


Abstract
mRNA expression involves transcription initiation, elongation and degradation. In cells, these dynamic processes are highly regulated. However, experimental characterization of the dynamic processes in vivo is difficult due to the paucity of methods capable of direct measurements. We present a highly sensitive and versatile method enabling direct characterization of the dynamic processes. Our method is based on single-molecule fluorescence in situ hybridization (smFISH) and quantitative analyses of hybridization signals. We hybridized multiple probes labelled with spectrally distinct fluorophores to multiple sub-regions of single mRNAs, and visualized the kinetics of synthesis and degradation of the sub-regions. Quantitative analyses of the data lead to absolute quantification of the lag time of mRNA induction (the time it takes for external signals to activate transcription initiation), transcription initiation rate, transcription elongation speed (i.e. mRNA chain-growth speed), the rate of premature termination of transcripts and degradation rates. Applying our method to three different biological problems, we demonstrated how our method may be applicable to reveal dynamics of mRNA expression that was difficult to study previously. We expect such absolute quantification can greatly facilitate understanding of gene expression and its regulation working at the levels of transcriptional initiation, elongation and degradation.
INTRODUCTION
Gene expression is a process by which genetic information is transcribed into mRNAs and translated into proteins. Living cells adapt to their surrounding environments by regulating this process and, especially, regulation of mRNA expression is the primary means (1). Thus, cellular adaptation strategies to environments are reflected in changes of mRNA expression and the characterization of mRNA expression is central in a wide range of biological research activities.
mRNA expression is dynamic, involving transcription initiation, elongation and degradation, and each process is tightly regulated (2–6). Recent biophysical and biochemical studies in vitro have revealed intricate, molecular-level events in the processes, emphasizing that mRNA abundance in cells are determined by a complex interplay of these dynamic processes (7–10). It is becoming increasingly clear that to better understand mRNA expression in cells, in vivo characterization of these dynamic processes is important. For such characterization, absolute quantification of kinetic parameters, such as transcription initiation rate, elongation speed, premature termination rate and degradation rate, is needed. With technological developments, it became straightforward to measure mRNA abundance in vivo, e.g. using qPCR, northern blot, microarray, RNAseq, etc. (11,12). However, application of these methods to measure kinetic parameters related to mRNA expression is limited. Previously, the Northern blot and its variations (such as dot blots) have been used to measure transcription elongation speed (13,14). This method has poor detection limits, requiring considerable amounts of target mRNAs, and involves the use of hazardous radioactive isotopes (12). Moreover, absolute quantification of other kinetic parameters is not possible. There have been efforts to deduce such kinetic parameters from quantitative modelling of mRNA expression kinetics (15,16), but the values obtained are model-dependent.
Here, we introduce a highly sensitive and versatile method enabling absolute quantification of these kinetic parameters in vivo. Our method builds on recent development of single-molecule fluorescence in situ hybridization (smFISH) that allows for detection of individual mRNA molecules of interest (17–20). We hybridized multiple probes labelled with spectrally distinct fluorophores to multiple sub-regions of single mRNAs, and analysed the hybridization signals quantitatively. Revealing the kinetics of mRNA expression, the method allowed for absolute quantification of various kinetic parameters with unprecedented accuracy. Using three biological problems as examples, we demonstrated the usefulness of this method.
MATERIALS AND METHODS
Bacterial strain and growth conditions
Escherichia coli K-12 strain NCM3722 (NMK1) or its derivative (NMK80) were used. In the NMK80 strain, the expression of the lacZ gene was controlled by the synthetic promoter PLtet-O1 (See Supplementary Methods for the details of strain construction). TetR repressor proteins were constitutively expressed in the strain. Inducers, anhydrotetracycline (aTc) or chlortetracycline (cTc) inhibit the repression of the promoter by TetR, thereby activating the expression of the lacZ mRNA molecules. aTc has a high affinity for TetR; 3 ng/ml gives induction at half maximum. Hence, controlling low levels of expression using aTc is tricky. In comparison, cTc has a low affinity; 40 ng/ml gives induction at half maximum. Thus, to gradually change the expression in the dose response curve in Figure 2, we used cTc. But, aTc diffuses into cells rapidly (∼6 s as estimated below). Hence, to obtain the temporal induction curve after maximum induction (Figures 1B, C and 3), we used aTc.
Figure 2.

Comparison of the mean number of the head, body and tail of the lacZ mRNA molecules per cell (Nhead, Nbodyand Ntail) and LacZ protein amounts. A synthetic promoter PLtet-O1 drives the expression of the lacZ gene in our strain (23). We added various concentrations of the inducer, cTc, to exponentially-growing cultures (the cTc is preferable for induction at low levels due to its low affinity; see Materials and Methods). After several doublings, the expression of lacZ mRNAs and LacZ proteins reaches the steady state. We observed a good agreement between the mRNA copy number per cell (red circles for Nhead, green squares for Nbody and blue triangles for Ntail; left axis) and LacZ protein amounts (right axis). Note that the left and right axes have the same scale.
Figure 1.

Visualization of dynamics of mRNA expression. (A) The three sets of probes were hybridized to the head, body and tail of the lacZ mRNA molecules. They were labelled with red (for head), green (for body) and blue (for tail) fluorophores; note that these are pseudo-colours assigned for illustration. See Supplementary Table for the sequences of the probes. (B) At time zero, 100 μg/ml of aTc (inducer) was added to the culture. Red fluorescence foci (head), green foci (body) and blue foci (tail) appear sequentially in cells. The scale bar in the image of the tail at 90 s (black line) represents 1 μm and applies to all the images in Figure 1B. (C) Even after the mRNA levels reach their steady state (approximately after 10 min of induction), the colours of fluorescence foci in cells are not uniform, revealing the dynamics of synthesis and degradation of different sub-regions of mRNA molecules. The scale bar (black line) represents 1 μm. See the text for details.
Figure 3.

Quantification of mRNA expression and degradation dynamics. (A) Analysing hybridization signals from the head, body and tail of the lacZ mRNA (typical images used were shown in Figure 1B), we obtained Nhead (red circles), Nbody(green squares) and Ntail (blue triangles) as a function of time in cells growing exponentially in a minimal medium with glycerol and ammonium as the sole carbon and nitrogen sources. Thead, Tbody and Ttail indicate the times at which Nhead, Nbody and Ntail begin to rise; the actual values were obtained from the fit of the regions of the linear increase (dashed lines). From these values, we obtained the transcription elongation speed. From the speed and Thead, we estimated Tlag, the time it takes for the inducer to activate transcription initiation. From the slopes, we obtained the transcription rate, as well as the rate of premature termination of transcripts. See the text for the details of the analyses. (B) We inhibited the mRNA synthesis by adding rifampicin to an exponentially-growing culture; the addition of rifampicin marks the time zero. Nhead, Nbody and Ntail decrease exponentially (note a semi-log scale). Nhead begins to decrease at earlier time points than Ntail; see the insets.
Cells were grown in a 37°C water bath shaker shaking at 250 rpm. NMK80 cells were cultured in a Luria-Bertani (LB) medium (seed culture). For experiments using minimal media, cells were transferred to a N-C- minimal medium (21) with 20 mM glycerol as the carbon source and 20 mM NH4Cl as the nitrogen source, and grown overnight (pre-culture). The next morning, cells were transferred to a fresh medium with the same composition to the OD600 of 0.02 (experimental culture). To obtain the dose response curve in Figure 2, various concentrations of cTc were added to the fresh medium at the time of the transfer and samples for FISH were collected at OD600 of ∼0.5. To obtain the temporal induction curve (Figures 1B, C and 3), 100 ng/ml of aTc was added to the culture at an OD600 of ∼0.5 and samples for FISH were collected at various time points after the addition. For the diauxic shift experiment (yielding the data plotted in Figure 4A), NMK1 was cultured in a minimal medium with 2.5 mM of glucose and 10 mM of glycerol as carbon sources, and 20 mM of ammonium as the nitrogen source. For the ammonium starvation experiment (yielding the data shown in Table 1 and Figure 4B), NMK1 was cultured in a minimal medium with 3 mM of ammonium as the nitrogen source and 10 mM of glycerol.
Figure 4.

Dynamics of expression of glpK mRNA and amtB mRNA molecules in dynamically-changing environments. (A) Cells were cultured in a minimal medium with glucose and glycerol as carbon sources (ammonium as the sole nitrogen source). Cells initially grew on glucose (the shared region). At the time zero, their growth abruptly stopped (indicating depletion of glucose), and after some time, cells began to grow on glycerol (see Supplementary Figure S3A for growth curve). During this period, the expression of mRNA molecules of glycerol kinase glpK was measured. The two sets of probes were hybridized to the head and tail of glpK mRNA molecules. The sequences of the probes and probe-binding regions were provided in Supplementary Table. Nhead (red circles) increases before cell growth stops. Ntail (blue triangles) increases after a time delay. See the text for the details. (B) Cells were cultured with a limited amount of ammonium as the nitrogen source (and glycerol as the carbon source). As cells use ammonium to grow, the ammonium level in the medium decreases and eventually cell growth stops due to depletion of ammonium (See Supplementary Figure S3D for growth curve). The onset of the growth stop defines the time zero. The two sets of probes were hybridized to the head and tail of amtB mRNA molecules. The sequences of the probes and probe-binding regions were provided in Supplementary Table. We see that Nhead (red circles) increases before cell growth stops due to ammonium depletion, indicating that cells anticipate impending ammonium depletion. Ntail (blue triangles) increases after a time delay. The shaded area indicates the period of cell growth, adopted from Supplementary Figure S3D. See the text for the details.
Table 1. The kinetic parameters of mRNA expression in cells growing at different rates.
| Cells growing at the rate of | |||
|---|---|---|---|
| 1.9 /ha | 0.7 /hb | 0 /hc | |
| Transcription rate, head | 7.6 /min | 5.0 /min | 1.8 /min |
| Transcription rate, tail | 7.4 /min | 4.9 /min | 1.3 /min |
| Transcription elongation speed | 49.5 nt/s | 48.6 nt/s | 14.0 nt/s |
| Degradation rate, head | 0.31 /min | 0.47 /min | 0.53 /min |
| Degradation rate, tail | 0.4 /min | 0.5 /min | 0.55 /min |
aCells were cultured in LB. See Supplementary Figures S2A and S2C for the raw data.
bCells were cultured in the minimal medium with glycerol and ammonium as the carbon and nitrogen sources respectively. See Figure 3A and B for the raw data.
cCells were cultured in the minimal medium without ammonium (depleted of nitrogen) and with glycerol. See Supplementary Figures S2B and S2D for the raw data.
Fluorescence in situ hybridization
We followed the procedure described in (17); please see Supplementary Methods for the details of the procedure. Briefly, for the probe design, the software, Stellaris Probe Designer, from Biosearch Technologies was used. The sequences of the probes used were provided in Supplementary Table. The sets of probes binding to the head, body and tail regions were labelled with Atto 647N NHS ester (AD488-35, ATTO-TEC GmbH), Atto 488 NHS ester (AD647N-35, ATTO-TEC GmbH) and 6-TAMRA NHS ester (C6123, Invitrogen), respectively. Cell fixation and probe hybridization procedures are as follows. A total of 2.7 ml of the cell culture was collected at various time points (as noted in Figures) and mixed with 300 μl of 37% formaldehyde immediately. After incubating the culture in formaldehyde for 30 min, cells were spun down at 1000 g for 8 min. The supernatant was discarded and the cells were washed twice with 1x PBS. Finally cells were re-suspended in 300 μl of DEPC-treated water and 100% ethanol was added to the final volume of 1 ml. The cells were incubated either at room temperature for 1 h or stored at 4°C overnight. Then, cells were spun at 700 g for 7 min and re-suspended in 40% wash buffer (2x SSC buffer and 40% w/v formamide) and incubated at room temperature for 5 min. Cells were spun down again and re-suspended in the hybridization buffer (2x SSC, 40% formamide, 0.1% w/v dextran sulfate, 10 mg tRNA, 0.1% BSA and 0.5 mM ribonucleoside vanadyl complex). Fluorescent probes to a final concentration of 120 nM (total) were used to hybridize mRNAs in the cells and incubated overnight at 30°C. The following day, cells were re-suspended in 1 ml of wash buffer and spun down at 800 g for 7 min. The supernatant was removed and the cell pellet was washed three times using the wash buffer to remove un-hybridized excess probes. Then, cells were re-suspended in 10 μl of 2x SSC buffer before imaging.
Microscope imaging and analysis
The cells were placed on the microscope (Olympus IX83P2Z) and imaged using a cooled cSMOS camera (Andor Neo). The best focal plane for imaging (z- position) was first determined using phase contrast. Then, varying the z- position at a spacing 300 nm, nine fluorescence images were acquired. The image analysis was performed using a custom-built program (based on MATLAB). Briefly, cell boundaries were first determined using a cell segmentation program. Intensities of fluorescence foci within cells were quantified. The intensity values were converted to the copy number of mRNAs per cell as described in Supplementary Figure S1.
β-galactosidase assay
One hundred microlitres of cell culture were collected and fast-frozen on dry ice. Four samples were collected during exponential growth. The β-Galactosidase activity of each sample was measured at 37°C by the traditional Miller method (22). The activities obtained were plotted against the OD600 at which the sample was collected. The slope from the plot was taken to be the mean activity and was used in Figure 2.
RESULTS
Visualization of dynamics of mRNA expression
Using lacZ mRNAs in E. coli as an example, we describe how multicolour labelling of multiple sub-regions of single mRNAs can visualize the dynamics of mRNA expression. We designed three sets of oligonucleotide probes hybridized to the head, body and tail of the lacZ mRNA molecules; see Figure 1A, and Supplementary Table for the sequences of the probes. Each set is comprised of 25–28 probes and each probe is 20 nucleotides (nt) long. The three sets of the probes are labelled with red (for head), green (for body) and blue (for tail) fluorophores; note that these are pseudo-colours assigned for illustration. In our strain, the expression of the lacZ gene is driven by the synthetic promoter, PLtet-O1 (23). The repressor of the promoter, TetR is highly expressed in the strain. Inducers, aTc or cTc, inhibit the repression of the promoter by TetR, thereby activating the expression of the lacZ mRNA molecules. See Materials and Methods for details of the strain, probes labelling, etc. Cells were grown exponentially in a minimal medium using glycerol and ammonium as the sole carbon and nitrogen sources. We added a saturating level of aTc to the culture and activated the expression of the lacZ mRNAs; the addition of the inducer marks the time zero. Cells were collected at different time points and, after hybridization with the three sets of probes, imaged on a microscope. Figure 1B shows typical fluorescence images of cells collected at different time points. Cells exhibit red fluorescence foci (head) at 30 s after induction, red and green foci (head and body) at 60 s, and red, green and blue foci (head, body and tail) at 90 s. We note that after the mRNA levels reach their steady state levels (approximately after 10 min of induction), the colours of fluorescence foci in cells are not uniform; see Figure 1C. For example, some foci are white, indicating all head, body and tail are present. Some foci are yellow, indicating that the head, body, but no tail are present. Some foci are cyan, indicating only body and tail are present. Because the mRNA synthesis starts from 5′ end (head), the cyan colour means that the head was degraded after synthesis. Hence, the variation in the colours of fluorescence foci reflects the dynamic interplay of transcription initiation, elongation and degradation in mRNA expression.
Quantifying the transcription elongation speed
We wish to quantify various kinetic parameters related to mRNA expression from these images. To do so, we first converted the fluorescent foci intensities to mRNA counts using the procedure that was previously established (17,24), and obtained the mean copy number of the head, body and tail of lacZ mRNAs per cell, Nhead, Nbody and Ntail, respectively; see Supplementary Figure S1 for illustration. As a check of consistency, we plotted the steady state value of Nhead, Nbody and Ntail, together with the steady state levels of LacZ proteins (measured from a standard β-galactosidase assay) at various inducer concentrations in Figure 2, and observed a good agreement.
The sequential accumulation of hybridization signals from the head, body and tail of the mRNA in Figure 1B contains the information about the lag time of mRNA induction, transcription initiation rate, and transcription elongation speed. To quantify these parameters, we plotted Nhead, Nbody and Ntail as a function of time in Figure 3A (note that mRNA expression was fully induced by adding a saturating concentration of the inducer, aTc, to the medium at time zero). We see that Nhead, Nbody and Ntail begin to increase at different time points, revealing the duration of the time needed for cells to synthesize each segment of the mRNA. Fitting the linear portion of the increase, we obtained the X intercept of the head and tail, Thead = 25.8 (±0.5) s and Ttail = 69.8 (±2.9) s; the standard deviation is calculated from the measurement of three independent experiments. The time difference, Ttail – Thead, represents the ‘transit time’ of RNA polymerases on the body and tail segments of the lacZ mRNA molecules. From the difference, considering the known length of the sequence hybridized by the probes (Figure 1A), we obtain the transcription elongation speed of 48.6 nt/s [= (1079 + 1061 nt) / (69.8 – 25.8 s)]. This value agrees with a previous estimate of 45 nt/s in a similar growth condition (doubling time ∼60 min) using the Northern blots (Table 1 and in (25)).
The analyses of different combinations, e.g. Nhead and Nbody, can be performed in an identical manner and yield a similar result. However, we note that the body of the lacZ mRNA was hybridised with probes labelled with the Atto 488 that emits green fluorescence. This emission spectrum overlaps with auto-fluorescence spectrum of E. coli cells, resulting in a relatively high background fluorescence. Thus, we found it generally difficult to accurately determine the fluorescence intensities of green foci and, hence, Nbody.
Quantifying the lag time of mRNA induction
Next, we estimated the lag time of mRNA induction, Tlag, the time delay between the addition of the inducer to the medium and transcription initiation; see Figure 3A. Assuming that the transcription elongation speed remains constant, we deduced that it would take Tsynthesis = 19.2 s ( = 935/48.6) to synthesize the head of the lacZ mRNA molecule. This is shorter than Thead ( = 25.8 s), the time it takes for the head to appear after the addition of the inducer at the time zero, because Thead includes not only Tsynthesis, but also the time delay, Tlag. Hence, from the difference between Thead ( = 25.8 s) and Tsynthesis ( = 19.2 s), we obtain Tlag = 6.6 s ( = 25.8 – 19.2). When we calculate Tlag using the tail, we obtained a similar value; it would take 63.3 s ( = 3075/48.6) to synthesize the tail, and the difference of this value from Ttail gives Tlag = 6.5 s ( = 69.8 – 63.3), showing a good agreement with the estimate above. However, there are other factors contributing to this difference. First, our procedure involves formaldehyde fixation that may take some time (∼s). Also, as elaborated in Supplementary Note 1, there is an uncertainty in determination of a full-length segment of the head of lacZ mRNA molecules, though the error from this uncertainty is estimated to be small. Hence, the actual time delay may be a bit longer than 6.6 s. Note that these factors are expected to contribute equally to the rise of the head and tail, and hence, when the time difference (Thead – Ttail) is used to calculate the transcription elongation speed, the resultant value (of the speed) is not affected by these factors.
Quantifying the transcription rate and premature termination rate
Next, we deduced the transcription rate, i.e. how frequently each segment of mRNAs (head, body and tail) is synthesized. The law of mass action describes that the change of the mRNA concentration, m, is determined by the balance between the transcription rate, α, and the degradation rate, β, in the following way,
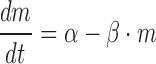 |
(1) |
Hence, immediately after induction, when the mRNA level is low (m ≈ 0), we have 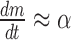 and
and 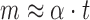 . Thus, the slope in the linear increase of the mRNA level after induction in Figure 3A (dashed lines) is equal to the transcription rate, α. Analysing the slopes, we obtained 5.0 (±0.3)/min and 4.9 (±0.2)/min for the transcription rate of the head and tail of the mRNA, respectively.
. Thus, the slope in the linear increase of the mRNA level after induction in Figure 3A (dashed lines) is equal to the transcription rate, α. Analysing the slopes, we obtained 5.0 (±0.3)/min and 4.9 (±0.2)/min for the transcription rate of the head and tail of the mRNA, respectively.
Comparison of the transcription rates of the head and tail can reveal premature termination of transcription. For example, consider that once RNA polymerases initiate transcription, they continue through head, body and tail segments, completing transcription. In that case, the transcription rate of the three segments will always match (after time delay determined by the transcription elongation speed). Hence, the close match of the transcription rates observed above (5.0 (±0.3)/min and 4.9 (±0.2)/min for the head and tail, respectively) indicates that premature termination does not occur or occurs at negligible levels during the synthesis of the body and tail of the mRNA molecules. Also, with the assumption that premature termination did not occur during the synthesis of the head, the transcription rate is equal to the transcription initiation rate (see Supplementary Note 2 for more details).
We note that, if necessary, the transcription initiation rate and premature termination rate can be more precisely estimated using a set of probes hybridized to narrower sub-regions in the head, and tail of mRNA molecules. For example, we have used as few as 12 probes that spanned approximately a region of 280 nt, and were able to detect fluorescence signals reliably.
Quantifying the degradation rate
Next, we characterized the degradation dynamics of the head, body and tail of the mRNA molecules. As described above, fluorescence foci at the steady state of mRNA expression in Figure 1C exhibit various colours, indicating the dynamics of synthesis and degradation of different sub-regions of mRNA molecules. Detailed analyses of such images can reveal degradation dynamics of mRNAs. Here, we took a different approach. To quantify the degradation rate, β, of the head, body and tail of the lacZ mRNAs (see Equation 1), we inhibited the mRNA synthesis by adding rifampicin to an exponentially-growing culture. We observed that Nhead, Nbody and Ntail decrease exponentially in Figure 3B (note a semi-log scale); the addition of rifampicin marks the time zero. Fitting the region of the exponential decrease, we obtained that for the cells growing at the rate of 0.7/h (in a medium with glycerol and ammonium as the sole carbon and nitrogen sources), the degradation rates, β, for the head, body and tail, are 0.47 (±0.05)/min, 0.49 (±0.06)/min and 0.5 (±0.02)/min; see Equation (1). From these values, we obtain the half-lives of the head, body and tail ( = ln2/β) are 1.5 min, 1.4 min and 1.4 min, respectively. We note a time delay between the onset of decrease of Nhead and that of Ntail; see the inset in Figure 3B (as described above, it is difficult to accurately determine Nbody due to a relatively high background fluorescence, which is why the data points of Nbody are more scattered and it is difficult to notice the time delay from Nbody). As will be shown later, such a delay is more pronounced in cells in a different physiological condition (Supplementary Figure S2). One factor contributing to the time delay may be the presence of on-going transcription. If rifampicin inhibits transcription initiation, but not elongation, immediately after the rifampicin treatment, there will be RNA polymerases that are already transcribing, and will continue to produce the mRNAs for some time. This could result in a time delay in the decrease of mRNA levels, and the delay will be more pronounced for the tail. Alternatively, this delay could result from distinct degradation kinetics of the different sub-regions of mRNAs.
Alternatively, we can deduce the degradation rate using the steady state level of mRNAs together with the transcription rate, α, obtained above; at the steady state, the Equation (1) yields 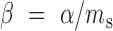 , where ms refers to the copy number of mRNAs in the steady state. When mRNA expression is fully induced, we observed that ms of the head is ∼6.7 (the value is very similar for the tail). The value, together with α = 5.0/min (as obtained above for the head and also similarly for the tail), yields the degradation rate of 0.75/min. This is less than 2-fold difference from the value obtained using rifampicin above, showing reasonable agreement.
, where ms refers to the copy number of mRNAs in the steady state. When mRNA expression is fully induced, we observed that ms of the head is ∼6.7 (the value is very similar for the tail). The value, together with α = 5.0/min (as obtained above for the head and also similarly for the tail), yields the degradation rate of 0.75/min. This is less than 2-fold difference from the value obtained using rifampicin above, showing reasonable agreement.
Having demonstrated how our method allows for absolute quantification of dynamics of mRNA expression, we now describe its potential use in different biological problems.
Effects of cell physiology on the dynamics of mRNA expression
The physiological state of cells exerts significant effects on gene expression, forming an additional layer of control of gene expression (26). There exist extensive studies to characterize these effects by growing cells at different rates in different steady-state conditions (25,27,28). However, because it was difficult to measure dynamics of mRNA expression, these studies have focused on the effects at the protein level and much less is known about the effects at the mRNA level. Here, repeating the experiments above in cells growing at different growth rates and characterizing the kinetic parameters of mRNA expression, we revealed the effects of physiology on mRNA expression. The results are summarized in the Table and the raw data are plotted in Supplementary Figure S2. The results show that the values of all the kinetic parameters are dependent on the rate of cell growth, revealing the effects of physiology on gene expression at the levels of transcriptional initiation, elongation and degradation. For example, as shown in the Table, the transcription rate and elongation speed increase at faster growth, while the degradation rate decreases marginally at faster growth. The results shed new light on previous findings in the literature. For example, a previous study characterizing several different promoters reported universal decrease of protein synthesis as cell growth is reduced to zero and deduced that the transcription rate decreased (28). Our results show that there are other factors contributing to the decrease in protein synthesis, in addition to the decrease in the transcription rate. For instance, as the cell growth is reduced to zero, transcription becomes inefficient as shown by decrease in the transcription elongation speed and increase of the premature termination rate (See Supplementary Note 2 for more details).
Dynamics of mRNA expression in changing environments
Natural environments change dynamically, and to survive and proliferate under such environments, organisms must regulate gene expression accordingly. Here, using changing nutrient availability as an example, we demonstrate the potential use of our method. When presented with two carbon sources, microbes exhibit diauxic growth (29,30). They first grow on the preferred carbon source. When the preferred source is depleted, their growth stops and after some period, they begin to grow on the less-preferred carbon source. In such conditions, the fitness of microbes depends on how rapidly they can adapt to the second carbon source after the depletion of the preferred carbon source, and the dynamics of the adaptation is determined by dynamics of expression of genes required to utilize the second carbon source, e.g. how early and how efficiently these genes are transcribed. However, such dynamics at the mRNA level is not well known. Here, we characterized the dynamics of mRNA expression of the glycerol kinase (glpk), an essential gene for glycerol metabolism, as an example.
We measured the kinetic parameters of its mRNA expression (driven by its own endogenous promoter), as cells undergo diauxic shift in the medium containing glucose (the preferred carbon source) and glycerol. We hybridized one set of probes labelled with Atto 647N to the first half of the glpk mRNA molecule (head) and the other set labelled with 6-TAMRA to the second half (tail); see Supplementary Table for the detail. The onset of the growth stop due to the depletion of glucose defines the time zero (Supplementary Figure S3A). As discussed above, because how rapidly cells can adapt to the second carbon source depends on how early and efficiently the required genes are expressed, we wish to determine when transcription of the glpK operon is activated, how fast it is transcribed (elongation speed), and if transcription terminates prematurely or not. The dynamics of Nhead and Ntail of glpK mRNA molecules plotted in Figure 4A reveals that Nhead begins to increase before cell growth stops at the time zero; see also Supplementary Figure S3B for the direct comparison of the dynamics of cell growth and Nhead of glpK mRNA molecules. The increase of Nhead is followed by the increase of Ntail with a time delay. The initial increase of the Nhead and Ntail can be approximated as linear and, fitting the first 4 points after the onsets of the increase with a linear line (dashed line), we obtained that the onset of the increase of Nhead and Ntail of the mRNA molecule is −4.6 min and −1.9 min, respectively. Using this delay, together with the known length of the sequence hybridized in the tail region (Supplementary Table), we obtain the transcription elongation speed of 4.5 nt/s [ = 723 nt/(4.6 – 1.9 min) × 1 min/60 s]. Assuming that the transcription elongation speed is constant within the gene, with the known length of the head (754 nt), we estimate that the transcription of the gene glpK is initiated 2.8 min [ = 754 nt/(4.5 nt/s) × 1 min/60s] before the onset of the increase of Nhead, i.e. 7.4 min (4.6 + 2.8 min) before cell growth stops due to the glucose depletion, revealing that cells anticipate the impending shift. The transcription rate obtained from the slopes of the dashed lines in Figure 4A are 0.23/min and 0.22/min for Nhead and Ntail, respectively. The match of the transcription rate indicates that premature termination of transcription is negligible. After the initial linear increase of Nhead and Ntail, however, the increase becomes steep, deviating from the linear dashed line. This steep increase indicates that the rate of the transcription increases. Indeed, when we estimated the rate from the increase of the mRNA copy numbers between consecutive time points, we see that the rate increases over time (Supplementary Figure S3C). In addition, cell-to-cell variability of protein expression during diauxic shift has attracted much attention in recent years (31–33). Our method based on smFISH can reveal the mRNA copy number with single-cell resolution (Figure 1C and Supplementary Figure S4) and, if needed, can be used for studies of cell-to-cell variation of mRNA expression during diauxic shift.
Lastly, when the amounts of nutrients in an environment are decreasing, it is important for bacteria to efficiently scavenge the limiting nutrients. The efficient scavenging depends on gene expression of nutrient transporters required for scavenging. AmtB is the ammonium transporter required to transport ammonium present at low levels (34,35), and here we examined dynamics of its mRNA expression (driven by its endogenous promoter), as the ammonium level in the environment is reduced. We hybridized one set of probes labelled with Atto 647N to the first half of the amtB mRNA molecule (head) and the other set labelled with 6-TAMRA to the second half (tail); see Supplementary Table for the details. The dynamics of Nhead and Ntail of the amtB mRNA is plotted in Figure 4B. Analysing the data similarly as described for the data from glnK above, we obtained that the transcription of the amtB gene is activated 16.6 min before cell growth stops due to ammonium depletion (see Supplementary Figure S3D for the growth curve) and proceeds at the elongation speed of 33.9 nt/s. The examples here demonstrate how our method may be used to characterize dynamics of mRNA expression in various conditions, including changing environments.
DISCUSSION
Regulation of mRNA expression according to external environments is crucial for organisms’ survival and proliferation. A growing number of biophysical and biochemical studies in vitro, revealing intricate molecular events during transcription, emphasizes that complex interplay of transcription initiation, elongation, premature termination and degradation governs the expression of mRNAs (7–10). Hence, to better understand the mRNA expression in vivo, absolute quantitative measurement of the kinetic parameters is crucial.
We have shown that our method to label sub-regions of single mRNAs with different fluorophores and quantitatively analyse the signals is applicable for such absolute quantification of the kinetic parameters. Importantly, our method based on smFISH is capable of identifying single mRNA molecules. Thus, it can be used to characterize kinetic parameters even when the expression of mRNAs is very low. Investigating the effects of cell physiology on mRNA expression, as well as dynamics of mRNA expression in changing environments as examples, we demonstrated how our method may be used to reveal new knowledge of gene expression and its regulation which was difficult to obtain previously. In addition, we expect various other applications of our method. For instance, a certain sub-region within a mRNA molecule can be particularly important for regulation of mRNA expression; a good example is the formation of hairpin structures of trp mRNAs that results in premature termination of transcription (36). In such cases, probes can be designed to hybridize the upstream and downstream of the sub-region to pinpoint the role of such regulations. Also, although we characterized expression kinetics of one gene here, with appropriate combinations of spectrally distinct fluorophores, the expression kinetics of more than one gene may be examined simultaneously. We note that there exist previous studies to use multiple-labelling of single mRNAs using FISH (37,38). However, in the studies, such multiple-labelling was used to uniquely determine the identities of mRNAs, and hence, different from our application.
Another advantage of our method based on smFISH is its low detection limits and sensitivity. Because a single mRNA molecule can be visualized (Figure 1), the dynamics of mRNA expression at very low levels can be characterized quantitatively. Furthermore, our method allows discernment of the copy number of mRNAs in individual cells. For example, analysing image sequences shown in Figure 1B, we plotted how the number of cells expressing mRNAs changes after induction, and how the distribution of the mRNA copy number per cell changes after induction in Supplementary Figure S4. Hence, our method can be applicable to measure the dynamics of mRNA expression at single-molecule and single-cell resolution.
Supplementary Material
Acknowledgments
The authors thank Hedia Maamar for her help during the early development of this method, and Ido Golding for kindly answering questions regarding the previously-published protocol of smFISH from his laboratory and providing us with test probes early on.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
MK's Emory start-up fund. Funding for open access charge: Emory start-up fund.
Conflict of interest statement. None declared.
REFERENCES
- 1.Cooper G.M., Hausman R.E. The Cell: A molecular approach. Washington D.C: ASM Press; 2004. [Google Scholar]
- 2.Browning D.F., Busby S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 3.Jonkers I., Lis J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015;16:167–177. doi: 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoenberg D.R., Maquat L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012;13:246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim H.D., Shay T., O'Shea E.K., Regev A. Transcriptional regulatory circuits: Predicting numbers from alphabets. Science. 2009;325:429–432. doi: 10.1126/science.1171347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dubnau D., Losick R. Bistability in bacteria. Mol. Microbiol. 2006;61:564–572. doi: 10.1111/j.1365-2958.2006.05249.x. [DOI] [PubMed] [Google Scholar]
- 7.Bai L., Santangelo T.J., Wang M.D. Single-molecule analysis of RNA polymerase transcription. Annu. Rev. Biophys. Biomol. Struct. 2006;35:343–360. doi: 10.1146/annurev.biophys.35.010406.150153. [DOI] [PubMed] [Google Scholar]
- 8.Forties R.A., Wang M.D. Discovering the power of single molecules. Cell. 157:4–7. doi: 10.1016/j.cell.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 9.McGary K., Nudler E. RNA polymerase and the ribosome: the close relationship. Curr. Opin. Microbiol. 2013;16:112–117. doi: 10.1016/j.mib.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X., Vukovic L., Koh H.R., Schulten K., Myong S. Dynamic profiling of double-stranded RNA binding proteins. Nucleic Acids Res. 2015;43:7566–7576. doi: 10.1093/nar/gkv726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morozova O., Hirst M., Marra M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genomics Hum. Genet. 2009;10:135–151. doi: 10.1146/annurev-genom-082908-145957. [DOI] [PubMed] [Google Scholar]
- 12.Reue K. mRNA quantitation techniques: Considerations for experimental design and application. J. Nutr. 1998;128:2038–2044. doi: 10.1093/jn/128.11.2038. [DOI] [PubMed] [Google Scholar]
- 13.Proshkin S., Rahmouni A.R., Mironov A., Nudler E. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science. 2010;328:504–508. doi: 10.1126/science.1184939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogel U., Sørensen M., Pedersen S., Jensen K.F., Kilstrup M. Decreasing transcription elongation rate in Escherichia Coli exposed to amino acid starvation. Mol. Microbiol. 1992;6:2191–2200. doi: 10.1111/j.1365-2958.1992.tb01393.x. [DOI] [PubMed] [Google Scholar]
- 15.Darzacq X., Shav-Tal Y., de Turris V., Brody Y., Shenoy S.M., Phair R.D., Singer R.H. In vivo dynamics of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zenklusen D., Larson D.R., Singer R.H. Single-RNA counting reveals alternative modes of gene expression in yeast. Nat. Struct. Mol. Biol. 2008;15:1263–1271. doi: 10.1038/nsmb.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skinner S.O., Sepúlveda L.A., Xu H., Golding I. Measuring mRNA copy number in individual Escherichia coli cells using single-molecule fluorescent in situ hybridization. Nat. Protoc. 2013;8:1100–1113. doi: 10.1038/nprot.2013.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trcek T., Chao J.A., Larson D.R., Park H.Y., Zenklusen D., Shenoy S.M., Singer R.H. Single-mRNA counting using fluorescent in situ hybridization in budding yeast. Nat. Protoc. 2012;7:408–419. doi: 10.1038/nprot.2011.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raj A., van den Bogaard P., Rifkin S.A., van Oudenaarden A., Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maamar H., Raj A., Dubnau D. Noise in gene expression determines cell fate in Bacillus Subtilis. Science. 2007;317:526–529. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Csonka L.N., Ikeda T.P., Fletcher S.A., Kustu S. The accumulation of glutamate is necessary for optimal growth of Salmonella typhimurium in media of high osmolality but not induction of the proU operon. J. Bacteriol. 1994;176:6324–6333. doi: 10.1128/jb.176.20.6324-6333.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller J.H. Experiments in molecular genetics. NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 23.Lutz R., Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.So L.-h., Ghosh A., Zong C., Sepulveda L.A., Segev R., Golding I. General properties of transcriptional time series in Escherichia coli. Nat. Genet. 2011;43:554–560. doi: 10.1038/ng.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bremer H., Dennis P. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus. 2008;3 doi: 10.1128/ecosal.5.2.3. doi:10.1128/ecosal.5.2.3. [DOI] [PubMed] [Google Scholar]
- 26.Klumpp S., Hwa T. Bacterial growth: global effects on gene expression, growth feedback and proteome partition. Curr. Opin. Biotechnol. 2014;28:96–102. doi: 10.1016/j.copbio.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klumpp S., Zhang Z., Hwa T. Growth rate-dependent global effects on gene expression in bacteria. Cell. 2009;139:1366–1375. doi: 10.1016/j.cell.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berthoumieux S., de Jong H., Baptist G., Pinel C., Ranquet C., Ropers D., Geiselmann J. Shared control of gene expression in bacteria by transcription factors and global physiology of the cell. Mol. Syst. Biol. 2013;9:634. doi: 10.1038/msb.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monod J. The growth of bacterial cultures. Annu. Rev. Microbiol. 1949;3:371–394. [Google Scholar]
- 30.Gorke B., Stulke J. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 2008;6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 31.Solopova A., van Gestel J., Weissing F.J., Bachmann H., Teusink B., Kok J., Kuipers O.P. Bet-hedging during bacterial diauxic shift. Proc. Natl. Acad. Sci. U.S.A. 2014;111:7427–7432. doi: 10.1073/pnas.1320063111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boulineau S., Tostevin F., Kiviet D.J., ten Wolde P.R., Nghe P., Tans S.J. Single-cell dynamics reveals sustained growth during diauxic shifts. PloS One. 2013;8:e61686. doi: 10.1371/journal.pone.0061686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Venturelli O.S., Zuleta I., Murray R.M., El-Samad H. Population diversification in a yeast metabolic program promotes anticipation of environmental shifts. PLoS Biol. 2015;13:e1002042. doi: 10.1371/journal.pbio.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim M., Zhang Z.G., Okano H., Yan D.L., Groisman A., Hwa T. Need-based activation of ammonium uptake in Escherichia coli. Mol. Syst. Biol. 2012;8:616. doi: 10.1038/msb.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soupene E., He L., Yan D., Kustu S. Ammonia acquisition in enteric bacteria: Physiological role of the ammonium/methylammonium transport B (AmtB) protein. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7030–7034. doi: 10.1073/pnas.95.12.7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yanofsky C. RNA-based regulation of genes of tryptophan synthesis and degradation, in bacteria. RNA. 2007;13:1141–1154. doi: 10.1261/rna.620507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lubeck E., Cai L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods. 2012;9:743–748. doi: 10.1038/nmeth.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levsky J.M., Shenoy S.M., Pezo R.C., Singer R.H. Single-cell gene expression profiling. Science. 2002;297:836–840. doi: 10.1126/science.1072241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.