

The past two decades of lymphoma research have uncovered the essential role of the B-cell antigen receptor (BCR) pathway in lymphoma biology. This resulted in the development of targeted inhibitors for the treatment of several B-cell malignancies, especially chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), and mantle cell lymphoma (MCL).1 However, response to BCR pathway inhibition is heterogeneous, possibly to entity-dependent varying degrees of BCR involvement in lymphoma biology.1,2 CLL BCRs, for instance, can be activated by complementarity determining region (CDR)-mediated interactions with a variety of autoantigens including BCR-intrinsic self-epitopes.3–5 In MCL, BCR involvement and antigenic interaction is still insufficiently understood. The MCL BCR seems to be antigen-experienced since it shows biased immunoglobulin heavy variable (IGHV) gene usage and suggestive patterns of clonal diversification despite low levels of somatic hypermutation.6,7 In addition, the BCR pathway is active in MCL cells in vitro and in vivo, and inhibiting the partially overexpressed key molecule Syk resulted in the induction of apoptosis in vitro.8,9 Moreover, clinical agents targeting downstream signaling molecules like the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib and the phosphoinositol 3-kinase δ (PI3Kδ) inhibitor idelalisib have shown marked anti-lymphoma activity in MCL patients,2,10 highlighting the importance of the BCR signaling pathway in vivo. However, it is currently unknown to what extent MCL depends on external signals through the BCR and what kind of antigens and epitopes are involved.
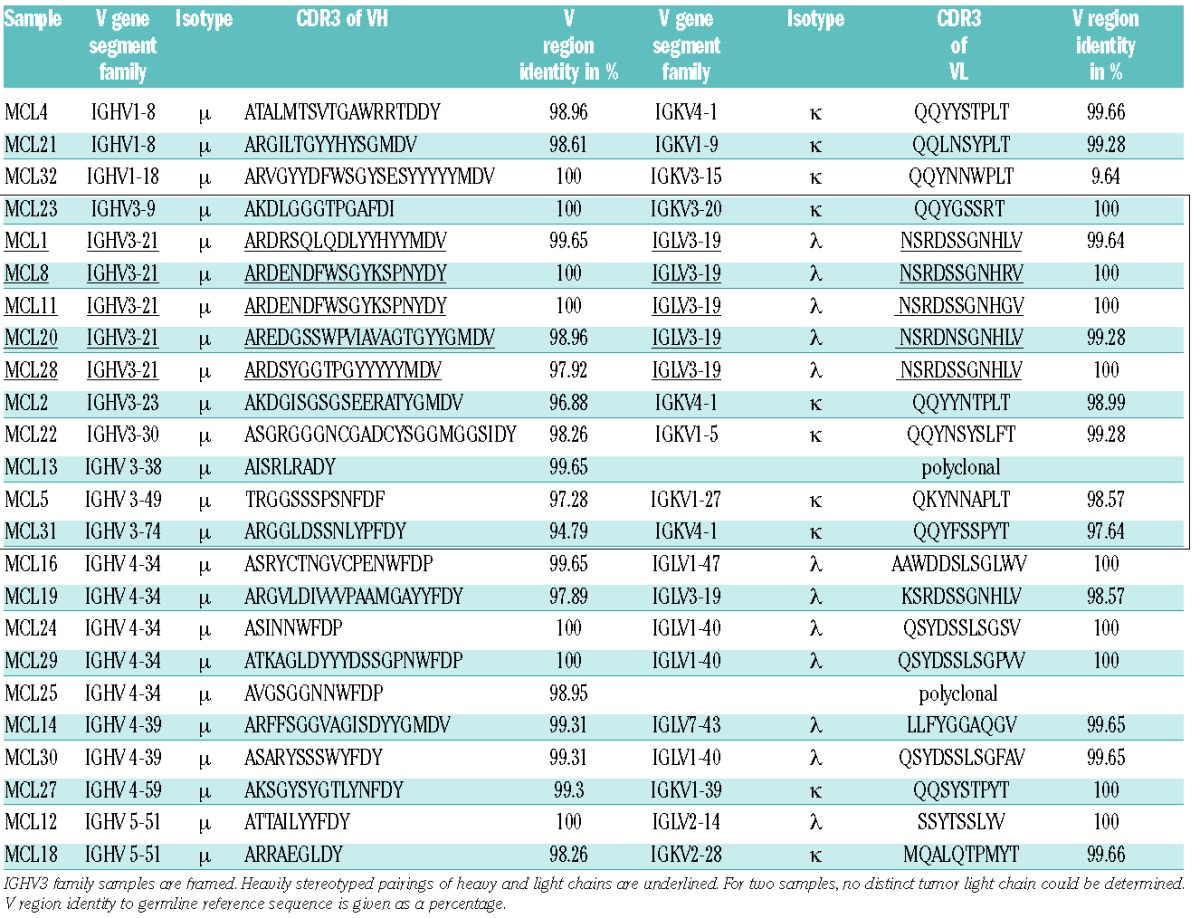
We intended to improve the understanding of BCR-antigen interactions in MCL. We therefore isolated RNA from 24 MCL samples and sequenced the variable heavy and light chain regions of the MCL BCR (Table 1). In line with previous data, the Ig heavy chains were considerably biased towards IGHV3 and IGHV4-34 gene segment usage.6 Together, Igs harboring IGHV3 and IGHV4-34 genes accounted for two-thirds (16/24) of all sequenced samples. All heavy chains were of the μ subtype and, in line with literature, the λ/κ ratio was biased towards the λ-isotype; 12 Igs harbored a λ- and 10 a κ-light chain. The ratio, however, was slightly lower than published for other cohorts (1.2:1 versus 2:1).11 This statistical deviation probably results from the relatively small cohort size. For two samples, no distinct MCL light chain was determined. Remarkably, all BCRs with an IGHV3-21 harbored λ-light chains of the IGLV3-19 family. This is in line with observations by Walsh et al., who proposed this MCL subpopulation as a new MCL entity.12 Within this subset, 4 out of 5 IGHV had similar CDR3 lengths, and in two cases the Igs were nearly identical (Table 1, underlined). The mutational load of the BCRs was very low; about 25% of the cohort expressed completely unmutated Igs, 67% were minimally/borderline mutated, and only two (~8%) BCR-Igs had germline deviations of more than 3%, which is compatible with previous reports.6
Table 1.
Overview of heavy and light chain pairings and CDR3 amino acid sequences of MCL immunoglobulins from different patient samples.
Next, we expressed eleven representative MCL-derived Igs as IgGs and used them for random peptide phage display library screenings. Only 3 out of 11 selections resulted in an enrichment of an epitope-mimicking phage (Online Supplementary Table S2). This low output contradicts findings from a previous work on epitope recognition profiling in CLL performed by our group, which showed strong enrichments of epitope mimics in CLL BCR-derived immunoglobulins known to interact with a number of well-defined auto-antigens.13 Furthermore, we tested the recombinant MCL-derived immunoglobulins for reactivity with autoantigens in a HEp2A-based immunofluorescence assay. We did not detect relevant signals (not shown), which implied that MCL BCRs do not react with potential HEp2A-expressed autoantigens as described for multiple CLL BCRs.3 Together, these data suggest that classical high affinity or CLL-like low affinity autoantigen recognition might not play a significant role in MCL biology in the majority of cases.
The largely unmutated Ig status, together with the difficulty identifying epitope-mimicking peptides, prompted us to search for Ig sequence features indicating CDR-independent antigen binding. Previous studies identified Staphylococcus aureus protein A (SpA) as a potent superantigen for immunoglobulins bearing family 3 heavy chains with a clearly defined binding motif.14,15 Due to the overrepresentation of this family in our cohort, we checked all BCRs harboring an IGHV3 for the existence of this motif. Interestingly, all MCL-, but only half of the control CLL- and FL-derived Ig heavy chains expressing this family presented the unmutated binding motif for the interaction with SpA (Figure 1A). In addition, we analyzed the IGHV3 gene sequences published by Hadzidimitriou et al.6 This dataset consisted of 127 mutated IGHV3-21 and IGHV3-23 genes from MCL patients. Only 31 samples (~24 %) had at least one mutation in the motif, and 9 samples (~7 %) had two or more. Additionally, about 40% of all Igs with the IGHV3-21 gene (IGHV3-23: ~17%) were completely unmutated and also presented the SpA motif. Since a single mutation did not impair the SpA binding abilities, this showed that more than 95 % of the published MCL Igs retained their affinity to SpA.
Figure 1.

IGHV3 expressing MCL BCRs bind to the staphylococcal protein A leading to BCR pathway activation. (A) Amino acid sequence alignment of the Staphylococcus aureus protein A (SpA) binding motif of all IGHV3-containing MCL-BCR- as well as some CLL-BCR- and FL-BCR-derived immunoglobulins. The motif is indicated in the top row. Deviations are highlighted with black background. (B) ELISA with different Fab immunoglobulin fragments derived from MCL, CLL and FL samples to determine their reactivity to SpA. Note that FL10 harbored a highly mutated SpA-motif and is completely inert. (C) Ca2+ flux analysis of Ramos cells expressing MCL-derived membrane-bound immunoglobulins (mIgs). Transduced cells were BCR-stimulated by adding either SpA (bottom row) or an anti-human-Fab antibody (top row). The time of addition is indicated by the gaps in the plots. Non-transduced cells served as control.
This raised the question as to whether SpA might act as a superantigen, triggering activation of the BCR signaling pathway in a substantial proportion of MCL patients.
Therefore, we explored the interaction of SpA with lymphoma BCR-derived immunoglobulins harboring the SpA binding motif. Since SpA has a high affinity to the Fc domain of human IgG, representative MCL Igs were expressed as Fab fragments. Six different MCL- (with and without SpA binding motif), two CLL- and one FL-derived Fab fragments were produced and tested for SpA reactivity using an ELISA with coated SpA. In fact, all Fab fragments exhibiting the SpA binding motif bound to SpA in this assay, whereas Fab fragments without SpA binding motif were non-reactive (Figure 1B).
Having established that SpA binds to a substantial proportion of MCL-derived immunoglobulins in vitro, we next investigated whether this interaction is sufficient to activate the BCR signaling pathway in human B-cells expressing SpA-reactive MCL BCR. We therefore lentivirally transduced IgM-negative Ramos lymphoma cells to constitutively express Ig heavy and light chain genes derived from either an SpA-reactive BCR (MCL11) or a non-SpA-reactive BCR (MCL19) (Online Supplementary Figure S2), treated the cells with soluble SpA, and used mobilization of the key second messenger Ca2+ as readout for BCR activation. While the MCL11-expressing Ramos cells showed Ca2+ flux upon stimulation with either SpA or anti-human-Fab antibodies alike, MCL19-expressing cells responded only to anti-human-Fab treatment but not to SpA (Figure 1C). Of note, the MCL11-derived Fab fragment has an intermediate affinity to SpA (Figure 1B) and, thus, is representative for this class of MCL BCRs.
To verify these results in MCL derived cell lines, we used MAVER-1 and Jeko-1 cells. According to Pighi et al.16 MAVER-1 cells express the unmutated IGHV3-9 gene and therefore present the SpA motif. Jeko-1 cells, which express the IGHV2-70 gene, are motif negative. As expected, only the motif positive MAVER-1 cells showed a Ca2+-flux after stimulation with SpA (Online Supplementary Figure S4). This also proved our Ramos-based cell system to be a reliable and versatile tool for BCR activation experiments.
In addition, we investigated the induction of tyrosine phosphorylation by SpA using the GRB2 SH2 domain as a phosphoprobe.17 In contrast to MCL19-expressing and untransduced Ramos cells, we observed a substantial increase of phosphorylation in MCL11-expressing cells, further demonstrating the activation of BCR downstream signaling by SpA (Online Supplementary Figure S5). These findings proved that SpA is able to crosslink the BCR and activate its downstream signaling cascade.
Taken together, our data indicate that many MCL BCRs do not recognize classical high-affinity antigens via conventional CDR-mediated contacts. Instead, a substantial proportion of MCL cases may be driven by BCR signaling through CDR-independent interactions. SpA might trigger such a scenario in vivo as previous studies have shown that systemically administered SpA can reach even remote sites of the body and may specifically target IGHV3 carrying BCRs.15
Taking into account the fact that up to 20 % of the population is persistently colonized by S. aureus,18 an encounter of B cells with SpA is a likely scenario. One might speculate that at some possibly distinctly vulnerable point in B-cell development, an infection with S. aureus might critically trigger proliferation of a B cell that may have already acquired oncogenic genetic alterations such as the t(11;14) translocation and is therefore predisposed to transformation. It remains open whether, in such a scenario, a persistent infection would be needed to drive tumor progression or if a single initial S. aureus contact might be sufficient to trigger transformation. Potential absence of the antigen in later stages of the disease might also be compensated by SpA-mimicking (auto-)antigenic structures leading to permanent exposition of the transformed B cell to antigenic stimulation. This might be sufficient, even at very low levels, to trigger a response in the genetically or epigenetically altered cell. Future studies will have to elucidate which (genetic or epigenetic) prerequisites are necessary, and at what critical point in time this interaction has to take place to direct the cell towards malignant transformation. Overall, our data indicate a significant role of SpA in MCL biology, and may provide the basis to further elucidate BCR involvement in the genesis or progression of this largely incurable disease.
Acknowledgments
The authors thank Arne Düsedau for the expert support in FACS measurements.
Footnotes
Funding: this work was supported by the German José Carreras Leukemia Foundation (grant R 12/08 to MT) as well as the Margarete Clemens Stiftung (endowed professorship to MT).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hervé M, Xu K, Ng Y-S, et al. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Invest. 2005;115(6):1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iacovelli S, Hug E, Bennardo S, et al. Two types of BCR interactions are positively selected during leukemia development in the Eμ-TCL1 transgenic mouse model of CLL. Blood. 2015;125(10):1578–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minden MD, Übelhart R, Schneider D, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309–312. [DOI] [PubMed] [Google Scholar]
- 6.Hadzidimitriou A, Agathangelidis A, Darzentas N, et al. Is there a role for antigen selection in mantle cell lymphoma¿ Immunogenetic support from a series of 807 cases. Blood. 2011;118(11):3088–3095. [DOI] [PubMed] [Google Scholar]
- 7.Xochelli A, Sutton L-A, Agathangelidis A, et al. Molecular Evidence for Antigen Drive in the Natural History of Mantle Cell Lymphoma. Am J Pathol. 2015;185(6):1740–1748. [DOI] [PubMed] [Google Scholar]
- 8.Pighi C, Gu T-L, Dalai I, et al. Phospho-proteomic analysis of mantle cell lymphoma cells suggests a pro-survival role of B-cell receptor signaling. Cell Oncol Dordr. 2011;34(2):141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol. 2006;132(3):303–316. [DOI] [PubMed] [Google Scholar]
- 10.Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schraders M, Oeschger S, Kluin PM, et al. Hypermutation in mantle cell lymphoma does not indicate a clinical or biological subentity. Mod Pathol. 2009;22(3):416–425. [DOI] [PubMed] [Google Scholar]
- 12.Walsh SH, Thorsélius M, Johnson A, et al. Mutated VH genes and preferential VH3-21 use define new subsets of mantle cell lymphoma. Blood. 2003;101(10):4047–4054. [DOI] [PubMed] [Google Scholar]
- 13.Binder M, Müller F, Jackst A, et al. B-cell receptor epitope recognition correlates with the clinical course of chronic lymphocytic leukemia. Cancer. 2011;117(9):1891–1900. [DOI] [PubMed] [Google Scholar]
- 14.Graille M, Stura EA, Corper AL, et al. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci USA. 2000;97(10):5399–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silverman GJ, Goodyear CS. Confounding B-cell defences: lessons from a staphylococcal superantigen. Nat Rev Immunol. 2006;6(6):465–475. [DOI] [PubMed] [Google Scholar]
- 16.Pighi C, Barbi S, Bertolaso A, Zamò A. Mantle cell lymphoma cell lines show no evident immunoglobulin heavy chain stereotypy but frequent light chain stereotype. Leuk Lymphoma. 2013;54(8):1747–1755. [DOI] [PubMed] [Google Scholar]
- 17.Machida K, Khenkhar M, Nollau P. Deciphering Phosphotyrosine-Dependent Signaling Networks in Cancer by SH2 Profiling. Genes Cancer. 2012;3(5–6):353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowy FD. Staphylococcus aureus Infections. N Engl J Med. 1998;339(8):520–532. [DOI] [PubMed] [Google Scholar]