

Abstract
This prospective observational cohort study aimed to explore the clinical features of incident immune thrombocytopenia in adults and predictors of outcome, while determining if a family history of autoimmune disorder is a risk factor for immune thrombocytopenia. All adults, 18 years of age or older, recently diagnosed with immune thrombocytopenia were consecutively recruited across 21 hospital centers in France. Data were collected at diagnosis and after 12 months. Predictors of chronicity at 12 months were explored using logistic regression models. The association between family history of autoimmune disorder and the risk of developing immune thrombocytopenia was explored using a conditional logistic regression model after matching each case to 10 controls. One hundred and forty-three patients were included: 63% female, mean age 48 years old (Standard Deviation=19), and 84% presented with bleeding symptoms. Median platelet count was 10×109/L. Initial treatment was required in 82% of patients. After 12 months, only 37% of patients not subject to disease-modifying interventions achieved cure. The sole possible predictor of chronicity at 12 months was a higher platelet count at baseline [Odds Ratio 1.03; 95%CI: 1.00, 1.06]. No association was found between outcome and any of the following features: age, sex, presence of either bleeding symptoms or antinuclear antibodies at diagnosis. Likewise, family history of autoimmune disorder was not associated with incident immune thrombocytopenia. Immune thrombocytopenia in adults has been shown to progress to a chronic form in the majority of patients. A lower platelet count could be indicative of a more favorable outcome.
Introduction
Immune thrombocytopenia (ITP) is an autoimmune disorder mediated by platelet antibodies thought to accelerate platelet destruction while inhibiting also their production,1 resulting in low platelet counts with potentially spontaneous bruising, petechial rash, mucosal bleeding or even life-threatening hemorrhage. ITP affects children and adults, with an incidence rate for the latter estimated between 2.8 and 3.9 per 100,000 person-years in Europe,2–6 and namely 2.9/100,000 in France.6 The onset of ITP is frequently insidious and low platelet counts often last beyond six months.7 A recent retrospective study based on administrative registers reported that about two-thirds of adult ITP patients are likely to develop a chronic form of the disease.6 The risk factors for chronicity were mainly explored in children8–10 and rarely in adults.11 Regarding the genetic risk, a few studies reported clusters of ITP incidence within families,12,13 but it is unknown whether a family history of autoimmune disorder can be a risk factor for developing ITP.
In France, a nationwide prospective cohort of adult patients presenting with a newly diagnosed episode of ITP was constituted primarily to explore the association between exposure to common vaccines and risk of developing ITP.14 In this context, the present study aimed at describing the clinical features of adult ITP and its evolution over a 12-month period and exploring the baseline predictors of chronicity. We also explored whether a history of autoimmune disorder in first-degree relatives could constitute a risk factor for developing ITP.
Methods
Source of data
This was a prospective observational cohort study using data from the Pharmacoepidemiologic General Research eXtension (PGRx) information system which is a set of population-based registries including case-patients (cases) with specific diseases recruited by their specialist physician and referent-patients (controls) routinely recruited by their general practitioner (GP).14,15 Inclusion and exclusion criteria are similar for cases and controls, except for the disease of interest. Medical information is collected by specialists (for cases) and GPs (for controls). All patients undergo the same standardized telephone interview to collect information on family medical history, lifestyle and use of medication.
Study population
Participating physicians in the PGRx-ITP registry were requested to consecutively include cases of ITP. All the patients met the following criteria: 1) age between 18 and 79 years; 2) diagnosis of primary ITP according to international consensus;16 3) delay between the first symptoms of ITP and inclusion of less than 365 days; 4) normal physical examination, except for bleeding symptoms; 5) domiciled in continental France; 6) able to understand and read French; and 7) agreement to participate. A strict procedure was used to confirm diagnosis (see Table 1 and Online Supplementary Appendix). In order to determine whether a history of autoimmune disorder in first-degree relatives constituted a risk factor for developing ITP or not, up to 10 controls with no lifetime history of ITP were identified from the PGRx-General Practice registry and matched to each case according to sex, age (±1 year), and inclusion date (±5 years).
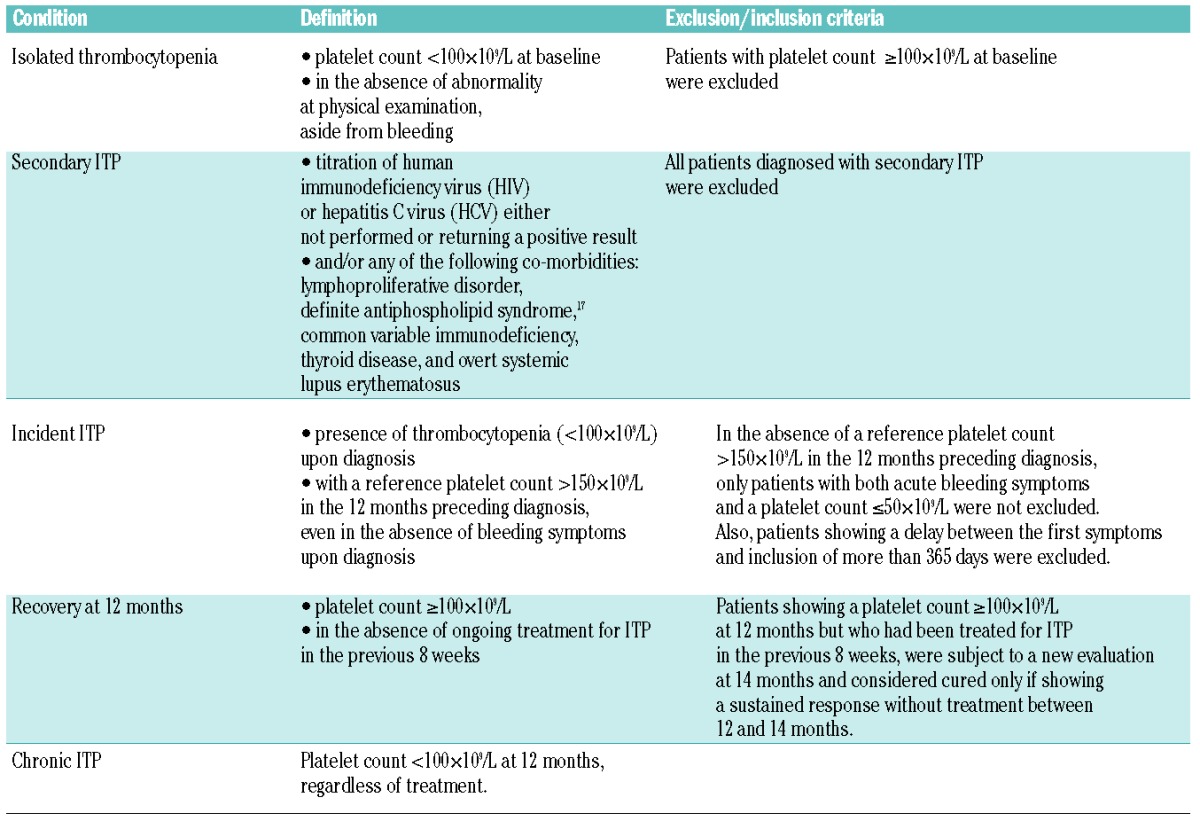
Table 1.
Definitions used for isolated thrombocytopenia, secondary immune thrombocytopenia (ITP), incident ITP and outcome.
Measures
All patient data were collected through dedicated case report forms (CRFs), similar in cases and controls regarding socio-demographics, personal medical history and family history of autoimmune disorder (in first-degree relatives): multiple sclerosis, lupus erythematosus, rheumatoid arthritis, Crohn disease, chronic ulcerative colitis, Hashimoto thyroiditis or Graves-Basedow disease. In cases, data were also collected on the ITP at baseline and after 12 months of follow up (Online Supplementary Appendix). The time elapsing between diagnosis and outcome measure at 12 months was preferred, given that first symptoms are often more difficult to specify. Recovery and chronicity were defined as in Table 1.
Statistical analysis
Statistical analysis was performed with SAS software version 9.18 and all comparative tests were two-sided with a type-1 error set at α=0.05. Bleeding symptoms were categorized into: a) no bleeding; b) cutaneous bleeding alone; and c) severe bleeding defined as mucocutaneous and/or visceral bleeding (epistaxis, or bleeding of the mucous membrane, or visceral bleeding). The titration of antinuclear antibodies (ANA) was considered positive if the titer was more than 1/80. Further details are provided in the Online Supplementary Appendix.
Ethics approval
This research was approved by the consultative committee for non-interventional research (Comité Consultatif sur le Traitement de l’Information en Matière de Recherche) and the French Data Protection Authority (Commission Nationale de l’Informatique et des Libertés) was informed that data were being collected for this purpose. All patients included in the PRGx database provided informed consent.
Results
Over a 28-month period, 188 patients diagnosed with ITP were consecutively included in the PGRx-ITP registry by hematologists and internists from 21 hospitals across France. Of these, 18 patients were excluded once diagnostic procedures had been completed and another 27 patients were excluded as a result of failure to confirm diagnosis of ITP at three months; this left 143 patients for analysis.
Characteristics of ITP at onset
A median of 35 days elapsed between the first symptoms and inclusion in the study, and 35 (24.5%) patients were recruited at 90 days or later after the onset of ITP symptoms. Table 2 and Figure 1 show the characteristics of the 143 patients and incident ITPs upon diagnosis. Median age was 50 years and on average there were more female than male, with a female/male ratio of 1.7 overall which dropped to 1 in patients over 50 years of age. Information on family history of autoimmune disorder was available in 109 (76.2%) patients. In 23 (16.1%) patients, bleeding symptoms were absent and the diagnosis was made fortuitously after a routine blood test. Visceral bleeding was present in 7 (4.9%) patients and none of the patients presented with intracerebral hemorrhage. The median platelet count was 10×109/L, and 99 (69.2%) patients showed values less than 20×109/L at presentation. A bone marrow aspiration was performed in 111 (77.6%) patients, namely in 93.5% of patients over 60 years of age (n=43). ANA was tested in 136 (95.1%) patients. A “watch and wait” strategy was decided for 18 (12.6%) patients. Among patients initiating treatment, 61 (48.8%) were administered corticosteroids and intravenous immunoglobulins (IVIg), 50 (40.0%) corticosteroids alone, and 14 (11.2%) IVIg alone. In 7 patients, additional therapy was necessary (vinblastine, vincristine or platelet transfusion).
Table 2.
Characteristics and initial medical management of 143 adult patients diagnosed with a first lifetime episode of immune thrombocytopenia (ITP).
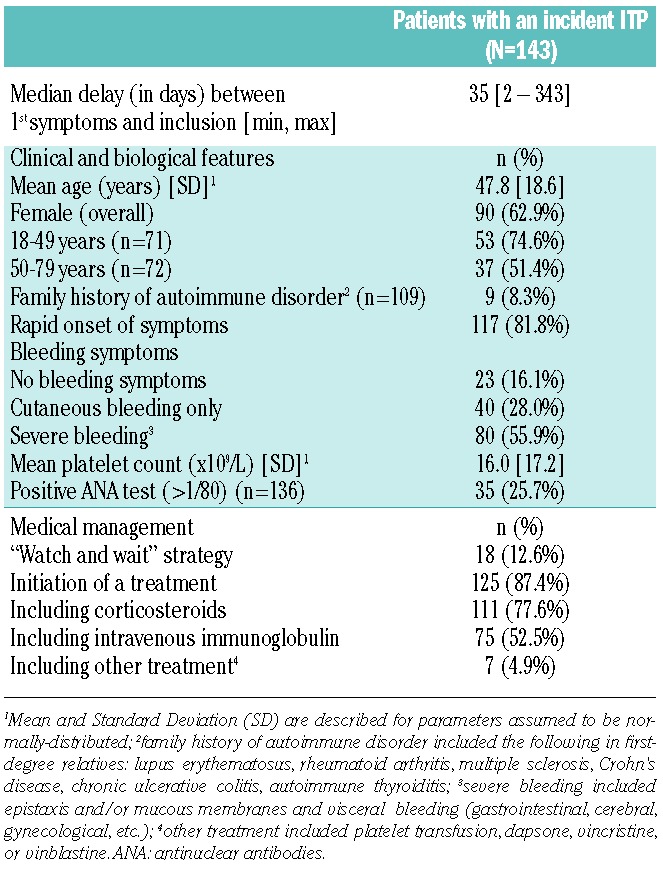
Figure 1.

Distribution of age and sex in 143 adults diagnosed with a first lifetime episode of immune thrombocytopenia.
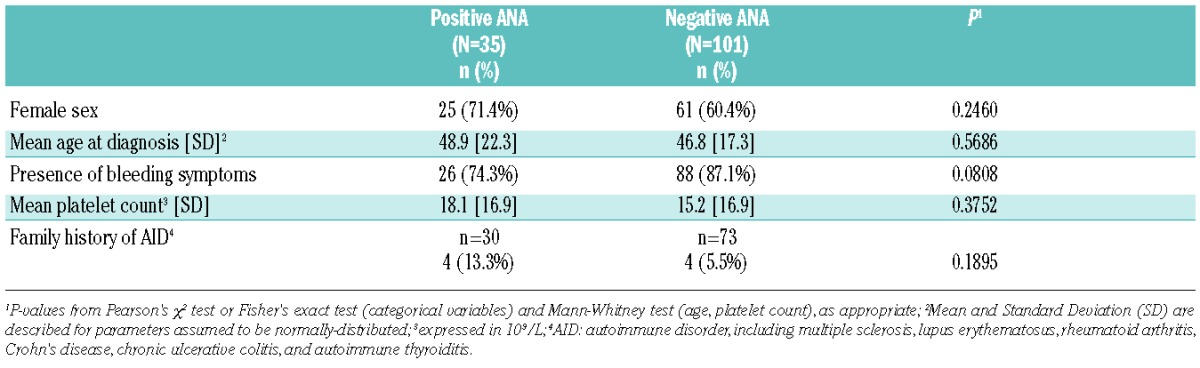
Table 3 provides comparisons among 35 patients testing positive for ANA (titer >1/80) and 101 patients with a negative ANA test. The two groups differed, albeit non-significantly, in terms of bleeding symptoms at diagnosis, the absence of such symptoms being more frequent in patients testing positive for ANA. The presence of a family history of autoimmune disorder was twice as frequent in patients with a positive ANA test, yet not statistically significant. There was no difference in other characteristics (age, sex, and platelet count) between groups.
Table 3.
Comparison of patients testing positive (>1/80) or negative for antinuclear antibodies (ANA) at baseline in 136 patients with a newly diagnosed episode of immune thrombocytopenia.
Twelve-month outcome
A total of 28 (19.6%) patients were lost to follow up at twelve months after ITP had been first diagnosed. Among the 115 patients with data available at one year, 58 (50.4%) showed progression to chronic ITP while 57 recovered: 43 (37.4%) did so spontaneously (i.e. with no disease-modifying treatment) and 14 (12.2%) achieved cure with a disease-modifying treatment: either rituximab alone (n=12) or rituximab plus splenectomy (n=2).
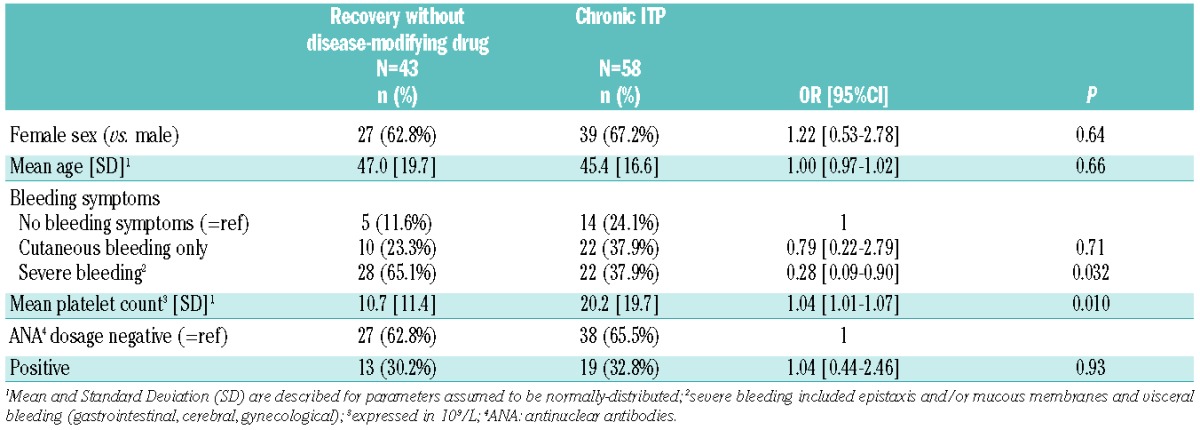
Table 4 provides the results from univariate logistic regression models exploring the baseline factors associated with 12-month outcome in patients showing progression to chronic ITP (n=58) and patients with spontaneous recovery (n=43). The presence of a higher platelet count or the absence of severe bleeding at baseline was found to be associated with increased odds of chronicity at 12 months. No difference was found among patients in terms of sex, age and testing positive for ANA. A bivariate model was fitted to explore the independent predictive effect of platelet counts (continuous variable) and severe bleeding (severe vs. no bleeding or cutaneous bleeding alone) at baseline. The model revealed that severe bleeding was not associated with chronicity [Odds Ratio (OR) 0.47; 95%CI: 0.19, 1.19; P=0.11], whereas a higher platelet count remained associated with increased odds of chronicity, despite not reaching the 0.05 significance level (OR 1.03; 95%CI: 1.00, 1.06; P=0.08). The interpretation of this result needs to take into account that “platelet counts” have been used as a continuous variable: for every 10×109/L increase in the platelet counts (e.g. from 10×109/L to 20×109/L, or from 20×109/L to 30×109/L), the equivalent increase in terms of odds of chronicity is about 34%. All results were similar when exploring the odds of chronicity versus recovery, which also included patients who recovered after disease-modifying treatment (data not shown).
Table 4.
Baseline factors associated with the 12-month outcome, in adults recently diagnosed with an immune thrombocytopenia (ITP); patients who recovered without any disease-modifying drug are compared to patients with chronic ITP; results of univariate logistic regression models providing Odds Ratio (OR) and 95% Confidence Intervals (95%CI).
Family history of autoimmune disorder
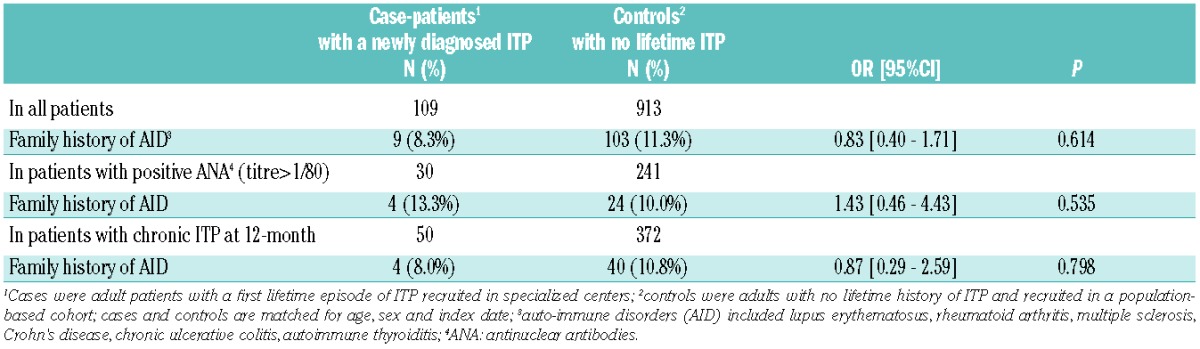
A familial history of autoimmune disorder was found in 9 (8.3%) out of the 109 case-patients explored. Table 5 provides the results from conditional logistic regression models comparing 109 case-patients to 913 controls matched on sex, age, and inclusion date. Overall, having a first-degree relative with a history of autoimmune disorder was not associated with increased odds of developing ITP. No association was found in either the subgroup of ITP patients with a positive ANA test at baseline or the subgroup of ITP patients progressing to a chronic form of the disease.
Table 5.
Association between family history of autoimmune disorder and risk of developing an immune thrombocytopenia (ITP) in adulthood; results of univariate conditional logistic regression models providing Odds Ratio (OR) and 95% Confidence Intervals (95%CI).
Discussion
Main results
Up till now, epidemiology data on ITP have been based on retrospective studies and/or administrative registers.6,19 Here we report for the first time results based on a prospective nationwide cohort of adults with incident ITP studied consecutively, using stringent diagnostic criteria, and followed up for at least one year. This one-year follow-up study provides interesting insights into characteristics of ITP as well as the risk factors predicting which patients will evolve to chronic forms of the disease. First, our results agree with earlier findings that ITP in adults is more frequently found in females, with a female/male ratio of 1.7 overall and a female/male ratio of 2.8 for patients between the 18 and 49 years of age. Both ratios agree with previous results.20,21 Regarding the clinical symptoms of ITP, only 16% of patients with ITP showed no signs of bleeding, a figure that is twice as low as that reported in the PARC-ITP Registry observational study.21 This discrepancy in results may be explained by the procedure used in our study to check diagnosis. In the present study, patients with no bleeding symptoms and no record of a normal platelet count in the preceding year were excluded, thus over-estimating the frequency of bleeding symptoms at diagnosis. It is interesting to note that only 4.9% of patients presented with visceral bleeding at baseline and that no patient had a fatal hemorrhage, thus confirming that ITP is rarely associated with life-threatening bleeding events at diagnosis.22
Second, our results show that a large majority of adults with newly diagnosed ITP are likely to develop a chronic form of the disease after one year. In our sample, only 37% of patients “spontaneously” recovered, i.e. without the need for disease-modifying treatment such as rituximab or splenectomy. This confirms previous data suggesting that ITP is more likely to follow a chronic course in affected adults, compared to children in whom cure is achieved within several weeks in most patients.10
In this context, the ability to predict the course of newly diagnosed ITP in adults is crucial for both patients and physicians, allowing for a potential reduction in healthcare costs, patient anxiety, and less need for aggressive initial treatment, such as splenectomy or immunosuppressant drugs. Our results confirm that sex and age are not risk factors for chronicity.23 Likewise, no association was found between bleeding symptoms at baseline and chronicity. Interestingly, a higher platelet count at baseline was found to be associated with increased odds of chronicity at 12 months, independent of confounders. However, it did not reach the 5% level of significance and further studies are needed to confirm such an association, which was also found when considering all patients recovering from ITP, regardless of disease-modifying treatment. Although of interest, this predictive association is insufficiently robust to be considered for clinical practice or to be taken into account by physicians when tailoring treatment decisions to individual patients. In our opinion, clinicians would only be able to reliably predict progression to chronic status by using a combination of clinical and biological factors. We previously reported that the presence of platelet antibodies detected by a MAIPA assay could help in predicting chronicity in ITP.11 However, the poor sensitivity of this test and its limited availability in many countries restricts its potential use. In the present study, the detection of platelet antibodies using a MAIPA assay was not systematically performed across all the participant hospitals, and, therefore, this measure was not taken into account, and other clinical and biological predictive factors should be identified.
Third, the present findings provide further insight into the frequency and prognostic value of autoimmune markers, particularly ANA at ITP onset, which have so far proved controversial.24,25 ANA has been found to be a possible predictor of chronicity in childhood ITP.25 In adults, this is most likely not the case. In a previous retrospective study focused on adults, we reported that the presence of autoimmune markers did not correlate with presenting features, response to treatments, or long-term outcome of ITP.26 These conflicting results explain why assays for ANA are not routinely recommended by an international panel of experts.27 In our sample, assays for ANA were performed at baseline in nearly all of the participants and the test was found positive (titer>1/80) in more than 25% of patients, a finding that is in line with previous reports.24,25 In patients with a positive ANA titer, the absence of bleeding symptoms was significantly more frequent, and unsurprisingly, the proportion of females was higher than that of males, and the presence of autoimmune disorder history in first-degree relatives was twice as frequent, although this was not significant, probably because of low sample size and insufficient statistical power. Unexpectedly, a positive ANA titer was not associated with ITP evolution. These results suggest that routine ANA assays appear non-mandatory in newly diagnosed ITP in adults. However, taking into account that the follow-up period for our study was limited to one year, we cannot exclude the possibility that the presence of ANA at the time of diagnosis may be a risk factor for late development of systemic lupus erythematosus.
Finally, using a case-control design, we explored whether a family history of autoimmune disorder was a risk factor of developing ITP in adulthood. Our results suggest that this is not the case.
Study limitations
The study setting used the entire network of French reference centers, which are highly specialized centers for ITP. Thus, more severe patients could have been included. However, there is no reason to believe that this selection bias confounded the predictive value of patients’ characteristics or modified the results regarding family history of autoimmune disorder.
Due to strict eligibility criteria for the analyses, the population used for the analyses consisted of 143 out of 188 adult patients initially identified and recruited. The choice to select patients with confirmed diagnosis of primary and incident ITP has consequently lowered the statistical power for some analyses, in particular regarding any association between ANA positivity and patients’ characteristics.
Conclusion
The study confirms that ITP is more common in women than men, and is frequently associated with cutaneous bleeding, even if life-threatening bleeding is rare. Initial treatment is required in more than 80% of patients and only 37% of patients achieve cure at one year of follow up with no need for disease-modifying treatment, indicating that ITP is a chronic disease in adults with a less favorable outcome compared to children. The predictors of chronicity were explored prospectively for the first time in adults, suggesting that higher platelet counts at diagnosis are negatively correlated with the risk of chronicity. However, further research is needed to confirm this finding and potentially to identify other predictive factors for chronicity at the time of diagnosis.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/9/1039
References
- 1.Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009;113(26):6511–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol. 2009;83(2):83–89. [DOI] [PubMed] [Google Scholar]
- 3.Schoonen WM, Kucera G, Coalson J, et al. Epidemiology of immune thrombocytopenic purpura in the General Practice Research Database. Br J Haematol. 2009;145(2):235–244. [DOI] [PubMed] [Google Scholar]
- 4.Michel M. Immune thrombocytopenic purpura: epidemiology and implications for patients. Eur J Haematol Suppl. 2009;(71):3–7. [DOI] [PubMed] [Google Scholar]
- 5.Bennett D, Hodgson ME, Shukla A, Logie JW. Prevalence of diagnosed adult immune thrombocytopenia in the United Kingdom. Adv Ther. 2011;28(12):1096–1104. [DOI] [PubMed] [Google Scholar]
- 6.Moulis G, Palmaro A, Montastruc JL, Godeau B, Lapeyre-Mestre M, Sailler L. Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood. 2014;124(22):3308–3315. [DOI] [PubMed] [Google Scholar]
- 7.Fogarty PF, Segal JB. The epidemiology of immune thrombocytopenic purpura. Curr Opin Hematol. 2007;14(5):515–519. [DOI] [PubMed] [Google Scholar]
- 8.Zeller B, Rajantie J, Hedlund-Treutiger I, et al. ; NOPHO ITP. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94(2):178–184. [DOI] [PubMed] [Google Scholar]
- 9.Bansal D, Bhamare TA, Trehan A, Ahluwalia J, Varma N, Marwaha RK. Outcome of chronic idiopathic thrombocytopenic purpura in children. Blood. 2014;124(22):3295–307. [DOI] [PubMed] [Google Scholar]
- 10.Heitink-Pollé KM, Nijsten J, Boonacker CW, de Haas M, Bruin MC. Clinical and laboratory predictors of chronic immune thrombocytopenia in children: a systematic review and meta-analysis. Blood. 2014;124(22):3295–3307. [DOI] [PubMed] [Google Scholar]
- 11.Grimaldi D, Canouï-Poitrine F, Croisille L, et al. Antiplatelet antibodies detected by the MAIPA assay in newly diagnosed immune thrombocytopenia are associated with chronic outcome and higher risk of bleeding. Ann Hematol. 2014;93(2):309–315. [DOI] [PubMed] [Google Scholar]
- 12.Patel AP. Idiopathic autoimmune thrombocytopenia and neutropenia in siblings. Eur J Haematol. 2002;69(2):120–121. [DOI] [PubMed] [Google Scholar]
- 13.Rischewski JR, Imbach P, Paulussen M, Kühne T. Idiopathic thrombocytopenic purpura (ITP): is there a genetic predisposition? Pediatr Blood Cancer. 2006;47(5 Suppl):678–680. [DOI] [PubMed] [Google Scholar]
- 14.Grimaldi-Bensouda L, Michel M, Aubrun E, et al. ; PGRx Immune Thrombocytopenia Study Group. A case-control study to assess the risk of immune thrombocytopenia associated with vaccines. Blood. 2012;120(25):4938–4944. [DOI] [PubMed] [Google Scholar]
- 15.Grimaldi-Bensouda L, Guillemot D, Godeau B, et al. ; PGRx-AID Study Group. Autoimmune disorders and quadrivalent human papillomavirus vaccination of young female subjects. J Intern Med. 2014;275(4):398–408. [DOI] [PubMed] [Google Scholar]
- 16.Rodeghiero F1, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386–2393. [DOI] [PubMed] [Google Scholar]
- 17.Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295–306. [DOI] [PubMed] [Google Scholar]
- 18.The SAS Institute Inc., SAS software: version 9.3. 2008: Cary, NC, USA. [Google Scholar]
- 19.Vianelli N, Valdrè L, Fiacchini M, et al. Long-term follow-up of idiopathic thrombocytopenic purpura in 310 patients. Haematologica. 2001;86(5):504–509. [PubMed] [Google Scholar]
- 20.Frederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood. 1999; 94(3):909–913. [PubMed] [Google Scholar]
- 21.Kühne T, Berchtold W, Michaels LA, et al. ; Intercontinental Cooperative ITP Study Group. Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica. 2011;96(12):1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michel M, Suzan F, Adoue D, et al. Management of immune thrombocytopenia in adults: a population-based analysis of the French hospital discharge database from 2009 to 2012. Br J Haematol. 2015;170(2):218–222. [DOI] [PubMed] [Google Scholar]
- 23.Andrès E, Mecili M, Fothergill H, Zimmer J, Vogel T, Maloisel F. Gender-related analysis of the clinical presentation, treatment response and outcome in patients with immune thrombocytopenia. Presse Med. 2012;41(9 Pt 1):e426–431. [DOI] [PubMed] [Google Scholar]
- 24.Abbasi SY, Milhem M, Zaru L. A positive antinuclear antibody test predicts for a poor response to initial steroid therapy in adults with idiopathic thrombocytopenic purpura. Ann Hematol. 2008;87(6):459–462. [DOI] [PubMed] [Google Scholar]
- 25.Altintas A, Ozel A, Okur N, et al. Prevalence and clinical significance of elevated antinuclear antibody test in children and adult patients with idiopathic thrombocytopenic purpura. J Thromb Thrombolysis. 2007;24(2):163–168. [DOI] [PubMed] [Google Scholar]
- 26.Vantelon JM, Godeau B, André C, Bierling P. Screening for autoimmune markers is unnecessary during follow-up of adults with autoimmune thrombocytopenic purpura and no autoimmune markers at onset. Thromb Haemost. 2000;83(1):42–45. [PubMed] [Google Scholar]
- 27.Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010; 115(2):168–186. [DOI] [PubMed] [Google Scholar]