

Abstract
DNA has been exploited as a biological target of chemotherapeutics since the 1940s. Traditional chemotherapeutics, such as cisplatin and DNA-alkylating agents, rely primarily on increased uptake by rapidly proliferating cancer cells for therapeutic effects, but this strategy can result in off-target toxicity in healthy tissue. Recently, research interests have shifted towards targeted chemotherapeutics, in which a drug targets a specific biological signature of cancer, resulting in selective toxicity towards cancerous cells. Here, we review a family of complexes, termed rhodium metalloinsertors, that selectively target DNA base pair mismatches, a hallmark of mismatch-repair (MMR) deficient cancers. These rhodium metalloinsertors, bind DNA mismatches with high specificity and display high selectively in killing MMR-deficient versus MMR-proficient cells. This cell selectivity is unique for small molecules that bind DNA. Current generations of rhodium metalloinsertors have shown nanomolar potency along with high selectivity towards MMR-deficient cells, and show promise as a foundation for a new family of chemotherapeutics for MMR-deficient cancers.
Keywords: Rhodium, Metalloinsertor, Mismatch Repair, DNA Probe
Graphical Abstract Synopsis
Rhodium metalloinsertors selectively target DNA base pair mismatches, leading to the ejection of the mismatched bases from the DNA π-stack. The selective binding of metalloinsertors to DNA mismatches provides the basis for targeting mismatch-repair deficient cells as a strategy for new chemotherapeutic design.
1. Introduction
DNA has proven to be a rich target for a large range of small-molecule therapeutic drugs. The first DNA-targeting compounds with therapeutic properties were discovered in the 1940s [1,2]. Nitrogen mustards and antifolate drugs were found to cause tumor regression in patients with non-Hodgkin's lymphoma and remission in children with lymphoblastic leukemia, respectively [2,3]. It was found that the anticancer properties of these drugs arise from their interactions with DNA; nitrogen mustards irreversibly alkylate DNA through an aziridinium intermediate to form inter-strand crosslinks and antifolates block DNA synthesis by inhibiting dihydrofolate reductase (DHFR), an enzyme necessary for the synthesis of purine bases [1,4]. Since the discoveries of these therapeutics, the versatility of DNA as a target has been significantly expanded. Therapeutics have been seen to bind covalently to DNA (alkylating agents, platinum drugs), non-covalently interact with DNA (actinomycin D, mitomycins, polyamides), to interfere with protein-DNA complexes (doxorubicin, etoposide), and even target DNA secondary structures such as G-quadruplexes (intrafloxin, in phase II clinical trials) [5–8]. These complexes and more DNA-targeting chemotherapeutics have been reviewed recently [5,9].
In the development of novel metallodrugs, DNA is consistently one of the most exploited targets. As seen in Figure 1, metal complexes can bind DNA through several different routes, involving both covalent and non-covalent interactions [10,11]. In the covalent binding mode, the small molecule, for example cisplatin, binds directly to DNA to form a covalent lesion, usually bonding with one or more bases and impeding DNA replication. In contrast, non-covalent interactions rely on thermodynamic stabilization through electrostatics, hydrogen bonding, hydrophobic interactions, and π-stacking interactions [12]. The majority of complexes that bind DNA non-covalently are either intercalators or groove binders. In intercalation, an aromatic, heterocyclic ligand slips indiscriminately between two adjacent base pairs. This process leads to a partial unwinding of the DNA, increasing the rise of the helix. Groove binding is another common non-covalent binding motif of small molecule therapeutics. In this binding mode, a small molecule that is generally crescent-shaped tightly binds the minor groove of DNA. Unlike intercalators, which generally lack sequence specificity, groove binders often target AT-rich regions. Moreover, sequence-specific intercalators and groove binders have been prepared [7,13]. Once bound to DNA, these non-covalent complexes primarily cause inhibition of proteins involved in DNA transcription and synthesis, which can lead to cytotoxicity [14–16]. Somewhat recently, a new non-covalent DNA binding mode, termed metalloinsertion, has been observed. In this mode, a large aromatic, heterocyclic ligand inserts into DNA at a destabilized site and ejects the destabilized bases from the helix, without causing an increase in base rise as is seen with intercalators [17]. In contrast to intercalators, metalloinsertors are highly specific for destabilized DNA mismatches, abasic sites, and single base bulges.
Figure 1.
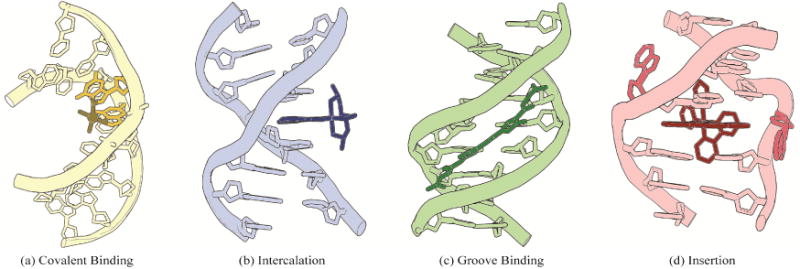
Four common binding modes of small molecules to DNA: (a) covalent binding, (b) intercalation, (c) groove binding, and (d) insertion. Structural data from PDB files 1AIO, 454D, 2LWH, and 3GSK.
Though DNA-targeting metallodrugs are a significant field of study for many researchers, few have had clinical success due to the general toxicity of heavy metals in the body [18–20]. The most noteworthy and well characterized metallodrug found to bind DNA is cis-diamminedichloroplatinum(II) (cisplatin). Cisplatin, the first inorganic chemotherapeutic, was discovered serendipitously by Barnett Rosenberg in 1965 while studying the effects of electric fields on E. coli using a platinum electrode [21]. Today, cisplatin and its derivatives, carboplatin and oxaliplatin, remain some of the most frequently used chemotherapeutics with over 50% of all cancer regimens containing one of these drugs [22]. Once within a cell, the chloride ligands of cisplatin are displaced by water [23]. This reactive intermediate binds the N7 position of purine bases to form inter- and intra-strand DNA crosslinks, with the biologically abundant adduct being 1,2-intrastrand crosslinks between two adjacent guanine bases [24]. This adduct was structurally characterized using X-ray crystallography in 1995 [25]. The 2.6 Å resolution structure shows the bending of the DNA duplex by 40° towards the major groove, accompanied by the widening of the minor groove. This lesion is recognized intracellularly by DNA-binding proteins, eventually leading to the apoptotic death of affected cells [23].
Despite its success in the clinic, cisplatin is not without its drawbacks. Patients treated with cisplatin often experience severe, dose-limiting side effects such as nausea, vomiting, nephrotoxicity, ototoxicity, and neurotoxicity [26]. These side effects occur because cisplatin, like many chemotherapeutics, is not selective towards cancer cells—it binds DNA inside healthy and cancerous tissues alike. Instead, cisplatin appears primarily to rely on increased uptake by rapidly dividing cancer cells for effect [5]. Targeted therapy, in which a specific biological signature of cancer drives preferential drug action on cancerous cells over healthy cells, is a clear alternative to these non-specific chemotherapeutics. For example, proteins that are upregulated or expressed exclusively in cancer cells may be exploited as cancer-selective targets [27]. For such protein targets, kinase inhibitors and monoclonal antibodies have found clinical use in the treatment of a variety of cancers [28,29]. As an example, cetuximab, a monoclonal antibody, targets and inhibits the epidermal growth factor receptor (EGFR), which is upregulated in several cancers in order to maintain rapid proliferation [30].
In addition to targeting specific proteins, it is also possible to target specific DNA lesions associated with cancer, such as single base-pair mismatches. Over the last 30 years, our laboratory has focused on targeting DNA mismatches using a unique family of complexes called rhodium metalloinsertors. These complexes bind single base-pair mismatches in vitro and have been shown to be selectively cytotoxic towards mismatch repair deficient cell lines [17,31]. This review is not an exhaustive examination of small molecules designed to target DNA mismatches as this topic has been recently reviewed elsewhere [32]. Instead, this article concentrates on the work of our own laboratory on this unique family of DNA-binding complexes termed rhodium metalloinsertors.
2. Mismatch Repair Machinery
2.1 DNA Damage and Errors in Replication
The DNA within cells is constantly subject to damage by exogenous agents, such as UV light and ionizing radiation, and endogenous modifications, such as depurination, methylation, and errors in replication [33]. Some estimates suggest cells experience up to 105 such lesions each day [34]. This damage can lead to interruptions in cellular processes, cell death, and mutations if uncorrected. High fidelity of DNA is essential, and therefore cells have evolved complicated systems to repair many types of DNA damage, known collectively as the DNA damage response. The DNA damage response consists of several processes that identify or correct a broad range of damage, including base excision repair, nucleotide excision repair, mismatch repair (MMR), and double strand break repair. These processes have all been reviewed recently [35–38].
MMR machinery is primarily responsible for identifying and correcting replication errors in the form of DNA base-pair mismatches and small insertions and deletions (indels). These lesions result from failed proofreading of replication polymerases and polymerase slippage during replication, respectively. The high fidelity of polymerases and their proofreading exonuclease activities result in a low error rate of ∼10-7 mismatches per base pair per replication, and this is improved upon by the MMR machinery, which increases fidelity an additional 50-1000-fold [39]. Indels are generated more frequently, especially in repetitive sequences [35,40].
2.2 Mechanisms of the MMR Machinery
The MMR machinery is responsible for identifying and correcting mismatches and indels in newly synthesized DNA, as depicted in Figure 2. This process involves several major steps. In the first step MutSα (heterodimer of MSH2 and MSH6) or MutSβ (heterodimer of MSH2 and MSH3) recognize and bind the mismatched region. MutSα, which contains 85% of cellular MSH2, is responsible for recognizing and binding all base pair mismatches and 1-2 base pair indels while Mutsβ can only efficiently repair indels [35,41]. Next, though the mechanism is not well understood, MutSα undergoes a mismatch and ATP-dependent conformational change that allows for the binding of MutLα, a heterodimer containing MLH1 and PMS2. It is believed that MutLα, which has endonuclease activity when activated by proliferating cell nuclear antigen (PCNA), identifies and nicks the nascent strand of DNA, initiating excision of the DNA strand containing the replication error [42]. Several models exist that explain the removal of the mismatch in both the 5′-to-3′ and 3′-to-5′ directions. Excision is followed by resynthesis of DNA and ligation. A detailed review on current models in MMR has recently been published [35].
Figure 2.
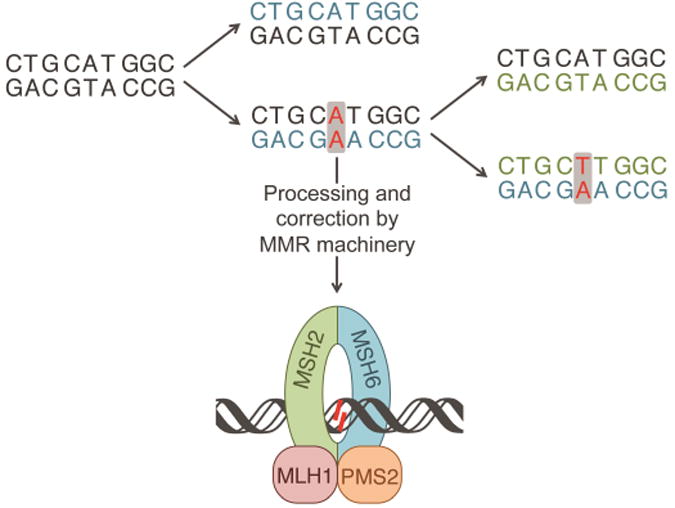
Propagation of an AA mismatch through multiple replications, resulting in a TA mutation. The first iteration of replication results in an AA mismatch, shown in red. The mismatch can be processed and repaired by the MMR machinery shown at the bottom. If unprocessed, upon a second iteration of replication the mismatch will result in a mutation, shown in red. Newly synthesized DNA from the first iteration and second iterations are shown in blue and green, respectively.
Deficiencies in either MutSα or MutLα have been associated with a loss of MMR proficiency. MMR deficiencies result in a 50-1000 fold increase in mismatches within the cell. When these mismatches are left unrepaired, they can propagate to form potentially catastrophic mutations in future generations of cells. As such, deficiencies in MMR machinery are associated with many forms of cancer, including nearly 80% of hereditary non-polyposis colon cancers and 15-20% of all solid tumors [43,44]. Additionally, these cancers often show resistance to traditional chemotherapeutics, such as cisplatin and alkylating agents, making them excellent candidates for targeted therapy [45].
3. Binding of Rhodium Metalloinsertors to DNA Mismatches
3.1 Designing a mismatch-targeting molecule
Our laboratory has explored complexes that can non-covalently target specific DNA sequences (such as 5′-TGCA-3′ and 5′-py-py-pu-3′ sites) as well as the non-conventional A- and Z- forms of DNA, but these targets are not implicated in disease and thus lack therapeutic potential [13,46–48]. DNA mismatches, however, are generic DNA targets that are involved in many types of cancer, as described above. Due to imperfect hydrogen bonding and π-stacking, DNA base pair mismatches are thermodynamically destabilized compared to well-matched DNA [49]. This slight destabilization has been successfully targeted through the use of rhodium metalloinsertors, which contain the sterically expansive 5,6-chrysenequinone diimine (chrysi) ligand.
The chrysi ligand was designed to be larger than traditional intercalating ligands and more akin in size to a well-matched base pair, making it too bulky to simply intercalate into DNA (Figure 3) [16]. Instead, chrysi interacts with DNA through insertion at a destabilized site. Insertion, which was originally proposed by L. S. Lerman in 1961, is a DNA binding mode in which a DNA base pair is separated and ejected from the π-stack by the inserting molecule [50]. Rhodium(III) was chosen to be a substitutionally inert metal anchor for the chrysi ligand due to its photophysical properties; the rhodium complexes promote DNA strand scission in related metallointercalators with photoexcitation [51]. The rhodium center also anchors two ancillary ligands, which add bulk to the complexes and limit how the chrysi ligand can interact with DNA, largely preventing indiscriminant intercalation [52]. These ancillary ligands can also be selected to tune DNA binding and cellular uptake properties, as discussed below.
Figure 3.
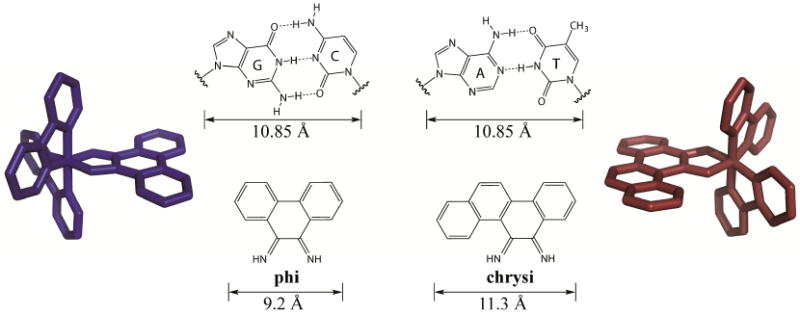
Comparison of the well matched GC and AT mismatches to the non-specific intercalating ligand, phi, and the mismatch-specific inserting ligand, chrysi. Representative rhodium complexes, [Rh(bpy)2(phi)]3+ (blue) and [Rh(bpy)2(chrysi)]3+ (red) are shown. Chrysi is 0.5 Å wider than well-matched base pairs and 2.1 Å larger than phi.
3.2 Targeting DNA mismatches with Rhodium Metalloinsertors
The first generation metalloinsertor, [Rh(bpy)2(chrysi)]3+ (bpy = 2,2′-bipyridine), is easily synthesized via a base-catalyzed condensation reaction between [Rh(bpy)2(NH3)2]3+ and 5,6-chrysene quinone [53]. As predicted, [Rh(bpy)2(chrysi)]3+ can selectively bind DNA base pair mismatches with nanomolar affinities and photocleave the DNA backbone adjacent to the mismatch when irradiated with UV-light [17]. This process is enantioselective, meaning only the right handed Δ-enantiomer can bind right handed, B-form DNA. A series of binding affinity assays were employed to determine the specificity of [Rh(bpy)2(chrysi)]3+. For instance, when incubated with a 2725 base pair linearized plasmid containing a single CC mismatch, [Rh(bpy)2(chrysi)]3+ selectively binds and photocleaves the DNA solely at the mismatched site [54]. No photocleavage is observed with the analogous well-matched plasmid. [Rh(bpy)2(chrysi)]3+ was also incubated with DNA segments containing all possible DNA mismatches and multiple different sequence contexts in which the base pairs flanking the mismatch were varied [55]. Through these experiments, it was determined that [Rh(bpy)2(chrysi)]3+ has >1000-fold preference for targeting mismatched sites over well-matched sites. Additionally, the complex binds and cleaves 80% of all DNA mismatches upon irradiation, irrespective of sequence context. Not surprisingly, the binding affinity of [Rh(bpy)2(chrysi)]3+ towards a mismatch correlates strongly with the thermodynamic destabilization of the mismatch; highly destabilized mismatches (such as CC, CA, and CT mismatches) are easily recognized by [Rh(bpy)2(chrysi)]3+ whereas more stabilized mismatches, specifically mismatches containing guanine, are not preferentially bound by the complex.
A crystal structure at 1.1 Å resolution of [Rh(bpy)2(chrysi)]3+ with a 12-mer oligonucleotide containing two AC mismatches further elucidated the binding mode of the complex to be metalloinsertion [56]. Unlike classical metallointercalators, the metalloinsertor binds DNA via the minor groove and results in little distortion to the DNA backbone. Instead, the DNA accommodates the inserting ligand through the ejection of the mismatched bases out of the π-stack and into the major and minor grooves. This binding mode was verified with an additional crystal structure of the complex bound to an AA mismatch (Figure 4), as well as a solution NMR structure of the complex with DNA containing a CC mismatch [57,58]. This structure provides additional insight into why G-containing mismatches are not detected by metalloinsertors; these highly stable mismatches are not easily ejected from the base-stack, so chrysi cannot displace mismatches at these sites.
Figure 4.
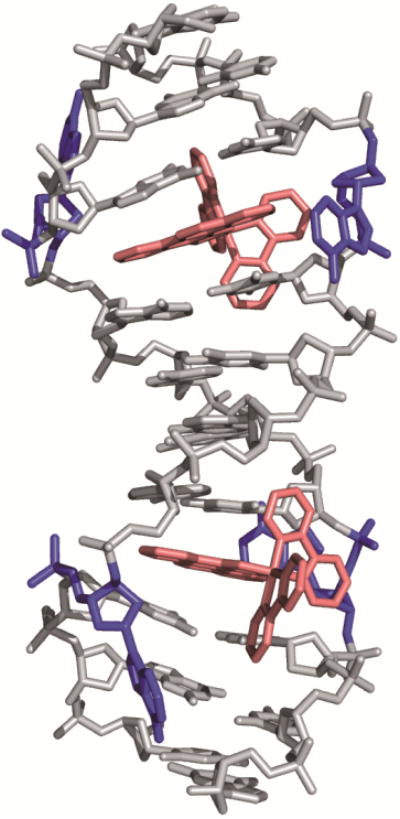
Crystal structure of [Rh(bpy)2(chrysi)]3+ bound to an AA mismatch. The metalloinsertor approaches the mismatch from the minor groove and ejects the mismatched bases from the DNA π-stack. Structural data from PDB 3GSK.
4. Biological Activity of Rhodium Metalloinsertors
4.1 Targeting MMR-deficiencies with Rhodium Metalloinsertors
The therapeutic potential of rhodium metalloinsertors was explored after experiments showed their ability to bind DNA base pair mismatches selectively. It was hypothesized that metalloinsertors would have increased toxicity towards cells that contain an increased number of DNA mismatches, as is present in MMR-deficient cells. To test this hypothesis, an enzyme-linked immunosorbent assay (ELISA) for cellular proliferation was performed with two colorectal cancer cell lines, the HCT116O cell line, which is MMR-deficient, and the MMR-proficient HCT116N cell line. These cell lines, which originate from the MLH1 deficient HCT116 parent cell line, are isogenically matched; the HCT116N cell line is transfected with human chromosome 3, which encodes for a functioning MLH1 gene, and the HCT116O cell line is transfected with human chromosome 2, leaving it MMR-deficient [59]. [Rh(bpy)2(chrysi)]3+ was found to have increased potency in the MMR-deficient cell line [60]. These complexes have also been seen to bind abasic sites and single-base bulges in DNA [61,62].
As with the DNA binding studies, only the Δ enantiomer was biologically active, suggesting the compounds do not decompose or racemize within the cell. This unique activity, the ability to selectively kill MMR-deficient cells over their MMR-proficient counterparts, has been found to be common to many rhodium metalloinsertor [31,63,64]. Importantly, while general to metalloinsertors, these are the only complexes known to exhibit this type of selectivity, with common chemotherapeutics such as cisplatin and the DNA-alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) showing the opposite trend (Figure 5) [45]. These results have been additionally verified in a MSH2-deficient cell line, showing that the characteristic activity of these complexes towards MMR-deficient cell lines is dependent on the MMR-deficient phenotype rather than a unique feature of the HCT116 cell lines [65].
Figure 5.
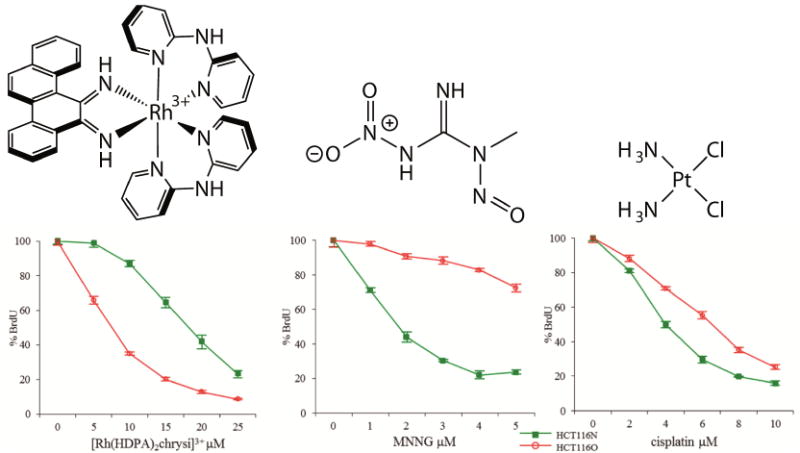
ELISA showing the biological activity of (left) [Rh(HDPA)2(chrysi)]Cl3, a rhodium metalloinsertor, (center) MNNG, a DNA-alkylating agent, and (right) cisplatin, a DNA-binding chemotherapeutic. The metalloinsertor preferentially targets the MMR-deficient HCT116O cell line (red) over the MMR-proficient HCT116N cell line (green). MNNG and cisplatin show the opposite activity by preferentially targeting the MMR-proficient cell line. Adapted from [64]
That the complexes show cell-selective inhibitory activity is somewhat surprising, however, considering that metalloinsertors interact only non-covalently with DNA and even MMR-deficient cells contain relatively few mismatches. Like other non-covalent DNA intercalators and groove binders, it is possible that rhodium metalloinsertors bind DNA mismatches and disrupt transcription or replication processes, leading to selective cytotoxicity in MMR-deficient cells. Ongoing research in our lab aims to better understand the activity of rhodium metalloinsertors and the mechanisms that lead to this selective cell death.
4.2 DNA-binding affinity and subcellular localization of metalloinsertors
As discussed previously, the design of the inserting chrysi ligand was central to obtaining mismatch specificity. It is important to note, however, that the design of the ancillary ligands has proven also to be important. The metalloinsertion binding mode places the ancillary ligands of the rhodium complexes in close proximity to the DNA bases and backbone. For this reason, several structure-function studies have been performed to determine the effect of the ancillary ligands on biological activity. In one study, the ancillary ligands were varied in size from small ammine groups to bulky 4,7-diphenyl-phenanthroline (DIP) groups [31]. The binding affinities to mismatched DNA spanned over 3 orders of magnitude and correlated well to biological activity, with higher affinity mismatch-binding complexes being more selective than their low-affinity counterparts. Again, none of the complexes showed selective inhibition of the MMR-proficient cells.
Differences in binding affinity, however, are not the sole predictor of biological activity. The activities of two structurally similar metalloinsertors, [Rh(DPAE)2(chrysi)]3+ and [Rh(PrDPA)2(chrysi)]3+ (DPAE = 2-(di(pyridin-2-yl)amino)ethanol, PrDPA =N-propyl-N-(pyridin-2-yl)pyridin-2-amine), were examined to explore in more detail the importance of uptake and subcellular localization (Figure 6) [66]. These two complexes have similar mismatch binding affinities and differ only in the presence of either ethanol or propyl modified HDPA (2,2′-dipyridylamine) ligands. The addition of these ligands does lead to a difference in lipophilicity, however, as measured by the partition constant. As expected, the more lipophilic complex, [Rh(PrDPA)2(chrysi)]3+, showed significantly higher cellular uptake than [Rh(DPAE)2(chrysi)]3+, making its non-selective biological activity initially surprising. However, upon subcellular fractioning and analysis using inductively coupled plasma spectrometry (ICP-MS), it became clear that the subcellular localization of these two complexes provided an explanation for their activities. [Rh(PrDPA)2(chrysi)]3+ showed a 10-fold increase in mitochondrial uptake over [Rh(DPAE)2(chrysi)]3+. Although more [Rh(PrDPA)2(chrysi)]3+ was also found in the nucleus of the cells, the percentage of total Rh found within the nucleus is higher for [Rh(DPAE)2(chrysi)]3+. These results suggest that localization of complexes to the mitochondria eradicates their biological selectivity for MMR-deficient cells and leads to MMR-independent death of both cell lines. Interestingly, these findings also support the hypothesis that metalloinsertors achieve their cell selectivity through binding nuclear DNA mismatches, not mitochondrial DNA.
Figure 6.
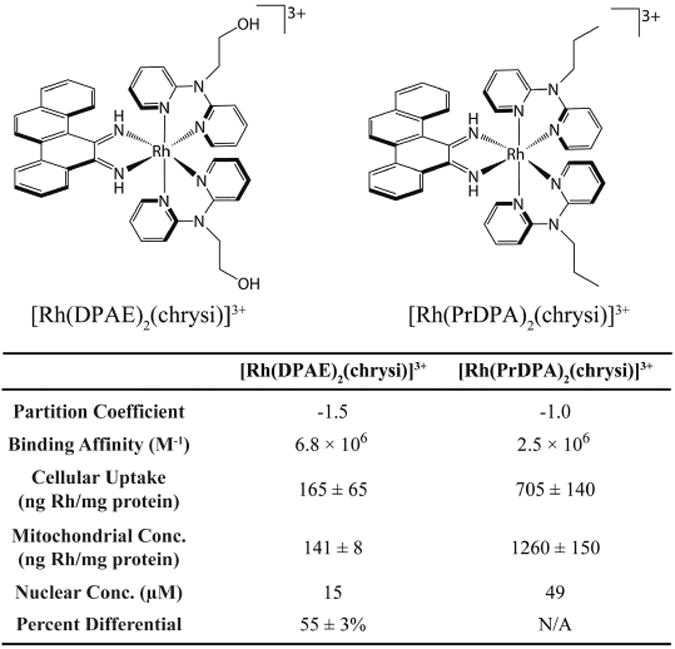
Structures of two closely related metalloinsertors, [Rh(DPAE)2(chrysi)]3+ and [Rh(PrDPA)2(chrysi)]3+, and a table of relevant structural and biological parameters. Data from [66].
The effects of subcellular localization were further examined with a larger family of complexes that differed primarily in lipophilicity [63]. With the exception of [Rh(DIP)2(chrysi)]3+, all complexes display similar binding affinities to mismatched DNA in the 106 to 107 M-1 range. Once again, it was found that biological selectivity correlated not with overall cellular uptake or nuclear localization, but instead with mitochondrial localization. As expected, the more lipophilic, greasy cations showed the highest mitochondrial localization, which correlated with elimination of biological selectivity [67]. The more hydrophilic complexes had significantly lower localization to the mitochondria, which correlated with higher cell selectivity. These studies highlighted the importance not only of cellular uptake but also considerations of subcellular localization. These studies furthermore suggested that off-target effects, such as seen with mitochondrial targeting, are detrimental to the biological function of metalloinsertors.
4.3 Conjugation of Metalloinsertors to cytotoxic agents
In an effort to increase the potency of metalloinsertors and obtain a covalent complex with DNA, our laboratory synthesized several new complexes that conjugate a selective rhodium metalloinsertor to a potent cytotoxic agent [68–70]. Three conjugates were designed that combine a reactive platinum moiety with a rhodium metalloinsertor (Figure 7) [69,71,72]. The conjugates were constructed to work through three different mechanisms. RhPt1 permanently tethers a Pt moiety to the metalloinsertor through a long flexible chain, which allows for concurrent binding of both the Pt center and the metalloinsertor. In theory, the metalloinsertor would bind a mismatch and guide the Pt moiety to a nearby GG on the same DNA strand. In experiments monitoring binding to oligonucleotides containing mismatches, increased platination of DNA containing the mismatch was observed, but significant platination of well-matched DNA was still present. In a different approach, RhPt2 was designed to contain an oxaliplatin moiety that can dissociate from the metalloinsertor. Ideally, after the metalloinsertor binds mismatched DNA, the oxaliplatin moiety would dissociate and bind the same DNA strand nearby. Once again, increased platination was observed for mismatched DNA, but the complex was not biologically selective. Finally, RhPt3 contains a short tether to a monofunctional Pt moiety. In contrast to RhPt1, this short tether would force Pt to bind DNA at the mismatched site following metalloinsertion. Although significantly more potent than cisplatin, this complex was also not selective towards MMR-deficient cells.
Figure 7.
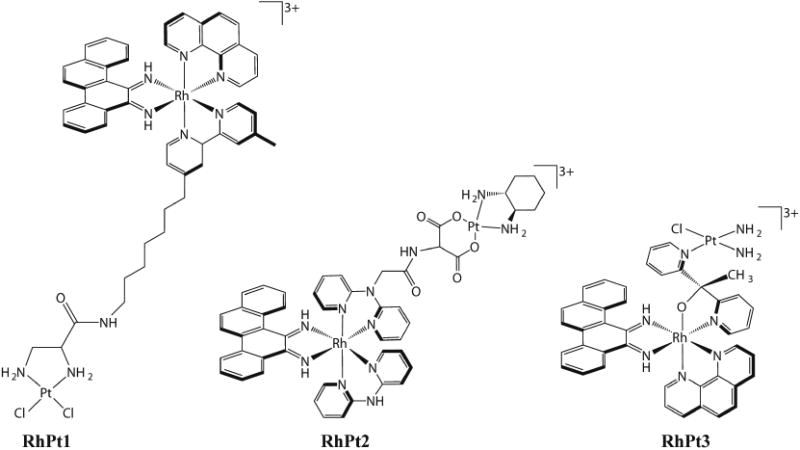
Structures of three metalloinsertor conjugates that combine a potent cisplatin-moiety with a selective rhodium metalloinsertor.
In all three experiments, the selective metalloinsertor was designed to bind a mismatch and therefore preferentially guide the covalent DNA-binding platinum moiety to mismatched DNA. However, these complexes do not exhibit selective cytotoxicity towards MMR-deficient cells. Even RhPt2, which showed increased platination of mismatched DNA in vitro, is not specific for MMR-deficient cells. It is likely that the differences between traditional rhodium metalloinsertors and RhPt conjugates are caused by the mode of cell death initiated by these complexes. RhPt conjugates undergo apoptotic cell death, similar to their parent complexes cisplatin and oxaliplatin. This result is in stark contrast to the distinctly necrotic cell death caused by traditional rhodium metalloinsertors. These observations suggest that initiation of necrotic cell death may be essential to the selective activity of metalloinsertors. It is possible that RhPt conjugates promote both modes of cell death, but the apoptotic pathway overrides the necrotic pathway, leading to biological activity reminiscent of traditional platinum drugs.
5. Current Design of Rhodium Metalloinsertors
Recently, a new family of potent rhodium metalloinsertors based on [Rh(DPE)(chrysi)(phen)]2+(DPE=1,1-di(pyridine-2-yl)ethan-1-ol, phen=1,10-phenanthroline) has been examined (Figure 8) [64]. Unlike previous generations of metalloinsertors, these complexescontain an unusual Rh-O bond through the coordination of their pyridyl-ethanol ligands. These Rh-O containing metalloinsertors are more potent than cisplatin and display optimal differential cellular activity in the nanomolar range, as much as two orders of magnitude more potent than earlier generations of complexes. Surprisingly, this scaffold is robust to many substitutions of the oxygen-containing ligand: replacing the dangling pyridyl group of DPE with a small methyl group, a phenyl group, or a greasy hexyl group all lead to improved, nanomolar cytotoxicity in MTT assays. Furthermore, and surprisingly, both the Δ and Λ enantiomers of these new complexes bind DNA with similar affinity in vitro and both show differential cell-selective activity in MTT assays.
Figure 8.
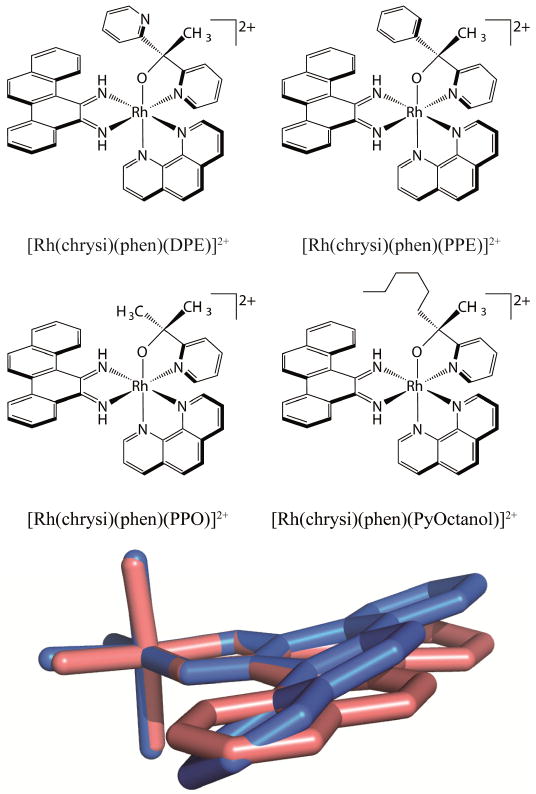
A family of potent metalloinsertors. (top) Structure of four metalloinsertors containing an unusual Rh-O coordination. (bottom) Overlay of the inserting chrysi ligands of planar [Rh(bpy)2(chrysi)]3+ (pink) and buckled [Rh(DPE)(chrysi)(phen)]2+ (blue). DPE, bpy, and phen ligands have been removed for clarity. Adapted from [64]
Remarkably, the increased potency and selectivity of [Rh(DPE)(chrysi)(phen)]2+ and its derivatives is not a result of increased DNA binding or localization. For example, in comparison to the complex [Rh(phzi)(NH3)4]3+, a selective metalloinsertor that utilizes the expansive benzo[α]phenazine quinone diimine (phzi) ligand, [Rh(DPE)(chrysi)(phen)]2+ possesses an order of magnitude lower mismatch binding affinity, similar cellular uptake and mitochondrial concentrations, and slightly lower nuclear concentration. Despite these differences, [Rh(DPE)(chrysi)(phen)]2+ shows comparable selectivity towards MMR-deficient cells as the tetrammine complex but is nearly 4-fold more potent than [Rh(phzi)(NH3)4]3+. Initially, it seemed possible that the increased potency and racemic binding could be due to a labile Rh-O bond, leading to covalent DNA binding and racemization within the cell. However, these possibilities were eliminated using several in vitro tests of stability. It appears that the enantiomeric activity of these complexes is authentic, with both Δ and Λ enantiomers being able to kill MMR-deficient cells selectively without racemization.
One apparent difference between the Rh-O containing metalloinsertors and previous generations are dramatic changes in the pKa of the chrysi imine protons. The pKa of the first generation complex, [Rh(bpy)2(chrysi)]3+, is 5.6 ± 0.2; thus the complex is deprotonated at cellular pH. The deprotonation of the chrysi ligand relieves steric clashes between its imine proton and aromatic ring system protons, allowing the chrysi ligand to lay planar (Figure 8). This planarity can be seen in the crystal structure of [Rh(bpy)2(chrysi)]3+ bound to a DNA mismatch. The Rh-O containing metalloinsertors have significantly higher pKa values of 8.3-8.9, meaning the chrysi ligand cannot deprotonate at cellular pH. To relieve steric clashing, the chrysi ligand must instead buckle relative to [Rh(bpy)2(chrysi)]3+ (Figure 8). This buckling can be seen in the crystal structure of [Rh(DPE)(chrysi)(phen)]2+. While we still believe the binding mode of these complexes to be metalloinsertion, it appears that this significant structural change in the inserting ligand of these complexes must create a slightly different DNA lesion than [Rh(bpy)2(chrysi)]3+. The new lesion or lesions must accommodate both the Δ and Λ enantiomers. Within the cell, this lesion may be more easily recognized by proteins activating necrotic cell death than lesions caused by previous metalloinsertors, leading to the increased potency and selectivity of these complexes. Clearly much needs to be done to elucidate the basis of the high potency and cell selectivity of these new complexes, both from a structural standpoint and with regard to understanding the biological fate of the complexes.
6. Future Outlook
Rhodium metalloinsertors constitute a unique family of transition metal complexes that selectively bind DNA base pair mismatches and preferentially inhibit proliferation and survival of MMR-deficient cells. Over the years, the design of these complexes has improved significantly, traversing from micromolar toxicities now into the nanomolar range. Thus a unique family of rhodium metalloinsertors with nanomolar potencies and high selectivity for MMR-deficient cell lines has now been characterized, bringing these complexes into the realm of therapeutic interest [64].
Despite improvements to the design of these complexes, however, relatively little is still understood with respect to their mechanism of action within the cell. The complexes possess similar, if not identical binding properties and localization patterns to previous generations of metalloinsertors, yet they are significantly more potent and selective. Additionally, from a biological standpoint, it seems incredible that the modest increase in DNA lesions associated with MMR-deficiencies can be so selectively targeted by these complexes. Clearly future studies must be carried out to better understand the selective activity of these complexes, specifically to identify biological pathways or specific proteins that may recognize the metalloinsertor-DNA lesion.
Based on the metalloinsertive binding mode of [Rh(bpy)2(chrysi)]3+, elucidated through solid and solution state structures, as well as in vitro and in cellulo work performed on the extensive family of metalloinsertors, we hypothesize that these complexes bind DNA mismatches within MMR-deficient cells, and that this Rh-DNA lesion is recognized by proteins and cellular machinery that eventually lead the cell to necrosis. Unlike cisplatin, these complexes do not appear to form covalent adducts with DNA. Instead, it is possible that the non-covalent binding of metalloinsertors inhibits proteins involved in DNA processes such as transcription or replication, similar to other non-covalent DNA groove binders and intercalators. In such a case, their activity may only be evident in MMR-deficient cells containing increased concentrations of mismatches, leading to their observed selectivity. It is possible that the lesion formed by the more potent and selective complexes of the current generation, that can accommodate the Δ and Λ enantiomer, may also be more recognizable in the cell, or processed more efficiently to produce necrosis. Future work on metalloinsertors will emphasize characterizing the structures of metalloinsertors bound to their target mismatches, understanding the biological mechanisms of metalloinsertors, identifying proteins and pathways involved in their processing, and developing future generations of metalloinsertors with improved potency or selectivity for use as a foundation for a new family of chemotherapeutics.
Indeed what has been clear from these studies at the outset is that transition metal chemistry offers a rich variety of means to target DNA sites along with novel methods to characterize their fates inside cells. Cisplatin, despite its simplicity in structure and relative lack of specificity in cellular targeting, has proven to be a powerfully important therapeutic. We expect that greater specificity in cellular targeting along with greater specificity in DNA targeting can only aid us in the development of new strategies upon which to build and potentially new potent and selective metal compounds as cancer therapeutics.
Highlights.
Cancers associated with MMR deficiencies are often resistant to chemotherapeutics
Rhodium metalloinsertors selectively bind DNA base pair mismatches
Rhodium metalloinsertors are preferentially cytotoxic to MMR deficient cell lines
Recent rhodium metalloinsertors have improved nanomolar potency and selectivity
Improved metalloinsertors show therapeutic promise for MMR deficient cancers
Acknowledgments
We are grateful to our coworkers and collaborators in elucidating this chemistry and to the NIH for their support of this research (GM33309). We also thank the United States Department of Defense for an NDSEG research fellowship to K.M.B.
Abbreviations
- MMR
mismatch repair
- DHFR
dihydrofolate reductase
- EGFR
epidermal growth factor receptor
- PCNA
proliferating cell nuclear antigen
- chrysi
5,6-chrysene quinone diimine
- bpy
2,2′-bipyridine
- ELISA
enzyme-linked immunosorbent assay
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
- DIP
4,7-diphenyl-phenanthroline
- DPAE
2-(di(pyridine-2-yl)amino)ethanol
- PrDPA
N-propyl-N(pyridine-2-yl)pyridine-2-amine
- HDPA
2,2′-dipyridylamine
- ICP-MS
inductively coupled plasma-mass spectrometry
- DPE
1,1-di(pyridine-2-yl)ethan-1-ol
- phen
1,10-phenanthroline
- phzi
benzo[a]phenazine quinone diimine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gilman A, Philips FS. The biological actions and therapeutic applications of the B-chloroethyl amines and sulfides. Science (80-) 1946;103:409–436. doi: 10.1126/science.103.2675.409. [DOI] [PubMed] [Google Scholar]
- 2.Farber S, Diamond LK, Mercer RD, Sylvester RF, Jr, Wolff JA. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (aminopterin) N Engl J Med. 1948;238:787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 3.Goodman LS, Wintrobe MM, Dameshek W, Goodman MJ, Gilman A, McLennan MT. Nitrogen mustard therapy: Use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin's disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J Am Med Assoc. 1946;132:126–132. doi: 10.1001/jama.1946.02870380008004. [DOI] [PubMed] [Google Scholar]
- 4.Li MC, Hertz R, Bergenstal DM. Therapy of choriocarcinoma and related trophoblastic tumors with folic acid and purine antagonists. N Engl J Med. 1958;259:66–74. doi: 10.1056/NEJM195807102590204. [DOI] [PubMed] [Google Scholar]
- 5.Hurley LH. DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- 6.Palchaudhuri R, Hergenrother PJ. DNA as a target for anticancer compounds: methods to determine the mode of binding and the mechanism of action. Curr Opin Biotechnol. 2007;18:497–503. doi: 10.1016/j.copbio.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 7.White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB. Recognition of the four Watson|[ndash] | Crick base pairs in the DNA minor groove by synthetic ligands. Nature. 1998;391:468–471. doi: 10.1038/446391a. [DOI] [PubMed] [Google Scholar]
- 8.Shalaby T, Fiaschetti G, Nagasawa K, Shinya K, Baumgartner M, Grotzer M. G-quadruplexes as potential therapeutic targets for embryonal tumors. Molecules. 2013;18:12500–12537. doi: 10.3390/molecules181012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Komor AC, Barton JK. A path for metal complexes to a DNA target. Chem Commun. 2013;49:3617–3630. doi: 10.1039/c3cc00177f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeglis BM, Pierre VC, Barton JK. Metallointercalators and Metalloinsertors. Chem Commun. 2007;44:4565–4579. doi: 10.1016/j.biotechadv.2011.08.021.Secreted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul A, Bhattacharya S. Chemistry and biology of DNA-binding small molecules. Curr Sci. 2012;102:212–231. [Google Scholar]
- 13.Krotz AH, Hudson BP, Barton JK. Assembly of DNA recognition elements on an octahedral rhodium intercalator: predictive recognition of 5′-TGCA-3′ by D-[Rh(R,R)-Me2trien]phi]3+ J Am Chem Soc. 1993;115:12577–12578. doi: 10.1021/ja00079a044. [DOI] [Google Scholar]
- 14.Baraldi PG, Bovero A, Fruttarolo F, Preti D, Tabrizi MA, Pavani MG, et al. DNA minor groove binders as potential antitumor and antimicrobial agents. Med Res Rev. 2004;24:475–528. doi: 10.1002/med.20000. [DOI] [PubMed] [Google Scholar]
- 15.Wheate NJ, Brodie CR, Collins JG, Kemp S, Aldrich-Wright JR. DNA Intercalators in Cancer Therapy: Organic and Inorganic Drugs and Their Spectroscopic Tools of Analysis. Mini Rev Med Chem. 2007;7:627–648. doi: 10.2174/138955707780859413. [DOI] [PubMed] [Google Scholar]
- 16.Zeglis BM, Pierre VC, Barton JK. Metallo-intercalators and metallo-insertors. Chem Commun. 2007:4565–4579. doi: 10.1039/b710949k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson BA, Barton JK. Recognition of DNA base mismatches by a rhodium intercalator. J Am Chem Soc. 1997;119:12986–12987. [Google Scholar]
- 18.Barbier O, Jacquillet G, Tauc M, Cougnon M, Poujeol P. Effect of heavy metals on, and handling by, the kidney. Nephron - Physiol. 2005;99:105–110. doi: 10.1159/000083981. [DOI] [PubMed] [Google Scholar]
- 19.Shi H, Hudson LG, Liu KJ. Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radic Biol Med. 2004;37:582–593. doi: 10.1016/j.freeradbiomed.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 20.Domingo JL. Metal-induced developmental toxicity in mammals: A review. J Toxicol Environ Health. 1994;42:123–141. doi: 10.1080/15287399409531868. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg B, Van Camp L, Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 22.Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalt Trans. 2010;39:8113–8127. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 23.Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 24.Eastman A. Reevaluation of interaction of cis-dichloro (ethylenediamine) platinum (II) with DNA. Biochemistry. 1986;25:3912–3915. doi: 10.1021/bi00361a026. [DOI] [PubMed] [Google Scholar]
- 25.Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. 1995;377:649–652. doi: 10.1038/377649a0. [DOI] [PubMed] [Google Scholar]
- 26.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 27.Weidmann AG, Komor AC, Barton JK. Targeted Chemotherapy with Metal Complexes. Comments Inorg Chem. 2014;34:1–10. doi: 10.1080/02603594.2014.890099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12:5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 30.Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 31.Ernst RJ, Song H, Barton JK. DNA mismatch binding and antiproliferative activity of rhodium metalloinsertors. J Am Chem Soc. 2009;131:2359–2366. doi: 10.1021/ja8081044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Granzhan A, Kotera N, Teulade-Fichou MP. Finding needles in a basestack: Recognition of mismatched base pairs in DNA by small molecules. Chem Soc Rev. 2014;43:3630–3665. doi: 10.1039/c3cs60455a. [DOI] [PubMed] [Google Scholar]
- 33.Dexheimer TS. DNA Repair Cancer Stem Cells. Springer; 2013. DNA repair pathways and mechanisms; pp. 19–32. [Google Scholar]
- 34.Hoeijmakers JHJ. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 35.Kunkel TA, Erie DA. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu Rev Genet. 2015;49 doi: 10.1146/annurev-genet-112414-054722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schärer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5:a012609. doi: 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dianov GL, Hübscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013;41:3483–3490. doi: 10.1093/nar/gkt076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 39.Kunkel TA. DNA replication fidelity. J Biol Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 40.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLα in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 43.Kolodner RD. Mismatch repair: mechanisms and relationship to cancer susceptibility. Trends Biochem Sci. 1995;20:397–401. doi: 10.1016/s0968-0004(00)89087-8. [DOI] [PubMed] [Google Scholar]
- 44.Arzimanoglou II, Gilbert F, Barber HRK. Microsatellite instability in human solid tumors. Cancer. 1998;82:1808–1820. doi: 10.1002/(sici)1097-0142(19980515)82:10<1808::aid-cncr2>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 45.Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998;4:1–6. [PubMed] [Google Scholar]
- 46.Sitlani A, Long EC, Pyle AM, Barton JK. DNA photocleavage by phenanthrenequinone diimine complexes of rhodium (III): shape-selective recognition and reaction. J Am Chem Soc. 1992;114:2303–2312. [Google Scholar]
- 47.Mei HY, Barton JK. Tris (tetramethylphenanthroline) ruthenium (II): a chiral probe that cleaves A-DNA conformations. Proc Natl Acad Sci. 1988;85:1339–1343. doi: 10.1073/pnas.85.5.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barton JK, Raphael AL. Site-specific cleavage of left-handed DNA in pBR322 by lambda-tris (diphenylphenanthroline) cobalt (III) Proc Natl Acad Sci. 1985;82:6460–6464. doi: 10.1073/pnas.82.19.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan S, Sun X, Lee JK. Stability of complementary and mismatched DNA duplexes: Comparison and contrast in gas versus solution phases. Int J Mass Spectrom. 2006;253:238–248. [Google Scholar]
- 50.Lerman LS. Structural considerations in the interaction of DNA and acridines. J Mol Biol. 1961;3:18–IN14. doi: 10.1016/s0022-2836(61)80004-1. [DOI] [PubMed] [Google Scholar]
- 51.Uchida K, Pyle AM, Morii T, Barton JK. High resolution footprinting of EcoRI and distamycin with Rh (phi) 2 (bpy) 3+, a new photofootprinting reagent. Nucleic Acids Res. 1989;17:10259–10279. doi: 10.1093/nar/17.24.10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suri K, Asif, Mao B, Amin S, Geacintov NE, Patel DJ. Solution Structure of the (+)-trans-anti-benzo[g]chrysene-dA Adduct Opposite dT in a DNA Duplex. J Mol Biol. 1999;292:289–307. doi: 10.1006/jmbi.1999.2974. [DOI] [PubMed] [Google Scholar]
- 53.Zeglis BM, Barton JK. DNA base mismatch detection with bulky rhodium intercalators: Synthesis and Applications. Nat Protoc. 2007;2:357–371. doi: 10.1038/nprot.2007.22.DNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jackson BA, Alekseyev VY, Barton JK. A versatile mismatch recognition agent: specific cleavage of a plasmid DNA at a single base mispair. Biochemistry. 1999;38:4655–4662. doi: 10.1021/bi990255t. [DOI] [PubMed] [Google Scholar]
- 55.Jackson BA, Barton JK. Recognition of base mismatches in DNA by 5, 6-chrysenequinone diimine complexes of rhodium (III): a proposed mechanism for preferential binding in destabilized regions of the double helix. Biochemistry. 2000;39:6176–6182. doi: 10.1021/bi9927033. [DOI] [PubMed] [Google Scholar]
- 56.Pierre VC, Kaiser JT, Barton JK. Insights into finding a mismatch through the structure of a mispaired DNA bound by a rhodium intercalator. Proc Natl Acad Sci. 2007;104:429–434. doi: 10.1073/pnas.0610170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeglis BM, Pierre VC, Kaiser JT, Barton JK. A bulky rhodium complex bound to an adenosine-adenosine DNA mismatch: general architecture of the metalloinsertion binding mode. Biochemistry. 2009;48:4247–4253. doi: 10.1021/bi900194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cordier C, Pierre VC, Barton JK. Insertion of a bulky rhodium complex into a DNA cytosine-cytosine mismatch: an NMR solution study. J Am Chem Soc. 2007;129:12287–12295. doi: 10.1021/ja0739436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maida M, Aebi S, Fink D, Howell SB, Los G. Loss of DNA mismatch repair: effects on the rate of mutation to drug resistance. J Natl Cancer Inst. 1997;89:1537–1541. doi: 10.1093/jnci/89.20.1537. [DOI] [PubMed] [Google Scholar]
- 60.Hart JR, Glebov O, Ernst RJ, Kirsch IR, Barton JK. DNA mismatch-specific targeting and hypersensitivity of mismatch-repair-deficient cells to bulky rhodium (III) intercalators. Proc Natl Acad Sci. 2006;103:15359–15363. doi: 10.1073/pnas.0607576103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ernst RJ, Komor AC, Barton JK. Selective cytotoxicity of rhodium metalloinsertors in mismatch repair-deficient cells. Biochemistry. 2011;50:10919–10928. doi: 10.1021/bi2015822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeglis BM, Boland JA, Barton JK. Targeting abasic sites and single base bulges in DNA with metalloinsertors. J Am Chem Soc. 2008;130:7530–7531. doi: 10.1021/ja801479y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Komor AC, Schneider CJ, Weidmann AG, Barton JK. Cell-selective biological activity of rhodium metalloinsertors correlates with subcellular localization. J Am Chem Soc. 2012;134:19223–19233. doi: 10.1021/ja3090687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Komor AC, Barton JK. An Unusual Ligand Coordination Gives Rise to a New Family of Rhodium Metalloinsertors with Improved Selectivity and Potency. J Am Chem Soc. 2014;136:14160–14172. doi: 10.1021/ja5072064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bailis JM, Gordon ML, Gurgel JL, Komor AC, Barton JK, Kirsch IR. An inducible, isogenic cancer cell line system for targeting the state of mismatch repair deficiency. 2013;8:e78726. doi: 10.1371/journal.pone.0078726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weidmann AG, Komor AC, Barton JK. Biological effects of simple changes in functionality on rhodium metalloinsertors. Philisophocal Trans R Soc. 2013;371:20120117. doi: 10.1098/rsta.2012.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Modica-Napolitano JS, Aprille JR. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv Drug Deliv Rev. 2001;49:63–70. doi: 10.1016/s0169-409x(01)00125-9. [DOI] [PubMed] [Google Scholar]
- 68.Schatzschneider U, Barton JK. Bifunctional rhodium intercalator conjugates as mismatch-directing DNA alkylating agents. J Am Chem Soc. 2004;126:8630–8631. doi: 10.1021/ja048543m. [DOI] [PubMed] [Google Scholar]
- 69.Petitjean A, Barton JK. Tuning the DNA reactivity of cis-platinum: Conjugation to a mismatch-specific metallointercalator. J Am Chem Soc. 2004;126:14728–14729. doi: 10.1021/ja047235l. [DOI] [PubMed] [Google Scholar]
- 70.Lim MH, Lau IH, Barton JK. DNA strand cleavage near a CC mismatch directed by a metalloinsertor. Inorg Chem. 2007;46:9528–9530. doi: 10.1021/ic701598k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weidmann AG, Barton JK. Construction and Application of a Rh--Pt DNA Metalloinsertor Conjugate. Inorg Chem. 2014;53:7812–7814. doi: 10.1021/ic501509x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weidmann AG, Barton JK. A Monofunctional Platinum Complex Coordinated to a Rhodium Metalloinsertor Selectively Binds Mismatched DNA in the Minor Groove. Inorg Chem. 2015;54:9626–9636. doi: 10.1021/acs.inorgchem.5b01722. [DOI] [PMC free article] [PubMed] [Google Scholar]