

Abstract
Recently, we developed 2nd generation backbone degradable N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer-drug conjugates which contain enzymatically cleavable sequences (GFLG) in both polymeric backbone and side-chains. This design allows using polymeric carriers with molecular weights above renal threshold without impairing their biocompatibility, thereby leading to significant improvement in therapeutic efficacy. For example, 2nd generation HPMA copolymer-epirubicin (EPI) conjugates (2P-EPI) demonstrated complete tumor regression in the treatment of mice bearing ovarian carcinoma. To obtain a better understanding of the in vivo fate of this system, we developed a dual-labeling strategy to simultaneously investigate the pharmacokinetics and biodistribution of the polymer carrier and drug EPI. First, we synthesized two different types of dual-radiolabeled conjugates, including 1) 111In-2P-EPI-125I (polymeric carrier 2P was radiolabeled with 111In and drug EPI with 125I), and 2) 125I-2P-EPI-111In (polymeric carrier 2P was radiolabeled with 125I and drug EPI with 111In). Then, we compared the pharmacokinetics and biodistribution of these two dual-labeled conjugates in female nude mice bearing A2780 human ovarian carcinoma. There was no significant difference in the blood circulation between polymeric carrier and payload; the carriers (111In-2P and 125I-2P) showed similar retention of radioactivity in both tumor and major organs except kidney. However, compared to 111In-labeled payload EPI, 125I-labeled EPI showed lower radioactivity in normal organs and tumor at 48 h and 144 h after intravenous administration of conjugates. This may be due to different drug release rates resulting from steric hindrance to the formation of enzyme-substrate complex as indicated by cleavage experiments with lysosomal enzymes (Tritosomes). A slower release rate of EPI(DTPA)111In than EPI(Tyr)125I was observed. It may be also due to in vivo catabolism and subsequent iodine loss as literature reported. Nevertheless, tumor-to-tissue uptake ratios of both radionuclides were comparable, indicating that drug-labeling strategy does not affect the tumor targeting ability of HPMA copolymer conjugates.
Keywords: N-(2-hydroxypropyl)methacrylamide (HPMA), Epirubicin, Polymer-drug conjugates, Dual-radiolabeling, Pharmacokinetics and biodistribution
1. Introduction
Conjugation of anticancer drugs to water-soluble polymers offers a possibility to improve their solubility, decrease adverse effects, modify pharmacokinetics, favorably change their biodistribution, and improve therapeutic efficacy by enhanced permeability and retention (EPR) effect [1-4]. Numerous synthetic polymers have been used such as poly(-ethylene glycol) (PEG) [5,6], [N-(2-hydroxypropyl)methacrylamide] (HPMA) copolymers [7-11], poly(amino acids) [12], polyoxazoline [13], poly(malic acid) [14], etc. Among the various polymeric carriers, the most commonly used is PEG, which has been approved by FDA for clinical application and is commercial available in a wide range of molar masses, end-functionalities as well as different architectures. However, the use of PEG can be problematic in some instances, with recent results indicating PEG-containing therapeutics can elicit complement activation, rapid clearance after repeated injections, and may suffer from peroxidation [15,16]. Moreover, the inability to effectively functionalize the polyether backbone mitigates the utility of many PEG drug delivery systems [17]. HPMA copolymers have comparable biocompatibility and advantages over PEG on non-immunogenicity and established bioconjugation strategies. Its favorable properties have been validated by diverse preclinical and clinical studies [18-22]. Recently, higher molecular weight (Mw) biodegradable HPMA copolymer-drug conjugates have been designed with enzymatically degradable tetrapeptide sequences (GFLG) in both polymer backbone and side chains to prolong plasma circulation and enhance tumor accumulation while preserving biocompatibility [23-28].
The knowledge of the pharmacokinetics and biodistribution of macromolecular therapeutics is a prerequisite for the understanding of the mechanism of their action and ultimate translation into clinical use. The fate of HPMA copolymers after administration to animals has been intensively studied. Initially, drug concentration in various organs was determined by direct extraction of drug from lyophilized tissue samples followed by HPLC analysis or fluorescence assay [29,30]. This approach needs large groups of animals and tedious work; it was replaced by radiolabeling strategy, which has high sensitivity and was widely applied in preclinical studies and clinical investigations [31-33]. Among different radionuclides single-step iodination (124I, 125I and 131I) and two-step radiometal labeling (90Y, 111In and 177Lu, etc.) are often performed. Iodination has been frequently used due to low cost and simple radiochemistry. For example, it was reported that radioiodination of drugs (daunomycin and doxorubicin) was achieved by mixing drug (or conjugate) solution with iodide in an iodogen reaction vial under ambient condition for a few minutes [19,34]. In the majority cases, however, tyrosine moiety was typically incorporated into polymer carrier via copolymerization followed by iodine labeling [35-41]. In these cases, the radioactive signals were correlated to the polymer carrier rather than drug. To examine the circulation and accumulation of real drug molecules, 14C-labeled drug might be an option. Nevertheless, such isotope-labeled drugs are expensive; the synthesis of conjugates will be complicated because 14C has a long half-life and will cause large level of irradiation. Recently, dual-labeling strategies have been developed in which one probe aims to track the polymer carrier, while the other one monitor the fate of drug (modified or model drug) [26, 42-43]. For example, dual-fluorescent conjugates were studied using Fluorescence resonance energy transfer (FRET) as a tool to track chain scission of the conjugates and drug release from the carrier [28], or using noninvasive multispectral optical imaging to real time monitor the distribution and tumor accumulation of polymer carrier and a cleavable model drug [44]. Recently we designed dual-isotope-labeled 2nd generation HPMA copolymer-drug model conjugate, whose HPMA copolymer backbone was labeled with 125I, whereas 111In-DTPA complex was bound at GFLG side-chain termini and served as the drug model [26].
We have reported the pharmacokinetics and therapeutic efficacy of 2nd generation diblock HPMA copolymer-epirubicin (EPI) conjugates (2P-EPI) in the treatment of experimental ovarian cancer [28]. Notably, treatment with 2P-EPI resulted in complete tumor remission and long-term inhibition of tumorigenesis (>100 days), whereas the tumor recurrence was observed in mice treated with the 1st generation HPMA copolymer-EPI conjugate (P-EPI, with Mw < 50 kDa). To demonstrate the different pharmacologies between these two generations of conjugates, we designed and synthesized a series of dual radiolabeled HPMA copolymer-EPI conjugates in which 125I and 111In were used to label polymer carrier and drug (EPI), respectively, or vice versa. This paper is devoted to the study of pharmacokinetics and biodistribution of 2nd generation HPMA copolymer-EPI conjugates in nude mice. In one approach, we labeled the polymer carrier with 125I and used DTPA-111In to modify the EPI structure. In the second design, we used DTPA-111In to label the polymer carrier and 125I for drug modification. The issues we addressed are: A) How does modification of the conjugate structure influence its fate? Is there a difference between the two labeling designs? B) How does the modification of drug structure impact the formation of the enzyme-substrate complex, rate of enzymatic drug release and the biodistribution of the drug? C) How does the data obtained differ from the behavior of the unlabeled conjugate that would be used as the macromolecular therapeutics?
2. Experimental section
2.1. Abbreviations
| APMA | N-(3-aminopropyl)methacrylamide hydrochloride |
| Boc-GFLG-OMe | methyl9-benzyl-2,2-dimethyl-4,7,10,13-tetraoxo-12-propyl-3-oxa-5,8,11,14-tetraazahexadecan-16-oate |
| Boc-GFLG-NH2 | tert-butyl(14-amino-4-benzyl-7-isobutyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazatetradecyl)carbamate |
| CTA | chain transfer agent (4-cyanopentanoic acid dithiobenzoate) |
| CTA-GFLG-CTA | 10-benzyl-2,25-dicyano-13-isobutyl-5,8,11,14,17,22-hexaoxo-6,9,12,15,18,21-hexaazahexacosane-2,25-diyl dibenzodithioate |
| DCC | N,N′-dicyclohexylcarbodiimide |
| DMAP | 4-(dimethylamino) pyridine |
| EPI | epirubicin |
| Fmoc-Abu(N3)-OH | (S)-2-(Fmoc-amino)-4-azidobutanoic acid |
| HPMA | N-(2-hydroxypropyl)methacrylamide |
| HATU | 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxidhexafluorophosphate |
| HOBt | 1-hydroxybenzotriazole |
| 111In-P-EPI-125I | frist generation HPMA copolymer-EPI conjugate with 111In-DTPA labeled polymer backbone and 125I-Tyr labeled EPI |
| 111In-2P-EPI-125I | second generation HPMA copolymer-EPI conjugate with 111In-DTPA labeled polymer backbone and 125I-Tyr labeled EPI |
| 125I-2P-EPI-111In | second generation HPMA copolymer-EPI conjugate with 125I-Tyr labeled polymer backbone and 111In-DTPA labeled EPI |
| MA-Tyr-NH2 | N-methacryloyltyrosinamide |
| MA-GFLG-OH | N-methacryloylglycylphenylalanylleucylglycine |
| MA-GFLG-Abu(N3)-OH |
N-(2-((4-azido-1-(((2S,3R,4S,6R)-3-hydroxy-2-methyl-6-((3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl)oxy) tetrahydro-2H-pyran-4-yl)amino)-1-oxobutan-2-yl)amino)-2-oxoethyl)-2-(2-(2-methacrylamidoacetamido)-3-phenylpropanamido)-4-methylpentanamide |
| MA-GFLG-Abu(N3)-EPI |
N-(2-((4-azido-1-(((2S,3R,4S,6R)-3-hydroxy-2-methyl-6-((3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl) oxy)tetrahydro-2H-pyran-4-yl)amino)-1-oxobutan-2-yl)amino)-2-oxoethyl)-2-(2-(2-methacrylamidoacetamido)-3-phenylpropanamido)-4-methylpentanamide |
| MA-GFLG-Abu(Tyr)-EPI | 2-(4-(1-(12-benzyl-3-(((2S,3R,4S,6R)-3-hydroxy-2-methyl-6-((3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6, 11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl)oxy)tetrahydro-2H-pyran-4-yl)carbamoyl)-9-isobutyl-18-methyl-5, 8,11,14,17-pentaoxo-4,7,10,13,16-pentaazanonadec-18-en-1-yl)-1H-1,2,3-triazol-5-yl)butanamido)-3-(4-hydroxyphenyl)propanoic acid |
| MA-GFLG-NHBoc | tert-butyl(11-benzyl-8-isobutyl-17-methyl-4,7,10,13,16-pentaoxo-3,6,9,12,15-pentaazaoctadec-17-en-1-yl)carbamate |
| MA-GG-EPI |
N-(2-((2-(((2S,3R,4S,6R)-3-hydroxy-2-methyl-6-((3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6, 11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl)oxy)tetrahydro-2H-pyran-4-yl)amino)-2-oxoethyl)amino)-2-oxoethyl)methacrylamide |
| NH2-GFLG-NH2 | 2-(2-(2-aminoacetamido)-3-phenylpropanamido)-N-(2-((2-aminoethyl)amino)-2-oxoethyl)-4-methylpentanamide |
| p-SCN-Bn-DTPA | 2,2′-((1-((2-((2-carboxyethyl)(carboxymethyl)amino)ethyl)(carboxymethyl)amino)-3-(4-thiocyanatophenyl)propan-2-yl)azanediyl)diacetic acid |
| P-EPI | first generation HPMA and MA-GFLG-EPI copolymer conjugate |
| P-EPI-Tyr | first generation HPMA and MA-GFLG-Abu(Tyr)-EPI copolymer conjugate |
| P-EPI-N3 | first generation HPMA, APMA and MA-GFLG-Abu(N3)-EPI copolymer conjugate |
| P-DTPA-EPI(Tyr) | first generation HPMA copolymer-EPI conjugate with DTPA pendent on polymer backbone and tyrosine moiety attachment with EPI |
| 2P-EPI-N3 | second generation HPMA, APMA and MA-GFLG-Abu(N3)-EPI copolymer conjugate |
| 2P-DTPA-EPI(Tyr) | second generation HPMA copolymer-EPI conjugate with DTPA pendent on polymer back bone and tyrosine moiety attachment with EPI |
| 2P-Tyr-EPI-N3 | second generation HPMA, MA-Tyr-NH2 and MA-GFLG-Abu(N3)-EPI copolymer conjugate |
| 2P-Tyr-EPI(DTPA) | second generation HPMA copolymer-EPI conjugate with tyrosine moiety pendent on polymer backbone and DTPA attachment with EPI |
| RAFT | reversible addition-fragmentation chain transfer |
| TBTA | tris(benzyltriazolylmethyl)amine |
| TFA | trifluoroacetic acid |
| Hexynoic-Tyr | 2-(hex-5-ynamido)-3-(4-hydroxyphenyl)propanoic acid |
| V-65 | 2,2′-azobis(2,4-dimethylvaleronitrile) |
| V-501 | 4,4-azobis (4-cyanopentanoic acid) |
| VA-044 | 2,2′-azobis[2-(2-imidazolin-2-yl) propane]dihydrochloride |
2.2. Materials
Common reagents were purchased from Sigma-Aldrich and used as received unless otherwise specified. 2,2′-Azobis(2,4-dimethylvaleronitrile) (V-65), 2,2′-azobis[2-(2-imidazolin-2-yl) propane]dihydrochloride (VA-044) were from Wako; 4,4-azobis (4-cyanopentanoic acid) (V-501) was from Fisher Scientific. N,N′-Dicyclohexylcarbodiimide (DCC), Fmoc-Abu(N3)-OH (S)-2-(Fmoc-amino)-4-azidobutanoic acid and 4-(dimethylamino) pyridine (DMAP) were obtained from AAPPTEC. Iodine-125 [125I] was obtained from Perkin-Elmer. 111InCl3 was from Intermountain Radiopharmacy. 1-Hydroxybenzotriazole (HOBt) and N-Boc-ethylenediamine were purchased from AnaSpec. p-SCN-Bn-DTPA was purchased from Macrocyclics. Reversible addition-fragmentation chain transfer (RAFT) agent, 4-cyanopentanoic acid dithiobenzoate (chain transfer agent, CTA), was synthesized according to the literature [45].
2.3. Methods
2.3.1. Synthesis of monomers
N-(2-Hydroxypropyl)methacrylamide (HPMA) was synthesized by acylating 1-aminopropan-2-ol with methacryloyl chloride in acetonitrile, as previously described [46]. Melting point: 69–71 °C; N-methacryloyltyrosinamide (MA-Tyr-NH2) [47], N-methacryloylglycylphenylalanylleucylglycine (MA-GFLG-OH), Boc-GFLG-OMe [48] and MA-GG-EPI [49] were synthesized as previously described.
2.3.1.1. Synthesis of Hexynoic-Tyr
Fmoc-Tyr(Bu)-OH (230 mg, 0.5 eq.) and DIPEA (313 μL, 4 eq.) were dissolved in 9 mL dry dichloromethane (DCM). The solution was added to the resin and shook for 2 h at room temperature (r.t.). The solution was drained and washed with 5 × DCM/MeOH/DIPEA (17/2/1, 9 mL), 3 × DCM and 3 × DMF. Kaiser Test showed a negative result. Piperidine/DMF (1/4, 9 mL) was added and shook for 5 min, a positive Kaiser Test result showed by dark blue solution and beads. Hex-5-ynoic acid (140 mg, 132 μL, 2.5 eq.), DIPEA (391 μL, 5 eq.) and HATU (456 mg, 2.4 eq.) in 5 mL DMF were added and shook for about 2 h. The solution was drained and washed with 3 × DMF, 3 × DCM and 3 × MeOH. TFA/DCM (3/7, 9 mL) was added and shook for 2 h. After filtration and removing the solvent, 97 mg of product was obtained (35%) as yellow oil. 1H NMR (300 MHz, CD3OD) δ 1.67–1.74 (m, 2H), 2.09–2.11 (m, 2H), 2.2 (t, 1H, J = 2.1), 2.26–2.29 (m, 2H), 2.83 (dd, 1H, J = 10.5, 6.9), 3.11 (dd, 1H, J = 10.5, 6.9), 4.59 (dd, 1H, J = 6.9, 3.6), 6.68–6.70 (m, 2H), 7.03 (dd, 2H, J = 4.3, 1.5).
2.3.1.2. Synthesis of MA-GLFG-Abu(N3)-OH
Fmoc-Abu(N3)-OH (73 mg, 0.5 eq.) and DIPEA (139 μL, 4 eq.) were dissolved in 4 mL dry DCM. The solution was added to the resin and shook for 2 h at r.t. The solution was drained and the resin was washed with 5 × DCM/MeOH/DIPEA (17/2/1, 6 mL), 3 × DCM and 3 × DMF. Kaiser Test was negative. The solvent was drained and piperidine/DMF (1/4, 2.5 mL) was added. After shaking for 5 min, a positive Kaiser Test was obtained. MA-GFLG-OH (230 mg, 2.5 eq.), DIPEA (208 μL, 5 eq.) and HBTU (182 mg, 2.4 eq.) in 2 mL DMF were added and shook for 2 h. The solution was drained and washed with 3 × DMF, 3 × DCM, 3 × MeOH. TFE/DCM (3/7, 4 mL) was added and shook for 2 h. After filtration and removing the solvent, 95 mg of product was obtained by precipitation in ethyl ether (81%) as white powder.
2.3.1.3. Synthesis of MA-GFLG-Abu(N3)-EPI
Under N2 atmosphere, MA-GFLG-Abu(N3)-OH (30 mg, 0.05 mmol), DIPEA (23 μL, 0.13 mmol) and HATU (19 mg, 0.05 mmol) were dissolved in 400 μL DMF. 2 min later, EPI (27 mg, 0.05 mmol) was added and the solution was stirred at r.t. for 1 h. Ethyl acetate (100 mL) was added and the solution was washed with NaHCO3 (0.1 M, 50 mL), HCl (0.1 M, 50 mL) and NaCl (sat., 50 mL) three times, respectively. The organic layer was dried with anhydrous MgSO4 and the solvent was removed under vacuum. The residue was precipitated into ethyl ether to yield 30 mg of product as red powder (yield 54%). The structure and purity of the product were confirmed by MALDI-TOF-MS and HPLC (Agilent ZORBAX, 5 μm, 300SB-C18 column 4.6 × 250 mm, using flow rate 1.0 mL/min and gradient elution from 2% to 90% of buffer B within 30 min. Buffer A: DI H2O with 0.1% TFA, Buffer B: acetonitrile with 0.1% TFA) analysis. MS (MALDI-TOF) m/z: 1134.46 [M + Na]+, 1150.43 [M + K]+.
2.3.1.4. Synthesis of MA-GFLG-Abu(Tyr)-EPI
MA-GFLG-Abu(N3)-EPI (36 mg, 32 μmol) was dissolved in 600 μL DMF and purged with N2 for 20 min. Hexynoic-Tyr (40 mg, 145 μmol), CuBr (4.5 mg, 32 μmol) and tris(benzyltriazolylmethyl)amine (TBTA) (15 mg, 32 μmol) were dissolved in 400 μL DMF (purged with N2 for 20 min) and added into the MA-GFLG-Abu(N3)-EPI solution. The mixture was stirred at r.t. and purged with N2 for 20 min. Ascorbic acid (100 mg/mL × 54 μL, in H2O purged with N2 for 20 min) was added and stirred at room temperature overnight. After working up, added 150 mL of ethyl acetate and washed with HCl (2 × 50 mL, 0.5 M). The aqueous phase turned red after washing with NaHCO3 (sat., 50 mL). The pH value was adjusted to about 1 with HCl (1 M) and the aqueous layer was extracted with ethyl acetate (3 × 50 mL). The organic phase was combined and dried with anhydrous MgSO4. The solvent was removed by rotary evaporation below 35 °C, the residue was precipitated into ethyl ether to yield 19 mg of product as red powder (yield 42%). The structure and purity of the product were confirmed by MALDI-TOF-MS and HPLC analysis. MS (MALDI-TOF) m/z: 1409.60 [M + Na]+, 1425.59 [M + K]+.
2.3.2. Synthesis of diarm chain transfer agent CTA-GFLG-CTA (Fig. S3)
Boc-GFLG-OMe (505 mg, 1 mmol) was dissolved in CH3OH (5 mL), NH2(CH2)2NH2 (647 μL, 10 mmol) was added and stirred at r.t. for 3 h. After working up, the solvent and part of NH2(CH2)2NH2 were removed by rotary evaporation below 35 °C; then 150 mL ethyl acetate were added. After washing with H2O (3 × 50 mL) and NaCl (sat. 3 × 50 mL), the organic phase was dried with MgSO4, the solvent was removed by rotary evaporation to yield 361 mg of product as white solid (Boc-GFLG-NH2, 68%).
Boc-GFLG-NH2 (390 mg, 0.73 mmol) was dissolved in DCM (10 mL), 4 mL TFA was added and stirred at r.t. for 4 h and at 4 °C overnight. After removing the solvent, the product was dried under reduced pressure to yield NH2-GFLG-NH2·2TFA as white solid (315 mg, 98%). CTA (217 mg, 0.78 mmol), DCC (167 mg, 0.8 mmol) and HOBt (105 mg, 0.78 mmol) were dissolved in DMF/acetonitrile (2 mL/10 mL). The solution was stirred for 30 min at r.t. NH2-GFLG-NH2·2TFA (173 mg, 0.26 mmol) was dissolved in 5 mL acetonitrile with 104 μL DIPEA (0.6 mmol). After 30 min, the solution (containing NH2-GFLG-NH2) was added to the CTA solution and stirred at room temperature overnight. After working up, filtered and removed solvent, 600 mL ethyl acetate was added and washed with HCl (0.5 M, 3 × 100 mL), NaHCO3 (sat., 3 × 100 mL) and NaCl (sat. 1 × 100 mL). The organic phase was combined and dried by MgSO4, the solvent was removed by rotary evaporation below 35 °C. The residue was purified by silica gel column chromatography (acetone: petroleum ether = 1: 5–5: 1) to yield CTA-GFLG-CTA as pink solid (260 mg, 50%). The purity and structure of CTA-GFLG-CTA were confirmed by HPLC, NMR and MALDI-TOF-MS analysis (Fig. S9). 1H NMR (400 MHz, CD3OD) δ 0.71–0.79 (m, 3H), 0.88–0.92 (m, 3H), 1.43–1.48 (m, 1H), 1.50–1.68 (m, 1H), 1.93 (s, 6H), 2.36–2.43 (m, 2H), 2.51–2.59(m, 6H), 2.93–3.03 (m, 2H), 3.16–3.23 (m, 1H), 3.70–3.91 (m, 4H), 4.07–4.20 (m, 1H), 4.45–4.86 (m, 1H), 7.22–7.26 (m, 5H), 7.44–7.44 (m, 4H), 7.59 (s, 2H), 7.91–7,92 (m, 4H). MS (MALDI-TOF) m/z: 957.35 [M + H]+, 979.33 [M + Na]+.
2.3.3. Synthesis of HPMA copolymer-EPI conjugates
2.3.3.1. Synthesis of first generation HPMA copolymer-EPI conjugate P-DTPA-EPI(Tyr)
2.3.3.1.1. Synthesis of P-EPI-N3
An ampoule containing HPMA (92 mg, 0.69 mmol, 92%), APMA (5.1 mg, 0.027 mmol, 4%) and MA-GFLG-Abu(N3)-EPI (30 mg, 0.027 mmol, 4%) were attached to the Schlenk-line. After three vacuum-nitrogen cycles to remove oxygen, 150 μL degassed MeOH was added and stirred at r.t. CTA (2.3 mg/mL × 150 μL, 0.000622 mmol, in degassed MeOH) and VA-044 (1 mg/mL × 145 μL, 0.000201 mmol, in degassed H2O) were added via syringe under magnetic stirring and bubbled with N2 for 10 min in ice bath. The ampoule was sealed, and polymerization was performed at 40 °C for 22 h. The copolymer was obtained by precipitation into acetone/ethyl ether (3:1) and purified by redissolving in methanol and precipitation in acetone/ethyl ether (3:1) two more times. The copolymer was isolated as red powder and dried under vacuum. Yield: 24 mg (19%).
The average molecular weight (Mw) and the polydispersity (PDI) of the conjugates were determined by size-exclusion chromatography (SEC) on an AKTA FPLC system equipped with a UV detector (GE Healthcare), mini DAWN TREOS, and Optilab rEX (refractive index) detector (Wyatt Technology) using a Superose 6 HR10/30 column with sodium acetate buffer containing 30% (vol/vol) acetonitrile (pH 6.5) as mobile phase. HPMA homopolymer fractions were used as molecular weight standards.
The red powder (24 mg) was further reacted with V-65 (8.4 mg, 0.034 mmol, over 40-times excess with respect to the polymer end groups) in 0.3 mL MeOH at 55 °C for 2 h, purified by precipitation into acetone/ethyl ether (3:1) twice, resulting in intermediate P-EPI-N3. The polymer was analyzed by ninhydrin assay [50] to determine amino content. The EPI content of copolymers was determined spectrophotometrically with free EPI standard working curve. ([NH2]: 216 nmol/mg polymer; [EPI]: 10.5 wt%).
2.3.3.1.2. Attachment of tyrosine moiety and DTPA chelator
The polymer precursor (P-EPI-N3, 23 mg) was dissolved in 200 μL DMF and purged with N2 for 20 min. Hexynoic-Tyr (3 eq.), CuBr (1 eq.) and TBTA (1 eq.) were dissolved in 200 μL DMF (purged with N2 for 20 min) and added into the polymer precursor solution. The mixture was stirred at room temperature and purged with N2 for 20 min. Ascorbic acid (10 eq., in H2O purged with N2 for 20 min) was added and the mixture was stirred at room temperature overnight. After purification by ultrafiltration (10,000 Da cut-off, Millipore) with DI H2O three times and freeze drying, the product was obtained as red powder (16 mg, 71%). The red powder (15 mg) was dissolved in 400 μL NaHCO3 buffer (0.2 M, containing 10 mM EDTA, pH 8.5) and mixed with p-SCN-Bn-DTPA (10 mg) in 200 μL DMSO. After stirring at room temperature overnight, the sample was applied to a pre-equilibrated PD-10 Sephadex G25 column (GE Healthcare) with DI H2O for primary purification. The fraction of 2.5–4.5 mL was collected and further purified by ultrafiltration (10,000 Da cut-off) with NaHCO3 buffer three times and DI H2O three times. The final product in DI H2O was freeze dried to remove solvent (13 mg, P-DTPA-EPI(Tyr), 87%). The DTPA content was determined using spectrophotometric method [51]. The content of DTPA in P-DTPA-EPI(Tyr) was calculated as 140 nmol/mg polymer. The conversion was 64%.
2.3.3.2. Synthesis of second generation HPMA copolymer-EPI conjugate 2P-DTPA-EPI(Tyr)
2.3.3.2.1. Synthesis of 2P-EPI-N3
An ampoule containing HPMA (80 mg, 0.56 mmol, 93%), APMA (3.4 mg, 0.02 mmol, 4%) and MA-GFLG-Abu(N3)-EPI (20 mg, 0.018 mmol, 3%) was attached to the Schlenk-line. After three vacuum-nitrogen cycles to remove oxygen, 136 μL degassed MeOH was added and the reaction mixture stirred at r.t. CTA-GFLG-CTA (10 mg/mL × 38 μL, 0.0004 mmol, in degassed MeOH) and VA-044 (2 mg/mL × 36 μL, 0.000223 mmol, in degassed H2O) were added via syringe under magnetic stirring and bubbled with N2 for 10 min in ice bath. The ampoule was sealed, and polymerization was performed at 40 °C for 44 h. The copolymer was obtained by precipitation into acetone/ethyl ether (3:1) and purified by redissolving in methanol and precipitation in acetone/ethyl ether (3:1) two more times. The copolymer was isolated as red powder and dried under vacuum. Yield: 35 mg (34%). The red powder (35 mg) was further reacted with V-65 (5.3 mg, 0.034 mmol, over 40-times excess with respect to the polymer end groups) in 0.4 mL MeOH at 55 °C for 2 h, purified by precipitation into acetone/ether (3:1) twice, resulting in intermediate 2P-EPI-N3. EPI and amino group content were determined by the same methods as P-EPI-N3 (see Section 2.3.3.1.1). ([EPI]: 9.7 wt%; [NH2]: 320 nmol/mg polymer).
2.3.3.2.2. Attachment of tyrosine moiety and DTPA chelator
The synthetic method was the same as in Section 2.3.3.1.2. Obtained 15 mg of final product (yield 87%). The content of DTPA in 2P-DTPA-EPI(Tyr) was determined as 83 nmol/mg polymer.
2.3.3.3. Synthesis of second generation conjugate 2P-Tyr-EPI-DTPA
2.3.3.3.1. Synthesis of 2P-Tyr-EPI-N3
An ampoule containing HPMA (80 mg, 0.56 mmol, 94.8%), MA-Tyr-NH2 (3 mg, 0.013 mmol, 2.2%) and MA-GFLG-Abu(N3)-EPI (20 mg, 0.018 mmol, 3%) was attached to the Schlenk-line. After three vacuum-nitrogen cycles to remove oxygen, 46 μL degassed MeOH was added and the mixture was stirred at r.t. CTA-GFLG-CTA (3 mg/mL × 128 μL, 0.4 μmol, in degassed MeOH) and VA-044 (2 mg/mL × 36 μL, 0.223 μmol, in de-gassed H2O) were added via syringe under magnetic stirring and bubbled with N2 for 10 min in ice bath. The ampoule was sealed, and polymerization was performed at 40 °C for 44 h. The copolymer was obtained by precipitation into acetone/ethyl ether (3:1) and purified by redissolving in methanol and precipitation in acetone/ethyl ether (3:1) two more times. The copolymer was isolated as red powder and dried under vacuum. Yield: 40 mg (39%). The red powder (40 mg) was further reacted with V-65 (5.3 mg, 0.034 mmol, over 40-times excess with respect to the polymer end groups) in 0.4 mL MeOH at 55 °C for 2 h, purified by precipitation into acetone/ethyl ether (3:1) twice, resulting in 2P-Tyr-EPI-N3. The EPI content was 5.5 wt%.
2.3.3.3.2. Attachment of DTPA chelator
2P-Tyr-EPI-N3 (30 mg, 1 eq.), propargylamine (1.2 mg, 3 eq.), CuBr (1.0 mg, 1 eq.) and TBTA (3.0 mg, 1 eq.) were dissolved in 400 μL DMF and purged with N2 for 20 min. The mixture was stirred at room temperature and purged with N2 for 20 min. Ascorbic acid (100 mg/mL × 12 μL, in H2O which was purged with N2 for 20 min) was added and stirred at room temperature overnight. After working up, the reaction mixture was purified by ultrafiltration (30,000 Da cut-off) with DI H2O three times. The remnant was precipitated in 14 mL acetone/ethyl ether (4:1) twice. 24 mg of red powder was obtained (73%). Amino content was determined as 75 nmol/mg polymer. Red powder (20 mg) was dissolved in 0.8 mL of NaHCO3 (0.2 M) containing 10 mM EDTA (pH 8.5), then mixed with p-SCN-Bn-DTPA (10 mg) in 200 μL of DMSO. After stirring at room temperature overnight, the sample was applied to a pre-equilibrated PD-10 Sephadex G25 column (GE Healthcare) with DI H2O for primary purification. The fraction of 2.5–4.5 mL was collected and further purified by ultrafiltration (30,000 Da cut-off) with NaHCO3 buffer three times and DI H2O three times. The final sample in DI H2O was freeze dried to remove solvent (16 mg, 80%). The content of DTPA was determined spectrophotometrically (16 nmol/mg) [51]. The conversion was 22%.
2.3.3.4. Synthesis of conjugates P-EPI and P-EPI(Tyr) for Tritosomes cleavage kinetic study
An ampoule containing 50 mg (or 73 mg) of HPMA and 10 mg of MA-GFLG-EPI (or 7 mg MA-GFLG-Abu(Tyr)-EPI) was attached to the Schlenk-line. After three vacuum-nitrogen cycles to remove oxygen, 100 μL (or 120 μL) degassed MeOH was added and stirred at r.t. CTA, 4 mg/mL × 50 μL (or 2 mg/mL × 70 μL) in degassed MeOH and 2 mg/mL × 39 μL (or 2 mg/mL × 27 μL) of VA-044 in degassed MeOH were added via syringe under magnetic stirring and bubbled with N2 for 10 min in ice bath. The ampoule was sealed, and polymerization was performed at 44 °C for 22 h. The copolymer was obtained by precipitation into acetone/ethyl ether (3:1) and purified by redissolving in methanol and precipitation in acetone/ethyl ether (3:1) two more times. The copolymer was isolated as red powder and dried under vacuum. Yield: 47 mg of P-EPI (57%) and 26 mg of P-EPI(Tyr) (46%). After end-modification with 40-times excess of V-65, the EPI content of copolymers was determined spectrophotometrically as 86 nmol/mg polymer for P-EPI and 120 nmol/mg polymer for P-EPI(Tyr).
2.3.4. Radiolabeling
125I labeling of tyrosine moiety and 111In labeling of DTPA were conducted immediately before use by the method as previously described [26]. For single labeling of 125I, HPMA copolymer-EPI conjugates, containing tyrosine amide in the side chains or pendant on the backbones, were reacted with Na125I (Perkin-Elmer) at r.t. in 0.01 M phosphate buffer containing chloramine-T for 30 min. After purification with ultrafiltration (10,000 Da cut-off tube for first generation conjugate and 30,000 Da cut-off for second generation conjugates), the copolymer with 125I labeling in 0.1 M sodium acetate solution (pH 5.2) was mixed with an aqueous solution of 111InCl3 at r.t. for 30 min and then purified by ultrafiltration (10,000 Da cut-off for first generation conjugate and 30,000 Da cut-off for second generation conjugates) to produce dual radiolabeled conjugates.
2.3.5. Tritosomes cleavage of P-EPI, P-EPI(Tyr)
2.3.5.1. Tritosomes preparation and activity toward Bz-Phe-Val-Arg-NAp substrate
Rat liver Tritosomes were prepared according to the method of Trouet [52]. Briefly, 20% Triton WR-1339 in NaCl (0.15 M) solution was intraperitoneally injected into rats (1 mL per 100 g of rat weight). After 4 days, the rats were sacrificed, liver isolated and placed into 0.25 M sucrose solution at 4 °C. The liver was diced into little pieces and pushed through a strainer. The mixture was homogenized gently and centrifuged at 2400 rpm for 10 min at 4 °C. The supernatants were collected and the pellet was washed with 0.25 M sucrose solution and centrifuged at 2400 rpm for 10 min at 4 °C. The combined supernatants were centrifuged at 22,700 rpm for 10 min at 4 °C. The supernatant and the pink fluffy layer were removed. The pellets were centrifuged at 22,700 rpm for 10 min at 4 °C and the supernatant and pink fluffy layer were removed again. The pellets and 45% sucrose solution were thoroughly mixed gently. A 34.5% w/w sucrose solution was added on the top of the 45% sucrose/pellet mixture and 14.3% w/w sucrose solution was added on the top of 34.5% sucrose solution. The mixture was centrifuged at 24,000 rpm for 2 h at 4 °C. The lysosomal fraction (Tritosomes) was the layer between the 34.5% and 14.3% sucrose solutions.
2.3.5.2. Tritosomes activity toward Bz-Phe-Val-Arg-NAp substrate
Tritosomes (0.1 mL) were mixed with 0.84 mL sodium phosphate buffer (213 mM sodium phosphate, 44 mM citrate, pH 5.5), 0.02 mL glutathione (0.25 M) in sodium phosphate buffer and 0.02 mL 10% Triton-X-100 in sodium phosphate buffer. The mixture was incubated at 37 °C for 5 min. The substrate (Bz-Phe-Val-Arg-NAp) solution (0.00102 g in 0.138 mL DMSO) was added and incubated at 37 °C for 15 min. The absorption of cleaved p-nitroaniline was measured at 410 nm. Within 10 min, 4.7% of p-nitroaniline was released from Bz-Phe-Val-Arg-NAp.
2.3.5.3. Tritosomes cleavage of P-EPI, P-EPI(Tyr)
Tritosomes (0.48 mL) were mixed with 0.40 mL buffer B-GSH (27 mM citrate, 20 mM sodium phosphate, 2 mM EDTA, 5 mM glutathione, pH 4.6) and 0.02 mL 10% Triton-X-100 (0.1 g/mL in B-GSH buffer). After incubation for 5 min at 37 °C, conjugate (35 mg/mL × 0.1 mL of P-EPI or 25 mg/mL × 0.1 mL of P-EPI(Tyr) in H2O) was added into the mixture and incubated at 37 °C. At predetermined time intervals samples (0.1 mL) were withdrawn, added 0.05 mL of iodoacetic acid sodium (1 mg/mL in H2O, inhibitor) and 0.05 mL of daunomycin (0.1 mg/mL in H2O, internal standard for P-EPI and P-EPI(Tyr)). After centrifugation for 5 min (13,500 rpm), 80 μL of supernatant was analyzed by HPLC. Gradient elution: 20% B to 35% B in 25 min, 35% B to 60% B in 15 min, 60% B to 90% B in 15 min for P-EPI and P-EPI(Tyr). Time intervals: 1 min, 1, 2, 4, 6, 8, 23, and 48 h.
2.3.6. Cleavage of 2P-DTPA-EPI(Tyr) and 2P-Tyr-EPI(DTPA) by Tritosomes
Tritosomes (200 μL) were buffer changed with phosphate buffer-A (200 mM, 0.2% Triton-X-100, EDTA 2 mM, pH 5.5) and mixed with 5 μL of glutathione (100 mM). After incubation at 37 °C for 5 min, 2P-DTPA-EPI(Tyr) or 2P-Tyr-EPI(DTPA), 50 mg/mL × 10 μL in H2O) was added and incubated at 37 °C. After 45 h, to 200 μL of reaction mixture was added 20 μL of iodoacetic acid sodium (2 mg/mL in H2O, inhibitor) and daunomycin (2 mg/mL × 4 μL in MeOH; internal standard for 2P-DTPA-EPI(Tyr) and 2P-Tyr-EPI(DTPA)). The mixture was centrifuged for 5 min (13,500 rpm). The supernatant was analyzed by HPLC. Tritosomes cleavage products were collected and determined by MALDI-TOF-MS. The Tritosomes cleavage products of 2P-DTPA-EPI(Tyr) include [G-EPI-Tyr + Na]+,1024.39 and [EPI-Tyr + Na]+, 967.37. The Tritosomes cleavage products of 2P-Tyr-EPI(DTPA) include [LG-EPI-DTPA + Ca + 2 K]+, 1549.39, [G-EPI-DTPA + Ca + 3 K]+, 1492.28 and [EPI-DTPA + Zn + 2 K + 2Na]+, 1447.22.
2.3.7. Cell culture
A2780 human ovarian carcinoma cells (ATCC) were maintained at 37 °C in a humidified atmosphere containing 5% CO2 in RPMI-1640 medium (Gibco) supplemented with 10% FBS and a mixture of antibiotics (100 units/mL penicillin, 0.1 mg/mL streptomycin).
2.3.8. Tumor model
All animal studies were carried out in accordance with the University of Utah Institutional Animal Care and Use Committee guidelines under approved protocols. A2780 human ovarian cancer cells (5 × 106) in 100 μL of PBS were subcutaneously inoculated in the right flank of 6- to 8-wk-old syngeneic female nude mice (22–25 g; Charles River Laboratories).
2.3.9. Pharmacokinetics and biodistribution study
For pharmacokinetic study, 6–8 week-old healthy female nude mice (22–25 g; Charles River Laboratories) (n = 5) were intravenously injected with dual-radiolabeled HPMA copolymer-EPI conjugates (1 mg, 20 μCi per mouse). At predetermined intervals, blood samples (10 μL) were taken from the tail vein, and the radioactivity of each sample was measured with Gamma Counter (Packard). The 125I activity was counted in a channel with windows set for 15–85 keV and 111In activity was counted in a channel having windows set for 237–257 keV [26]. Cross-counts in the 125I channel were 5% and in the 111In channel were 1.5%. Gross cpm values were corrected to compensate for cross-counting. The blood pharmacokinetic parameters for the radiotracer were analyzed using a noncompartmental model with WinNonlin 5.0.1 software (Pharsight).
For biodistribution study, 6–8 week-old female nude mice bearing s.c. A2780 tumors (22–25 g; Charles River Laboratories) received intravenous injection of dual-radiolabeled copolymer-EPI conjugates (1 mg, 20 μCi per mouse). At 48 h and 144 h after administration, the mice were sacrificed. Various tissues (heart, liver, spleen, lung, stomach, intestine, muscle, bone, brain and tumor) were harvested, weighed, and counted for radioactivity with Gamma Counter (Packard) with the aforementioned 111In/125I dual-isotope protocol. Uptake of the conjugate was calculated as the percentage of the injected dose per gram of tissue (%ID/g). Data are presented as mean ± standard deviation (n = 5).
2.3.10. Statistics
Statistical analyses were done using a two-tailed unpaired Student’s t-test, with p values of <0.01 indicating statistically significant differences.
3. Results and discussion
3.1. Design and synthesis of dual-radiolabeled HPMA copolymer-EPI conjugates
HPMA-based polymer drug conjugates have been investigated for decades; most of studies focused on the treatment of solid tumors. It has been demonstrated in preclinical studies that polymer-drug conjugates have numerous advantages compared to current marketed chemotherapeutical agents as a result of enhanced permeability and retention (EPR) effect. To accelerate the translation process from bench to bed, we propose a general approach to the synthesis of dual-labeled polymer-drug conjugates; one radiolabel is bound to polymeric carrier and the other is attached to drug (or model drug), in order to concurrently investigate two different molecular functions via multiple imaging modalities. As depicted in Scheme 1, the key component in our design is an azide-containing drug monomer MA-GFLG-Abu(N3)-Drug. By copolymerization of MA-GFLG-Abu(N3)-Drug with HPMA and APMA, a heterobifunctional polymer precursor will be obtained. The pendant amino groups can be used to incorporate a fluorescence dye containing N-hydroxysuccinimide (NHS) ester reactive group to polymer backbone. The second fluorophore will be introduced via azide-alkyne click reaction that has high specificity. The resulting dual-fluorophore labeled conjugates could be used for optical imaging studies such as bioluminescence imaging or fluorescence tomography (FMT). This approach can also be used to synthesize a dual-radiolabeled polymer-drug conjugate for PET/CT imaging or pharmacokinetics and biodistribution studies due to high sensitivity of radionuclides. For example, Bolton-Hunter reagent can be used to modify amino group to tyrosine-like structure for backbone iodination, while copper-free approach could be applied for attachment of a chelator such as DTPA followed by incorporation of a radiometal ion.
Scheme 1.

Illustration of a general strategy to synthesis of dual-fluorophore labeled and/or dual-radiolabeled HPMA copolymer conjugates.
In this study, we focused on dual-radiolabeling strategy. First generation polymer conjugate P-DTPA-EPI(Tyr) (Mw < 50 kDa, i.e. below the renal threshold, see SI Fig. S6) and backbone degradable HPMA copolymer-EPI conjugates 2P-DTPA-EPI(Tyr) (diblock with Mw > 50 kDa; above the renal threshold) were synthesized to evaluate the impact of molecular weight and degradability of the conjugate on its fate in the organism. We slightly modified the synthetic approach in Scheme 1: Instead of using Bolton-Hunter reagent, we attached p-SCN-Bn-DTPA to the pendant amino group on the polymer backbone, while a tyrosine moiety was coupled with N3 group via Cu(I) assisted alkyne-azide click reaction. To further investigate how radiolabel type impacts properties of the conjugates, 2P-Tyr-EPI(DTPA) containing an 125I labeling site on the polymer backbone and an 111In labeling site at the modified EPI was synthesized. The overall synthetic process for 2nd generation conjugates is described in Scheme 2, more detailed synthetic schemes including monomers, RAFT agent and 1st generation conjugate are listed in Supplementary Information. The conjugates to be evaluated were summarized in Table 1 (I, III and V). They were radiolabeled by 125I and 111In consecutively, yielded dual-labeled products II, IV and VI for pharmacokinetic and biodistribution studies.
Scheme 2.

Synthesis of HPMA copolymer-EPI conjugates and dual-radiolabeling strategy. (A) Synthesis of 2nd-generation conjugate 2P-Tyr-EPI(DTPA) by RAFT copolymerization of MA-GFLG-Abu(N3)-EPI with HPMA and MA-Tyr-NH2 using a cleavable diarm RAFT agent CTA-GFLG-CTA. Propargylamine was coupled with N3 group and chelator DTPA was attached for drug labeling with 111In. The polymer precursor was consecutively labeled with 125I (polymer backbone) and 111In (EPI). (B) Synthesis of 2nd-generation conjugate 2P-DTPA-EPI(Tyr) by copolymerization of MA-GFLG-Abu(N3)-EPI with HPMA and APMA. Tyrosine residue was incorporated via Cu(I) assisted alkyne-azide click reaction. DTPA was attached to polymer backbone via pendent amino group modification with p-SCN-Bn-DTPA. The polymer precursor was then labeled with 125I and 111In, consecutively.
Table 1.
Summary of dual labeled HPMA copolymer conjugates.
| No | Polymer precursor | Dual-labeled conjugate | Mw kDa | Mw/Mn | EPI (wt%) | DTPA |
Label isotode |
|
|---|---|---|---|---|---|---|---|---|
| No./per chain | Backbone | EPI | ||||||
|
I
II |
P-DTPA-EPI(Tyr) | 111In-P-EPI-125I | 28.2 | 1.07 | 10.5 | 3.7 | 111In | 125I |
|
III
IV |
2P-DTPA-EPI(Tyr) | 111In-2P-EPI-125I | 65.8 | 1.20 | 9.7 | 4.5 | 111In | 125I |
|
V
VI |
2P-Tyr-EPI(DTPA) | 125I-2P-EPI-111In | 75.2 | 1.25 | 5.5 | 1 | 125I | 111In |
3.2. Enzymatic cleavage study of HPMA copolymer-EPI conjugates
As a payload tracer, the cleavability of modified EPI is important for interpretation of pharmacokinetic profile and biodistribution result. In the ideal case, the radioisotope will be bound to the drug molecule and cleaved together without change of drug release rate. If the label and drug separate, then the radioactivity level will not be relevant to the drug concentration. To examine whether the modified EPI keeps intact when it is cleaved in the lysosomes, we synthesized model conjugate P-EPI(Tyr) and determined its stability in the presence of lysosome enzyme. Unmodified P-EPI was synthesized and served as control. Unlike the synthetic procedure described above (Scheme 2B), in P-EPI(Tyr), the tyrosine moiety was first incorporated to the monomer MA-GFLG-Abu(N3)-EPI via click reaction, then purified and polymerized to ensure there is no unmodified EPI left. The structure and physicochemical characterization of two conjugates are listed in Table 2 and Fig. S10.
Table 2.
Model conjugates for enzyme cleavage.
| Conjugate | Comonomer | Mw, kDa | Mw/Mn | EPI (wt%) |
|---|---|---|---|---|
| P-EPI (Tyr) |
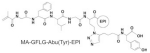
|
50.7 | 1.09 | 6.5 |
| P-EPI |
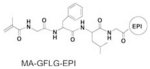
|
55.6 | 1.20 | 4.7 |
For cleavage, we have chosen to use Tritosomes, a mixture of lysosomal enzymes isolated from liver of rats, to mimic in vivo enzyme environment. The conjugates containing 300 nmol equivalent EPI were incubated with 0.48 mL Tritosomes at 37 °C for 48 h. Hydrolysis rate was monitored using HPLC. The results are shown in Fig. 1.
Fig. 1.

Tritosomes cleavage of P-EPI and P-EPI(Tyr). HPLC analysis of P-EPI (A) and P-EPI(Tyr) (B) at different time intervals (1 min, 1, 2, 4, 6, 8, 23, and 48 h). Daunomycin was used as internal standard. MALDI-TOF-MS analysis of cleaved products from P-EPI(Tyr) (C, D).
When incubated with Tritosomes, HPLC showed that the wide peak of conjugate P-EPI was decreased with incubation time, while free EPI peak emerged. Eventually conjugate was not detectable, indicating EPI was released from P-EPI gradually, and almost 100% release was reached within 48 h. In contrast, a new unknown peak occurred in P-EPI(Tyr) and increased slowly with incubation time (Fig. 1B). During 48 h incubation, a very small EPI peak also emerged. The fractions were collected and determined using MALDI-TOF mass spectroscopy. The result suggested that the cleavage mainly yielded two products, glycine-EPI-Tyr ([G-EPI-Tyr + Na]+, 1024.39) and EPI-Tyr ([EPI-Tyr + Na]+, 967.37) (Fig. 1C, D). The free drug EPI was only 2.5% compared to the modified EPI.
These findings are agreement with our previous studies. It was well established that the most important enzyme in the lysosomal compartment (and in Tritosomes) in the cleavage of oligopeptide sequences attached to HPMA copolymers is cathepsin B. Other enzymes participate in the cleavage to a lesser extent [9,53-54]. Cathepsin B (EC 3.4.22.1) is an important lysosomal enzyme active in the degradation of internalized proteins and peptides. It is a cysteine protease and its active site accommodates at least four amino acid residues toward the NH2 end of the substrate (positions P4–P1; subsites S1–S4; nomenclature of Schechter-Berger [55]) and two amino acid residues toward the COOH end of the substrate (positions P1′ and P2′). The GFLG sequence was designed to match the specificity of cathepsin B in the lysosomal compartment [53] and is now widely used as an attachment/release moiety in the design of lysosomotropic nanomedicines [9].
Comparison of cleavage of polymer-drug conjugates containing the GFLG spacer revealed that other enzymes participate in the cleavage within the lysosomal compartment [54]. In addition, the rate of enzymatically-catalyzed hydrolysis depends on the structure of the ligand (drug) bound to the GFLG sequence. Since the GFLG spacer aligns into S4-S1 subsites of cathepsin B, the different rate of drug release depends on the fit of the drug (ligand) into subsites S1′ and S2′ [56]. The formation of G-EPI-Tyr indicates that part of the substrate bound to the active site with phenylalanine in the (preferred [57]) P2 position. Probably, based on the bulkiness of EPI-Tyr, the cleavage of G-EPI-Tyr by cathepsin B occurred first and other enzymes present in Tritosomes subsequently cleaved the bond between glycine and EPI. In addition, the results indicated that the bond between the tyrosine moiety and EPI was stable since only a very small amount of free EPI was detected.
The two different dual label 2nd generation conjugates, 2P-Tyr-EPI(DTPA) and 2P-DTPA-EPI(Tyr), were also cleaved with Tritosomes. 2P-DTPA-EPI(Tyr) showed similar cleavage pattern to P-EPI(Tyr), mainly yielded EPI(Tyr) with little G-EPI(Tyr) and tiny free EPI. However, 2P-Tyr-EPI(DTPA) resulted in more complicated cleavage products, as shown in MALDI-TOF-MS (Fig. 2C). It revealed the presence of LG-EPI(DTPA), G-EPI-DTPA and EPI-DTPA. This indicates the due to the size of EPI-DTPA the substrate aligns to the cathepsin B active site with GFLG in subsites S2–S2′. Other lysosomal enzymes participate in the cleavage of LG-EPI(DTPA) to G-EPI(DTPA) and EPI(DTPA) (Table 3).
Fig. 2.

Tritosomes cleavage of 2nd-generation HPMA copolymer-EPI conjugates. HPLC analysis of 2P-DTPA-EPI(Tyr) (A) and 2P-Tyr-EPI(DTPA) (B) following incubation with Tritosomes for 45 h. MALDI-TOF-MS analysis of cleaved products from 2P-Tyr-EPI(DTPA) (C).
Table 3.
Structure of the conjugates and enzyme cleavage products.

|
The results of enzymatic cleavage of the conjugates clearly indicate the impact of the steric hindrance of the bulky payload on the S1′–P1′ and S2′–P2′ interactions. This needs to be taken into account when interpreting biodistribution data.
3.3. Pharmacokinetics and biodistribution study of dual-radiolabeled copolymer-PEI conjugates
Dual-labeled products II, IV and VI (see Table 1) were administered to nude mice via tail veins for pharmacokinetics and biodistribution studies. The dual-radiolabel strategy allowed to simultaneously track the fate of the payload (EPI- 125I or EPI- 111In) and of the polymeric carriers (111In-P/111In-2P or 125I-2P). Fig. 3A shows blood radioactivity-time profiles of 1st and 2nd generation conjugates in which polymer backbone was labeled with 111In and drug with 125I. For each conjugate, both 111In and 125I showed very similar pattern indicating the linker GFLG between drug and polymeric carrier is stable in blood circulation during transport. Moreover, it clearly demonstrated the superior property of 2nd generation conjugate (111In-2P-EPI-125I) with higher molecular weight on enhanced plasma concentration and elongation of circulation time as compared with 111In-P-EPI-125I. Consequently, the tumor-uptake of 111In-2P-EPI-125I was 2–3-folds greater than 111In-P-EPI-125I as illustrated in Fig. 3B.
Fig. 3.

Comparison of pharmacokinetic profiles and tumor-uptake of dual-labeled 1st generation conjugate 111In-P-EPI-125I and 2nd generation conjugate 111In-2P-EPI-125I in nu/nu mice. (A) Blood activity-time profiles of 111In-labeled polymer carriers and 125I labeled drug EPI in mice. (B) Radioactivity level in tumors for the mice bearing subcutaneous A2780 xenografts 48 h after intravenous injection of dual radiolabeled conjugates. Here 111In corresponds to polymer carrier (1st generation P- and 2nd generation 2P-) and 125I to drug EPI, respectively. The data represent the mean radioactivity expressed as a percentage of the injected dose per gram of blood (n = 5). p < 0.001.
This observation provides a direct proof that backbone degradable diblock HPMA copolymer carriers can more effectively deliver drugs to the tumor, resulting in the improvement of the treatment efficacy and decrease of non-specific adverse effect, in agreement with our previous report [28].
Fig. 4 shows the blood radioactivity-time profiles of different isotope-labeled polymer carriers (A) and payload (modified EPI) (B). Conjugate IV has similar Mw as conjugate VI. Their backbones were labeled with 111In and 125I, respectively, and side chain drug EPI was labeled with 125I and 111In; similar pharmacokinetics for polymer carrier and for payload was observed. Combined with results from Fig. 3, this suggests that for water-soluble polymer-drug conjugates, the molecular weight plays a decisive role on its circulation time and body clearance, while side-chain modification has a minor impact. This result also suggests that the modification of EPI structure does not influence the PK of the conjugate.
Fig. 4.

Effect of radiolabeling strategy on pharmacokinetic profiles of the polymer carrier (A) and payload (EPI) (B) of backbone degradable diblock HPMA copolymer-EPI conjugates in tumor-bearing nu/nu mice. 2P-DTPA-EPI(Tyr) with average Mw 66 kDa and 2P-Tyr-EPI(DTPA) with Mw 75 kDa were used. Data obtained using the radioactivity count method are plotted as percentage of injected dose per gram of tissue (%ID/g). All data are expressed as mean ± standard deviation (n = 5).
Pharmacokinetic parameters are summarized in Table 4. Both 2nd generation conjugates, 111In-2P-EPI-125I and 125I-2P-EPI-111In, showed similar half-life, which was much longer than that of the 1st generation conjugate 111In-P-EPI-125I. For example, the terminal half-lives of pay-load EPI(Tyr)125I and EPI(DTPA)111In in 2nd generation conjugates were 29.79 h and 28.40 h, respectively; they are very close to the terminal half-lives of polymeric carriers 111In-2P (31.52 h) and 125I-2P (31.11 h). However, terminal half-life of payload EPI(Tyr)125I in 1st generation conjugates and the polymeric carrier 111In-P were 9.63 h and 9.20 h, respectively. The total area under the blood concentration versus time curve (AUC) of 111In-2P-EPI-125I (1135% injected dose per mL (ID/mL) for 125I and 1474% ID/mL for 111In) and of 125I-2P-EPI-111In (1604% ID/mL for 111In and 1628% ID/mL for 125I) were significantly higher than that of 111In-P-EPI-125I (261% ID/mL for 125I and 324% ID/mL for 111In) (P < 0.001) (Table 4). Comparing with 1st generation conjugate 111In-P-EPI-125I, the increased exposure of 2nd generation conjugates 111In-2P-EPI-125I and 125I-2P-EPI-111In resulted mainly from a significantly slower mean systemic clearance (CL) (P < 0.001).
Table 4.
Comparison of pharmacokinetic parameters of dual-labeled (125I- and 111In-based) 1st generation conjugate and 2nd generation conjugates in mice.
|
|
1st generation |
2nd generation |
||||
|---|---|---|---|---|---|---|
| Conjugate |
111In-P-EPI-125I |
111In-2P-EPI-125I |
125I-2P-EPI-111In |
|||
| Component | EPI(Tyr)-125I | P(DTPA)111In | EPI(Tyr)125I | 2P(DTPA)111In | EPI(DTPA)111In | 2P(Tyr)125I |
| T1/2,α (h) | 0.57 ± 0.07 | 0.50 ± 0.08 | 1.51 ± 0.20 | 1.58 ± 0.19 | 0.49 ± 0.13 | 0.47 ± 0.16 |
| T1/2,β (h) | 9.63 ± 11.47 | 9.20 ± 1.68 | 29.79 ± 1.62 | 31.52 ± 2.01 | 28.40 ± 2.03 | 31.11 ± 2.19 |
| AUC (% ID h/mL) | 260.72 ± 24.64 | 324.25 ± 36.98 | 1135.00 ± 43.81 | 1473.97 ± 65.83 | 1604.36 ± 95.44 | 1628.32 ± 97.28 |
| CL (mL/h) | 0.38 ± 0.03 | 0.30 ± 0.03 | 0.08 ± 0.003 | 0.06 ± 0.003 | 0.06 ± 0.003 | 0.06 ± 0.004 |
| MRT (h) | 13.01 ± 1.96 | 12.46 ± 2.25 | 41.85 ± 2.21 | 43.95 ± 2.73 | 40.71 ± 2.89 | 44.69 ± 13.15 |
| Vss (mL) | 4.99 ± 0.31 | 3.84 ± 0.29 | 3.68 ± 0.08 | 2.98 ± 0.08 | 2.53 ± 0.05 | 2.74 ± 0.06 |
T1/2,α = initial half-life; T1/2,β = terminal half-life; AUC = total area under the blood concentration versus time curve; %ID = percentage of injected dose; CL = total body clearance; MRT = mean residence time; Vss = steady-state volume of distribution. Data are presented as mean ± standard deviation (n = 5).
We also analyzed biodistribution of payload and carrier in female nude mice bearing A2780 human ovarian carcinoma. A high and lasting radioactivity level of polymer carrier labeled with 111In (111In-2P) in kidney was observed, in contrast to the polymer carrier labeled with 125I (125I-2P, Fig. 5A & B). Two phenomena may contribute to this observation: First, the incorporation of chelator DTPA for 111In labeling induces negative charges to the polymer carrier; it has been observed that this enhances the accumulation in the cortex and renal tubules after intravenous administration in mice [32,58,59]. Second, it is more likely due to 111In strong kidney accumulation and slow clearance as noted by others [60-62]. Other than 111In-2P in kidney, at 48 h after intravenous injection of 2nd generation conjugates, the highest concentration of conjugates (both payload EPI and polymer carrier) was still in blood stream, indicating the advantage of high molecular weight conjugates. Until 144 h, the concentrations of polymer carrier and payload EPI(DTPA)111In in tumor were clearly higher than the levels in the normal organs except liver, and spleen, demonstrating the targeting ability of polymer conjugates to tumor (Fig. 5A & B). However, EPI(Tyr)125I showed lower uptake than EPI(DTPA)111In in major organs and tumor at both 48 h and 144 h (Fig. 5A & B); at same time points payload EPI(DTPA)111In showed similar uptake as polymeric carrier 125I-2P in major organs and tumor. There are several factors that may contribute to this result: different drug release rates resulting from steric hindrance of EPI modification, and lower retention of radioiodine. It has been reported that after endocytosis and proteolytic degradation in lysosomes, the iodine-labeled conjugates generate iodotyrosine, which is rapidly excreted from cells and shows poor retention in normal organs and tumors [63,64]. In addition, radioiodide could be liberated from iodotyrosine by dehalogenase enzymes and rapidly sequestered by thyroid [32]. Future in vivo experiments using d-amino acids could evaluate the importance of this pathway [65-67].
Fig. 5.

Effect of radiolabeling strategy on biodistribution of the polymer carrier and payload (EPI) of backbone degradable diblock HPMA copolymer-EPI conjugates in tumor-bearing mice at 48 h (A) and 144 h (B) after intravenous administration. Data obtained using the radioactivity count method are plotted as percentage of injected dose per gram of tissue (%ID/g). All data are expressed as mean ± standard deviation (n = 5).
We compared the tumor-to-tissue uptake ratios of payload and polymeric carriers and found that the tumor-to-tissue uptake ratios of both radionuclides were similar (Fig. S15). The tumor-to-tissue uptake ratios of 2nd generation conjugates were higher than that of 1st generation conjugate for both payloads and polymeric carriers.
Taking together with the results we previously obtained [26] and other literatures [19,68,69], pharmacokinetics and biodistribution results indicated: 1) GFLG linker between payload and polymeric carrier remained stable in the bloodstream during transport. 2) After internalization via endocytosis and localization in the lysosomes, the payload EPI(Tyr)125I of 111In -2P-EPI-125I was slowly cleaved from the polymeric carrier; its excretion from cells resulted in low retention in normal organs and tumor. 3) Payload EPI(DTPA)111In of 125I-2P-EPI-111In was cleaved more slowly from the carrier compared to payload EPI(Tyr)125I. 4) Different radiolabel designs do not influence tumor-to-tissue uptake ratios of payload and polymeric carrier. 5) The pharmacokinetics of an unlabeled conjugate that would be used as the macromolecular therapeutics should be similar with that of labeled conjugate, but the drug retention of unlabeled conjugate would be shorter as a result of faster cleavage of payload from the GFLG spacer.
4. Conclusions
We developed a unique and general route to synthesize dual-isotope labeled HPMA copolymer-EPI conjugates for evaluation of the fate of conjugates in animal bodies. Tritosomes hydrolysis study demonstrated that the cleavage site and hydrolysis rate depend on the structure of substrate—it can be cleaved within the GFLG linker, or between the linker and payload. The bulky group used for drug modification (EPI(Tyr) or EPI(DTPA)) may cause steric hindrance on the formation of the enzyme-substrate complex, therefore slow down the drug release. Pharmacokinetics and biodistribution study illustrated that the labeling by viable radionuclides does not change the blood-clearance behavior, but may have impact on biodistribution. Iodine-based biodistribution may differ significantly from that obtained using a radiometal label. The tumor-to-tissue uptake ratios of both radionuclides were similar, indicating that drug-labeling strategy does not affect the tumor targeting ability of HPMA copolymer conjugates. We anticipate that this practical and efficient dual radiolabeling method offers a reliable method to study different polymer-based water-soluble drug delivery systems. Numerous anticancer drugs, including doxorubicin, paclitaxel, and gemcitabine bound to carriers of different structures (PEG, HPMA, poly(amino acid)s, polyacetals, polyamidoamines, etc.) could be evaluated and the mechanism of their action revealed. The double-labeled conjugates have a potential in dual-isotope imaging, radiochemotherapy, and pre-clinical evaluation and optimization of nanoconjugates.
Supplementary Material
Acknowledgements
The research was supported in part by U.S. Department of Defense grant W81XWH-13-1-0160 and NIH grant CA156933.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2016.06.004.
References
- [1].Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug. Chem. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- [2].Zhou Y, Kopeček J. Biological rationale for the design of polymeric anti-cancer nanomedicines. J. Drug Target. 2013;21:1–26. doi: 10.3109/1061186X.2012.723213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Duncan R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- [4].Yang J, Kopeček J. Design of smart polymer-based nanomedicines. J. Control. Release. 2015 doi: 10.1016/j.jconrel.2015.10.003. http://dx.doi.org/10.1016/j.jconrel.2015.10.003. [DOI] [PMC free article] [PubMed]
- [5].Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- [6].Pasut G, Veronese FM. PEG conjugates in clinical development or use as anticancer agents: an overview. Adv. Drug Deliv. Rev. 2009;61:1177–1188. doi: 10.1016/j.addr.2009.02.010. [DOI] [PubMed] [Google Scholar]
- [7].Ulbrich K, Subr V. Structural and chemical aspects of HPMA copolymers as drug carriers. Adv. Drug Deliv. Rev. 2010;62:150–166. doi: 10.1016/j.addr.2009.10.007. [DOI] [PubMed] [Google Scholar]
- [8].Duncan R, Seymour LW, O’Hare KB, Flanagan PA, Wedge S, Hume IC, Ulbrich K, Strohalm J, Subr V, Spreafico F, Grandi M, Ripamonti M, Farao M, Suarato A. Preclinical evaluation of polymer-bound doxorubicin. J. Control. Release. 1992;19:331–346. [Google Scholar]
- [9].Kopeček J, Kopečková P. HPMA copolymers: origins, early developments, present, and future. Adv. Drug Deliv. Rev. 2010;62:122–149. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Říhová B, Kovář M. Immunogenicity and immunomodulatory properties of HPMA-based polymers. Adv. Drug Deliv. Rev. 2010;62:184–191. doi: 10.1016/j.addr.2009.10.005. [DOI] [PubMed] [Google Scholar]
- [11].Shiah J-G, Sun Y, Kopečková P, Peterson CM, Straight RC, Kopeček J. Combination chemotherapy and photodynamic therapy of targetable N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin/mesochlorin e6-OV-TL16 antibody immunoconjugates. J. Control. Release. 2001;74:249–253. doi: 10.1016/s0168-3659(01)00325-x. [DOI] [PubMed] [Google Scholar]
- [12].Paz-Ares L, Ross H, O’Brien M, Riviere A, Gatzemeier U, Von Pawel J, Kaukel E, Freitag L, Digel W, Bischoff H, Garcia-Campello R, Iannotti N, Reiterer P, Bover I, Prendiville J, Eisenfeld AJ, Oldham FB, Bandstra B, Singer JW, Bonomi P. Phase III trial comparing paclitaxel poliglumex vs docetaxel in the second-line treatment of non-small-cell lung cancer. Br. J. Cancer. 2008;98:1608–1613. doi: 10.1038/sj.bjc.6604372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Viegas TX, Bentley MD, Harris JM, Fang Z, Yoon K, Dizman B, Weimer R, Mero A, Pasut G, Veronese FM. Polyoxazolines: chemistry, properties, and applications in drug delivery. Bioconjug. Chem. 2011;22:976–986. doi: 10.1021/bc200049d. [DOI] [PubMed] [Google Scholar]
- [14].Ding H, Inoue S, Ljubimov AV, Patil R, Portilla-Arias J, Hu J, Konda B, Wawrowsky KA, Fujita M, Karabalin N, Sasaki T, Black KL, Holler E, Ljubimova JY. Inhibition of brain tumor growth by intravenous poly(β-L-malic acid) nanobioconjugate with pH-dependent drug release. Proc. Natl. Acad. Sci. U. S. A. 2010;107:18143–18148. doi: 10.1073/pnas.1003919107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Abu Lila AS, Kiwada H, Ishida T. The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage. J. Control. Release. 2013;172:38–47. doi: 10.1016/j.jconrel.2013.07.026. [DOI] [PubMed] [Google Scholar]
- [16].Barz M, Luxenhofer R, Zentel R, Vicent MJ. Overcoming the PEG-addiction: well-defined alternatives to PEG, from structure-property relationships to better defined therapeutics. Polym. Chem. 2011;2:1900–1918. [Google Scholar]
- [17].Tucker BS, Sumerlin BS. Poly(N-(2-hydroxypropyl)methacrylamide)-based nanotherapeutics. Polym. Chem. 2014;5:1566–1572. doi: 10.1039/C7PY00196G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Minko T, Kopečková P, Kopeček J. Comparison of the anticancer effect of free and HPMA copolymer-bound adriamycin in human ovarian carcinoma cells. Pharm. Res. 1999;16:986–996. doi: 10.1023/a:1018959029186. [DOI] [PubMed] [Google Scholar]
- [19].Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, Thomson AH, Murray LS, Hilditch TE, Murray T, Burtles S, Fraier D, Frigerio E, Cassidy J. Phase I clinical and pharmacokinetic study of PK1 (HPMA copolymer doxorubicin): first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Clin. Cancer Res. 1999;5:83–94. [PubMed] [Google Scholar]
- [20].Julyan PJ, Seymour LW, Ferry DR, Daryani S, Boivin CM, Doran J, David M, Anderson D, Christodoulou C, Young AM, Hesslewood S, Kerr DJ. Preliminary clinical study of the distribution of HPMA copolymers bearing doxorubicin and galactosamine. J. Control. Release. 1999;57:281–290. doi: 10.1016/s0168-3659(98)00124-2. [DOI] [PubMed] [Google Scholar]
- [21].Meerum Terwogt JM, ten Bokkel Huinink WW, Schellens JH, Schot M, Mandjes IA, Zurlo MG, Rocchetti M, Rosing H, Koopman FJ, Beijnen JH. Phase I clinical and pharmacokinetic study of PNU166945, a novel water-soluble polymer-conjugated prodrug of paclitaxel. Anti-Cancer Drugs. 2001;12:315–323. doi: 10.1097/00001813-200104000-00003. [DOI] [PubMed] [Google Scholar]
- [22].Rademaker-Lakhai JM, Terret C, Howell SB, Baud CM, De Boer RF, Pluim D, Beijnen JH, Schellens JH, Droz JP. A phase I and pharmacological study of theplatinum polymer AP5280 given as an intravenous infusion once every 3 weeks in patients with solid tumors. Clin. Cancer Res. 2004;10:3386–3395. doi: 10.1158/1078-0432.CCR-03-0315. [DOI] [PubMed] [Google Scholar]
- [23].Yang J, Luo K, Pan H, Kopečková P, Kopeček J. Synthesis of biodegradable multiblock copolymers by click coupling of RAFT-generated heterotelechelic polyHPMA conjugates. React. Funct. Polym. 2011;71:294–302. doi: 10.1016/j.reactfunctpolym.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Luo K, Yang J, Kopečková P, Kopeček J. Biodegradable multiblock N-(2-hydroxypropyl)methacrylamide copolymers via reversible addition-fragmentation chain transfer polymerization and click chemistry. Macromolecules. 2011;44:2481–2488. doi: 10.1021/ma102574e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pan H, Yang J, Kopečková P, Kopeček J. Backbone degradable multiblock N-(2-hydroxypropyl)methacrylamide copolymer conjugates via reversible addition fragmentation chain transfer polymerization and thiol-ene coupling reaction. Biomacromolecules. 2011;12:247–252. doi: 10.1021/bm101254e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang R, Yang J, Sima M, Zhou Y, Kopeček J. Sequential combination therapy of ovarian cancer with degradable N-(2-hydroxypropyl)methacrylamide copolymer paclitaxel and gemcitabine conjugates. Proc. Natl. Acad. Sci. U. S. A. 2014;111:12181–12186. doi: 10.1073/pnas.1406233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang J, Kopeček J. Polymeric biomaterials and nanomedicines. J. Drug Deliv. Sci. Technol. 2015;30:318–330. doi: 10.1016/j.jddst.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yang J, Zhang R, Radford DC, Kopeček J. FRET-trackable biodegradable HPMA copolymer-epirubicin conjugates for ovarian carcinoma therapy. J. Control. Release. 2015;218:36–44. doi: 10.1016/j.jconrel.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shiah J, Dvořák M, Kopečková P, Sun Y, Peterson CM, Kopeček J. Biodistribution and antitumour efficacy of long-circulating N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates in nude mice. Eur. J. Cancer. 2001;37:131–139. doi: 10.1016/s0959-8049(00)00374-9. [DOI] [PubMed] [Google Scholar]
- [30].Shiah J, Sun Y, Peterson CM, Kopeček J. Biodistribution of free and N-(2-hydroxypropyl)methacrylamide copolymer-bound mesochlorin e6 and adriamycin in nude mice bearing human ovarian carcinoma OVCAR-3 xenografts. J. Control. Release. 1999;61:145–157. doi: 10.1016/s0168-3659(99)00113-3. [DOI] [PubMed] [Google Scholar]
- [31].Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, Doran J, Young AM, Burtles S, Kerr DJ. Hepatic drug targeting: phase I evaluation of polymer-bound doxorubicin. J. Clin. Oncol. 2002;20:1668–1676. doi: 10.1200/JCO.2002.20.6.1668. [DOI] [PubMed] [Google Scholar]
- [32].Lammers T, Kühnlein R, Kissel M, Subr V, Etrych T, Pola R, Pechar M, Ulbrich K, Storm G, Huber P, Peschke P. Effect of physicochemical modification on the biodistribution and tumor accumulation of HPMA copolymers. J. Control. Release. 2005;110:103–118. doi: 10.1016/j.jconrel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- [33].Low SA, Galiford CV, Yang J, Low PS, Kopeček J. Biodistribution of fracture-targeted GSK3b inhibitor-loaded micelles for improved fracture healing. Biomacromolecules. 2015;16:3145–3153. doi: 10.1021/acs.biomac.5b00777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Říhová B, Vereš K, Fornůsek L, Ulbrich K, Strohalm J, Větvička V, Bilej M, Kopeček J. Action of polymeric prodrugs based on N-(2-hydroxypropyl)methacrylamide copolymers. II. Body distribution and T-cell accumulation of free and polymer-bound [125I] daunomycin. J. Control. Release. 1989;10:37–49. [Google Scholar]
- [35].Wang D, Sima M, Mosley RL, Davda JP, Tietze N, Miller SC, Gwilt PR, Kopečková P, Kopeček J. Pharmacokinetic and biodistribution studies of a bone-targeting drug delivery system based on N-(2-hydroxypropyl)methacrylamide copolymers. Mol. Pharm. 2006;3:717–725. doi: 10.1021/mp0600539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yu Q, Dong C, Zhang J, Shi J, Jia B, Wang F, Gan Z. Synthesis of poly(ethylene glycol)-b-poly(N-(2-hydroxypropyl)methacrylamide) block copolymers with well-defined structures and their influence on in vivo circulation and biodistribution. Polym. Chem. 2014;5:5617–5627. [Google Scholar]
- [37].Pan H, Sima M, Kopečková P, Wu K, Gao S, Liu J, Wang D, Miller SC, Kopeček J. Biodistribution and pharmacokinetic studies of bone-targeting N-(2-hydroxypropyl)methacrylamide copolymer-alendronate conjugates. Mol. Pharm. 2008;5:548–558. doi: 10.1021/mp800003u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang R, Luo K, Yang J, Sima M, Sun Y, Janát-Amsbury MM, Kopeček J. Synthesis and evaluation of a backbone biodegradable multiblock HPMA copolymer nanocarrier for the systemic delivery of paclitaxel. J. Control. Release. 2013;166:66–74. doi: 10.1016/j.jconrel.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pan H, Sima M, Miller SC, Kopečková P, Yang J, Kopeček J. Efficiency of high molecular weight backbone degradable HPMA copolymere prostaglandin E1 conjugate in promotion of bone formation in ovariectomized rats. Biomaterials. 2013;34:6528–6538. doi: 10.1016/j.biomaterials.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lammers T, Subr V, Ulbrich K, Peschke P, Huber PE, Hennink WE, Storm G. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using proto-typic polymeric drug carriers. Biomaterials. 2009;30:3466–3475. doi: 10.1016/j.biomaterials.2009.02.040. [DOI] [PubMed] [Google Scholar]
- [41].Quan L, Yuan F, Liu X, Huang J, Alnouti Y, Wang D. Pharmacokinetic and biodistribution studies of N-(2-hydroxypropyl)methacrylamide copolymer-dexa-methasone conjugates in adjuvant-induced arthritis rat model. Mol. Pharm. 2010;7:1041–1049. doi: 10.1021/mp100132h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hoffmann S, Vystrčilová L, Ulbrich K, Etrych T, Caysa H, Müller T, Mäder K. Dual fluorescent HPMA copolymers for passive tumor targeting with pH-sensitive drug release: synthesis and characterization of distribution and tumor accumulation in mice by noninvasive multispectral optical imaging. Biomacromolecules. 2012;13:652–663. doi: 10.1021/bm2015027. [DOI] [PubMed] [Google Scholar]
- [43].Chytil P, Hoffmann S, Schindler L, Kostka L, Ulbrich K, Caysa H, Müller T, Mäder K, Etrych T. Dual fluorescent HPMA copolymers for passive tumor targeting with pH-sensitive drug release II: impact of release rate on biodistribution. J. Control. Release. 2013;172:504–512. doi: 10.1016/j.jconrel.2013.05.008. [DOI] [PubMed] [Google Scholar]
- [44].Kunjachan S, Gremse F, Theek B, Koczera P, Pola R, Pechar M, Etrych T, Ulbrich K, Storm G, Kiessling F, Lammers T. Noninvasive optical imaging of nanomedicine biodistribution. ACS Nano. 2013;7:252–262. doi: 10.1021/nn303955n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules. 2001;34:2248–2256. [Google Scholar]
- [46].Kopeček J, Bazilová H. Poly[N-(2-hydroxypropyl)methacrylamide]. 1. Radical polymerization and copolymerization. Eur. Polym. J. 1973;9:7–14. [Google Scholar]
- [47].Duncan R, Cable HC, Rejmanová P, Kopeček J, Lloyd JB. Tyrosinamide residues enhance pinocytic capture of N-(2-hydroxypropyl)methacrylamide copolymers. Biochim. Biophys. Acta. 1984;799:1–8. doi: 10.1016/0304-4165(84)90320-9. [DOI] [PubMed] [Google Scholar]
- [48].Kopeček J, Rejmanová P, Strohalm J, Ulbrich K, Říhová B, Chytrý V, Lloyd JB, Duncan R. US Patent 5,037,883 Synthetic polymeric drugs. 1991
- [49].Omelyanenko V, Kopečková P, Gentry C, Shiah J-G, Kopeček J. HPMA copolymer-anticancer drug-OV-TL16 antibody conjugates. 1. Influence of the methods of synthesis on binding affinity to OVCAR-3 ovarian carcinoma in vitro. J. Drug Target. 1996;3:357–373. doi: 10.3109/10611869608996827. [DOI] [PubMed] [Google Scholar]
- [50].Starcher B. A ninhydrin-based assay to quantitate the total protein content of tissue samples. Anal. Biochem. 2001;292:125–129. doi: 10.1006/abio.2001.5050. [DOI] [PubMed] [Google Scholar]
- [51].Pippin CG, Parker TA, McMurry TJ, Brechbiel MW. Spectrophotometric method for the determination of a bifunctional DTPA ligand in DTPA-monoclonal antibody conjugates. Bioconjug. Chem. 1992;3:342–345. doi: 10.1021/bc00016a014. [DOI] [PubMed] [Google Scholar]
- [52].Trouet A. Isolation of modified liver lysosomes. Methods Enzymol. 1974;31:323–329. doi: 10.1016/0076-6879(74)31034-8. [DOI] [PubMed] [Google Scholar]
- [53].Rejmanová P, Pohl J, Baudys M, Kostka V, Kopeček J. Polymers containing enzymatically degradable bonds. 8. Degradation of oligopeptide sequences in N-(2-hydroxypropyl)methacrylamide copolymers by bovine spleen cathepsin B. Makromol. Chem. 1983;184:2009–2020. [Google Scholar]
- [54].Subr V, Kopeček J, Pohl J, Baudys M, Kostka V. Cleavage of oligopeptide side-chains in N-(2-hydroxypropylmethacrylamide copolymers by mixtures of lysosomal enzymes. J. Control. Release. 1988;8:133–140. [Google Scholar]
- [55].Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- [56].Duncan R, Kopečková-Rejmanová P, Strohalm J, Hume I, Cable HC, Pohl J, Lloyd JB, Kopeček J. Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. I. Evaluation of daunomycin and puromycin conjugates in vitro. Br. J. Cancer. 1987;55:165–174. doi: 10.1038/bjc.1987.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hasnain S, Hirama T, Tam A, Mort JS. Characterization of recombinant rat cathepsin B and nonglycosylated mutants expressed in yeast. New insights into the pH dependence of cathepsin B-catalyzed hydrolyses. J. Biol. Chem. 1992;267:4713–4721. [PubMed] [Google Scholar]
- [58].Kodaira H, Tsutsumi Y, Yoshioka Y, Kamada H, Kaneda Y, Yamamoto Y, Tsunoda S-I, Okamoto T, Mukai Y, Shibata H, Nakagawa S, Mayumi T. The targeting of an ionized polyvinylpyrrolidone to the renal system. Biomaterials. 2004;25:4309–4315. doi: 10.1016/j.biomaterials.2003.10.097. [DOI] [PubMed] [Google Scholar]
- [59].Yu Q, Wei Z, Shi J, Guan S, Du N, Shen T, Tang H, Jia B, Wang F, Gan Z. Polymer-doxorubicin conjugate micelles based on poly(ethylene glycol) and poly(N-(2-hydroxypropyl)methacylamide): effect of negative charge and molecular weight on biodistribution and blood clearance. Biomacromolecules. 2015;16:2645–2655. doi: 10.1021/acs.biomac.5b00460. [DOI] [PubMed] [Google Scholar]
- [60].Borgman MP, Coleman T, Kolhatkar RB, Geyser-Stoops S, Line BR, Ghandehari H. Tumor-targeted HPMA copolymer-(RGDfK)-(CHX-A″-DTPA) conjugates show increased kidney accumulation. J. Control. Release. 2008;132:193–199. doi: 10.1016/j.jconrel.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tran TA, Rosik D, Abrahamsén L, Sandström M, Sjöberg A, Wållberg H, Ahlgren S, Orlova A, Tolmachev V. Design, synthesis and biological evaluation of a multifunctional HER2-specific affibody molecule for molecular imaging. Eur. J. Nucl. Med. Mol. Imaging. 2009;36:1864–1873. doi: 10.1007/s00259-009-1176-z. [DOI] [PubMed] [Google Scholar]
- [62].Laznickova A, Laznicek M, Trejtnar F, Maecke HR, Eisenweiner KP, Reubi JC. Biodistribution of two octreotate analogs radiolabeled with indium and yttrium in rats. Anticancer Res. 2010;30:2177–2184. [PubMed] [Google Scholar]
- [63].Williams LE, Wu AM, Kenanova VE, Olafsen T, Yazaki PJ. Numerical comparison of iodine-based and indium-based antibody biodistributions. Cancer Biother. Radiopharm. 2014;29:91–98. doi: 10.1089/cbr.2013.1564. [DOI] [PubMed] [Google Scholar]
- [64].Press OW, Shan D, Clark JH, Eary J, Appelbaum FR, Matthews D, King DJ, Haines AMR, Hamann P, Hinman L, Shochat D, Bernstein ID. Comparative metabolism and retention of iodine-125, yttrium-90, and indium-Ill radioimmunoconjugates by cancer cells. Cancer Res. 1996;56:2123–2129. [PubMed] [Google Scholar]
- [65].Foulon CF, Welsh PC, Bigner DD, Zalutsky MR. Positively charged templates for labeling internalizing antibodies: comparison of N-succinimidyl 5-iodo-3-pyridinecarboxylate and the D-amino acid peptide KRYRR. Nucl. Med. Biol. 2001;28:769–777. doi: 10.1016/s0969-8051(01)00239-6. [DOI] [PubMed] [Google Scholar]
- [66].Araujo RL, Carvalho DP. Bioenergetic impact of tissue-specific regulation of iodothyronine deiodinases during nutritional imbalance. J. Bioenerg. Biomembr. 2011;43:59–65. doi: 10.1007/s10863-011-9327-x. [DOI] [PubMed] [Google Scholar]
- [67].Martin EB, Kennel SJ, Richey T, Wooliver C, Osborne D, Williams A, Stuckey A, Wall JS. Dynamic PET and SPECT imaging with radioiodinated, amyloid-reactive peptide p5 in mice: a positive role for peptide dehalogenation. Peptides. 2014;60:63–70. doi: 10.1016/j.peptides.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yokoyama K, Carrasquillo JA, Chang AE, Coicher D, Roselli M, Sugarbaker P, Sindelar W, Reynolds JC, Perentesis P, Gansow OA, Francis B, Adams R, Finn R, Schiom J, Larson SM. Differences in biodistribution of indium-111- and iodine-131-labeled B72.3 monoclonal antibodies in patients with colorectal cancer. J. Nucl. Med. 1989;30:320–327. [PubMed] [Google Scholar]
- [69].Cohen R, Vugts DJ, Visser GWM, Walsum MS, Bolijn M, Spiga M, Lazzari P, Shankar S, Sani M, Zanda M, Dongen GAMS. Development of novel ADCs: conjugation of tubulysin analogues to trastuzumab monitored by dual radiolabeling. Cancer Res. 2014;74:5700–5710. doi: 10.1158/0008-5472.CAN-14-1141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.