

Abstract
Mitochondria contain multiple copies of the circular mitochondrial genome (mtDNA) that encodes ribosomal RNAs and proteins locally translated for oxidative phosphorylation. Loss of mtDNA integrity, both altered copy number and increased mutations, is implicated in cellular dysfunction with aging. Published data on mtDNA copy number and aging is discordant which may be due to methodological limitations for quantifying mtDNA copy number. Existing quantitative PCR (qPCR) mtDNA copy number quantification methods provide only relative abundances and are problematic to normalize to different template input amounts and across tissues/sample types. As well, existing methods cannot quantify mtDNA copy number in subcellular isolates, such as isolated mitochondria and neuronal synaptic terminals, which lack nuclear genomic DNA for normalization. We have developed and validated a novel absolute mtDNA copy number quantitation method that uses chip-based digital polymerase chain reaction (dPCR) to count the number of copies of mtDNA and used this novel method to assess the literature discrepancy in which there is no clear consensus whether mtDNA numbers change with aging in skeletal muscle. Skeletal muscle in old mice was found to have increased absolute mtDNA numbers compared to young controls. Furthermore, young Sod1 −/− mice were assessed and show an age-mimicking increase in skeletal muscle mtDNA. These findings reproduce a number of previous studies that demonstrate age-related increases in mtDNA. This simple and cost effective dPCR approach should enable precise and accurate mtDNA copy number quantitation in mitochondrial studies, eliminating contradictory studies of mitochondrial DNA content with aging.
Electronic supplementary material
The online version of this article (doi:10.1007/s11357-016-9930-1) contains supplementary material, which is available to authorized users.
Keywords: Mitochondria, Digital PCR, mtDNA, mtDNA copy number, Aging, Sod1, Synaptosomes
Introduction
Mitochondria, the cellular organelles responsible for the majority of ATP generation through oxidative phosphorylation, are critical to maintaining a proper homeostatic cellular environment (Taylor and Turnbull 2005). Each mitochondrion contains multiple circular copies of the maternally inherited mitochondrial genome (mtDNA), which encodes necessary subunits for oxidative phosphorylation and ribosome formation for proper mitochondria-localized mRNA translation (Taylor and Turnbull 2005). Many diseases (Dimauro and Davidzon 2005) have been linked to genetically and biochemically dysfunctional mitochondria including cancer (Dickinson et al. 2013), diabetes (Sharma 2015), and central nervous system disorders (Chaturvedi and Flint Beal 2013). More subtly, damage to mitochondria is hypothesized to drive the aging process, termed the mitochondrial free-radical theory of aging (Barja 2013; Benz and Yau 2008). Additionally, cumulative damage to mitochondria is hypothesized to perpetuate disease phenotypes in cancer (Chatterjee et al. 2006) and diabetes (Kowluru 2013).
A critical aspect of mitochondrial and subsequently cellular homeostasis is the regulation of mtDNA copy number (Clay Montier et al. 2009; Shmookler Reis and Goldstein 1983; Veltri et al. 1990). Mitochondria can undergo fission and fusion events as part of mitochondrial biogenesis and turnover, respectively, in order to regulate mtDNA copy number. When cells are under demand for energy production, replication of mtDNA is induced and mitochondria undergo fission to meet the energy demands of the cell. Disruption of this process or the failure to adapt can lead to cellular dysfunction (Clay Montier et al. 2009; Ekstrand et al. 2004). Therefore, quantifying mtDNA ploidy/copy number is an important surrogate of tissue and cellular dysfunction and represents a unique form of copy number variation differing from nuclear DNA copy number analysis. A survey of the literature identifies studies showing both increases and decreases in mtDNA copy number as indications of mitochondrial dysfunction (Malik and Czajka 2013). Current approaches to quantifying mtDNA only provide an analog relative quantitation between samples and require the presence of an endogenous control, typically a nuclear-encoded gene, or a standard curve (D’Erchia et al. 2015; Fernandez-Vizarra et al. 2011; Miller et al. 2003; Nicklas et al. 2004; Phillips et al. 2014). In studies where no endogenous reference is present, for example subcellular fractions that do not contain nuclear DNA, these methods cannot be applied. Furthermore, the literature is marred by contradictory findings of changes in mtDNA copy number stemming from these inadequate existing methods (Malik and Czajka 2013). Therefore, we have developed a method to accurately and precisely count mtDNA copy number from genomic DNA (gDNA) samples. This method uses fluorogenic copy number assays designed against mtDNA and chip-based digital PCR (dPCR).
While dPCR was first proposed over a decade ago (Vogelstein and Kinzler 1999), only recently have instruments been optimized to make dPCR a practical reality (Baker 2012; Hindson et al. 2013). dPCR has a number of advantages over conventional PCR including being less sensitive to reaction efficiency differences, great precision, and the ability to absolutely quantify, i.e., count molecules (Hindson et al. 2013). The basic concept of dPCR is that thousands of micro-PCR reactions, in which there are either one or no copies of the template of interest, are performed for each sample. By counting the number of reactions that are positive and negative for reaction products and then performing a Poisson correction, the number of molecules of templet in the original sample can be determined. Initial versions of dPCR chips/plates contained a few thousand wells (Morrison et al. 2006; Warren et al. 2006) or through emulsion PCR (Hindson et al. 2011) in which up to 20,000 droplet reactions were generated. Both of these approaches are valid, though limitations include a low number of reactions for the microwell formats and the added complexity and instrument costs of emulsion PCR. More recently, an ultra-high microwell format with 20,000 wells has been developed which seeks to combine the ease of use of a chip-based assay with the high reaction number of emulsion PCR. The increase in reaction number increases the precision of quantitation.
Here, we report absolute quantification of mtDNA using an ultra-high density chip-based format of dPCR. This method has been shown to have utility in quantifying mtDNA copy number, however, without proper validation (Wang et al. 2014). We validated this method using in vitro-generated standards developed for human, rat, and mouse mtDNA. This new approach was then used to confirm previous findings in the literature of tissue differences in mtDNA. We then applied it in a novel manner to the skeletal muscle and the brain from young and old wild-type mice and young Sod1 −/− mice in order to address the contradictory results in the literature. We found an age-related increase in mtDNA copy number in old wild-type mice and in the skeletal muscle but not the brain from young Sod1 −/− mice, a mouse model of increased oxidative stress.
Material and methods
Animals
Female and male mice lacking CuZn superoxide dismutase (Sod1 −/− mice) were bred and maintained as previously described (Jang et al. 2010; Muller et al. 2006; Wanagat et al. 2015). Wild-type male C57B6 were used for aging comparisons. Tissues were collected and frozen in liquid nitrogen from euthanized mice at 8 (Sod1 −/− and C57B6) or 28 (C57B6) months of age as noted. All procedures were approved by the Institutional Animal Care and Use Committee at the Oklahoma Medical Research Foundation. Male and female Sod1 −/− mice were used for these studies as no differences in mitochondrial content and activity have been shown in the C57B6 strain (Sanz et al. 2007). Studies on differences between the retina and the heart were carried out on 3-month-old male C57B6 mice maintained according to the Institutional Animal Care and Use Committee at the University of Oklahoma Health Sciences Center. Mice were euthanized by decapitation and the retinas and the heart tissue were rapidly excised and frozen in liquid nitrogen. Retinal synaptosomes were isolated from fresh retinas dissected from 3-month-old Sprague Dawley rats euthanized with CO2 followed by cervical dislocation according to the Institutional Animal Care and Use Committee guidelines at the University of Oklahoma Health Sciences Center.
Assay design
Custom fluorogenic copy number assays were designed using GeneAssist Copy Number Assay Workflow Builder (TaqMan, Life Technologies) online tool (https://www.lifetechnologies.com/order/custom-genomic-products/tools/copy-number-variation/). Human (NC_012920.1), rat (NC_001665.2), and mouse (NC_005089.1) mitochondrial genomes were used in designing species-specific copy number assays for mtDNA (Table 1). Two assays (A and B) were designed for each species’ genome. The designs of these assays were targeted for regions of the mtDNA not considered to be in the “common deletion” (Phillips et al. 2014). Human and mouse TERT fluorogenic copy number assays were used as the nuclear reference for human and mouse samples (Life Technologies). A rat nuclear reference assay was designed against Actb on chromosome 12 (NC_005111.3). Each custom assay passed Life Technologies’ internal quality control standards. Additionally, standard PCR and PAGE which were used to size and verify the primers produced a single product of appropriate size from the gDNA of the corresponding species (Fig. S1). Amplicons also matched the targeted sequence as verified by Sanger sequencing.
Table 1.
mtDNA assays and genomic locations
| Assay | Genome accession number | Location (bp, relative to nucleotide 1 of respective reference) | |
|---|---|---|---|
| Hs A | HsMtDNA_CC6RNZN | NC_012920.1 | 678–978 |
| Hs B | HsMtDNA_CCHS0I8 | 15,591–15,891 | |
| Rn A | RnMtDNA_CCI1MPG | NC_001665.2 | 666–966 |
| Rn B | RnMtDNA_CCS0728 | 13,716–14,016 | |
| Mm A | Mm_CCQJBRT | NC_005089.1 | 257–557 |
| Mm B | Mm_CCWR2MX | 14,927–15,227 | |
Assay order numbers from Life Technologies. The genome accession number of each species mtDNA genome where each assay was designed. mtDNA genome location where the primer/probe set was designed
Synaptosome isolation
Synaptosomes were isolated by differential centrifugation in sucrose buffer as described previously (VanGuilder et al. 2008). Synaptosomal and combined nuclear and cytoplasmic, or somatic, cellular fractions were used to isolate total DNA by silica spin column purification (Qiagen). Western blotting on retinal fractions, 3 μg protein per lane, was carried out as previously described (VanGuilder et al. 2008). The following primary antibodies were used; Lamin B1 (abcam ab133741) 1:5000 in 5 % non-fat dry milk in PBST, synaptophysin (abcam ab8049–500) 1:500 in 5 % BSA in PBST, and COXIV (abcam ab14744) 1:1000 in 5 % BSA in PBST. Secondary HRP-conjugated antibodies against mouse (TrueBlot 18–8817-33) and rabbit (TruBlot 18–8816–33), 1:1000 in 5 % BSA or non-fat dry milk in PBST, respectively, were used.
Digital PCR
The workflow for absolute quantitation is described in Fig. 1. Total genomic DNA (gDNA) (both nuclear and mitochondrial) was extracted from rat and mouse tissues using the Qiagen AllPrep DNA/RNA mini column-based prep according to the manufacturer’s instructions. Tissues were homogenized in 2-ml round-bottom microcentrifuge tubes with 600 μl RLT buffer and one 5-mm stainless steel ball using the Retsch TissueLyser II prior to gDNA extraction and elution in 50 μl of H2O. Human gDNA was isolated from saliva using Oragene-Discover OGR-500 (DNAgenotek) genomic DNA isolation kit according to the manufacturer’s instructions. DNA quality was determined by spectrophotometry and quantification was carried out using fluorescent-based PicoGreen assay (Life Technologies). Digital PCR was carried out according to the manufacturer’s instructions by mixing 3.33 μl diluted template gDNA (concentrations of DNA are described below and varied by experiment) with 16.5 μl Quantstudio 3D master mix, 3.33 μl TaqMan assay, and 9 μl water (32.16 μl [enough for two chips with excess]) (Life Technologies). Reactions were loaded onto Quantstudio 3D digital PCR chips using the Quantstudio 3D chip loader according to manufacturer’s instructions (Life Technologies). Chips were then sealed and cycled on a GeneAmp PCR system 9700 with a flatblock attachment using the following conditions: stage 1, 96 °C for 10 min; stage 2, 60 °C for 2 min; then 98 °C for 30 s; repeat stage 2 39 times; stage 3, 60 °C for 2 min; and an infinite 10 °C hold. Chips were then read in the Quantstudio 3D chip reader to obtain raw fluorescent values (Life Technologies). Quality check of the chips and counting of positive and negative wells in order to determine copies/μl were carried out on the Quantstudio 3D AnalysisSuite cloud software (https://apps.lifetechnologies.com/quantstudio3d/index.html; LifeTechnologies) using the Absolute Quantification module. Data from two to three chips per input amount were combined to generate the standard curves.
Fig. 1.

Absolute mtDNA quantitation dPCR workflow. a Total genomic DNA is isolated from tissue/cells and mixed with proper assay components. The reactions are distributed across a chip with 20,000 856-pl wells. Template is diluted to a point where there is either 0 or 1 copies per well. Reactions are then cycled to end-point and fluorescence is read in each well. Those wells that were loaded with template are positive wells, while those wells that were filled without template are negative. Based on the count of fluorescent positive and negative wells and using a Poisson distribution, the number of target copies can be calculated per microliter. b Representative raw fluorescence data from a mtDNA assay (left) and a nuclear DNA reference assay (right) showing chips with positive and negative wells (top) and corresponding well counts of fluorescent intensities showing a bimodal distribution of positive and negative wells
Quantitative PCR
Quantitative PCR was carried out as previously described (Masser et al. 2013; Masser et al. 2014) using 1 ng input gDNA. The same TaqMan assays from dPCR experiments were used for qPCR. Relative mtDNA copy number was calculated by 2-ΔΔct method using the species-specific nuclear assay as the reference.
Standard generation
In vitro standards were needed in order to determine the quantitative accuracy of dPCR; however, no such standards exist; therefore, standards from human, rat, and mouse DNA were created. mtDNA standards were generated by individually cloning the amplicon from assays Hs A, Rn A, and Mm A into TOPO TA plasmids (Life Technologies) and transforming into One Shot TOP10 competent cells (Life Technologies). Transformed cells were plated on kanamycin (50 μg/ml) positive agar plates and incubated overnight at 37 °C. Two colonies were picked per plate and grown in 3 ml Hanahan’s broth (Sigma) with kanamycin (1:1000 of 50 μg/ml) overnight with shaking at 37 °C. Plasmid DNA was isolated from grown cultures using the Zyppy Plasmid Miniprep (Zymo Research) according to manufacturer’s instructions with a final elution volume of 30 μl. Purified plasmid DNA was quality checked by spectrophotometry and quantification was carried out using fluorescent-based PicoGreen assay (Life Technologies). Copy number concentrations of standards (copies/μl) were determined by multiplying the concentration (in ng/μl) of each standard by the inverse of their mass in nanogram. Each plasmid was checked for the correct mtDNA insert sequence by Sanger sequencing using the T3 primer on an ABI 3730 capillary sequencer. Expected copy number concentrations were then determined based on the mass of the plasmid.
Statistics
Statistical tests were carried out with SigmaPlot 12.5 software. Parametric t test was used to determine the difference in two group comparisons, and One-way and Two-way analysis of variance were used for multi group comparisons with α = 0.05 for factors and Student Newman Kuels (SNK) pairwise post hoc testing.
Results
dPCR validation
In vitro-derived standards with either human, rat, or mouse mtDNA amplicons were subjected to dPCR (Fig. 1) in order to validate the quantitative accuracy of the dPCR method. Standards were diluted to reaction input amounts to yield 600, 1000, or 1600 copies/μl based on the mass and concentration of plasmids. Two to three chips per dilution were used and copies/μl were plotted for each of the species-specific standard (Fig. 2). As shown, each of the plasmid concentrations were accurately and precisely quantified using dPCR based on target dilutions. Positive (gDNA from the species of interest) and negative controls (gDNA from another species) show specificity and lack of cross-reactivity of the mtDNA mtDNA assays, as well as the nuclear copy number reference assays (Fig. S2).
Fig. 2.

mtDNA standard validation of dPCR. Plasmids containing either human, rat, or mouse mtDNA were diluted to 600, 1000, or 1600 copies per microliter based on mass. Each of these plasmid dilutions were subjected to dPCR. Measured copies per microliter versus expected copies per microliter are plotted. Points represent 2–3 chips per dilution. Error bars = SEM
A novel application of this method is to quantify mtDNA in subcellular isolates, such as synaptic terminals from retinal neural tissue, or retinal synaptosomes (Fig. 3a). Retinal somatic and synaptic fractions from male 3-month-old Sprague Dawley rats were subjected to dPCR to quantify mtDNA and nucDNA copies. In the somatic fraction, there was at least a tenfold enrichment for nucDNA compared to the synaptic fraction. Both fractions contained mtDNA with more mtDNA copies per microgram of input gDNA (as can be expected given that there is very little nucDNA). Representative western blots for nuclear (Lamin B1), synaptic (synaptophysin), and mitochondrial (COXIV) markers are also shown, demonstrating enrichment of synaptosomes (Fig. 3b). Importantly, these results would be impossible to obtain using existing relative methods. mtDNA was also absolutely quantified in order to confirm a known tissue difference in mtDNA quantity between neural tissue (retina) and heart muscle from male 3-month-old mice (n = 5–7/tissue) (Fig. 4) (Fernandez-Vizarra et al. 2011). In agreement with previously published relative quantitation results, the heart tissue contained more mtDNA copies compared to the neural tissue (***p < 0.001). These results are represented as both per nanogram reaction input DNA (Fig. 4a) and normalized to number of haploid genomes (Fig. 4b).
Fig. 3.

mtDNA absolute quantitation from rat retinal synaptosomes. a Somatic and synaptic retinal cell fractions were used to determine the absolute quantitation of mtDNA. Somatic cell fraction contained both nuclear and mtDNA molecules. Synaptic fraction only contained mtDNA molecules with an insignificant amount of nuclear. b Representative western blots on nuclear, cytoplasmic, and synaptosome fractions probed for Lamin B1 (66–70 kDa), a nuclear marker, Synaptophysin (38 kDa), a pre-synaptic terminal marker, and COXIV (15–16 kDa), a mitochondrial marker
Fig. 4.

Mouse heart muscle contains more mtDNA compared to neural (retina) tissue. dPCR was used to absolutely quantify mtDNA copies in mouse heart muscle and retinal neural tissue. Data is represented as either normalized per nanogram input DNA (a) or normalized on a per haploid genome basis (b). Both demonstrate that heart muscle contains more mtDNA when compared to retinal neural tissue (***p < 0.001, parametric t test, n = 5–7/tissue). Break interval—30,000–100,000 (right)
mtDNA quantitation from Sod1−/− mice
Skeletal muscle (gastrocnemius) and brain tissue from young male wild-type (8 month), old male wild-type (28 month), and young female and male (8 month) Sod1 −/− mice were subjected to mtDNA absolute quantitation by dPCR and relative quantitation by qPCR. Using dPCR, there was a statistically significant age-related increase in mtDNA copy number in skeletal muscle (**p < 0.01) (Fig. 5a). Similarly, mtDNA copy number increased in the skeletal muscle from Sod1 −/− mice compared to the muscle from age-matched young wild type mice (*p < 0.05) (Fig. 5a). In the brain, there was no age-related effect on mtDNA copy number and no effect of Sod1 deletion (Fig. 5b). In addition to the within tissue comparison, a cross tissue comparison of mtDNA copy number was carried out (Fig. 5c). Independent of the sample group, the skeletal muscle was measured to have significantly more mtDNA copies compared to the brain (***p < 0.001) (Fig. 5c). Duplicate measurements using the same assays were carried out using qPCR with the nuclear assay as the reference (Fig. 5d). In skeletal muscle, there was a trending but not significant increase in median mtDNA copies between young and old mice, and between young wild type and young Sod1 −/− mice (Fig. 5d). In the brain, similar to the dPCR results, there was no effect of age or Sod1 − deletion on mtDNA copy number (Fig. 5e). Similar to the dPCR results, relative mtDNA copy number was significantly higher in skeletal muscle compared to brain (***p < 0.001) (Fig. 5F).
Fig. 5.
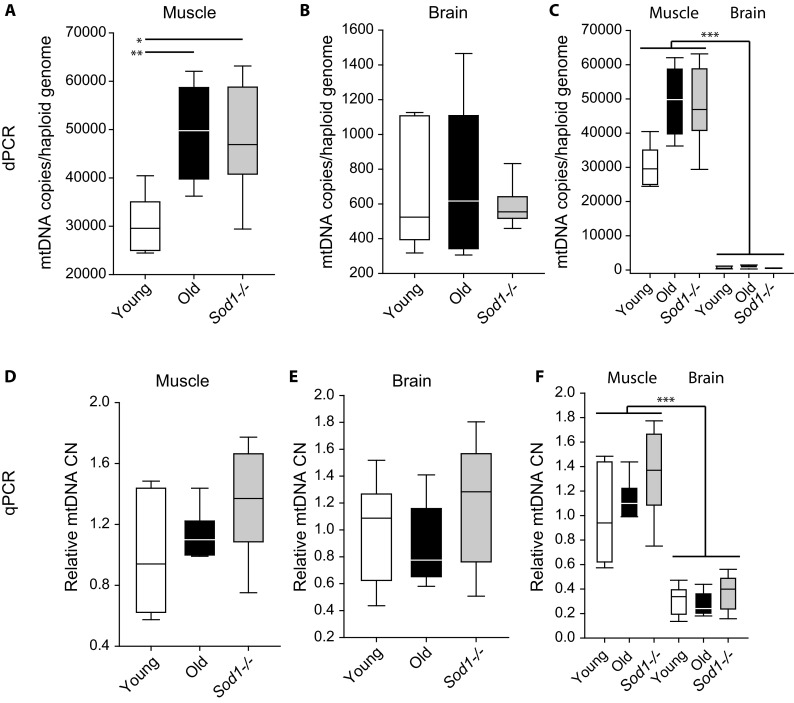
Increase in mtDNA copies from skeletal muscle but not brain from aged wild type and Sod1 −/− mice. Absolute mtDNA copy numbers in a skeletal muscle and b brain from young (8 months) , old (28 months), and young (8 months) Sod1 −/− mice. Aging and deletion of Sod1 −/− result in an increase in mtDNA copies compared to young animals in skeletal muscle, but not brain. c Skeletal muscle has higher mtDNA copies compared to brain (*p < 0.05, **p < 0.01, one-way ANOVA on group, SNK post hoc, n = 5–7/group; ***p < 0.001, two-way ANOVA on group and tissue, n = 5–7/group). Relative mtDNA copy number from young, old, and Sod1 −/− mouse skeletal muscle (d) and brain (e). (F) Relative mtDNA copy number is higher in the muscle compared to the brain (***p < 0.001, two-way ANOVA on group and tissue, SNK post hoc, n = 5–6/group)
Discussion
Age-associated changes in mitochondrial function and mitochondrial content have been implicated in the aging process and have been proposed to be a driving factor for the onset of sarcopenia (Anson and Bohr 2000; Jang et al. 2010). An important aspect of mitochondrial health and homeostasis is the mtDNA content and copy number. Previous reports of age-related changes in mtDNA copy number have been inconclusive as both increases and decreases have been reported. A number of confounding factors could lead to these discrepancies in the literature, and these have been highlighted previously (Malik and Czajka 2013). We sought to use dPCR as a robust way to absolutely quantify mtDNA copy number in order to clarify these discrepancies. Here we have validated this method to absolutely quantify mtDNA copy number, a much needed tool in the field of mitochondrial biology (Malik and Czajka 2013). With this tool, we have shown for the first time that the age-related increase in mtDNA copy number is mimicked by the age-accelerated Sod1 −/− mouse model in the skeletal muscle, but not the brain.
Multiple studies have shown changes in mtDNA copy number in skeletal muscle with age. A majority of these studies showed increases in mtDNA copy number with age in muscle (Bai et al. 2004; Barrientos et al. 1997a; Barrientos et al. 1997b; Dimmock et al. 2010; Gadaleta et al. 1992; Masuyama et al. 2005; Pesce et al. 2001). However, studies also have shown no change or an age-related decline in mtDNA copy number with age in muscle (Lanza et al. 2008; Miller et al. 2003; Short et al. 2005; Welle et al. 2003). Our data supports those studies showing an increase in mtDNA copy number with age. The discrepancies in these reports are likely due to methodological differences (Malik and Czajka 2013). The majority of reports use a relative method, such as qPCR, for quantifying mtDNA. As we have shown, relative methods of quantifying mtDNA content are suitable for detecting large differences in mtDNA copy number, such as between tissues. However, for more sensitive applications such as age-related differences absolute quantitative methods, such as dPCR, more robustly and reproducibly quantify mtDNA copy number.
This is the first study to show an age-related increase in muscle mtDNA copy number that is mimicked by Sod1 deletion. This data supports the Sod1 −/− mouse as a model of accelerated sarcopenia building off previous characterization studies of this model and its use in aging and sarcopenia research (Jang et al. 2010; Muller et al. 2006; Nagahisa et al. 2016; Wanagat et al. 2015). It has been shown that the muscle fiber type most affected in the Sod1 −/− mice is the Type IIbx fiber, and these fibers have been shown to contribute to the increase in reactive oxygen species, or oxidative stress (Nagahisa et al. 2016). In human fibroblasts, a concomitant increase in mtDNA copy number with increased oxidative stress has been reported (Lee et al. 2000). This supports our findings that the skeletal muscle from Sod1 −/− mice or aged wild-type mice contains more mtDNA. An important aspect of this finding is that these increases in oxidative stress in Sod1 −/− mice are not associated with increased heteroplasmy or mtDNA deletions (Wanagat et al. 2015). Therefore, in response to age-related oxidative stress in the muscle, mtDNA damage may occur, but it may be diluted out by the response of increased mtDNA copy number in a positively adaptive manner that nonetheless is insufficient to maintain mitochondrial function. Furthermore, in a longevity study, relative mtDNA copy numbers were reported to decrease in blood of a long-lived human population compared to controls (van Leeuwen et al. 2014). This supports our hypothesis that the induction of mtDNA in aged and Sod1 −/− mice is either maladaptive of an insufficient homeostatic response to restore normal mitochondrial function. However, these age-related and Sod1 −/−-associated changes are not seen in the brain. Relative mtDNA content has been previously reported across several CNS regions with age; however, no reproducible trends in increase or decrease in mtDNA copy number were shown (McInerny et al. 2009). Because there are both increases and decreases in mtDNA copy number in various brain regions with age, a likely result of quantifying mtDNA copy number in the whole brain would yield no change, as we report here. Therefore, future studies should address CNS subregion-specific or cell type-specific alterations in mtDNA copy number with age using absolute quantitative methodology such as dPCR.
A unique aspect of muscle mitochondria is that they are present in two distinct subcellular populations, subsarcolemmal and intermyofibrillar (Ferreira et al. 2010). These two populations of mitochondria have been shown to react differently with aging (Huang and Hood 2009). Furthermore, the subsarcolemmal has been shown to be the mitochondria that contribute most to increases in reactive oxygen species. An intriguing future direction will be to quantify mtDNA in subsarcolemmal mitochondria from Type IIbx fibers from Sod1 −/− and aged mice. This is only possible using absolute quantitation of mtDNA as presented here as isolates would lack nuclear DNA used for normalization with standard mtDNA qPCR methods.
Quantifying mtDNA using dPCR has a number of benefits compared to relative methods. This includes the use of fluorogenic primer/probe sets without the need to address or correct for differing assay reaction efficiencies, and the ability to quantify mtDNA without an endogenous control, which sometimes may not be present. With any PCR method, however, caveats persist. As with qPCR, heteroplasmies in the binding regions of the primers and fluorogenic probes could affect quantitation. For this reason, we generated two primer/probe sets per mtDNA reference genome for human, rat, and mouse, and designed in regions without frequent damage or deletion. Performing dPCR quantitation with two independent sets of primers would control for the possible effects of heteroplasmies. A benefit of qPCR is the high sample throughput capacity. Chip-based dPCR is limited in its throughput by the number of chips which can be cycled simultaneously, currently 24, in a standard flatblock thermalcycler. Additionally, with any dPCR approach, specific instrumentation is needed. A critical component of dPCR experiments is proper and precise quantitation of starting template material. We suggest the use of DNA-specific fluorescent quantitation assays to quantify template.
In conclusion, we have demonstrated dPCR as a useful method for absolute quantitation of mtDNA. This is the first report to validate this methodology and use it to quantify mtDNA in the skeletal muscle and the brain in the Sod1 −/− mouse model. We were able to identify increases in mtDNA copy number in the skeletal muscle in aging mice and the Sod1 −/−-accelerated sarcopenia model. Because the previous literature shows increases and decreases in mtDNA copy number with age in the skeletal muscle, we propose future studies use this or a similar absolute quantitative method of quantifying mtDNA copy number in order to robustly and reproducibly quantify changes in mtDNA copy number. Furthermore, our method is the only method with capabilities of quantifying mtDNA in subcellular isolates, such as subsarcolemmal and intermyofibrillar mitochondria, highlighting its potential as the go-to method for quantifying mtDNA.
Electronic supplementary material
(PDF 424 kb)
(PDF 318 kb)
Acknowledgments
The authors wish to thank the Laboratory for Molecular Biology and Cytometry Research at OUHSC for providing Sanger sequencing assistance and Peter Eckhart for assistance with figure generation. This work was supported by the National Institutes of Health [National Eye Institute R01EY02176, R21EY024520 to WMF, T32EY023202 to DRM]; National Institute on Aging Oklahoma Nathan Shock Center (P30AG050911) to HVR, WMF; the Donald W. Reynolds Foundation [DRM & WMF]; The University of Oklahoma Health Sciences Center Graduate Student Association research grant to [DRM]; and in part by an award from Harold Hamm Diabetes Center at the University of Oklahoma [DRM].
Compliance with ethical standards
All procedures were approved by the Institutional Animal Care and Use Committee at the Oklahoma Medical Research Foundation.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Dustin R. Masser, Email: dmasser@ouhsc.edu
Nicholas W. Clark, Email: Nicholas-clark@ouhsc.edu
Holly Van Remmen, Email: Holly-vanremmen@omrf.org.
Willard M. Freeman, Phone: 1-405-271-8000, Email: wfreeman@ouhsc.edu
References
- Anson RM, Bohr VA. Mitochondria, oxidative DNA damage, and aging. J Am Aging Assoc. 2000;23:199–218. doi: 10.1007/s11357-000-0020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai RK, Perng CL, Hsu CH, Wong LJ. Quantitative PCR analysis of mitochondrial DNA content in patients with mitochondrial disease. Ann N Y Acad Sci. 2004;1011:304–309. doi: 10.1007/978-3-662-41088-2_29. [DOI] [PubMed] [Google Scholar]
- Baker M. Digital PCR hits its stride. Nat Methods. 2012;9:541–544. [Google Scholar]
- Barja G. Updating the mitochondrial free radical theory of aging: an integrated view, key aspects, and confounding concepts. Antioxid Redox Signal. 2013;19:1420–1445. doi: 10.1089/ars.2012.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos A, et al. Qualitative and quantitative changes in skeletal muscle mtDNA and expression of mitochondrial-encoded genes in the human aging process. Biochem Mol Med. 1997;62:165–171. doi: 10.1006/bmme.1997.2647. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Casademont J, Cardellach F, Estivill X, Urbano-Marquez A, Nunes V. Reduced steady-state levels of mitochondrial RNA and increased mitochondrial DNA amount in human brain with aging. Brain Res Mol Brain Res. 1997;52:284–289. doi: 10.1016/s0169-328x(97)00278-7. [DOI] [PubMed] [Google Scholar]
- Benz CC, Yau C. Ageing, oxidative stress and cancer: paradigms in parallax nature reviews. Cancer. 2008;8:875–879. doi: 10.1038/nrc2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- Chaturvedi RK, Flint Beal M. Mitochondrial diseases of the brain. Free Radic Biol Med. 2013;63:1–29. doi: 10.1016/j.freeradbiomed.2013.03.018. [DOI] [PubMed] [Google Scholar]
- Clay Montier LL, Deng JJ, Bai Y. Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics. 2009;36:125–131. doi: 10.1016/S1673-8527(08)60099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Erchia AM, et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion. 2015;20:13–21. doi: 10.1016/j.mito.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Dickinson A, et al. The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ. 2013;20:1644–1653. doi: 10.1038/cdd.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–232. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- Dimmock D, Tang LY, Schmitt ES, Wong LJ. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem. 2010;56:1119–1127. doi: 10.1373/clinchem.2009.141549. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Enriquez JA, Perez-Martos A, Montoya J, Fernandez-Silva P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion. 2011;11:207–213. doi: 10.1016/j.mito.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Vitorino R, Alves RM, Appell HJ, Powers SK, Duarte JA, Amado F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics. 2010;10:3142–3154. doi: 10.1002/pmic.201000173. [DOI] [PubMed] [Google Scholar]
- Gadaleta MN, Rainaldi G, Lezza AM, Milella F, Fracasso F, Cantatore P. Mitochondrial DNA copy number and mitochondrial DNA deletion in adult and senescent rats. Mutat Res. 1992;275:181–193. doi: 10.1016/0921-8734(92)90022-h. [DOI] [PubMed] [Google Scholar]
- Hindson BJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson CM, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10:1003–1005. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JH, Hood DA. Age-associated mitochondrial dysfunction in skeletal muscle: contributing factors and suggestions for long-term interventions. IUBMB Life. 2009;61:201–214. doi: 10.1002/iub.164. [DOI] [PubMed] [Google Scholar]
- Jang YC, et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010;24:1376–1390. doi: 10.1096/fj.09-146308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA. Mitochondria damage in the pathogenesis of diabetic retinopathy and in the metabolic memory associated with its continued progression. Curr Med Chem. 2013;20:3226–3233. doi: 10.2174/09298673113209990029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza IR, et al. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57:2933–2942. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Yin PH, Lu CY, Chi CW, Wei YH. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem J. 2000;348(Pt 2):425–432. [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion. 2013;13:481–492. doi: 10.1016/j.mito.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Masser DR, Berg AS, Freeman WM. Focused, high accuracy 5-methylcytosine quantitation with base resolution by benchtop next-generation sequencing. Epigenetics Chromatin. 2013;6:33. doi: 10.1186/1756-8935-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser DR, et al. Hippocampal subregions exhibit both distinct and shared transcriptomic responses to aging and nonneurodegenerative cognitive decline. J Gerontol A Biol Sci Med Sci. 2014;69:1311–1324. doi: 10.1093/gerona/glu091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuyama M, Iida R, Takatsuka H, Yasuda T, Matsuki T. Quantitative change in mitochondrial DNA content in various mouse tissues during aging. Biochim Biophys Acta. 2005;1723:302–308. doi: 10.1016/j.bbagen.2005.03.001. [DOI] [PubMed] [Google Scholar]
- McInerny SC, Brown AL, Smith DW. Region-specific changes in mitochondrial D-loop in aged rat CNS. Mech Ageing Dev. 2009;130:343–349. doi: 10.1016/j.mad.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW, Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31 doi: 10.1093/nar/gng060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison T, et al. Nanoliter high throughput quantitative PCR. Nucleic Acids Res. 2006;34 doi: 10.1093/nar/gkl639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Nagahisa H, Okabe K, Iuchi Y, Fujii J, Miyata H. Characteristics of skeletal muscle fibers of SOD1 knockout mice. Oxidative Med Cell Longev. 2016;2016:9345970. doi: 10.1155/2016/9345970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas JA, Brooks EM, Hunter TC, Single R, Branda RF. Development of a quantitative PCR (TaqMan) assay for relative mitochondrial DNA copy number and the common mitochondrial DNA deletion in the rat. Environ Mol Mutagen. 2004;44:313–320. doi: 10.1002/em.20050. [DOI] [PubMed] [Google Scholar]
- Pesce V, et al. Age-related mitochondrial genotypic and phenotypic alterations in human skeletal muscle. Free Radic Biol Med. 2001;30:1223–1233. doi: 10.1016/s0891-5849(01)00517-2. [DOI] [PubMed] [Google Scholar]
- Phillips NR, Sprouse ML, Roby RK. Simultaneous quantification of mitochondrial DNA copy number and deletion ratio: a multiplex real-time PCR assay. Sci Rep. 2014;4:3887. doi: 10.1038/srep03887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz A, et al. Evaluation of sex differences on mitochondrial bioenergetics and apoptosis in mice. Exp Gerontol. 2007;42:173–182. doi: 10.1016/j.exger.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K. Mitochondrial hormesis and diabetic complications. Diabetes. 2015;64:663–672. doi: 10.2337/db14-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmookler Reis RJ, Goldstein S. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J Biol Chem. 1983;258:9078–9085. [PubMed] [Google Scholar]
- Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen N, Beekman M, Deelen J, van den Akker EB, de Craen AJ, Slagboom PE, ‘t Hart LM. Low mitochondrial DNA content associates with familial longevity: the Leiden longevity study. Age (Dordr) 2014;36:9629. doi: 10.1007/s11357-014-9629-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGuilder HD, Brucklacher RM, Patel K, Ellis RW, Freeman WM, Barber AJ. Diabetes downregulates presynaptic proteins and reduces basal synapsin I phosphorylation in rat retina. Eur J Neurosci. 2008;28:1–11. doi: 10.1111/j.1460-9568.2008.06322.x. [DOI] [PubMed] [Google Scholar]
- Veltri KL, Espiritu M, Singh G. Distinct genomic copy number in mitochondria of different mammalian organs. J Cell Physiol. 1990;143:160–164. doi: 10.1002/jcp.1041430122. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanagat J, Ahmadieh N, Bielas JH, Ericson NG, Van Remmen H. Skeletal muscle mitochondrial DNA deletions are not increased in CuZn-superoxide dismutase deficient mice. Exp Gerontol. 2015;61:15–19. doi: 10.1016/j.exger.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Sha H, Ji D, Zhang HL, Chen D, Cao Y, Zhu J. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell. 2014;157:1591–1604. doi: 10.1016/j.cell.2014.04.042. [DOI] [PubMed] [Google Scholar]
- Warren L, Bryder D, Weissman IL, Quake SR. Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc Natl Acad Sci U S A. 2006;103:17807–17812. doi: 10.1073/pnas.0608512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welle S, Bhatt K, Shah B, Needler N, Delehanty JM, Thornton CA. Reduced amount of mitochondrial DNA in aged human muscle. J Appl Physiol (1985) 2003;94:1479–1484. doi: 10.1152/japplphysiol.01061.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 424 kb)
(PDF 318 kb)