparin while minimizing heterogeneity and desulfation in vivo. These include various polysacharides,56 sulfonated dextrans,57,58 sulfonated and sulfated polymers,59,60 anionic polymers,61 peptides,62 sulfated glycopolymers63 and ionomers.64,65 These alternatives have been used for protein stabilization,66 anticoagulation63 and stimulating cellular processes.67 We previously reported that conjugating a heparin-mimicking polymer, poly(sodium 4-styrenesulfonate-co-poly(ethylene glycol) methyl ether methacrylate) p(SS-co-PEGMA) (molecular weight 23.0 kDa), stabilized FGF2 to various stressors including heat and long-term storage.66 Although stabilization of FGF2 improves the outlook for its therapeutic use, we found that unlike heparin, the conjugate was not able to facilitate the dimerization of FGF receptors (FGFRs) in cells lacking heparin sulfate proteoglycans.66 As a result, we subsequently screened various sulfonated polymers to identify other heparin-mimicking polymers that could facilitate receptor binding and dimerization.60 We identified that poly(vinylsulfonate) (pVS) exhibited heparin-like activity by enabling the binding of FGF2 to its high affinity receptors when added as an excipient.60 Herein, we describe the combination of these two polymer types into a block copolymer containing both stabilizing and FGF2 binding sequences, namely FGF2-p(SS-co-PEGMA)-b-VS (Figure 1). The conjugation of this new heparin mimicking block copolymer to FGF2 and evidence of the resulting increased stability and growth factor activity is discussed herein.
Figure 1.
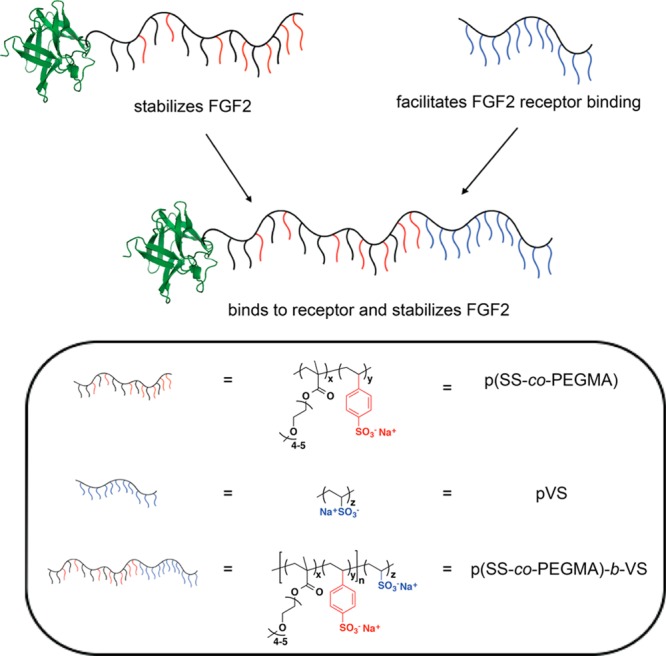
Polymer p(SS-co-PEGMA) stabilizes FGF2 as a conjugate and pVS facilitates FGF2-receptor binding when added as an excipient. When combined into a block copolymer, p(SS-co-PEGMA)-b-VS, the new conjugate both stabilizes FGF2 and increases protein activity. Protein structure modified from PDB 1CVS using PyMOL software.
Experimental Section
Materials
Chemicals and reagents were purchased from Sigma-Aldrich and used as received unless otherwise indicated. Silica gel column chromatography was performed using Merck 60 (230–400 mesh) silica gel. Prior to polymerizations 4-styrene sulfonic acid and vinyl sulfonic acid monomers were pretreated with Na+ and dried to produce the sodium salt. Before polymerization azobis(isobutyronitrile) (AIBN) was recrystallized twice from ethanol and dried, and V501 initiator was dried prior to use. Protein was expressed and purified from the plasmid pET29c(+)hFGF-2, which was kindly provided by Professor Thomas Scheper from the Helmholtz Centre for Infection Research (Braunschweig, Germany) according to Chen et al.68 HiTrap Heparin HP columns were purchased from GE Healthcare. ELISA was performed using the ELISA Development DuoSet kit purchased from R&D Systems. Recombinant human FGFR1α(IIIc) Fc chimera, and ELISA Development DuoSet kits were purchased from R&D Systems. Blot antibodies were purchased from CALBIOCHEM (rabbit antifibroblast growth factor basic) and Bio-Rad (goat antirabbit IgG-HRP conjugate). Normal human dermal fibroblasts and human umbilical vein endothelial cells were purchased from ATCC. BaF3-FR1C expressing FGFR1 were kindly provided by Professor David Ornitz (Washington University, Saint Louis).69 Cell medium was purchased from ATCC or Invitrogen unless otherwise indicated. CellTiter-Blue Cell Viability Assay was purchased from Promega. Polystyrene standards for gel permeation chromatography (GPC) calibration were purchased from Polymer Laboratories.
Methods
Analytical Techniques
1H NMR and 13C NMR spectroscopy were performed on Avance DRX 400 or 500 MHz instruments. UV–vis spectrophotometry was performed on a Biomate 5 Thermo Spectronic spectrometer. Dimethylformamide (DMF) GPC was conducted in DMF containing 0.10 M LiBr (40 °C, 0.8 mL/min) on a Shimadzu HPLC system equipped with a refractive index detector RID-10A, one Polymer Laboratories PLgel guard column, and two Polymer Laboratories PLgel 5 μm mixed D columns. Calibration was performed using near-monodisperse polystyrene standards. Chromatograms were processed using the EZStart 7.2 chromatography software. Fast protein liquid chromatography (FPLC) was performed on a Bio-Rad BioLogic DuoFlow chromatography system equipped with a GE Healthcare Life Sciences Superdex 75 10/300 column and was run in Dulbecco's phosphate-buffered saline (D-PBS) + 1 mM ethylenediaminetetraacetic acid (EDTA). Gel electrophoresis was performed using Any kDTM Mini-PROTEAN TGXTM precast gels with Tris-glycine as running buffer (Biorad, Hercules). ELISA assays were read on an ELX800 Universal Microplate Reader (Bio-Tek Instrument Inc., Winooski) with λ = 450 nm for signal and 630 nm for background. Western blot was developed on a FluorChem FC2 System version 3.2 (Cell Biosciences, Santa Clara). CellTiter-Blue assays were read on a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale). Cell images for cell viability/cytotoxicity, cell migration and angiogenesis were taken on an Axiovert 200 microscope equipped with an AxioCam MRm camera and FluoArc mercury lamp (Carl Zeiss, Thornwood). NIH ImageJ software was used to assist cell counting, to measure distances of cell-free paths and to measure degree of cord-like structure formation according to the literature.23
Synthesis of 2-((Ethoxycarbonothioyl)thio)-2-methylpropanoic Acid (1)
A three-neck round-bottom was purged with argon followed by 40 mL of 1:1 water:acetone v:v. The solvents were degassed with argon for 1 h prior to use. The round-bottom was submerged in an ice bath to cool to 0 °C and then NaOH (1.26 g, 31.5 mmol) was added to the round-bottom and the mixture was stirred for 20 min. Degassed EtOH (1.24 g, 27.0 mmol) was added dropwise over 10 min followed by the dropwise addition of carbon disulfide (1.90 mL, 31.4 mmol). The reaction turned yellow and was stirred for 30 min at 0 °C. After 30 min, 2-bromo-2-methylpropanoic acid (1.50 g, 8.98 mmol) was added and the reaction was slowly warmed to 23 °C and stirred for 48 h, over which time the reaction turned orange. After 48 h the acetone was removed in vacuo, and the aqueous layer was checked for basicity, then extracted three times with dichloromethane (DCM). The aqueous layer was then acidified using 1 M HCl and extracted three times with DCM. The combined DCM layers were dried over MgSO4, filtered, and the solvent was removed in vacuo. Next, 3 mL of water were added to the orange oil, and then the mixture was heated until all solid dissolved. The mixture was slowly cooled to room temperature 12 h. Light yellow to white crystals were observed in the water and were filtered and rinsed with cold water. The crystals were dried in vacuo (538 mg, 35% yield). δ 1H NMR (400 MHz, CDCl3): 9.21–10.64 (1H, br s, COOH), 4.63–4.58 (2H, q, J = 8 Hz), 1.63 (6H, s), 1.41–1.37 (3H, t, J = 7.12 Hz). δ 13C NMR (400 MHz, CDCl3): 210.32, 118.43, 70.01, 53.86, 25.51, 13.32. FT- IR (cm–1): 2986, 2867, 2653, 2553, 1704, 1466, 1416, 1362, 1284, 1249, 1175, 1108, 1037, 999, 922, 850, 806, 691, 631, 610, 590, 561, 538, 519, 509, 485, 471, 458. HRMS-ESI (expected, observed): [MH+] = (209.0307, 209.0289).
Synthesis of 2-(pyridin-2-yldisulfanyl)ethyl 2-((ethoxycarbonothioyl)thio)-2-methylpropanoate (CTA). A two-neck round-bottom flask was purged with argon, and pyridyl disulfide (PDS)-alcohol (670 mg, 3.61 mmol) and 1 (500 mg, 2.40 mmol) were added and dissolved in 5 mL of dry DCM. The round-bottom was submerged in an ice bath for 20 min. (1-Ethyl-3-(3′-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) (450 mg, 2.36 mmol) and 4-dimethylaminopyridine (DMAP) (58.7 mg, 0.48 mmol) were added to the reaction mixture, and the reaction was yellow. The reaction was slowly warmed to room temperature and stirred for 6 h. After 6 h, the solvent was removed in vacuo and a silica gel column was run in 2:1 hexane:EtOAc. Fractions were collected and solvent removed to obtain a yellow oil (407 mg, 45% yield). δ 1H NMR (400 MHz, CDCl3): δ 1H NMR (400 MHz, CDCl3): 8.45–8.43 (1H, dq, J = 4.85, 0.92 Hz), 7.68–7.60 (2H, m), 7.08–7.05 (1H, ddd, J = 6.61, 4.82, 1.32 Hz), 4.56–4.51 (2H, q, J = 7.15 Hz), 4.36–4.33 (2H, t, J = 6.42 Hz), 3.02–2.99 (2H, t, J = 6.40 Hz), 1.58 (6H, s), 1.34–1.30 (3H, t, J = 7.08 Hz). δ 13C NMR (400 MHz, CDCl3): 210.80, 172.82, 159.53, 149.74, 137.15, 120.94, 119.88, 69.84, 63.18, 54.08, 37.11, 25.74, 13.42. FT- IR (cm–1): 2960, 2870, 1736, 1572, 1447, 1416, 1366, 1274, 1224, 1154, 1116, 1056, 1029, 986, 950, 931, 905, 806, 759, 731, 716, 616, 579, 565, 529, 508, 497, 492, 487, 471, 457. HRMS-ESI (expected, observed): [MH+] = (377.0248, 377.0301).
Synthesis of p(SS-co-PEGMA) macroCTA
Reversible addition–fragmentation chain transfer (RAFT) polymerization was performed with initial feed ratio of [SS]:[PEGMA]:[CTA]:[AIBN] = 35:10:1:0.2 and a monomer concentration of 1.0 M. Styrenesulfonate (382 mg, 1.85 mmol) and PEGMA Mn 300 (159 mg, 0.53 mmol) were dissolved in 1.2 mL of degassed water in a Schlenk tube. CTA (20 mg, 0.053 mmol) was dissolved in 1.2 mL of degassed DMF along with AIBN (1.74 mg, 0.011 mmol) and then transferred to the Schlenk tube. The Schlenk tube was sealed and subjected to four freeze–pump–thaw cycles before immersion in an oil bath set to 60 °C. After 4 h, the polymerization was stopped by cooling to room temperature (23 °C) and bubbling air through the Schlenk tube. The polymer was purified by dialysis in 1000 MWCO tubing against 1:1 v/v water:MeOH followed by 100% Milli-Q water. The resulting contents of the dialysis tubing were lyophilized to dryness. 1H NMR 500 MHz (D2O) δ: 8.40 (1H, s), 8.0–6.2 (NaSO3C6H4 side chains), 4.2–2.8 (PEGMA side chains), 2.8–0.0 (polymer backbone). The 1H NMR spectrum of the resulted polymer was calibrated to the peak at 8.40 ppm, and the Mn of the polymers were calculated using the formula: Mn = [(integral of 8.2–6.2 ppm/4)*MW SS monomer] + [(integral of 4.2–2.8 ppm/20.4)*MW PEGMA monomer] + MW CTA. The Mn = 33.0 kDa by NMR. Mn = 15.1 kDa by GPC (DMF), Đ of 1.13.
Synthesis of p(SS-co-PEGMA)-b-VS
RAFT polymerization was performed with initial feed ratio of [VS]:[CTA]:[V501] = 200:1:0.5 and a monomer concentration of 1 M. The CTA (50 mg, 0.003 mmol), VS monomer (65 mg, 0.50 mmol) and V501 (0.4 mg, 0.001 mmol) were weighed and added to a 1 mL Schlenk tube and dissolved in 0.5 mL of degassed water. The Schlenk tube was sealed and subjected to 4 freeze–pump–thaw cycles before immersion in an oil bath set to 60 °C. After 3 h, the polymerization was stopped by opening the Schlenk tube to atmosphere. The polymer was purified by dialysis in 6000 MWCO tubing against water over 72 h. The resulting contents of the dialysis were lyophilized to dryness. 1H NMR 500 MHz (D2O) δ: 8.72 (1H, s), 8.2–6.0 (SS side chains), 4.3–2.8 (PEGMA side chains and CH2CHSO3Na polymer backbone), 2.8–0.0 (SS and PEGMA polymer backbone and CH2CHSO3Na polymer backbone). The Mn = 50.7 kDa by NMR, 28.3 kDa by GPC (DMF), Đ of 1.20 by GPC. All cell studies, except endothelial cell migration were performed using this polymer. Cell migration studies were performed using a different batch with a Mn of 57.2 kDa by 1HNMR and Mn of 31.6 by GPC (DMF) with Đ = 1.46)
Preparation of FGF2 Conjugates
FGF2 (100 μg, 5.9 × 10–3 μmol) was diluted in 500 μL of D-PBS + 1 mM EDTA and loaded onto a hand-packed 0.5 mL-heparin Sepharose column. Next, 100 equiv of PDS-p(SS-co-PEGMA), PDS-VS or PDS-p(SS-co-PEGMA)-b-VS were dissolved in 500 μL of D-PBS + 1 mM EDTA and loaded onto the column. The column was incubated at 4 °C for 16 h. After the incubation, the column was first washed with 5 column volumes of D-PBS + 1 mM EDTA to remove unreacted polymer. Next, the column was washed with 10 column volumes of D-PBS + 2 M NaCl to remove conjugated protein. Conjugates were purified using a CentriPrep centrifugal membrane MWCO 3000 against D-PBS 10 times at 12.0 rcf for 10 min/cycle. When needed, fast protein liquid chromatography (FPLC) in D-PBS + 1 mM EDTA was performed to further purify conjugates. The collected conjugate was then characterized by gel electrophoresis, and the concentration was determined by ELISA prior to in vitro studies.
Stability Study
Samples of protein and conjugates were prepared at a concentration of 0.05 ng/μL in sterile D-PBS and then stored at either 4 or 23 °C for 7 days. After 7 days an aliquot was removed from each sample and diluted to 1 ng/mL in Ultraculture cell medium. Fresh FGF2 samples were prepared the day of each cell experiment at 1 ng/mL in Ultraculture medium. Each sample was used in the HDF proliferation assay described below. Plates were prepared with 6 repeats per sample.
BaF3 Cell Proliferation
BaF3-FR1C cells were grown in RPMI 1640 medium containing 2 mM l-glutamine, 10% newborn bovine calf serum, 0.5 ng/mL IL-3, 50 nM 2-mercaptoethanol, 600 μg/mL G418, 100 μg/mL penicillin and 100 μg/mL streptomycin. Before seeding cells for experiments, the cells were collected and washed twice with culture medium without IL-3. Cells were plated at a concentration of 20 000 cells/well/50 μL in the internal wells of a 96 well plate in culture medium without IL-3. Samples were prepared in culture medium without IL-3 to contain double the final concentration and then 50 μL of each sample was added to the corresponding well. The external wells were blocked in 100 μL of DPBS and then incubated at 37 °C and 5% CO2. After 48 h incubation CellTiter-Blue assay was performed to determine extent of cell growth. All samples were normalized to the control group, which contained only culture medium without IL-3. Each group contained six replicates.
ELISA-Based FGFR Binding Assay
A 96-well plate was incubated with rhFGFR1α(IIIc) (100 uL per well at a concentration of 0.5 ug/mL in D-PBS) for 16 h at 23 °C. After 16 h, the wells were blocked with 1% bovine serum albumin (BSA) in D-PBS (2 h). Next, solutions of protein or conjugate were prepared at a concentration of 1 ng/mL, and 100 uL was plated in the wells and then incubated for 2 h. After 2 h, 100 uL of FGF2 antibody–biotin conjugate was added and incubated for an additional 2 h before streptavidin–horseradish peroxidase solution was incubated for 20 min. The plate was developed by incubating with 100 uL of 1-Step Ultra 3,3',5,5'-tetramethylbenzidine (TMB) solution (Pierce Biotechnology, Rockford) for 8 min. The assay was terminated by the addition of 50 uL of 1 M H2SO4. Absorbance was read at λ = 450 nm. Each sample was plated in triplicate.
Fibroblast Proliferation
Normal human dermal fibroblasts (HDFs) were grown in ATCC Fibroblast Basal Medium with 100 μg/mL penicillin and 100 μg/mL streptomycin supplemented. Cells were trypsinized and resuspended in Lonza UltraCULTURE serum-free medium supplemented with 2 mM l-glutamine, 100 unit/mL penicillin, and 100 μg/mL streptomycin then plated in the internal wells of a 96 well plate at a concentration of 2000 cells/well/100 μL. The external wells were blocked with D-PBS, and the plate was incubated at 37 °C, 5% CO2 for 16 h to allow cells to adhere. After the 16-h incubation period the medium was aspirated out of the wells and replaced with 100 μL of samples diluted in the supplemented UltraCULTURE medium. The cells with samples were incubated for 72 h at 37 °C, 5% CO2 and then CellTiter-Blue assay was performed to determine extent of cell proliferation. All groups were normalized to the control, which contained only UltraCULTURE medium. Each group contained six repeats.
Endothelial Cell Proliferation
Human umbilical vein endothelial cells (HUVECs) were grown in ATCC endothelial cell medium supplemented with 100 unit/mL penicillin and 100 μg/mL streptomycin. Cells were trypsinized and resuspended in the growth medium without bovine brain extract (−BBE) then plated in the internal wells of a 96 well plate at a concentration of 1000 cells/well/100 μL. The external wells were blocked with D-PBS and the plate was incubated at 37 °C, 5% CO2 for 16 h to allow cells to adhere. After the 16-h incubation period, the medium was aspirated out of the wells and replaced with 100 uL of samples diluted in the supplemented UltraCULTURE medium. The cells with samples were incubated for 72 h at 37 °C, 5% CO2 and then CellTiter-Blue assay was performed to determine extent of cell proliferation. All groups were normalized to the control, which contained only growth medium −BBE. Each group contained six repeats.
Cell Migration Assay
HUVECs were seeded in the internal wells of a 24 well plate and grown to 90–95% confluency. After reaching confluency the cell monolayers were washed once with PBS, and then the medium was replaced with starvation medium (endothelial growth medium (EGM) (−) BBE (−) rhEGF). The cells were starved for 24 h at 37 °C, 5% CO2. The medium was then aspirated out of the wells and replaced with PBS. A scratch was made in the center of each well using a standard P1000 pipet tip, and a marker line perpendicular to the scratch was drawn on the bottom of each well. The PBS was aspirated out of the wells, and the wells were washed once more with PBS to remove cell debris. The cell monolayers were allowed to incubate with 400 μL of starvation medium or samples in starvation medium at 37 °C, 5% CO2. Immediately after treatment (T = 0), two pictures of the scratch/well (one above and one below the marker) were obtained using a 5X objective on the Axiovert 200 microscope equipped with an AxioCam MRm camera, n = 4–6. At the end of the 18-hour incubation (T = 18), pictures of the scratches were taken again in the same manner. CellTiter-Blue assay was then used to quantify the extent of cell growth. The NIH ImageJ software was used to analyze the cell images: two parallel lines were drawn to outline each scratch, then the distance between them was measured using the “Measure Length” option. Percent migration was calculated using the formula: 100% – (distance T = 18/distance T = 0) × 100. The experiment was blinded and repeated three times.
Coculture Angiogenesis Assay
Experimental for coculture angiogenesis assay and staining of cord-like structures were adapted from known literature procedures.70,71 HDFs were trypsinized and resuspended in endothelial growth medium then plated at a concentration of 12 500 cells/well/250 μL in the internal 48-well plate. The external wells were blocked and the cells were incubated at 37 °C, 5% CO2 for 72 h or until cells reached confluency. The fibroblasts were then starved for 18 h in EGM (−)BBE (−)rhEGF. After the starvation period, the starvation medium was replaced with HUVECs at a concentration of 10 000 cells/well/125 μL in EGM (−BBE) (−EGF). Samples were prepared in EGM (−)BBE (−)rhEGF to contain double the concentration and then 125 μL of each sample was placed in the wells containing HUVECs. Sample solutions were refreshed after 72 and 144 h by aspirating out the medium and replacing with sample solutions prepared fresh. Ten days after the addition of HUVECs, the medium was removed from each well, and the cells were fixed with 70% EtOH (at −20 °C) for 30 min. The wells were then rinsed with 0.5 mL of 1% BSA in D-PBS three times. Endogenous alkaline phosphatase was removed by incubating the cells in 0.3% H2O2 in MeOH for 15 min at room temperature before washing the wells again with 1% BSA. The wells were incubated with mouse antihuman PECAM1/CD31 (R&D Systems) at a concentration of 1 mg/mL in 1% BSA for 1 h at 37 °C, 5% CO2. The wells were again rinsed three times with 1% BSA and then incubated with goat antimouse IgG alkaline phosphatase (Sigma-Aldrich) at 3 mg/mL in 1% BSA for 1 h at 37 °C, 5% CO2. The wells were then washed three times with Milli-Q water and then incubated with BCIP/NBT solution (1 tablet in 10 mL Milli-Q water, sterile filtered) for 15 min at room temperature. The cord-like structures were visually stained, and the wells were rinsed three times with Milli-Q water and allowed to dry. Images were taken using 5× magnification (five images per well). Plates were stored for up to 2 months at −80 °C. Cord-like structures were analyzed using NIH ImageJ Software while blinded. The values for each of the five images per well were summed, and the sums from each well were averaged (a total of three wells per sample).
Results and Discussion
In our recent report, pVS was added (not conjugated) to FGF2 and promoted proliferation in cells lacking native heparan sulfate proteoglycans; remarkably, the activity of FGF2 with pVS was the same as the positive control with added heparin.60 We then wanted to investigate whether conjugating pVS to FGF2 would additionally stabilize the protein to storage. To explore this, a protein-reactive pVS polymer was prepared with a pyridyl disulfide end group for protein conjugation (Scheme S1, see the Supporting Information (SI) for details). The FGF2-pVS conjugate was examined for its ability to stabilize FGF2 to storage. The conjugate or FGF2 was incubated at a concentration of 0.05 ng/μL at either 4 or 23 °C for 7 days. After 7 days, protein activity was evaluated through a cell proliferation assay in human dermal fibroblasts (HDFs). The results were compared to FGF2-p(SS-co-PEGMA) and pristine FGF2 as positive controls (Figure 2). Although FGF2-pVS showed stabilization effects, it was not to the same extent as our previously reported conjugate, FGF2-p(SS-co-PEGMA); the pVS conjugate exhibited statistically higher proliferation when compared to stressed FGF2; however, it did not maintain activity as high as pristine FGF2, especially at 23 °C.
Figure 2.
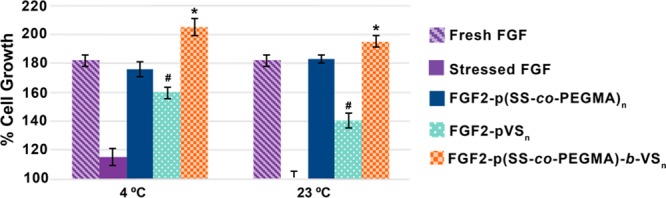
Stability study of conjugates in human dermal fibroblasts at both 4 and 23 °C after 7 days. Protein and conjugates were stressed at 0.5 ng/μL and plated for proliferation study in HDFs at 1 ng/mL. Each sample was plated with six repeats. Error bars represent standard deviation. Statistical analysis was done using Student’s t test. * p < 0.01 compared to fresh FGF2. # p < 0.005 compared to fresh FGF2.
To overcome this issue, we hypothesized that by combining the stabilizing polymer, p(SS-co-PEGMA) and the receptor binding polymer, pVS, into a single block copolymer conjugate, we could fabricate a new conjugate that would not only stabilize FGF2, but also facilitate protein-receptor binding, thus increasing protein activity. One of the difficulties with this approach was that the first block contains activated monomers and the second block deactivated monomers. Thus, a CTA was designed to balance the requirements of both activated (styrenesulfonate and PEGMA) and deactivated (vinyl sulfonate) monomers (Scheme 1a). Specifically, the CTA contained dimethyl substituents at the fragmentation site (R group) to better match the rate of addition and fragmentation during SS and PEGMA polymerization, and the xanthate group was retained to accommodate the deactivated VS monomer through a RAFT/MADIX type mechanism.72 The CTA was first used in the RAFT polymerization of styrenesulfonate and PEGMA with initial feed ratio of [SS]:[PEGMA]:[CTA]:[AIBN] = 35:10:1:0.2 to afford the p(SS-co-PEGMA) macro-CTA with a ratio of monomer incorporation of 2.2:1 SS:PEGMA (Scheme 1b). This macro-CTA was used in a subsequent RAFT polymerization with VS monomer to afford p(SS-co-PEGMA)-b-VS (50.7 kDa by 1H NMR, molecular weight dispersity Đ = 1.20) as shown in Scheme 1c. Incorporation of the VS monomer was observed by 1H NMR by an increase in backbone hydrogens between 0 and 2.8 ppm when compared to styrenesulfonate protons. 1H NMR analysis also revealed a loss in PDS end group (approximately 30% overall), most likely due to hydrolysis of the ester linkage during polymerization. Yet, the remaining end group was still sufficient for conjugation since an excess of polymers was utilized in the reaction. Additional evidence for VS incorporation was obtained through GPC analysis, which showed a molecular weight increase from 15.1 kDa for the macro-CTA to 28.3 kDa for the block copolymer.
Scheme 1. (a) Synthesis of CTA, (b) RAFT Polymerization of p(SS-co-PEGMA)-b-VS to Yield Macro CTA, (c) Subsequent RAFT Polymerization to Yield Block Copolymer p(SS-co-PEGMA)-b-VS, and (d) Protein Polymer Conjugation to FGF2 Conducted on a Heparin Column.

Protein structure modified from PDB 1CVS using PyMOL software.
Toxicity tests of the polymer revealed that the block resulted in no loss of cell viability up to 1 mg/mL in both HDFs and HUVEC lines, with a slight loss in activity above that concentration (Figure S12). However, even 1 mg/mL is more than 1000 times over the concentration that the polymer would be used in the conjugate. The resulting polymer was then conjugated to FGF2 by incubation on a heparin resin column as previously described, in phosphate buffer at pH 7.4 (Scheme 1d).66 Successful conjugation was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Figure S13a). Comparison of the nonreducing and reducing lanes in the SDS-PAGE provided evidence the disulfide linkage could be cleaved between protein and polymer. The conjugate then was purified by centrifugation filtration to yield FGF2-p(SS-co-PEGMA)-b-VS conjugate. Western Blot of the native-PAGE confirmed that the conjugate was still intact and immunologically active (Figure S13b). There are two surface exposed free cysteines in FGF2, and we have found that by performing polymer conjugation on a heparin resin, only one of the two cysteines is modified.66 Elman’s assay was employed to determine the amount of free thiols and confirmed that approximately one cysteine was modified.
After successfully making the FGF2-p(SS-co-PEGMA)-b-VS conjugate, stability studies were performed to determine whether the new block copolymer conjugate could stabilize FGF2 at a concentration of 0.05 ng/μL at either 4 or 23 °C for 7 days. As shown in Figure 2, FGF2-p(SS-co-PEGMA)-b-VS completely stabilized FGF2. In fact, there appeared to be a slight, but significant increase in cell growth which indicated an enhancement of activity, suggesting that the polymer may increase growth factor activity. It should also be noted that the conjugate linkages during storage in buffer at 4 °C were observed by native PAGE to be intact out to at least 1 year and 9 months (data not shown). However, if in the future greater chemical stability is required for in vivo use, the polymer end group could be readily altered to contain an amide rather than an ester, and a thiol ether rather than a disulfide bond.
To investigate whether FGF2-p(SS-co-PEGMA)-b-VS facilitated receptor binding, we first tested the conjugate for its effects on cell proliferation in BaF3-FR1C cells. This cell line lacks native heparan sulfate proteoglycans on the cell surface and requires added heparin to observe significant FGF2-induced proliferation.73 Thus, this cell line allows for indirect determination of the effect of heparin mimicking polymers on FGF/FGFR binding. The conjugate was compared to FGF2 alone, as well as FGF2 plus an excess of heparin. At a protein concentration of 0.5 ng/mL, the FGF2-p(SS-co-PEGMA)-b-VS conjugate increased cell growth more than native FGF2, with percent proliferation values of 352 ± 45% for the block copolymer and 151 ± 9% for FGF2 (Figure 3). Furthermore, the block copolymer conjugate FGF2-p(SS-co-PEGMA)-b-VS increased cell proliferation to the same extent as the positive control sample incubated with added 1 μg/mL heparin. The same trend was observed with protein concentrations of 1.5 ng/mL and 10 ng/mL. This data suggests that the FGF2-p(SS-co-PEGMA)-b-VS conjugate successfully bound to, and activated the FGF receptors. This further suggested that the block copolymer could facilitate binding of FGF to its receptors.
Figure 3.
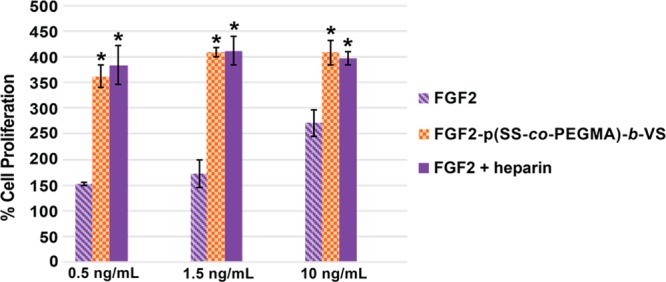
Cell growth of heparin-mimicking polymer conjugates in BaF3-FR1C cells. Incubation of 20 000 cells/well in 96-well plate with FGF2 or the heparin-mimicking polymer conjugates in the absence or presence of 1 μg/mL of heparin was carried out for 48 h. CellTiter-Blue assay was performed to quantify the extent of cell growth. Data was normalized to the blank medium group, which was set at 100%. Each sample contained four replicates and the experiment was repeated three times. Error bars represent standard error of the mean (SEM). Statistical analysis was done using Student’s t test. * p < 0.01 compared to FGF2.
To verify that the block copolymer in FGF2-p(SS-co-PEGMA)-b-VS was capable of facilitating binding of FGF2 to the receptor, we performed a receptor based enzyme linked immunosorbent assay. In this assay, FGF receptor 1a was plated in the wells of a 96 well plate and then either free protein (negative control) or conjugate were plated to assay degree of binding. Since heparin is known to facilitate receptor binding, 1 ng/mL FGF2 plus excess heparin (1 μg/mL) was used as a benchmark positive control to determine desired binding. As shown in Figure 4, p(SS-co-PEGMA)-b-VS in the conjugate facilitated FGF2/FGFR binding similarly to heparin. The results for the block copolymer conjugate correlate to the cell-based results above and demonstrate that the polymer facilitates FGF2 binding to the receptor.
Figure 4.
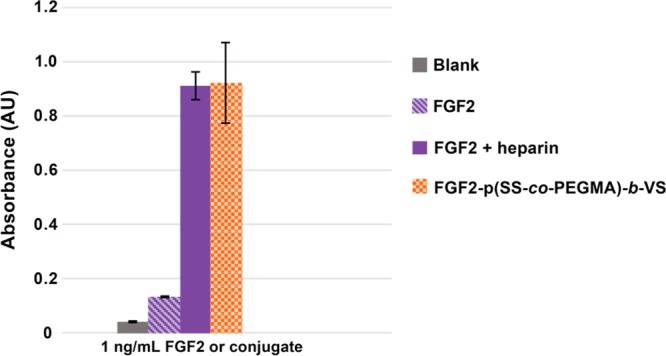
FGF Receptor based ELISA. Samples were incubated with 1 ng/mL FGF2, 1 ng/mL conjugate, or 1 ng/mL FGF2 plus 1 μg/mL heparin added as excipient. The experiment was repeated twice with n = 3. Error bars represent standard deviation.
Since the conjugate was active in the BaF3-FR1C cell line, FGF2-p(SS-co-PEGMA)-b-VS was also tested in normal human cell lines for the effect on proliferation. In human dermal fibroblasts the conjugates were tested at increasing concentrations and were compared to FGF2 alone. FGF2-p(SS-co-PEGMA)-b-VS performed marginally better than FGF2 at concentrations of 1.5 ng/mL, 3 ng/mL, and 5 ng/mL (Figure S14a). At lower concentrations of 0.5 ng/mL and 1 ng/mL, the protein–polymer conjugate and FGF2 exhibited statistically similar proliferation. The experiment was repeated in human umbilical vein endothelial cells the and the results showed that FGF2-p(SS-co-PEGMA)-b-VS conjugate stimulated cell proliferation only slightly better than FGF2 alone at all concentrations tested, 0.5 ng/mL, 1.0 ng/mL, 1.5 ng/mL, 3 ng/mL, and 5 ng/mL (Figure S14b). These experiments suggest that FGF2-p(SS-co-PEGMA)-b-VS does not greatly increase cell proliferation in normal cell lines containing heparin sulfate proteoglycans. In our previous study whereby FGF2 was dimerized by PEG, we noticed that cell migration and angiogenesis were significantly affected by the architecture, greater than HUVEC proliferation.23 Thus, we went on to further examine the effects of FGF2-p(SS-co-PEGMA)-b-VS on in vitro migration and tubulogenesis assays.
FGF2 is known to stimulate both migration and vasculogenesis of endothelial cells.74−76 First we performed a scratch migration assay to determine if FGF2-p(SS-co-PEGMA)-b-VS stimulates endothelial cell migration. As shown in Figure 5, 10 ng/mL FGF2-p(SS-co-PEGMA)-b-VS induced migration of endothelial cells better than 10 ng/mL FGF2 after 18 h, with a migration percentage of 214 ± 6% for the conjugate compared to 139 ± 4% for the native protein (blank media set at 100%). It also induced migration better than the addition of 1 μg/mL of heparin to FGF2. The heparin control did not induce cell migration, which was not unexpected, since it has been previously reported that added heparin at high concentrations has little effect on endothelial cell migration in combination with FGF2, and sometimes even displays inhibitory effects.77 To ensure the observed effect of migration was not due to the ability of the conjugate to induce greater HUVEC proliferation, we quantified cell proliferation after the 18-h incubation period. The percent of cell growth induced by the conjugate was not statistically different than native protein (Figure S15) because of the shorter incubation period (18 h versus 72 h in Figure S14b) as has been previously reported.75
Figure 5.
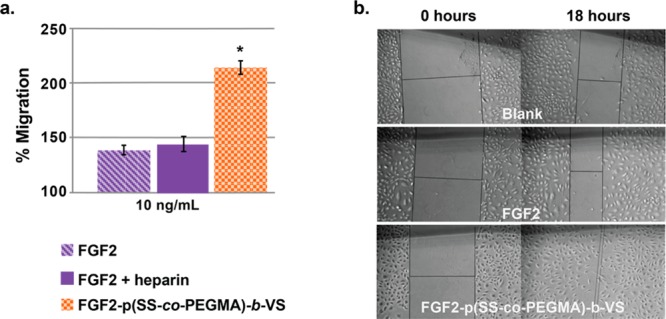
HUVEC migration after 18 h. (a) Percent migration in the presence of FGF2, FGF2 plus 1 μg/mL of heparin, or FGF2-p(SS-co-PEGMA)-b-VS. Percent migration was calculated using the formula: 100% – (distance at T0/distance at T18) and blank medium was set at 100%. Each sample contained four to six replicates and the experiment was repeated three times, with each repeat being blinded. Error bars represent SEM. (b) Representative images of HUVEC migration taken at 0 and 18 h. Statistical analysis was done using Student’s t test. * p < 0.01 compared to FGF2.
FGF2 also induces angiogenesis.76 To begin to determine whether the diblock copolymer conjugate had similar effects on angiogenesis as FGF2, a simple in vitro coculture assay was performed.70,71,78 In this assay, fibroblasts are first grown to confluency. During this incubation period, the fibroblasts produce a layer of collagen, which provides a matrix for endothelial cells to burrow in. After the fibroblasts reach confluency, a layer of endothelial cells are plated along with sample solutions in starvation medium. This assay provides information on the two-dimensional aspects of angiogenesis, but does not provide any information about the three-dimensional aspects, such as tubular structure and perfusion of vessels.
The endothelial cells were allowed to grow over 14 days with medium/sample changes every 2–3 days. Cord-like structures could be observed after antibody staining for CD-31 (or PECAM-1), a glycoprotein present at cell–cell junctions of vascular endothelial cells.79 In wells incubated with starvation medium alone, no cord-like structures were formed; however, when FGF2 was added to the starvation medium, cord-like structures became apparent at concentrations as low as 0.5 ng/mL. Samples were measured for an increase in length of cord-like structures, total number of cord-like structures and the number of nodes compared to FGF2 alone. In wells containing the FGF2-p(SS-co-PEGMA)-b-VS conjugate, the number and length of cord-like structures, as well as the nodes were significantly increased compared to FGF2 alone (Figure 6a), suggesting that the protein–polymer conjugate induced cord-like structures better than the native protein. Control wells containing FGF2 plus 1 μg/mL heparin induced cord-like structure formation to a significantly lesser extent than FGF2-p(SS-co-PEGMA)-b-VS, and in some cases even showed a slight inhibitory effect. It has previously been reported that large concentrations of heparin have an inhibitory effect on angiogenesis; thus these data are not unexpected.80 In order to determine whether excess block copolymer as an excipient would act similarly to heparin, control wells containing FGF2 plus 1 μg/mL free polymer were tested, and indeed the results were similar to added heparin. These results highlight the benefits of conjugating the polymer to the protein, and thus using the polymer at a small concentration, rather than as an additive in excess. Representative images of cord like structure formation are shown in Figure 6b. These results also indicate that the block copolymer conjugate could potentially induce angiogenesis better than FGF2 alone. In the future, more in depth three-dimensional assays will be carried out to fully determine the success of FGF2-p(SS-co-PEGMA)-b-VS as an inducer of angiogenesis.
Figure 6.
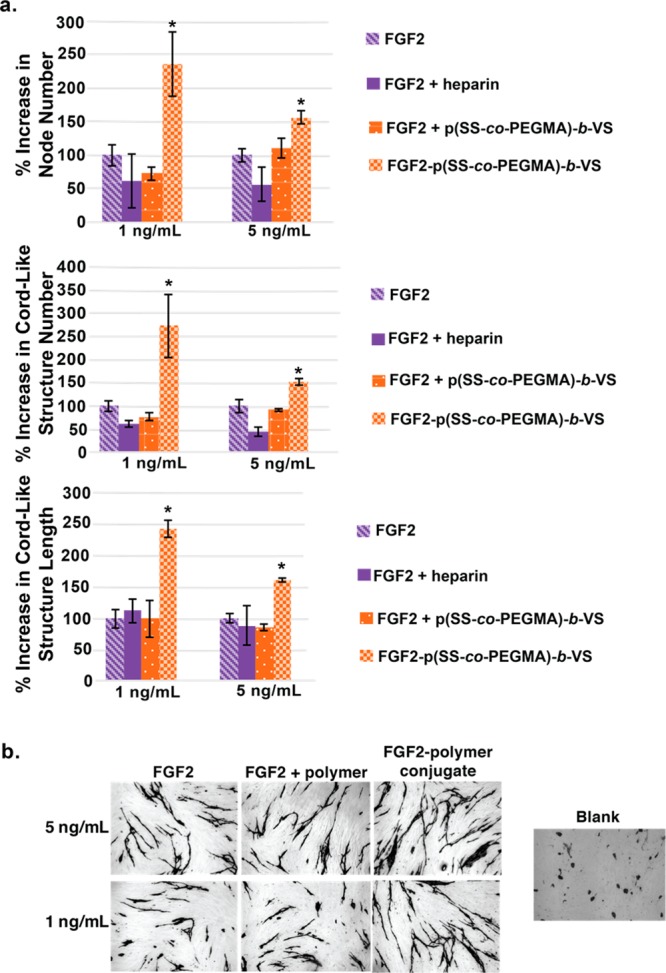
Coculture angiogenesis of HUVEC and HDF. (a) Node number, cord-like structure length and cord-like structure number. (b) Representative images of cord-like structures stained for CD31. Three replicates per sample, error bars represent SEM. Statistical analysis was done using Student’s t test. * p < 0.01 compared to FGF2.
Taken together, these data suggest that FGF2-p(SS-co-PEGMA)-b-VS is a promising candidate for new wound healing and pro-angiogenic treatments. Besides an increase in cellular proliferation, migration, two-dimensional angiogenesis, and stability, the growth factor polymer conjugate presented here has potential advantages over heparin added to growth factors in treatments. Lower FGF2 polymer conjugate concentration is needed to achieve similar cellular response compared to when just FGF2 is added. In addition, the use of a heparin mimicking polymer allows for well-defined biomaterials for more predictable cell response. The conjugate is also more stable to storage in the refrigerator and at room temperature than the native protein.
Conclusion
We have developed a novel protein–polymer conjugate, FGF2-p(SS-co-PEGMA)-b-VS that is composed of a heparin-mimicking polymer that imparts superagonist activity and stability to FGF2. This new polymer, containing both stabilization and receptor binding segments was conjugated to FGF2 through disulfide exchange with a pyridyl disulfide end group on the polymer and a surface exposed cysteine on the protein. The FGF2-p(SS-co-PEGMA)-b-VS conjugate was tested in multiple in vitro cell based assays and compared to FGF2 alone and addition of heparin. FGF2-p(SS-co-PEGMA)-b-VS, facilitated receptor binding in both cellular and ELISA assays similar to addition of heparin. Additionally, FGF2-p(SS-co-PEGMA)-b-VS accelerated migration and endothelial cell cord-like structure formation when compared to native FGF2 and addition of heparin. It also completely stabilized the protein to standard storage conditions, both refrigeration and room temperature. Together, these data suggest that this conjugate is promising and should be studied further for applications in tissue regeneration and wound healing. In the future, a switchable RAFT agent similar to the one described by Rizzardo and co-workers may be utilized to prepare these types of sulfonated block copolymer with better control, which is important for biomedical applications.81 Additionally, in vitro and in vivo studies and further optimization of polymer design will be carried out to further determine the efficacy and potential of FGF2-p(SS-co-PEGMA)-b-VS as a therapeutic for healing chronic and ischemic wounds.
Acknowledgments
This work was supported by the National Institute of Health (NIH R01EB013674). S.J.P. thanks the NIH Chemistry Biology Interface Training Fellowship (T32 GM 008496) and UCLA Graduate Division for funding. Additionally, the authors thank the Helmholtz Centre for Infection Research in Braunschweig, Germany for the pET29c(+)hFGF-2 plasmid, and Prof. David Ornitz (Washington University) for providing the BaF3 cell line. The authors thank Prof. Samir Zard (École Polytechnique, Paris, France) for his helpful discussion regarding the chain transfer agent.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.6b01182.
Experimental and methods sections not shown in the paper, NMR data, conjugate characterization, polymer toxicity, ELISA results, and cell proliferation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Agarwal S.; Sud K.; Shishehbor M. H. J. Am. Coll. Cardiol. 2016, 67, 1901. 10.1016/j.jacc.2016.02.040. [DOI] [PubMed] [Google Scholar]
- Sen C. K.; Gordillo G. M.; Roy S.; Kirsner R.; Lambert L.; Hunt T. K.; Gottrup F.; Gurtner G. C.; Longaker M. T. Wound Repair and Regen. 2009, 17, 763. 10.1111/j.1524-475X.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer A. J.; Clark R. A. F. N. Engl. J. Med. 1999, 341, 738. 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Doctor N.; Yang S.; Maerzacker S.; Watkins P.; Dissanaike S. J. Burn Care Res. 2016, 37, E56. 10.1097/BCR.0000000000000327. [DOI] [PubMed] [Google Scholar]
- Driver V. R.; Fabbi M.; Lavery L. A.; Gibbons G. J. Am. Podiatr. Med. Assoc. 2010, 100, 335. 10.7547/1000335. [DOI] [PubMed] [Google Scholar]
- Greenhalgh D. G. J. Trauma Injury Infect. Crit. Care 1996, 41, 159. 10.1097/00005373-199607000-00029. [DOI] [Google Scholar]
- Demidova-Rice T. N.; Hamblin M. R.; Herman I. M. Adv. Skin Wound Care 2012, 25, 304. 10.1097/01.ASW.0000416006.55218.d0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells L. A.; Valic M. S.; Lisovsky A.; Sefton M. V. Isr. J. Chem. 2013, 53, 637. 10.1002/ijch.201300053. [DOI] [Google Scholar]
- Rufaihah A. J.; Seliktar D. Adv. Drug Delivery Rev. 2016, 96, 31. 10.1016/j.addr.2015.07.003. [DOI] [PubMed] [Google Scholar]
- Itoh N.; Ornitz D. M. J. Biochem. 2011, 149, 121. 10.1093/jb/mvq121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D. M.; Itoh N. Genome Biol. 2001, 2, 3005. 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikfalvi A.; Klein S.; Pintucci G.; Rifkin D. B. Endocr. Rev. 1997, 18, 26. 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- Werner S.; Grose R. Physiol. Rev. 2003, 83, 835. [DOI] [PubMed] [Google Scholar]
- Fu X. B.; Shen Z. Y.; Guo Z. R.; Zhang M. L.; Sheng Z. Y. Chin. Med. J. 2002, 115, 331. [PubMed] [Google Scholar]
- Whitaker M. J.; Quirk R. A.; Howdle S. M.; Shakesheff K. M. J. Pharm. Pharmacol. 2001, 53, 1427. 10.1211/0022357011777963. [DOI] [PubMed] [Google Scholar]
- Presta M.; Dell’Era P.; Mitola S.; Moroni E.; Ronca R.; Rusnati M. Cytokine Growth Factor Rev. 2005, 16, 159. 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Richard J. L.; Bringer J.; Parerrichard C.; Rodier M.; Daures J. P.; Jacob C.; Clouet S.; Comtebardonnet M.; Vannereau D. Diabetes Care 1995, 18, 64. 10.2337/diacare.18.1.64. [DOI] [PubMed] [Google Scholar]
- Frykberg R. G.; Banks J. Adv. Wound Care 2015, 4, 560. 10.1089/wound.2015.0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer B. A.; Pantoliano M. W.; Sharp C. M.. Analogs of human basic fibroblast growth factor mutated at one or more of the positions glutamute 89, aspartate 101 or leucine 137. U.S. Patent US 6274712 B1, Aug. 14, 2001.
- Bogin O.; Yayon A. Fgf variants and methods for use thereof. WIPO Patent WO 2003094835 A3, Nov. 20, 2003.
- Yayon A.; Rom E.; Chumakov I.; Blumenstein S. FGF-2 variants having N-terminal deletions and increased receptor selectivity and uses thereof. U.S. Patent US 8962556 B2, Feb. 24, 2015.
- Kwan C. P.; Venkataraman G.; Shriver Z.; Raman R.; Liu D. F.; Qi Y. W.; Varticovski L.; Sasisekharan R. J. Biol. Chem. 2001, 276, 23421. 10.1074/jbc.M010786200. [DOI] [PubMed] [Google Scholar]
- Decker C. G.; Wang Y.; Paluck S. J.; Shen L.; Loo J. A.; Levine A. J.; Miller L. S.; Maynard H. D. Biomaterials 2016, 81, 157. 10.1016/j.biomaterials.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran M.; Eisenstein M.; Aviezer D.; Yayon A. Biochem. J. 2000, 345, 107. 10.1042/bj3450107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko O.; Tkach V.; Berezin V.; Bock E. Neurosci. Res. 2010, 68, 35. 10.1016/j.neures.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Anderson A. A.; Kendal C. E.; Garcia-Maya M.; Kenny A. V.; Morris-Triggs S. A.; Wu T.; Reynolds R.; Hohenester E.; Saffell J. L. J. Neurochem. 2005, 95, 570. 10.1111/j.1471-4159.2005.03417.x. [DOI] [PubMed] [Google Scholar]
- Nur-E-Kamal A.; Ahmed I.; Kamal J.; Babu A. N.; Schindler M.; Meiners S. Mol. Cell. Biochem. 2008, 309, 157. 10.1007/s11010-007-9654-8. [DOI] [PubMed] [Google Scholar]
- Choi K.-C.; Yoo D.-S.; Cho K.-S.; Huh P.-W.; Kim D.-S.; Park C.-K. J. Korean Neurosurg. Soc. 2008, 44, 375. 10.3340/jkns.2008.44.6.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson T. P.; Peters M. C.; Ennett A. B.; Mooney D. Nat. Biotechnol. 2001, 19, 1029. 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- Wu J.; Mao Z.; Hong Y.; Han L.; Gao C. Bioconjugate Chem. 2013, 24, 1302. 10.1021/bc300670t. [DOI] [PubMed] [Google Scholar]
- Gospodarowicz D.; Cheng J. J. Cell. Physiol. 1986, 128, 475. 10.1002/jcp.1041280317. [DOI] [PubMed] [Google Scholar]
- Plotnikov A. N.; Schlessinger J.; Hubbard S. R.; Mohammadi M. Cell 1999, 98, 641. 10.1016/S0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Caldwell M. A.; Garcion E.; terBorg M. G.; He X. L.; Svendsen C. N. Exp. Neurol. 2004, 188, 408. 10.1016/j.expneurol.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Ishihara M.; Ono K. Trends Glycosci. Glycotechnol. 1998, 10, 223. 10.4052/tigg.10.223. [DOI] [Google Scholar]
- Theroux P.; Ouimet H.; McCans J.; Latour J. G.; Joly P.; Levy G.; Pelletier E.; Juneau M.; Stasiak J.; Deguise P.; Pelletier G. B.; Rinzler D.; Waters D. D. N. Engl. J. Med. 1988, 319, 1105. 10.1056/NEJM198810273191701. [DOI] [PubMed] [Google Scholar]
- Fareed J.; Hoppensteadt D. A.; Bick R. L. Clin. Appl. Thromb./Hemostasis 2003, 9, 101. 10.1177/107602960300900202. [DOI] [PubMed] [Google Scholar]
- Carter N. J.; McCormack P. L.; Plosker G. L. Drugs 2008, 68, 691. 10.2165/00003495-200868050-00012. [DOI] [PubMed] [Google Scholar]
- Saliba M. J. Burns 2001, 27, 349. 10.1016/S0305-4179(00)00130-3. [DOI] [PubMed] [Google Scholar]
- Serra R.; Buffone G.; Molinari V.; Montemurro R.; Perri P.; Stillitano D. M.; Amato B.; de Franciscis S. Int. Wound J. 2015, 12, 150. 10.1111/iwj.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niers T. M. H.; Klerk C. P. W.; DiNisio M.; Van Noorden C. J. F.; Buller H. R.; Reitsma P. H.; Richel D. J. Crit. Rev. Oncol. Hematol. 2007, 61, 195. 10.1016/j.critrevonc.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Tyrrell D. J.; Horne A. P.; Holme K. R.; Preuss J. M. H.; Page C. P. Adv. Pharmacol. 1999, 46, 151. 10.1016/S1054-3589(08)60471-8. [DOI] [PubMed] [Google Scholar]
- Shafiq N.; Malhotra S.; Pandhi P.; Sharma N.; Bhalla A.; Grover A. Pharmacology 2006, 78, 136. 10.1159/000096484. [DOI] [PubMed] [Google Scholar]
- Edelman E. R.; Mathiowitz E.; Langer R.; Klagsbrun M. Biomaterials 1991, 12, 619. 10.1016/0142-9612(91)90107-L. [DOI] [PubMed] [Google Scholar]
- Whalen G. F.; Shing Y.; Folkman J. Growth Factors 1989, 1, 157. 10.3109/08977198909029125. [DOI] [PubMed] [Google Scholar]
- Cummings C.; Murata H.; Koepsel R.; Russell A. J. Biomacromolecules 2014, 15, 763. 10.1021/bm401575k. [DOI] [PubMed] [Google Scholar]
- Keefe A. J.; Jiang S. Nat. Chem. 2012, 4, 59. 10.1038/nchem.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini R. J.; Lee J.; Maynard H. D. J. Am. Chem. Soc. 2012, 134, 8474. 10.1021/ja2120234. [DOI] [PubMed] [Google Scholar]
- Lucius M.; Falatach R.; McGlone C.; Makaroff K.; Danielson A.; Williams C.; Nix J. C.; Konkolewicz D.; Page R. C.; Berberich J. A. Biomacromolecules 2016, 17, 1123. 10.1021/acs.biomac.5b01743. [DOI] [PubMed] [Google Scholar]
- Averick S.; Mehl R. A.; Das S. R.; Matyjaszewski K. J. Controlled Release 2015, 205, 45. 10.1016/j.jconrel.2014.11.030. [DOI] [PubMed] [Google Scholar]
- Pelegri-O’Day E. M.; Lin E.-W.; Maynard H. D. J. Am. Chem. Soc. 2014, 136, 14323. 10.1021/ja504390x. [DOI] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Nat. Chem. 2016, 8, 103. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Cobo I.; Li M.; Sumerlin B. S.; Perrier S. Nat. Mater. 2015, 14, 143. 10.1038/nmat4106. [DOI] [PubMed] [Google Scholar]
- Sasisekharan R.; Moses M. A.; Nugent M. A.; Cooney C. L.; Langer R. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 1524. 10.1073/pnas.91.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver Z.; Capila I.; Venkataraman G.; Sasisekharan R. Handb. Exp. Pharmacol. 2012, 207, 159. 10.1007/978-3-642-23056-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M.; Demers C.; Gurfinkel E. P.; Turpie A. G. G.; Fromell G. J.; Goodman S.; Langer A.; Califf R. M.; Fox K. A. A.; Premmereur J.; Bigonzi F.; et al. N. Engl. J. Med. 1997, 337, 447. 10.1056/NEJM199708143370702. [DOI] [PubMed] [Google Scholar]
- Papy-Garcia D.; Barbier-Chassefiere V.; Rouet V.; Kerros M. E.; Klochendler C.; Tournaire M. C.; Barritault D.; Caruelle J. P.; Petit E. Macromolecules 2005, 38, 4647. 10.1021/ma048485p. [DOI] [Google Scholar]
- Mauzac M.; Jozefonvicz J. Biomaterials 1984, 5, 301. 10.1016/0142-9612(84)90078-4. [DOI] [PubMed] [Google Scholar]
- Tardieu M.; Gamby C.; Avramoglou T.; Jozefonvicz J.; Barritault D. J. Cell. Physiol. 1992, 150, 194. 10.1002/jcp.1041500126. [DOI] [PubMed] [Google Scholar]
- Liekens S.; Leali D.; Neyts J.; Esnouf R.; Rusnati M.; Dell’Era P.; Maudgal P. C.; De Clercq E.; Presta M. Mol. Pharmacol. 1999, 56, 204. 10.1124/mol.56.1.204. [DOI] [PubMed] [Google Scholar]
- Nguyen T. H.; Paluck S. J.; McGahran A. J.; Maynard H. D. Biomacromolecules 2015, 16, 2684. 10.1021/acs.biomac.5b00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan J. R.; Bruno J. G.; Chang M. N.; Sabatino R.; D'Alisa R.; Bensasson S. A.; Eilat D. J. Bioact. Compat. Polym. 1993, 8, 317. 10.1177/088391159300800402. [DOI] [Google Scholar]
- Mammadov R.; Mammadov B.; Guler M. O.; Tekinay A. B. Biomacromolecules 2012, 13, 3311. 10.1021/bm3010897. [DOI] [PubMed] [Google Scholar]
- Sun X. L.; Grande D.; Baskaran S.; Hanson S. R.; Chaikof E. L. Biomacromolecules 2002, 3, 1065. 10.1021/bm025561s. [DOI] [PubMed] [Google Scholar]
- Grasel T. G.; Cooper S. L. J. Biomed. Mater. Res. 1989, 23, 311. 10.1002/jbm.820230304. [DOI] [PubMed] [Google Scholar]
- Ito Y.; Iguchi Y.; Imanishi Y. Biomaterials 1992, 13, 131. 10.1016/0142-9612(92)90060-2. [DOI] [PubMed] [Google Scholar]
- Nguyen T. H.; Kim S.-H.; Decker C. G.; Wong D. Y.; Loo J. A.; Maynard H. D. Nat. Chem. 2013, 5, 221. 10.1038/nchem.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra M.; Ishai-Michaeli R.; Ben-Sasson S. A.; Vlodavsky I. J. Cell. Physiol. 2002, 192, 276. 10.1002/jcp.10136. [DOI] [PubMed] [Google Scholar]
- Chen R.; John J.; Lavrentieva A.; Mueller S.; Tomala M.; Zhao Y.; Zweigerdt R.; Beutel S.; Hitzmann B.; Kasper C.; Martin U.; Rinas U.; Stahl F.; Scheper T. Eng. Life Sci. 2012, 12, 29. 10.1002/elsc.201100045. [DOI] [Google Scholar]
- Ornitz D. M.; Leder P. J. Biol. Chem. 1992, 267, 16305. [PubMed] [Google Scholar]
- Hetheridge C.; Mavria G.; Mellor H. Biochem. Soc. Trans. 2011, 39, 1597. 10.1042/BST20110738. [DOI] [PubMed] [Google Scholar]
- Bishop E. T.; Bell G. T.; Bloor S.; Broom I. J.; Hendry N. F.; Wheatley D. N. Angiogenesis 1999, 3, 335. 10.1023/A:1026546219962. [DOI] [PubMed] [Google Scholar]
- Perrier S.; Takolpuckdee P. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 5347. 10.1002/pola.20986. [DOI] [Google Scholar]
- Ornitz D. M.; Yayon A.; Flanagan J. G.; Svahn C. M.; Levi E.; Leder P. Mol. Cell. Biol. 1992, 12, 240. 10.1128/MCB.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A.; AnandApte B.; Zetter B. R. Growth Factors 1996, 13, 57. 10.3109/08977199609034566. [DOI] [PubMed] [Google Scholar]
- Seghezzi G.; Patel S.; Ren C. J.; Gualandris A.; Pintucci G.; Robbins E. S.; Shapiro R. L.; Galloway A. C.; Rifkin D. B.; Mignatti P. J. Cell Biol. 1998, 141, 1659. 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano R.; Vassalli J. D.; Baird A.; Guillemin R.; Orci L. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 7297. 10.1073/pnas.83.19.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraux J. L.; Matou S.; Bros A.; Tapon-Bretaudiere J.; Letourneur D.; Fischer A. M. Eur. J. Cell Biol. 1998, 77, 352. 10.1016/S0171-9335(98)80094-0. [DOI] [PubMed] [Google Scholar]
- Donovan D.; Brown N. J.; Bishop E. T.; Lewis C. E. Angiogenesis 2001, 4, 113. 10.1023/A:1012218401036. [DOI] [PubMed] [Google Scholar]
- DeLisser H. M.; ChristofidouSolomidou M.; Strieter R. M.; Burdick M. D.; Robinson C. S.; Wexler R. S.; Kerr J. S.; Garlanda C.; Merwin J. R.; Madri J. A.; Albelda S. M. Am. J. Pathol. 1997, 151, 671. [PMC free article] [PubMed] [Google Scholar]
- Jung S. P.; Siegrist B.; Wade M. R.; Anthony C. T.; Woltering E. A. Angiogenesis 2001, 4, 175. 10.1023/A:1014089706107. [DOI] [PubMed] [Google Scholar]
- Benaglia M.; Chiefari J.; Chong Y. K.; Moad G.; Rizzardo E.; Thang S. H. J. Am. Chem. Soc. 2009, 131, 6914. 10.1021/ja901955n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.