

SUMMARY
In ellipsoid-shaped ovococcus bacteria, such as the pathogen Streptococcus pneumoniae (pneumococcus), side-wall (peripheral) peptidoglycan (PG) synthesis emanates from midcells and is catalyzed by the essential class B penicillin-binding protein PBP2b transpeptidase (TP). We report that mutations that inactivate the pneumococcal YceG-domain protein, Spd_1346 (renamed MltG), remove the requirement for PBP2b. ΔmltG mutants in unencapsulated strains accumulate inactivation mutations of class A PBP1a, which possesses TP and transglycosylase (TG) activities. The “synthetic viable” genetic relationship between Δpbp1a and ΔmltG mutations extends to essential ΔmreCD and ΔrodZ mutations that misregulate peripheral PG synthesis. Remarkably, the single MltG(Y488D) change suppresses the requirement for PBP2b, MreCD, RodZ, and RodA. Structural modeling and comparisons, catalytic-site changes, and an interspecies chimera indicate that pneumococcal MltG is the functional homologue of the recently reported MltG endo-lytic transglycosylase of Escherichia coli. Depletion of pneumococcal MltG or mltG(Y488D) increases sphericity of cells, and MltG localizes with peripheral PG synthesis proteins during division. Finally, growth of Δpbp1a ΔmltG or mltG(Y488D) mutants depends on induction of expression of the WalRK TCS regulon of PG hydrolases. These results fit a model in which MltG releases anchored PG glycan strands synthesized by PBP1a for crosslinking by a PBP2b:RodA complex in peripheral PG synthesis.
Keywords: MltG YceG-domain proteins, ovococcus peptidoglycan biosynthesis, pneumococcus class A PBP1a, pneumococcal MreCD and RodA functions, WalRK TCS activation
INTRODUCTION
Streptococcus pneumoniae (pneumococcus) is a common commensal bacterium that colonizes the human nasopharynx as biofilms (Chao et al., 2014, Donkor, 2013, Hakansson et al., 2015, Short & Diavatopoulos, 2015). S. pneumoniae can become an opportunistic pathogen in individuals recovering from influenza or with compromised immune systems, causing a number of serious respiratory and invasive diseases, such as pneumonia, otitis media, bacteremia, and meningitis (Ferreira & Gordon, 2015, Gratz et al., 2015, Henriques-Normark & Tuomanen, 2013, Oliver & Swords, 2015, Vernatter & Pirofski, 2013). S. pneumoniae cells are shaped like prolate-ellipsoids, referred to as ovococci, that divide perpendicularly to their long axis and often form chains of cells, especially when capsule is present (Fig. S1A) (Barendt et al., 2009, Massidda et al., 2013, Pinho et al., 2013, Tsui et al., 2014, Zapun et al., 2008b). Pneumococcal cell shape and chaining are important to colonization and infection (Weiser, 2013, Rodriguez et al., 2012, Dalia & Weiser, 2011). The ellipsoid shape of S. pneumoniae and other ovococcus bacteria is mainly determined by the thick peptidoglycan (PG) cell wall surrounding these Gram-positive bacteria (Gisch et al., 2015, Massidda et al., 2013). PG is composed of glycan chains of β-1-4-linked N-acetylmuramic acid (MurNAc) and N-acetylglucosamine (GlcNAc) sugars that are cross-linked by PG peptides (Egan & Vollmer, 2013, Lovering et al., 2012, Turner et al., 2014, Typas et al., 2012). Pneumococcal PG provides the major scaffold for the covalent attachment of wall-teichoic acid (WTA), capsule, and surface proteins linked by sortases, many of which are virulence factors (Eberhardt et al., 2012, Gisch et al., 2015, Schneewind & Missiakas, 2013, Tomasz & Fischer, 2006).
PG is synthesized by bitopic, high-molecular-weight penicillin-binding (PBP) proteins, whose catalytic domains are extracellular (Fig. S1B) (Egan et al., 2015, Lovering et al., 2012, Typas et al., 2012). Class A PBPs contain separate transglycosylase (TG) and transpeptidase (TP) domains that catalyze glycan chain synthesis from external Lipid II substrate and PG peptide crosslinking, respectively. Class B PBPs contain TP domains and additional regulatory domains of unknown function. S. pneumoniae contains three class A (PBP1a, PBP2a, and PBP1b) and two class B (PBP2x and PBP2b) enzymes (Gisch et al., 2015, Massidda et al., 2013). As implied by their name, TP domains of PBPs are the targets of covalent inactivation by β-lactam antibiotics (Kocaoglu et al., 2015, Kocaoglu & Carlson, 2015, Hakenbeck et al., 2012, Zapun et al., 2008a). Pneumococcal β-lactam resistance has been increasing at an alarming rate worldwide (Philippe et al., 2015, Hakenbeck, 2014), and S. pneumoniae clinical strains resistant to multiple antibiotics are considered by the CDC to be a serious threat to public health in the U.S.A. (CDC, 2013). Unlike many other bacterial species that pick up genes for β-lactamases by horizontal gene transfer, S. pneumoniae acquires resistance to β-lactam antibiotics by mutations in its core PBPs (Hakenbeck et al., 2012, Zapun et al., 2008a). These resistance mutations can arise spontaneously and can be transferred by natural competence from related Streptococcus species to form mosaic pbp genes (Todorova et al., 2015, Hakenbeck et al., 2012). High-level β-lactam resistance is caused by combinations of mutations in pbp1a, pbp2x, and pbp2b (Philippe et al., 2015, Chewapreecha, et al., 2014, Hakenbeck, 2014, Hakenbeck et al., 2012).
Given its potential as a target for future antibiotic discovery and its role in resistance development to current antibiotics, it is important to understand the mechanisms of PG biosynthesis in differently shaped pathogenic bacteria (den Blaauwen et al., 2014, Sham et al., 2012, Teo & Roper, 2015). To form ellipsoid-shaped cells, S. pneumoniae and other ovococcus bacteria require two modes of PG synthesis: septal PG synthesis to divide cells and a form of side-wall (peripheral) PG synthesis to elongate cells (Fig. S1A) (Gisch et al., 2015, Massidda et al., 2013, Pinho et al., 2013, Tsui et al., 2014, Zapun et al., 2008b). Peripheral PG synthesis distinguishes ovococci from cocci species that only carry out septal PG synthesis (Pinho et al., 2013, Zapun et al., 2008b). In contrast to rod-shaped bacteria, S. pneumoniae lacks MreB family proteins, and septal and peripheral PG synthesis are initially directed by the FtsZ ring from the midcells of dividing pneumococcal cells (Gisch et al., 2015, Massidda et al., 2013, Tsui et al., 2014, Zapun et al., 2008b). In this sense, pneumococcal peripheral PG synthesis resembles pre-septal PG side-wall synthesis in some rod-shaped bacteria (Potluri et al., 2012, Typas et al., 2012, van der Ploeg et al., 2013).
Recent work shows that class B PBP2x and PBP2b are essential for growth and required for septal and peripheral PG synthesis, respectively, in dividing S. pneumoniae cells (Berg et al., 2013, Berg et al., 2014, Fleurie et al., 2014, Land et al., 2013, Morlot et al., 2013, Peters et al., 2014, Tsui et al., 2014). pbp1a and pbp2a, encoding PBP1a and PBP2a, respectively, are synthetically lethal and cannot both be inactivated (Hoskins et al., 1999, Paik et al., 1999). However, PBP1a and PBP2a are not equivalent as indicated by the reduction of cell width and size of Δpbp1a mutants compared to those of Δpbp2a mutants or wild-type parent cells (see Results) (Land & Winkler, 2011). PBP1a may be subject to control by division regulators, such as MreCD (Land & Winkler, 2011), and mutant PBP1a contributes to high-level β-lactam resistance, suggesting a possible role in both septal and peripheral PG synthesis (Philippe et al., 2015, Chewapreecha, et al., 2014, Hakenbeck, 2014, Hakenbeck et al., 2012). On the other hand, the functions of class A PBP2a and PBP1b remain largely unknown in growing and stressed pneumococcal cells (Massidda et al., 2013). PBP2b, PBP2x and PBP1a follow a different localization pattern than FtsZ and remain at division septa after FtsZ reappears at the equators of daughter cells (Land et al., 2013, Tsui et al., 2014). In addition, PBP2x moves to the centers of septa surrounded by an adjacent constricting ring containing PBP2b, PBP1a, and regulators StkP and MreC, suggesting a separation of the septal and peripheral PG synthesis machines in mid-to-late divisional cells (Cadby & Lovering, 2014, Tsui et al., 2014).
Models have been proposed for interactions of PBP2x, PBP2b, and PBP1a with pneumococcal homologues of regulatory proteins that control PG synthesis in rod-shaped bacteria (Massidda et al., 2013, Philippe et al., 2014). Depletion of these regulatory proteins results in elongation or increased sphericity of cells, consistent with roles in septal or peripheral PG synthesis, respectively (Land & Winkler, 2011, Tsui et al., 2014). Corroborated interactions have been demonstrated with some regulatory proteins (e.g., PBP2x and the StkP protein kinase (Morlot et al., 2013) and PBP2b and RodA (Philippe et al., 2014)), but for the most part, the composition, chronology, and coordination between the septal and peripheral PG synthesis machines remains speculative (Gisch et al., 2015, Massidda et al., 2013, Philippe et al., 2014). The current model of the peripheral PG synthesis machine (Fig. S1B) includes class B PBP2b, class A PBP1a, regulatory proteins MreC, MreD, and RodZ, and RodA, whose role as a Lipid II flippase was recently called into question in E. coli (Sham et al., 2014). With the exception of pbp1a (PBP1a), all other known genes that encode peripheral PG synthesis proteins are essential for growth of S. pneumoniae (see below) (Berg et al., 2013, Land & Winkler, 2011, Massidda et al., 2013, Tsui et al., 2014, van Opijnen & Camilli, 2012). Missing from this model is a PG cleavage enzyme to release newly synthesized, Lipid II-tethered glycan strands for insertion and PG peptide crosslinking into existing PG. In this paper, we report the isolation of multiple suppressor mutations in the spd_1346 gene that relieve the requirement for essential PBP2b. We show that spd_1346, renamed mltG, encodes the structural and functional pneumococcal homologue of the membrane-bound endo-lytic transglycosylase of E. coli recently reported by the Bernhardt laboratory (Yunck et al., 2015). Localization studies and characterization of phenotypes of mltG mutations in combinations with mutations in genes that mediate peripheral PG synthesis support the hypothesis that MltG is the missing PG cleavage enzyme that releases newly synthesized glycan strands during peripheral PG synthesis. Moreover, combinations of mutations defective in peripheral PG synthesis are consistent with an interaction between PBP2b and RodA in pneumococcal cells.
RESULTS
Mutations in spd_1346 (mltG) suppress a Δpbp2b deletion mutation
Some reports of essential genes in Streptococcus pneumoniae did not list pbp2b, which encodes class B PBP2b in the peripheral PG synthesis machine (Fig. S1B), as essential (see Massidda et al., 2013). However, recent characterizations of pbp2b strongly support its essentiality in wild-type cells (Berg et al., 2013, Berg et al., 2014, Tsui et al., 2014). Consequently, we examined whether suppressors could arise that allow growth of Δpbp2b mutants lacking essential PBP2b. We transformed a Δpbp2b<>aad9 (spectinomycinR) amplicon into an unencapsulated isogenic derivative of serotype 2 wild-type strain D39 [IU1945 (D39 Δcps)] (Lanie et al., 2007) and incubated plates for different lengths of time (Tables 1 and 2). We performed initial selections in an unencapsulated mutant, because previous work showed that capsule synthesis often dampens phenotypes of pneumococcal strains defective in peptidoglycan biosynthesis or cell division (see below) (Barendt et al., 2009, Barendt et al., 2011). As a control, we transformed the same amount of Δpbp2b<>aad9 amplicon into a merodiploid strain (IU7337; Table S1) that expresses PBP2b from an ectopic site under the control of a fucose-regulated promoter. Hundreds of transformants of strain IU7337 appeared overnight (20 h) on each plate containing fucose, whereas no colonies were visible for transformations of strain IU1945 or IU7337 on plates lacking fucose (rows 1–3, Table 2). However, <10 Δpbp2b<>aad9 transformants of IU1945 appeared on each plate in 40 h (Table 1). By comparison, no suppressors of a Δpbp2x<>aad9 deletion mutation, which would lack essential Class B PBP2x involved in the septal PG synthesis machine (Land et al., 2013, Massidda et al., 2013, Tsui et al., 2014), were isolated in parallel experiments (data not shown).
TABLE 1.
Mutation in D39 Δcps Δpbp2b suppressor mutants determined by Illumina whole-genome sequencinga
| Strainb | Gene and mutationc | Function | nucleotide changed |
|---|---|---|---|
| 1. sup2 (IU7477) | spd_1346 (mltG) (Δ5 bp at Y403) | Hypothetical YceG-like family protein | TACTATAG→TAG (Δbp 1212–1216/1656, Y403stop) |
| 2. sup3 (IU7567) | spd_1346 (mltG) (Y488D) | Hypothetical YceG-like family protein | TAT→GAT (bp 1462/1656) |
| 3. sup4 (IU7570) | spd_1346 (mltG)( Δ488 bp at T54) | Hypothetical YceG-like family protein | Internal out-of-frame Δ488 bp at 165–652/1656 |
| groES (upstream) | Co-chaperonin GroES | G→T at −48 | |
| 4. sup5 (IU7765) | spd_1346 (mltG) (Ω45 bp)2 | Hypothetical YceG-like family protein | Out of frame 45 bp (bp 302–346) duplication, TCT→TGA (S101stop) at bp 301–303 |
| spd_0115 (G41C) | HlyD family secretion protein | GGC→TGC (bp 121/1253) |
Illumina whole-genome sequencing was performed as described in Experimental procedures.
Strains IU7477, IU7567, IU7570 and IU7765 were isolated from the small number of colonies that arose following independent transformations of unencapsulated D39 Δcps strain IU1945 with a Δpbp2b<>aad9 amplicon (see text and Table S1).
Besides the Δpbp2b<>aad9 insertion, the mutational changes shown are compared to the sequence of the IU1945 parent strain.
Nucleotide and codon changes are indicated by underlined and bold letters, respectively.
TABLE 2.
Appearance of colonies after transformation with a Δpbp2b<>aad9 amplicon into D39 strainsa
| Recipient strain and condition | Genotype | Number of colonies at 20 h after transformation with Δpbp2b<>aad9 amplicon |
|---|---|---|
| Strains of IU1945 (D39 Δcps) genetic background | ||
| 1. IU1945b | WT | 0 |
| 2. IU7337 | ΔbgaA::PfcsK-pbp2b | 0 |
| 3. IU7337 + fucosec | ΔbgaA::PfcsK-pbp2b | >500 |
| Strains of IU1824 (D39 Δcps rpsL1) genetic background | ||
| 4. IU1824 | pbp1a+ wild-type parent | 0 |
| 5. IU6741 | Δpbp1a | 0 |
| 6. IU7325 | Δpbp1a ΔmltG::Pc-[kan-rpsL+] | >500 |
| 7. IU7327 | Δpbp1a ΔmltG::Pc-erm | >500 |
| 8. IU8549 | Δpbp1a mltG(Δ5bp) (sup2) | >500 |
| 9. IU8551 | Δpbp1a mltG(Y488D) (sup3) | 0 |
| 10. IU9760d | pbp1a+ mltG(Y488D) (sup3) | >500 |
| 11. IU8553 | Δpbp1a mltG(Δ488bp) (sup4) | >500 |
| 12. IU8555 | Δpbp1a mltG(Ω45bp)2 (sup5) | >500 |
| 13. IU8873 | Δpbp1a mltG(E428Q) | >500 |
| 14.IU8982 | Δpbp1a mltG(E428A) | >500 |
| 15. IU8910 | Δpbp1a mltG(ΔDUF_1346) | 0 |
| 16. IU9025 | pbp1a+ mltG(ΔDUF_1346) | 0 |
| 17. IU10919 | pbp1a+ mltGSpn-Eco | >500 tiny colonies |
| 18. IU10965 | Δpbp1a mltGSpn-Eco | >500 tiny colonies |
| 19. IU11007e | pbp1a+ mltGEco | >500 tiny colonies |
| 20. IU11009e | Δpbp1a mltGEco | >500 tiny colonies |
| Strain of IU1690 (D39 cps+) genetic background | ||
| 21. IU1690 | WT | 0 |
Recipient strains were constructed as described in Table S1. Transformations and visualization of colonies normalized to 1 mL of transformation mixture were performed as described in Experimental procedures. The same results for each strain were obtained from two independent transformation experiments. Normal wild-type colony size was observed, except for the last two strains. Full MltG activity in a pbp1a+ or Δpbp1a strain gave 0 transformants, whereas no MltG activity in a Δpbp1a strain gave >500 normal sized colonies (see text for details).
<10 colonies were visible after 40 h of incubation. Transformants of IU1945 with the Δpbp2b<>aad9 amplicon that arose after 40 h of incubation were characterized as the original sup2-sup5 suppressor mutations described in Table 1 (see text and Table S1).
1% (wt/vol) L-fucose was added to all steps in the transformation procedure to induce PBP2b+ expression in merodiploid strain IU7337 (Table S1).
Strain IU9895 (mltG(Y488D)-Pc-erm, which was independently constructed in the IU1945 Δcps background (Table S1), showed the same suppression of the Δpbp2b mutation (data not shown).
mltGSpn reading frame in the pneumococcal chromosome is replaced with the intact mltGEco reading frame; the promoter, ribosome binding site, and downstream regions are from S. pneumoniae.
Δpbp2b suppressor strains were isolated from independent transformations, and the presence of the Δpbp2b<>aad9 deletion mutation was confirmed by PCR. Whole-genome sequencing showed that 4 of 5 suppressor mutants contain mutations at different positions in the spd_1346 gene (Fig. 1A), along with some dissimilar drift mutations (Table 1). In this study, we characterized the suppression of the Δpbp2b mutation by the four mutations (sup2-sup5) in spd_1346, three of which (sup2, sup4, and sup5) were predicted to truncate the spd_1346 gene product (Fig 1B, middle). The other suppressor mutation (sup3) caused a Y488D amino acid (aa) change in the Spd_1346 protein. Western blotting of FLAG-epitope tagged mutant Spd_1346 proteins expressed from their native chromosomal locus confirmed that only the sup3(Y488D) mutant produced Spd_1346 protein, which was not detectable in the other three suppressor mutants (Fig. S2A).
Fig. 1.

(A) Arrangement of genes surrounding mltG (spd_1346) in the S. pneumoniae D39 chromosome. The genes encode the following proteins: murC (UDP-N-acetylmuramate-alanine ligase); spd_1348, spd_1347, and spd_1343 (putative GNAT family acetyltransferases; greA (transcription elongation); and spd_1344 (hypothetical protein). spd_1346 is predicted to be in an operon with the two downstream genes greA and spd_1344 by the DOOR prediction program (Mao et al., 2009). (B) Domain architecture of pneumococcal MltG, predicted MltG peptide products from Δpbp2b suppressor strains (Table 1), and constructed mltG mutations. L indicates a conserved LysM-like structure with a β1α1α2β2α3 fold (see text). The MltG(Y488D), MltG(E428Q) and MltG(E428A) mutant proteins are expressed as shown in Western blots (Fig. S2A, S2B). MltG(ΔDUF_1346) and the MltGSpn-Eco chimera proteins are expressed, because they are functional (Fig. S9 and text).
Spd_1346 contains three predicted domains: a cytoplasmic region containing a domain of unknown function (called DUF_1346 here; 187 aa) that has weak homology to a segment of MinC (FtsZ placement), MCLC (chloride channel-like), and other DUF domains; a transmembrane domain (TM; 21 aa); and a YceG domain (275 aa) that also contains a LysM-like putative PG binding segment (L) (Fig. 1B). Concurrent with our study, the Bernhardt laboratory discovered that the YceG-domain protein of E. coli encodes a new endo-lytic transglycosylase (endo-LT), named MltG, which was described recently in a separate paper (Yunck et al., 2015). Evidence presented later in this paper shows that Spd_1346 is the MltG endo-LT homologue in S. pneumoniae, and hence we named the spd_1346 gene “mltG” to unify nomenclature. Proteins encoding extracellular YceG domains are distributed widely in bacteria (Yunck et al., 2015). In addition, there are two distinct architectures of YceG-domain proteins. In Streptococcus species and other ovoid bacteria, these proteins contain a cytoplasmic domain (150–200 aa) that varies among species in addition to the TM (≈24 aa) and YceG domains (Fig. S3). In contrast, the YceG-domain proteins of Gram-positive and -negative rod-shaped bacteria contain a short N-terminal cytoplasmic domain (<20 aa). Staphylococcus species lack detectable YceG-domain protein homologues (Fig. S3) (Yunck et al., 2015). Pneumococcal mltG(ΔDUF_1346) mutants are characterized below.
ΔmltG mutants acquire suppressor mutations that inactivate pbp1a, which encodes class A PBP1a
The 4 original Δpbp2b suppressor mutants grew surprisingly well, considering that they are missing the gene encoding essential class B PBP2b involved in peripheral PG synthesis (data not shown; see Fig. 2A for reconstructed strains). To confirm suppression of the Δpbp2b deletion mutation by the sup2-5 alleles of mltG (Table 1), we needed to re-construct the suppressor strains in a clean genetic background. We tried to replace an internal segment of mltG with the Janus (Pc-[kan-rpsL+]) cassette that allows allele replacement in an rpsL1 (streptomycinR) background (Table S1) (Sung et al., 2001). ΔmltG::Janus or comparable ΔmltG::Pc-erm transformants of unencapsulated strains appeared at 20 h as barely visible colonies, along with a few (<5) larger colonies (Fig. 3C, top row). Control transformations, in which a second copy of mltG+ was expressed ectopically under control of a zinc-inducible promoter, gave >500 uniform, normal sized colonies (Fig. 3C, middle row). Single colony isolation of the barely visible ΔmltG::Pc-erm colonies resulted in a mixture of barely visible and larger colonies, indicative of accumulation of suppressor mutations.
Fig. 2.

Growth curves and Boc-FL labeling of PBPs in reconstructed Δpbp2b sup2-5 strains and growth curve of the mltGSpn-Eco chimera mutant strain. The mutations are illustrated in the middle and at the bottom of Figure 1. Reconstruction of Δpbp2b sup 2-5 is diagramed in Figure S4 and described in Table S1. Reconstructed Δpbp2b sup2-5 strains are: IU9777 (mltG(Δ5bp) (sup2) Δpbp2b); IU9783 (mltG(Y488D) (sup3) Δpbp2b); IU9905 (mltG(Δ488bp) (sup4) Δpbp2b); and IU9907 (mltG(Ω45bp) (sup5) Δpbp2b). (A) Representative growth curves of isogenic pbp2b+ parent strain IU1824 and reconstructed Δpbp2b sup2-5 strains. Strains were grown as described in Experimental procedures. (B) Fluorescent-bocillin (Boc-FL) binding to PBPs in isogenic strains IU6741 (Δpbp1a), IU7850 (Δpbp1b), IU7852 (Δpbp2a), IU1824 (wild-type parent) and the reconstructed Δpbp2b sup2-sup5 strains listed above was performed as described in Experimental procedures. (C) Growth curves of S. pneumoniae strains IU1824 wild-type parent (D39 Δcps rpsL1), IU10919 (IU1824 mltGSpn-Eco), and IU11007 (mltGEco) (see Table S1). Growth curve determinations and Boc-FL binding experiments were performed three times independently with similar results. See text for additional details.
Fig. 3.

Mutations in pbp1a suppress the ΔmltG mutations. (A) Arrangement of genes surrounding pbp1a in Streptococcus pneumoniae D39 chromosome. recU encodes recombination protein U. (B) Domain architecture of PBP1a, TP active site motifs, and mapped mutations in pbp1a in ΔmltG suppressor strains. M, transmembrane domain (aa 13 to 35); TG, transglycosylase domain (aa 59 to 237); TP, transpeptidase domain aa (332 to 622). All pbp1a mutants, except pbp1a(S89F), could not be transformed with a Δpbp2a deletion and showed the small-cell phenotype characteristic of Δpbp1a mutants, whereas pbp1a(S89F) mutants showed an intermediate size (data not shown). Western blots of FLAG-tagged PBP1a(S89F) or PBP1a(G494E) showed a wild-type or significantly reduced (≈33%) amount of PBP1a, respectively (data not shown). (C) Appearance and number of colonies obtained after transformation with a ΔmltG amplicon into S. pneumoniae D39 Δcps strains. 50 ng of ΔmltG::Pc-erm amplicon obtained from strain IU7260 was transformed into the strains as described in Experimental procedures and examined at 20 h of incubation at 37°C in an atmosphere of 5% CO2. Strains and genotypes, upper half of table: IU1945 wild-type parent (D39 Δcps); K180 (Δpbp1b::Pc-[kan-rpsL+]); IU6680 (Δpbp2a::Pc-[kan-rpsL+]); IU8872 (pbp1a+ mltG+//ΔbgaA::tet-PZn-RBSmltG-mltG); and IU6662 (Δpbp1a::Pc-[kan-rpsL+]). MltG expression in merodiploid strain IU8872 was induced by 0.2 mM ZnCl2 + 0.02 mM MnSO4 (to counter zinc toxicity), which was added where indicated to growing cultures 1h before transformation and to all subsequent steps in transformations, including plates. Strains and genotypes, lower half of table: IU1824 wild-type parent (D39 Δcps rpsL1); IU6741 (Δpbp1a); IU7845 (pbp1a (T insertion at Phe33); IU7840 (pbp1a (S89F)); IU7843 (pbp1a (A deletion at Lys160); IU7839 (pbp1a (G deletion at Gly451); and IU7837 (pbp1a (G494E). Construction of strains is described in Supplemental experimental procedures and Table S1. (TG) or (TP), aa substitution in transglycosylase or transpeptidase domain; (FS), frameshift mutation.
Previously, we reported a “synthetic-viable” genetic relationship in which a ΔmreCD deletion mutation is suppressed by a Δpbp1a mutation that eliminates class A PBP1a from pneumococcal cells (Fig. 4A) (Land & Winkler, 2011). In that study, we also isolated a stop codon mutation near the middle of mltG(L354-Stop) that suppressed the ΔmreCD mutation, but this suppression was not studied further at that time (Land & Winkler, 2011). Since both PBP2b and MreCD are required for peripheral PG synthesis in S. pneumoniae (Fig. S1B) (Berg et al., 2013, Land & Winkler, 2011, Tsui et al., 2014), we tested whether faster growing ΔmltG::Janus or ΔmltG::Pc-erm mutants contained suppressor mutations that eliminated or reduced PBP1a activity (Fig. 3A and 3B). Indeed, 5/5 faster growing ΔmltG::Janus or ΔmltG::Pc-erm mutants that were characterized contained frame-shift or missense mutations in pbp1a (Fig. 3B). Additional experiments showed that the ΔmltG::Pc-erm deletion mutation could readily be transformed into a Δpbp1a mutant or into reconstructed strains containing the originally selected pbp1a suppressor alleles (Fig. 3C, bottom row). In contrast, the ΔmltG::Pc-erm amplicon could not be transformed stably into Δpbp1b or Δpbp2a mutants, which lack the other two pneumococcal class A PBPs (Fig. 3C, top row). We conclude that there is a synthetic-viable relationship between mltG and pbp1a, where ΔmltG mutations are only tolerated in unencapsulated strains when pbp1a is partially or wholly inactivated. This suppression precluded constructing a ΔmltG Δpbp2b pbp1a+ mutant. Complementation of the ΔmltG mutation was performed in MltG depletion experiments described below.
Fig. 4.

Summary of Δpbp1a synthetic-viable suppression patterns and viable and inviable mutation combinations with the mltG(Y488D) mutation. (A) Red arrows indicate direct suppression of ΔmltG, ΔmreCD, and ΔrodZ mutations by the Δpbp1a deletion. Blue arrows indicate suppression of Δpbp2b and ΔrodA mutation by the combination of ΔmltG Δpbp1a mutations (Tables 2 and 3). (B) mltG(Y488D) suppresses the requirement for each component of the peripheral PG synthesis machine, including essential proteins PBP2B, MreCD, RodZ, and RodA, as well as combinations of mutations that were tested in this study (Tables 2 and 3). Neither the mltG(Y488D) Δpbp1a Δpbp2b nor mltG(Y488D) Δpbp1a ΔrodA mutant could be constructed by multiple strategies. Construction of strains is described in Supplemental experimental procedures and Table S1. See text for additional details.
Reconstructed strains containing mutations in mltG suppress a Δpbp2b mutation
We took advantage of the stabilization of the ΔmltG mutation in Δpbp1a mutants to reconstruct strains to verify that the originally selected mltG mutations suppress the requirement for PBP2b (Table 1). To reconstruct the original suppressor strain, we constructed a Δpbp1a ΔmltG::Janus intermediate strain that allowed allele exchange of the mltG(Δ5bp) (sup2), mltG(Δ488bp) (sup4), or mltG(Ω45bp)2 (sup5) alleles into the chromosome (Fig. 1; Fig. S4; Table S1). We next crossed the Δpbp2b mutation into each strain and finally repaired the Δpbp1a mutation back to pbp1a+ (Fig. S4, Table S1). The resulting strains have only the ΔmltG(Δ5bp) (sup2), mltG(Δ488bp) (sup4), or mltG(Ω45bp)2 (sup5) allele and the Δpbp2b mutation, without any additional drift mutations found in the original suppressor strains (Table 1; Fig. S4; Table S1). During these constructions, we noticed that the mltG(Y488D) (sup3) allele could be crossed directly into a pbp1a+ strain without the need for a Δpbp1a intermediate (Fig. S4, right). We ascribe the special phenotypes of the mltG(Y488D) mutant to partial MltG activity (see below).
The reconstructed suppressed strains grew remarkably well, considering that they are missing essential PBP2b (Fig. 2A), similar to the original suppressor strains (Table 1; data not shown). We checked the relative activities of the pneumococcal PBPs by fluorescent-bocillin labeling of the parent and suppressed Δpbp2b strains (Fig. 2B). The reconstructed sup2-5 Δpbp2b mutants had comparable activities of PBP1a, PBP1b, PBP2x, PBP2a, and PBP3 to the wild-type parent strain (Fig. 2B). However, despite their relatively normal growth in culture (Fig. 2A), re-streaking and regrowth of the reconstructed mltG(Δ5bp) (sup2) Δpbp2b, mltG(Δ488bp) (sup4) Δpbp2b, or mltG(Ω45bp)2 (sup5) Δpbp2b suppressed mutants led to the appearance of colonies of different sizes and variability in growth rates, suggesting that these strains may be accumulating additional suppressor mutations (data not shown) (Fig. S5). In contrast, the mltG(Y488D) (sup3) Δpbp2b mutant exhibited consistently stable growth characteristics (see below; Fig. 2A and 4B).
To confirm that mltG mutations suppress the requirement for PBP2b, we stabilized mltG mutations in a Δpbp1a mutant background (see Fig. 3C and S5) and determined transformation frequency with a Δpbp2b<>aad9 amplicon (Table 2). Consistent with previous work (Berg et al., 2013, Tsui et al., 2014), pbp2b is essential in unencapsulated or encapsulated wild-type D39 strains (Table 2, lines 1, 4, and 21). As expected, the Δpbp2b amplicon could be transformed into a strain that expresses PBP2b ectopically under the control of a fucose-inducible promoter (Table 2, line 3). Notably, a Δpbp1a mutation alone does not suppress a Δpbp2b mutation directly (Table 2, line 5; Fig. 4A). Δpbp1a ΔmltG and the Δpbp1a [mltG(Δ5bp) (sup2), mltG(Δ488bp) (sup4), or mltG(Ω45bp)2 (sup5)] mutants were all readily transformable by the Δpbp2b amplicon (Table 2, lines 6–8, 11, and 12). Together, these results confirm that the absence of MltG obviates the requirement for PBP2b, and we conclude that Δpbp2b suppression by null mutations of mltG is only genetically stable when PBP1a activity is reduced or eliminated in unencapsulated strains (Table 2; Fig. 3 and S5). In contrast, unlike the pbp1a+ parent, the pbp1a+ mltG(Y488D) (sup3) mutant was directly transformable by the Δpbp2b amplicon (Table 2, lines 4 and 10), consistent with reduced activity of the MltG(Y488D) protein. However, the Δpbp1a mltG(Y488D) (sup3) mutant was unexpectedly not transformable by the Δpbp2b amplicon (Table 2, line 9). mltG(Y488D) (sup3) Δpbp1a Δpbp2b mutants also could not be constructed by other strategies. The inviability of the mltG(Y488D) Δpbp1a Δpbp2b can be accounted for by the roles played by these three proteins in peripheral PG synthesis (see Discussion).
S. pneumoniae MltG (MltGSpn) is the structural and functional homologue of the MltG endo-LT in E. coli (MltGEco)
Concurrent with these studies, the Bernhardt laboratory identified MltGEco as a novel inner membrane-bound, extracellular endo-LT (Yunck et al., 2015). MltGEco overexpression was not tolerated in an E. coli ΔponB mutant lacking class A PBP1b (Yunck et al., 2015). Note that PBP1bEco is functionally analogous to PBP1aSpn instead of PBP1bSpn, which plays a dispensable role in pneumococcal PG synthesis (Introduction). In contrast to ΔmltG mutants of S. pneumoniae (Fig. 3), ΔmltG mutants of E. coli do not show growth phenotypes (Yunck et al., 2015). MltGSpn is the only YceG-domain protein in S. pneumoniae, and MltGSpn shows aa similarities and identities in its TM and YceG domains to MltGEco and the MltG homologue of Listeria monocytogenes (MltGLmo) (Fig. S3 and S6). The crystal structures of the YceG-domains of MltGEco and MltGLmo were reported (see (Yunck et al., 2015)), and we threaded the MltGSpn sequence onto each structure using the Phyre2 program (see Experimental procedures) (Fig. 5 and S7A; Table S3). The predicted structural similarity of MltGSpn(YceG) with MltGEco(YceG) (Z-score = 31.4; RMSD = 3.7 A) and with MltGLmo (YceG) (Z score = 38.4; RMSD = 0.9 A) indicates that these three proteins are folded nearly identically (Fig. 5 and S7A; Table S3). Furthermore, the critical, conserved active-site Glu (E428/E218 in MltGSpn/MltGEco, respectively) overlaps spatially, as does Tyr (Y488/Y274), which is changed in the partially active mltG(Y488D) mutant (Table 2; Fig. S4 and S5; Discussion). The Glu and Tyr aa’s are highly conserved in MltGLmo and other MltG homologues (α10 and α13, Fig. S7). Finally, the predicted LysM-like putative PG binding domain found in the N-terminal proximal region of the MltGLmo (YceG) domain is predicted to be present in both MltGSpn(YceG) (Fig. S7A and S7B) and MltGEco(YceG) (Yunck et al., 2015). These nearly identical predicted structures suggest that MltGSpn is the homologue of the MltGEco endo-LT.
Fig. 5.
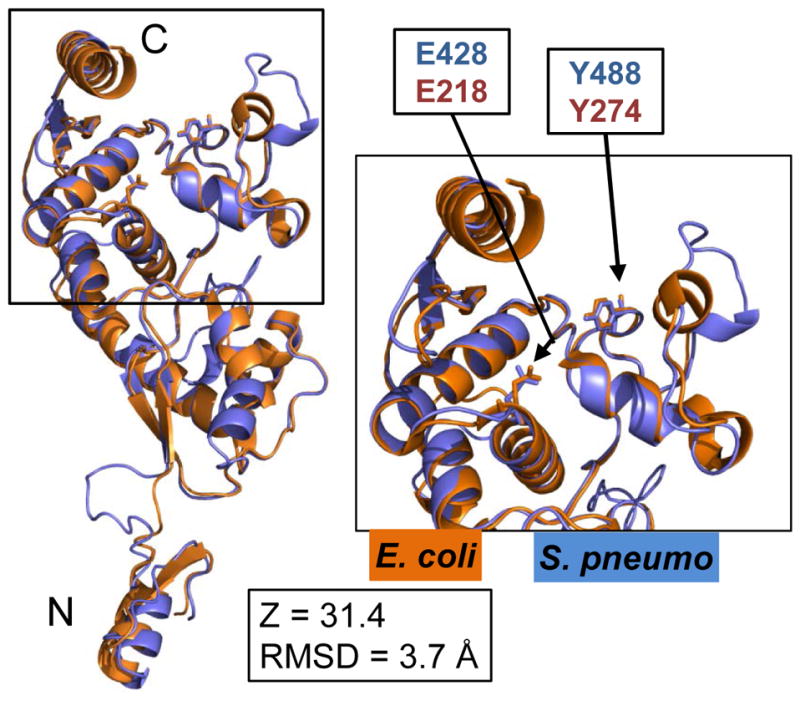
Superposition of the C-terminal YceG domains of MltGEco (orange, aa 81–340) and MltGSpn (blue, aa 266–547), including side chains of the conserved catalytic glutamate (E428 of MltGSpn; E218 of MltGEco) and tyrosine (Y488 of MltGSpn; Y274 of MltGEco). The MltGSpn structural model was generated by using the Phyre2 server as described in Experimental procedures, which listed aa 266–547 of MltGSpn alignment to aa 81–340 of MltGEco with 100% match confidence. The short N-terminal intracellular region and transmembrane of MltGEco (aa 1–80) were not included in the crystal structure analysis. The Phyre2-generated structural model of MltGSpn (blue) was overlaid and aligned with the crystal structure of the C-terminal of MltGEco (orange, PDB 2R1F) using PyMOL. Inset shows a close up of the fold with the conserved catalytic glutamate (E428/E218) and tyrosine (Y488/Y274). N and C indicate the amino- and carboxyl-ends of the aligned peptides. RMSD and Z-scores were obtained as described in Experimental procedures and Table S4.
To strengthen this conclusion, we introduced E428Q and E428A changes in what should be the catalytic Glu of MltGSpn (see (Yunck et al., 2015)), if indeed it functions as an endo-LT (Fig. 1, bottom, 5, and S7B; Table S1). As expected from attempts to construct ΔmltG mutants (Fig. 3), strains containing the mltG(E428Q) and mltG(E428A) mutations are unstable in the wild-type background, consistent with inactivation of MltG. Therefore, we again constructed the mltG(E428Q) and mltG(E428A) mutations in a Δpbp1a mutant to stabilize the expected null mutants. Both the Δpbp1a mltG(E428Q) and Δpbp1a mltG(E428A) mutants are transformed at high efficiency with the Δpbp2b amplicon, consistent with loss of MltG LT activity (Table 2, lines 13 and 14). Western-blot control experiments confirmed that the MltG(E428Q) and MltG(E428A) proteins can be detected in cells, although their amounts are somewhat lower (≈50–60%) compared to wild-type MltG (Fig. S2B). Titration control experiments showed that expression levels of wild-type MltG at <10% of its normal level is sufficient to restore growth to a ΔmltG mutant (data not shown). Taken together, these results support the conclusion that the MltG(E428Q) and MltG(E428A) mutant proteins are inactive.
The soluble MltGEco(YceG) domain was purified and shown to have weak, but detectable, endo-LT activity (Yunck et al., 2015). We purified the analogous soluble MltGSpn(YceG) domain, but we could not detect PG cleavage by sMltGSpn(YceG) under a variety of assay conditions (data not shown). We also could not detect PG cleavage activity of refolded MltGSpn from whole cell extracts in zymograms of ΔlytC mutants, which removed potential overlap of same-sized MltG and LytC N-acetylmuramidase (Fig. S8; Table S4). To further demonstrate that MltGSpn is an endo-LT, we constructed a chimera containing the DUF_1346 and TM domains of MltGSpn fused to the LysM-like (L) and YceG domains of MltGEco (Fig. 1B, bottom). Soluble MltGEco containing the LysM-like (L) and YceG domains was established enzymatically as an endo-LT (Yunck et al., 2015). The MltGSpn-Eco chimera expressed from the native mltG chromosomal locus in an unencapsulated strain (IU10919, Table S1) formed colonies and grew in culture similar to the mltG+ parent strain (Fig. 2C), rather than a ΔmltG mutant (Fig. 3C). A second chimera containing the DUF_1346, TM, and LysM-like domains of MltGSpn fused to the YceG endo-LT domain of MltGEco formed tiny colonies and was unstable when cultured (data not shown). Transformation of the pbp1a+ mltG(Y488D) or pbp1a+ mltGSpn-Eco mutant with Δpbp2b amplicon gave >500 normal-sized or >500 tiny colonies, respectively (Table 2, rows 10 and 17), suggesting that MltGSpn-Eco is more active than MltG(Y488D). Consistent with this interpretation, a mltGSpn-Eco Δpbp1a Δpbp2b mutant is impaired for growth, but viable, whereas a mltG(Y488D) Δpbp1a Δpbp2b is inviable (Table 2, lines 9 and 18). Finally, we found that the mltGEco reading frame could replace the mltGSpn reading frame, without the need to construct an interspecies chimera (Table 2, lines 19 and 20; Table S1). Pneumococcal pbp1a+ strains expressing MltGEco grow normally (Fig. 2C), and pbp1a+ and Δpbp1a strains expressing MltGEco form >500 tiny colonies when transformed with a Δpbp2b amplicon, similar to strains expressing the MltGSpn-Eco chimera (Table 2, lines 17 and 18). From these combined results, we conclude that MltGSpn is the structural and functional homologue of the MltGEco endo-LT.
The cytoplasmic MltGSpn(DUF_1346) domain is not required for endo-LT function
The YceG domain of MltGEco was definitively shown to be extracellular (Yunck et al., 2015). Given the structural and functional conservation of MltGEco(YceG) and MltGSpn(YceG) described above, we make the reasonable assumption that the MltGSpn(YceG) domain is extracellular. The MltGSpn contains an additional intracellular domain (DUF_1346) not present in MltGEco (Fig. S3). We deleted the large central region (133 aa’s) of DUF_1346 that shows weak homology to domains and DUFs in other proteins (Fig. 1B, bottom). The mltG(ΔDUF_1346) mutant did not show any growth, cell shape, or morphology defects (Fig. S9A and S9D) and could not be transformed with the Δpbp2b mutation (Table 2, lines 15 and 16), indicative of nearly full endo-LT activity of MltG(ΔDUF_1346). We examined whether growth of the mltG(ΔDUF_1346) mutant was affected by several stress conditions. Compared to the unencapsulated mltG+ parent strain, growth of the mltG(ΔDUF_1346) mutant was unaffected by addition of moderate salt (0.08 M NaCl), pH change (pH 7.2 or 5.8), a sublethal concentration of penicillin G (0.004 μg per mL) in BHI broth, or higher temperature (42°C) (Fig. S9A–S9C; data not shown). Only high salt (0.3 M NaCl) addition inhibited the growth of the mltG(ΔDUF_1346) mutant compared to the unencapsulated mltG+ parent (Fig. S9A). We did not study this condition further, because S. pneumoniae does not encounter high salt in the human host (Wilson, 2005). We conclude that the cytoplasmic DUF_1346 domain is not required for pneumococcal MltG endo-LT activity under normal culture conditions and several stress conditions tested.
MltG depletion or reduced activity causes spherical cell formation in unencapsulated and encapsulated D39 strains
We depleted MltG cellular amount in an unencapsulated D39 derivative to understand its role in cell division. A ΔmltG deletion was introduced into a merodiploid strain expressing MltG from an ectopic chromosomal location under the control of a zinc-inducible promoter (Fig. 6, Table S1). Growth of the ΔmltG/PZn-mltG+ merodiploid strain in BHI broth containing 0.2 mM Zn2+ (and 0.02 mM Mn2+ to reduce zinc toxicity) was similar to that of the wild-type parent strain (Fig. 6A and S10), and cells of both strains had similar dimensions and volumes within experimental error (Fig. 6B and 6C). Removal of Zn2+ at time = 0 depleted MltG expression, and growth started to slow down significantly (≈3-fold) after ≈3h. The MltG-depleted cells continued to grow slowly for at least 6 h longer. This residual growth is consistent with the appearance of slow-growing, barely visible colonies of ΔmltG transformants on plates (Fig. 3, top row). It may also reflect the time it takes to deplete MltG below a critical concentration. MltG depletion causes a decrease in cell length, but not width, and corresponding decreases in cell aspect ratio (to ≈1.1) and volume (to ≈67%) (Fig. 6B and 6C). We conclude that depletion of MltG significantly decreases cell growth rate and causes cells to become nearly spherical, suggesting a defect in peripheral PG synthesis (see (Berg et al., 2013, Land & Winkler, 2011, Tsui et al., 2014)).
Fig. 6.

Depletion of MltG in an unencapsulated derivative of strain D39 leads to slow growth and spherical cell shape. Parent strain IU1945 (D39 Δcps) and merodiploid IU9102 (ΔmltG// ΔbgaA::PZn-mltG) were grown overnight in BHI broth containing 0.2 mM ZnCl2 + 0.02 mM MnSO4, centrifuged to remove Zn2+ and Mn2+, and resuspended to OD620 ≈ 0.005 in BHI broth containing 0.2 mM ZnCl2 + 0.02 mM MnSO4 (IU9102 + Zn2+) or no Zn2+ + Mn2+. (IU1945 and IU9102; no Zn2+). (A) Growth curves. Titration of IU9102 growth with different concentrations of Zn+2 indicated maximal growth in 0.2 mM Zn+2 + 0.02 mM Mn+2, which was added to prevent zinc toxicity (see Fig. S10). Doubling times before MltG depletion (≈37 min) and after MltG depletion (≈121 min) are indicated. (B) Representaive phase-contrast images taken at the time points marked by arrows in (A). All micrographs are at the same magnification (scale bar = 1 μm). (C) Box-and-whisker plots (whiskers, 5 and 95 percentile) of cell lengths, widths, aspect ratios (cell length to width) and relative cell volumes of IU1945 and IU9102 grown with or without Zn2+ and Mn2+. Fifty or more cells from two independent experiments were measured as described in Experimental procedures for each strain. P values were obtained by one-way ANOVA analysis (GraphPad Prism, nonparametric Kruskal-Wallis test). *, **, and *** denote p<0.05, p<0.01, and p<0.001 respectively. Comparisons not marked by brackets are not significantly different.
We also observed increased sphericity of mltG(Y488D) pbp1a+ mutant cells (Fig. 7). The growth of the mltG(Y488D) pbp1a+, mltG(Y488D) Δpbp2b, and mltG(Y488D) Δpbp1a mutants is similar to that of the wild-type unencapsulated parent strain (Fig. 7A). However, mltG(Y488D) pbp1a+ mutant cells are more spherical and have smaller cell widths, aspect ratios (to ≈1.4), and relative volumes (to ≈80%) than the parent strain (Fig. 7B and 7C), and mltG(Y488D) Δpbp2b mutant cells were even more spherical than mltG(Y488D) pbp1a+ cells (aspect ratio to ≈1.2; Fig. 7C). We reported earlier that unencapsulated Δpbp1a mutant cells are shorter, narrower, and more elongated than wild-type parent cells, resulting in a drop in relative volume to ≈40% (Fig. S11B and S11C) (Land & Winkler, 2011). The decrease of cell width, in particular, caused mltG(Y488D) Δpbp1a mutant cells to have higher aspect ratios (≈1.8), similar to that of the parent strain (≈1.6) (Fig. 7B and 7C). Likewise, Δpbp1a ΔmltG and Δpbp1a ΔmltG Δpbp2b mutants form relatively small cells (volumes to ≈40%) with aspect ratios slightly larger than that of the parent strain (Fig. S11C). In addition, the Δpbp1a ΔmltG Δpbp2b mutant has a marked autolysis phenotype in early stationary phase compared to other strains (Fig. S11A).
Fig. 7.

The mltG(Y488D) mutant grows slightly slower and forms cells of shorter lengths and greater sphericity than the wild-type parent strain. Strains: parent IU1824 (1, D39 Δcps rpsL1); IU9760 (2, mltG(Y488D)); IU9783 (3, mltG(Y488D) Δpbp2b); and IU8551 (4, mltG(Y488D) Δpbp1a). Strains were grown overnight in BHI broth and resuspended to OD620 ≈0.003 in BHI broth to strart growth cultures (see Experimental procedures. (A) Representive growth curves from one experiment. Doubling times (DT, mean ± SEM) were determined from 1 to 3 independent experiments (number in parenthesis) for cultures growing exponentially between OD620 = 0.01 to 0.2. *denotes p<0.05 compared to the parent, analyzed by a paired t-test from three experiments. (B) Representaive phase-contrast images taken at mid-exponential growth phase (OD620 ≈0.15). (C) Box-and-whisker plots (whiskers, 5 and 95 percentile) of cell lengths, widths, aspect ratios (cell length to width ratio) and relative cell volumes. Fifty or more cells from two independent experiments were measured as described in Experimental procedures for each strain. P values were obtained by one-way ANOVA analysis (GraphPad Prism, nonparametric Kruskal-Wallis test). *** and ** denote p<0.001 and p<0.01, respectively. Only comparisons of mutant strains with the parent strain are shown. Comparisons that are not significantly different are not marked by brackets.
Previously we reported that the presence of exopolysaccharide capsule dampens division and shape phenotypes of pneumococcal mutants (Barendt et al., 2009, Barendt et al., 2011). Consistent with this trend, ΔmltG mutants of encapsulated strain D39 could be constructed without a concomitant accumulation of pbp1a suppressor mutations, although D39 ΔmltG mutants form tiny colonies (Fig. S12A) and show biphasic growth in BHI broth with slower doubling times (to ≈2.6X) than that of the parent strain (Fig. 8A and 8B). The mollifying effect of capsule on the stability of ΔmltG mutants was illustrated by transforming encapsulated D39 ΔmltG pbp1a+ to an unencapsulated strain. Δcps2E transformants of D39 ΔmltG pbp1a+ rapidly accumulated spontaneous pbp1a suppressor mutations (Fig. S12B). The presence of an additional Δpbp2b or Δpbp1a mutation substantially improves the growth of the D39 ΔmltG mutants (Fig. 8A and 8B), similar to what happened in unencapsulated D39 derivatives. Again, the median cell length and aspect ratio of the D39 ΔmltG and D39 ΔmltG Δpbp2b mutants were smaller than those of the D39 parent, and the cells appear more spherical (aspect ratio to ≈1.2) and somewhat smaller (to ≈70%) (Fig. 8C and 8D). Unlike the D39 parent, the chains of D39 ΔmltG and D39 ΔmltG Δpbp2b mutant cells contain frequent dead cells (Fig. 8C, arrows). Similar to the unencapsulated mltG(Y488D) mutant (Fig. 7B and 7C), the D39 ΔmltG Δpbp1a mutant is elongated with a greater aspect ratio than the D39 ΔmltG mutant (Fig. 8C and 8D). Together, these data support the conclusions that the primary effect of a decrease in MltG activity is formation of more spherically shaped cells and that loss of MltG severely impairs growth of encapsulated and unencapsulated S. pneumoniae strains.
Fig. 8.

Impaired growth and spherical cell shape of encapsulated D39 ΔmltG pbp1a+ strains (A) Representative growth curves of IU1690 (1, D39 cps+ wild-type parent); IU9771 (2, cps+ ΔmltG); IU9897 (3, cps+ ΔmltG Δpbp2b); and IU10021 (4, cps+ ΔmltG Δpbp1a). Strains were inoculated from ice stocks, grown in BHI broth for 11h, and diluted to OD620 ≈0.005 in BHI broth to tart growth cultures (see Experimental procedures). (B) Doubling times (mean ± SEM) of the above strains obtained from at least two independent experiments. IU9771, IU9897 and IU10021 grow biphasically with a change to a slower growth rate at OD620 = 0.05 to 0.1. (C) Representaive phase-contrast images taken at OD620 ≈0.1 to 0.15. Particularly spherical or lysed cells are marked by astericks (*) or arrows, respectively. Micrographs are at the same magnification (scale bar = 1 μm). (D) Box-and-whisker plots (whiskers, 5 and 95 percentile) of cell lengths, widths, aspect ratios (length to width ratio), and relative volumes. Fifty or more cells from two independent experiments were measured as described in Experimental procedures for each strain. P values were obtained by one-way ANOVA analysis (GraphPad Prism, nonparametric Kruskal-Wallis test). *** denotes p<0.001. Only comparisons of mutant strains with the parent strain are shown. Comparisons that are not significantly different are not marked by brackets.
MltG localizes separately from FtsZ with the peripheral PG synthesis machine
MltG inactivation causes cell shape to become more spherical, suggesting MltG acts in peripheral PG synthesis (Fig. S1B) (see (Tsui et al., 2014)). Consequently, we localized MltG relative to master septal organizer FtsZ and to MreC, a known regulator of peripheral PG synthesis (Land & Winkler, 2011, Massidda et al., 2013, Philippe et al., 2014). We constructed pneumococcal strains in which the wild-type copies of ftsZ and mltG were replaced in their native chromosome loci by ftsZ-L-rfp and gfp-L-mltG, where L refers to a linker peptide (see Experimental procedures; Table S1). The resulting single ftsZ-L-rfp and gfp-L-mltG and double ftsZ-L-rfp gfp-L-mltG strains did not cause detectable defects in cell growth or morphology compared to the unencapsulated parent strain (Fig. 9A, 9B, and 9C). We retrospectively sorted images of exponentially growing pneumococcal cells into different division stages and averaged the relative positions of FtsZ-L-RFP and GFP-L-MltG as described before (Fig. 9A, 9B, and 9D; see Experimental procedures) (Land et al., 2013, Tsui et al., 2014). FtsZ-L-RFP or GFP-L-MltG show the same localization patterns reported previously for epitope-tagged FtsZ constructs or peripheral PG synthesis proteins, respectively (Land et al., 2013, Tsui et al., 2014). FtsZ-L-RFP and GFP-L-MltG colocalize to the septum in early stage 1 divisional cells (Fig. 9B and 9D). In middle divisional stage 2 and 3 cells, the diameter of the average FtsZ ring is smaller than that of MltG, and FtsZ starts to assemble at the equators of the new daughter cells. The difference in ring sizes was confirmed by a statistical graphical method that compares septum widths in each measured cell (Fig. 9D) (Land et al., 2013, Tsui et al., 2014). By division stage 4, FtsZ migrates from the septum to the new equators of daughter cells, while a substantial amount of MltG remains at the septum, with some organizing at the new equators. This pattern of MltG localization relative to FtsZ matches that of the PBP1a, PBP2b, and MreC peripheral PG synthesis proteins reported previously (Land et al., 2013, Tsui et al., 2014).
Fig. 9.

GFP-L-MltG localize differently from FtsZ-L-RFP. (A) Representative phase and fluorescent images of strain IU10353 (ftsZ-L2-mKate2 gfp-L1-mltG) grown in BHI to mid-exponential phase (OD620 ≈0.15). Numbers on top of panels indicate stage of cell division (see text and Experimental details). The last row shows superimposed images from the two fluorescent channels, with overlapping signals shown as yellow. Scale bar (top left image) = 1 μm. (B) Averaged images and fluorescence intensity traces. Cells were binned into division stages 1–4, and images of the indicated number of cells (n) from two experiments were averaged and quantified using the graphical user interface program (GUI) described in Experimental procedures. Row 1, cell shapes from phase-contrast images; row 2, GFP-L-MltG fluorescent signal, row 3, FtsZ-L-RFP fluorescent signal and row 4, normalized average fluorescence intensity distributions along the horizontal cell axis for each channel (black, phase; green, MltG; red, FtsZ). (C) Representative growth curve of strains IU1824 (wild-type parent), IU9148 (ftsZ-L2-mKate2), IU10228 (gfp-L1-mltG), and IU10353 (ftsZ-L2-mKate2 gfp-L1-mltG). (D) Scatter plot of GFP-L-MltG versus FtsZ-L-RFP width at midcell equators and septa of cells in division stages 1 to 3 in (B). Labeled midcell widths were quantified using the GUI program (see Experimental procedures). The dotted reference line (same width of each protein) has slope = 1 and intercepts the origin. For statistical analysis, differences between the midcell widths of MltG and FtsZ were calculated for each cell in each division stage, and a one-sample t test (GraphPad Prism) was performed to determine if differences are significantly different from 0. ***, p<0.001. Data in (B) and (D) were from two independent biological replicates.
A similar conclusion was reached by comparing localization of epitope-tagged MreC-L-FLAG3 with MltG-HA by 2D and 3D-SIM immunofluorescence microscopy (IFM) (Fig. 10). In these experiments, MreC-L-FLAG3 and MltG-HA were expressed from their native chromosomal loci in the same cell (see Experimental procedures; Table S1), and the epitope-tagged proteins did not cause any detectable growth or cell morphology defects (Fig. 10B; Fig. S2C). Epitope-tagged MltG was detected as a single band with no obvious degradation products in Western blots (Fig. S2D). 2D images (Fig. 10A), averaged 2D images (Fig. 10B), and statistical comparison of midcell diameters (Fig. 10C) show that MreC and MltG co-localize at each stage of pneumococcal cell division. This interpretation is supported by 3D-SIM of stage 2 and 3 cells (Fig. 10D). Finally, we determined the localization of MltG relative to regions of cell surface labelling by fluorescent vancomycin (FL-V), which binds to the PG pentapeptide substrate where there is active PG synthesis (Daniel & Errington, 2003, Ng et al., 2004, Pinho & Errington, 2005). We previously reported that during septal ring constriction, proteins of the peripheral PG synthesis machine (PBP2b, PBP1a, and MreC) appear in a ring circling regions of FL-Van labelling (Tsui et al., 2014). By contrast, PBP2x, which mediates septal PG synthesis, forms rings and locates contiguously inside regions of FL-V labelling (Tsui et al., 2014). 2D and 3D-SIM IFM of cells labeled for MltG-HA and FL-V reveal that MltG, like PBP2b and PBP1a, locates at regions outside of nascent PG synthesis demarcated by FL-V staining (Fig. S13). Based on these combined results, we concluded that MltG is part of the peripheral PG synthesis machine in S. pneumoniae.
Fig. 10.

Dual-protein 2D and 3D-SIM IFM showing similar localization of MreC and MltG in equators and septa of dividing pneumococcal cells. Strain IU7580 (mreC-L-FLAG3 mltG-HA) was grown to mid-exponential phase in BHI broth and processed for dual-protein 2D and 3D-SIM IFM with DAPI labeling of DNA as described in Experimental procedures. (A) Representative field of 2D-IFM images with phase images. (B) Representative 3D-SIM IFM images of cells at division stages 2–4. DNA (DAPI) staining is pseudo-colored white or blue in column 1 or 5, respectively. MreC or MltG are pseudo-colored green or red respectively, and overlapping signal is colored yellow. Images in the first row of each panel were captured in the XY plane, and the second-row images were obtained by rotating a section of the midcell region around the X or Y axis. Images are representative of >20 examined cells in different division stages from one experiment. (scale bar = 1 μm). (C) Averaged 2D IFM images and fluorescence intensity traces. Cells were binned into division stages 1–4, and images from the indicated number of cells (n) were averaged and quantified as described for Figure 9, with the addition of the DNA nucleoid (DAPI) locations (row 2). (D) Scatter plot of labeled MreC width versus MltG width at midcell equators and septa of cells at division stages 1 to 4 in (C) (see Fig 9). NS, difference between the two widths was not significant (p>0.05). Data points in (C) and (D) were obtained from one experiment in which >100 cells were analyzed. Similar 3D-SIM IFM images and quantitation of 2D IFM images were obtained for strain IU7582 (mreC-L-FLAG3 mltG-Myc), which expresses MltG-Myc instead of MltG-HA (data not shown).
A synthetic-viable genetic relationship exists between Δpbp1a and ΔrodZ, but not ΔrodA
We showed previously that a Δpbp1a deletion mutation obviates the requirement for MreC or MreD (Land & Winkler, 2011), and we show here that Δpbp1a mutations suppress the requirement for MltG (Table 2; Fig. 3, 4 and S5). Consequently, we determined whether a Δpbp1a deletion suppresses the requirement for the RodZ and RodA proteins, which have been implicated in peripheral PG synthesis (Fig. S1B) (Gisch et al., 2015, Massidda et al., 2013, Philippe et al., 2014). Transformation assays into merodiploid strains ectopically expressing RodA or RodZ indicated that both of these proteins are essential for growth of unencapsulated and encapsulated D39 strains (Table 3, lines 1–5 and 14). The Δpbp1a deletion suppressed the requirement for RodZ, but surprisingly, did not suppress the requirement for RodA (Table 3, row 7; Fig. 4). The lack of a synthetic-viable genetic relationship between Δpbp1a and ΔrodA echoes the lack of Δpbp1a suppression of Δpbp2b (Table 2, line 5; Fig. 4). Furthermore, the mltG(Y488D) Δpbp1a ΔrodA mutant was inviable like the mltG(Y488D) Δpbp1a Δpbp2b mutant (Table 2, line 9; Table 3, line 8). In contrast, the ΔmltG(Δ488bp) Δpbp1a combination suppressed the requirement for RodA, MreCD, or RodZ separately (Table 3, line 11) or for MreCD and RodZ or PBP2b, MreCD, and RodA or RodZ together (Table 3, lines 12 and 13; Fig. 4A). Similarly, in a pbp1a+ strain, the mltG(Y488D) allele suppressed the requirement for the entire peripheral PG synthesis machine (PBP2b, RodA, MreCD, or RodZ) (Table 3, lines 9 and 10; Fig. 4B). mltG(Y488D) pbp1a+ and mltG(Y488D) [Δpbp2b; ΔmreCD; ΔrodZ; or ΔrodA] mutants form short chains of 6 to 10 rounded cells during exponential growth in BHI broth and grow with doubling times similar to that of the mltG+ parent strain (≈41 min) (Fig. 4B, 7; data not shown). Together, these complex genetic relationships point to a functional link between PBP2b and RodA and indicate separate pathways to cope with disruption of peripheral PG synthesis in Δpbp1a or pbp1a+ strains (see Discussion). Moreover, these results show that the peripheral PG synthesis machine of S. pneumoniae can be eliminated in a mutant containing a single acid change in MltG (Y488D) or in a Δpbp1a ΔmltG double mutant.
TABLE 3.
Appearance of colonies after transformation with ΔrodA, ΔmreCD, or ΔrodZ amplicons into D39 strainsa
| Recipient strain and condition | Genotype | Number of colonies at 22 h after transformation with deletion ampliconsb | ||
|---|---|---|---|---|
| ΔrodA | ΔmreCD | ΔrodZ | ||
| Strains of IU1945 (D39 Δcps) genetic background | ||||
| 1. IU1945 | WT | 0d | 0 | 0d |
| 2. IU10921 | ΔbgaA::PZn-rodA | 0d | ||
| 3. IU10921 + Znc | >500 | |||
| 4. IU9765 | ΔbgaA::PZn-rodZ | 0d | ||
| 5. IU9765 + Znc | >500 | |||
| Strains of IU1824 (D39 Δcps rpsL1) genetic background | ||||
| 6. IU1824 | pbp1a+ wild-type parent | 0 | 0 | 0 |
| 7. IU6741 | Δpbp1a | 0 | >500 | >500 |
| 8. IU8551 | Δpbp1a mltG(Y488D) | 0 | >500 | >500 |
| 9. IU9760e | pbp1a+ mltG(Y488D) | >500 | >500 | >500 |
| 10. IU9783 | pbp1a+ mltG(Y488D) Δ pbp2b | >500 | >500 | >500 |
| 11. IU8553 | Δpbp1a mltG(Δ488bp) | >500 | >500 | >500 |
| 12. IU10731 | Δpbp1a mltG(Δ488bp) ΔmreCD | >500 | >500 | |
| 13. IU10743 | Δpbp1a mltG(Δ488bp) Δpbp2b ΔmreCD | >500 | >500 | |
| Strain of IU1690 (D39 cps+) genetic background | ||||
| 14. IU1690 | WT | 0 | 0 | 0 |
Recipient strains were constructed as described in Table S1. Transformations and visualization of colonies normalized to 1 mL transformation mixture were performed as described in Experimental procedures. All transformants formed uniform colonies of nearly wild-type size, except for the quadruple and quintuple mutants (IU10731 and IU10743), which grew slower than the parent strain. The same results for each strain were obtained from two independent transformation experiments. Blank cells are non-applicable experiments.
rodZ was not named in the genome annotation and corresponds to spd_2050. Strains used to prepare amplicons are listed in Table S2.
0.4 mM ZnCl2 and 0.04 mM MnSO4 were added to all steps in the transformation procedure to induce RodA or RodZ expression in merodiploid strain IU10921 or IU9765, respectively (Table S1).
Fast-growing suppressors (<10 colonies per plate) were occasionally seen for IU1945 or the merodiploid strains under non-inducing condition.
Strain IU9895 (mltG(Y488D)-Pc-erm, which was independently constructed in the IU1945 Δcps background (Table S1), showed the same suppression of the ΔmreCD mutation (data not shown).
Suppression of ΔmltG mutations by Δpbp1a depends on induction of the WalRK two-component system (TCS) regulon
To gain insight into the mechanism of synthetic-viable genetic relationships involving Δpbp1a (Fig. 4A), we performed RNA-Seq analyses of Δpbp1a, Δpbp1a mltG(Δ488bp) (sup4), and Δpbp1a mltG(Δ488bp) (sup4) Δpbp2b mutants compared to the isogenic unencapsulated parent strain (Table 4). Unexpectedly, the Δpbp1a deletion causes an almost exclusive induction in relative transcript amounts of genes that are in the WalRK TCS regulon, which includes established and putative PG hydrolases and division proteins of unknown functions (Table 4, column 3) (Ng et al., 2003, Ng et al., 2005, Winkler & Hoch, 2008). Besides WalRK regulon genes, relative transcription is induced for only two other genes, recU and spd_0542. recU is located immediately upstream of pbp1a (Fig. 3A) and is co-transcribed with pbp1a (data not shown); therefore, induction of the relative amount of the recU transcript may reflect a change in transcript stability. spd_0452 encodes a prophage-like integrase/recombinase, and its transcription induction in the Δpbp1a mutant may be a low-level stress response. The WalRK regulon is induced even more in the Δpbp1a mltG(Δ488bp) (sup4) and Δpbp1a mltG(Δ488bp) (sup4) Δpbp2b mutants than in the Δpbp1a mutant (Table 4, columns 4 and 7). In addition, there are low level changes in relative transcript amounts of several other genes in the double and triple mutant, including several genes of unknown functions. We conclude that the absence of class A PBP1a causes induction of the WalRK TCS and its regulon.
Table 4.
Changes in relative transcript amounts in Δpbp1a, Δpbp1a mltG(Δ488bp), and Δpbp1a mltG(Δ488bp) Δpbp2b mutants compared to the wild-type (WT) parent strain grown exponentially in BHI brotha
| Δpbp1a vs WT | Δpbp1a mltG(Δ488bp) vs WT | Δpbp1a mltG(Δ488bp) Δpbp2b vs WT | |||||
|---|---|---|---|---|---|---|---|
| Gene tag | Encoded protein | Fold change | FDR | Fold change | FDR | Fold change | FDR |
| Increased relative expression | |||||||
| WalRK regulon | |||||||
| spd_0104 | LysM domain protein | 5.1 | 8.2 E-37 | 8.5 | 8.9 E-81 | 9.4 | 3.3 E-88 |
| spd_0703 | conserved hypothetical, putative SEDS protein | 1.9 | 8.7 E-15 | 2.1 | 8.6 E-21 | ||
| spd_1870b | degenerate pyrrolidone- carboxylate peptidase | 2.8 | 8.5 E-18 | 2.8 | 1.1 E-18 | ||
| spd_1871b | conserved hypothetical membrane protein | 3.4 | 8.0 E-4 | 5.6 | 4.2 E-12 | 5.3 | 1.8 E-11 |
| spd_1872b | degenerate MarR family protein | 3.2 | 9.6 E-8 | 5.4 | 3.3 E-24 | 6.0 | 6.9 E-28 |
| spd_1874b | LysM domain protein, putative N- acetylmuramidase | 7.0 | 2.7 E-33 | 14.6 | 8.9 E-81 | 14.7 | 2.2 E-81 |
| spd_2043 | PcsB, secreted putative N-acetylmuramoyl-l- alanine amidase/endopeptidase | 2.4 | 3.2 E-6 | 3.3 | 6.7 E-18 | 3.6 | 2.3 E-20 |
| Other genes | |||||||
| spd_0074 | phosphorylase, Pnp/Udp family protein | 1.8 | 6.8 E-06 | ||||
| spd_0093c | membrane protein, putative | 3.3 | 7.1 E-12 | ||||
| spd_0094c | conserved hypothetical protein | 3.3 | 6.2 E-13 | ||||
| spd_0095c | conserved hypothetical protein | 2.9 | 2.6 E-13 | ||||
| spd_0096c | transcriptional regulator, PadR family protein | 2.7 | 3.2 E-07 | ||||
| spd_0308 | ClpL, ATP-dependent Clp protease | 2.7 | 7.7 E-09 | 1.8 | 1.5 E-02 | ||
| spd_0337d | RecU, recombination protein U, upstream of pbp1a | 2.3 | 7.5 E-05 | 2.6 | 8.3 E-09 | 2.5 | 7.7 E-08 |
| spd_0450 | type I restriction- modification system | 2.7 | 8.1 E-03 | 3.5 | 2.9 E-05 | ||
| spd_0452 | integrase/recombinase | 2.9 | 6.7 E-08 | 4.5 | 1.2 E-22 | 4.4 | 1.1 E-21 |
| spd_1596 | TrpA, tryptophan synthase, alpha subunit | 1.8 | 1.3 E-02 | ||||
| spd_1831 | PTS system, IIC component | 2.0 | 3.5 E-03 | ||||
| spd_2012 | GlpO, alpha- glycerophosphate oxidase | 2.0 | 3.8 E-02 | 2.2 | 9.4 E-03 | ||
| spd_2013 | GlpK, glycerol kinase | 2.3 | 1.8E-03 | ||||
| Decreased relative expression | |||||||
| spd_0139 | glycosyl transferase, group 2 family protein | −2.7 | 1.0 E-05 | −2.6 | 4.1 E-05 | ||
| spd_0140 | ABC transporter, ATP- binding protein | −2.0 | 1.1 E-05 | ||||
| spd_0336e | PBP1a, penicillin- binding protein 1A | −84 | 1.5 E-39 | −97 | 3.6 E-52 | −77 | 1.0 E-47 |
| spd_0451 | type I restriction- modification system | −2.1 | 2.6 E-07 | −2.1 | 1.0 E-07 | ||
| spd_0646 | conserved hypothetical protein | −1.8 | 3.1 E-06 | ||||
| spd_1134 | PyrR, pyrimidine operon regulatory protein/uracil phosphoribosyltransfera se | −1.9 | 9.9 E-06 | ||||
| spd_1137 | ABC transporter, ATP- binding protein | −2.9 | 1.1 E-12 | −2.9 | 9.6 E-13 | ||
| spd_1346f | MltG, YceG domain protein | −1.9 | 3.7 E-14 | −1.9 | 4.4 E-15 | ||
| spd_1486g | PBP2b, penicillin- binding protein 2B | −414 | 3.7 E-88 | ||||
Strain constructions, growth conditions, and RNA-Seq analyses are described in
Experiment procedures, Supplemental experimental procedures, and Tables S1. RNA was prepared from cultures of strains IU6741 (Δpbp1a), IU8553 (Δpbp1a mltG(Δ488bp)), IU8567 (Δpbp1a mltG(Δ488bp) Δpbp2b) and IU1824 (pbp1a+ mltG+ pbp2b+ parent) grown exponentially in BHI media at 37°C to OD620 ≈0.15 to 0.2. Fold changes and false discovery rates (FDR) are based on three independent biological replicates. Cut-off limits were 1.8-fold and FDR value < 0.05. Empty boxes indicate no significant change in expression under at least one of the three comparisons.
spd_1874 is the first gene of an operon containing spd_1874 to spd_1870.
spd_0096 is the first gene of the operon containing spd_0096 to spd_0093.
recU is upstream of pbp1a (Fig. 1) and has a 4-bp overlap with the start of the PBP1a ORF.
pbp1a is deleted in these strains.
488 bps are deleted from mltG in IU8553 (Δpbp1a mltG(Δ488bp)) and IU8567 (Δpbp1a mltG(Δ488bp) Δpbp2b).
pbp2b sequence is deleted in this strain.
We tested whether induction of the WalRK regulon is required for growth of the Δpbp1a or Δpbp1a ΔmltG mutant. To this end, we deleted the dispensable walK gene, which encodes the WalK histidine kinase (Ng et al., 2003, Winkler & Hoch, 2008), in the parent and each mutant (Fig. 11; Table S1). The absence of WalK decreases growth of the parent and Δpbp1a mutant by about 25% (Fig. 11A and 11B). In contrast, the Δpbp1a ΔmltG ΔwalK mutant grew about 2.5X slower than the Δpbp1a ΔmltG, which grew at about the same rate as the Δpbp1a mutant and only slightly slower than the parent strain (Fig. 11C). We next tested whether individual genes in the WalRK regulon are required for growth of the Δpbp1a ΔmltG mutant (Fig. S14). In contrast to the ΔwalK mutation, growth of the Δpbp1a ΔmltG mutant is not slowed down by deletions in the following WalRK-regulon genes: spd_0104 (LysM protein); spd_1874 (LysM protein); lytB (N-acetyl-glucosaminidase); spd_0703 (novel division protein); spd_0126 (pspA); or double deletion of Δspd_0104 and Δspd_1874 (Fig. S14; Table S4). In addition, controlling expression of the essential pcsB+ gene from a promoter not regulated by the WalRK TCS does not improve the poor growth of the Δpbp1a ΔmltG ΔwalK mutant (data not shown). These results suggest that induction of expression of two or more genes in the WalRK regulon is needed to maintain growth of the Δpbp1a ΔmltG mutant.
Fig. 11.

A ΔwalK mutation is deleterious to the growth of Δpbp1a ΔmltG and mltG(Y488D) mutant strains. The panels contain the following growth curves: (A) IU1824 (wild-type parent) and IU9231 (ΔwalK); (B) IU6741 (Δpbp1a) and IU9233 (Δpbp1a ΔwalK); (C) IU7327 (Δpbp1a ΔmltG) and IU9235 (Δpbp1a ΔmltG ΔwalK); and (D) IU9760 (mltG(Y488D)) and IU10829 (mltG(Y488D) ΔwalK). Full genotypes of strains are listed in Table S1. Doubling times (DT, average ± SE) were obtained from two or more independent growths.
We also tested whether lack of other known or putative pneumococcal PG hydrolases impedes growth of the Δpbp1a ΔmltG mutant. Growth in BHI broth is similar for the Δpbp1a ΔmltG mutant containing the following additional mutations: Δpmp23 (putative LT) (Pagliero et al., 2008); ΔcbpD (fratricide CHAP-domain endopeptidase) (Eldholm et al., 2010); ΔlytC (fratricide N-acetylmuramidase) (Perez-Dorado et al., 2010); or Δspd_0873 (putative PG hydrolase) (Barendt et al., 2011) (Fig. S14; Table S4). Likewise, Δpbp1a ΔmltG mutants form normal-sized colonies when transformed with the following additional mutations: ΔdacA (D,D-carboxypeptidase) (Barendt et al., 2011); ΔdacB (L,D-carboxypeptidase) (Barendt et al., 2011); ΔlytA (stress-induced amidase autolysin) (Mellroth et al., 2012); or Δspd_0173 (putative PG hydrolase) (Barendt et al., 2011). We conclude that there are no pairwise synthetic-lethal genetic combinations between the Δpbp1a-suppressed ΔmltG mutation and mutations in genes for other pneumococcal PG hydrolases.
Finally, we determined whether the mltG(Y488D) allele induced expression of the WalRK TCS regulon. QRT-PCR experiments showed that the relative transcript amount of WalRK regulon gene spd_1874 (LysM protein) increases 3.9 ± 0.3 fold in the Δpbp1a mutant, 6.9 ± 1.2 fold in the Δpbp1a mltG(Δ488bp) mutant, 5.1 ± 0.7 fold in the Δpbp1a mltG(Δ488bp) Δpbp2b mutant, 6.2 ± 0.1 fold in the mltG(Y488D) mutant, and 3.6 ± 0.6 fold in the mltG(Y488D) Δpbp2b mutant compared to the isogenic parent strain (Fig. S15A). Similar induction patterns were found for relative transcript amounts of the spd_0104 and pcsB transcripts (Fig. S15B and S15C). These induction patterns of the relative amounts of the spd_1874, spd_0104, and pcsB transcripts confirm the patterns seen in RNA-Seq analysis of these strains (Table 4). The QRT-PCR results show that the mltG(Y488D) mutation induces the expression of the WalRK regulon as much as, if not more, than the Δpbp1a mutation (Fig. S15). One difference is that WalRK regulon induction consistently goes down in the mltG(Y488D) Δpbp2b mutant compared to the mltG(Y488D) mutant, whereas induction in the Δpbp1a mltG(Δ488bp) Δpbp2b and Δpbp1a mltG(Δ488bp) mutant remains elevated to about the same level (Fig. S15; Table 4). Consistent with this drop in WalRK regulon induction, growth of the mltG(Y488D) Δpbp2b mutant is not strongly dependent on WalK, whereas growth of the mltG(Y488D) ΔwalK mutant is much slower than that of the mltG(Y488D) mutant (Fig. 11D; data not shown). We conclude that the mltG(Y488D) allele, like the Δpbp1a deletion, leads to strong induction of the WalRK TCS regulon, and that WalRK TCS induction is required for optimal growth of the mltG(Y488D) and Δpbp1a ΔmltG mutants (Fig. 11C and 11D).
DISCUSSION
ΔmltG Δpbp1a and mltG(Y488D) mutations relieve the requirement for essential genes that mediate peripheral PG synthesis
We report here the discovery that essential class B PBP2b can be deleted in S. pneumoniae by decreasing or eliminating activity of the MltG protein (Tables 1–3; Fig. 1, 2, 4, 7, and 8), which is the structural and functional homologue of the MltG endo-LT in E. coli (Table 2; Fig. 5, S6, and S7). Deletion or inactivation of the mltG severely impairs growth of unencapsulated and encapsulated serotype 2 D39 strains of S. pneumoniae (Fig. 6 and 8), and mltG null alleles lead to accumulation of mutations deficient or lacking activity of PBP1a in unencapsulated strains (Fig. 3). Class A PBP1a is thought to participate in both septal and peripheral PG synthesis in S. pneumoniae (see Fig. S1). The “synthetic-viable” genetic relationship that allows pneumococcal ΔmltG Δpbp1a mutants to grow is similar to the previously reported suppression of the requirement for essential mreCD by Δpbp1a deletion (Land & Winkler, 2011). We also show that the Δpbp1a deletion suppresses the requirement for the essential rodZ gene, (Table 3; Fig. 4), which like mreCD mediates peripheral PG synthesis (Fig. S1). Thus, Δpbp1a deletion allows growth in combination with ΔmltG, ΔmreCD, or ΔrodZ single mutations or in combinations of ΔmltG Δpbp2b ΔmreCD ΔrodA or ΔmltG Δpbp2b ΔmreCD ΔrodZ mutations tested so far. (Table 3; Fig. 4). Notably, Δpbp1a mutations do not suppress Δpbp2b or ΔrodA mutations directly (Tables 2 and 3; Fig. 4). Remarkably, the single Y488D aa change that reduces MltG activity (see below) suppresses the requirement for essential PBP2b, MreCD, RodZ or RodA singly and PBP2b and RodA, MreCD, or RodZ combinations tested so far (Table 3). Thus, the mltG(Y488D) mutation alone obviates the requirement for the peripheral PG synthesis machine in S. pneumoniae (Table 3; Fig. 4 and 7). On the other hand, mltG(Y488D) Δpbp1a Δpbp2b and mltG(Y488D) Δpbp1a ΔrodA mutants are inviable (Tables 2 and 3; Fig. 4).
Suppression of the requirement for sidewall PG synthesis has previously been reported in E. coli (Bendezu et al., 2009, Kruse et al., 2005, Vinella et al., 1993); but it occurs by a mechanism different from that operating in pneumococcal mltG(Y488D) and Δpbp1a mutants (Tables 2 and 3; Fig. 4, 7 and S11). In E. coli, PBP2, MreB, MreC, MreD, and RodA are essential and depletion leads to formation of inviable spherical cells, packed with internal membranes and misplaced FtsZ (Bendezu et al., 2009, Kruse et al., 2005). FtsZ overexpression or induction of the stringent response suppresses the requirement for sidewall PG synthesis and allows the propagation of spherical E. coli cells that do not lyse (Bendezu et al., 2009, Kruse et al., 2005, Vinella et al., 1993). In contrast, pneumococcal Δpbp1a cells exhibit orderly division planes (Boersma et al., 2015, Land & Winkler, 2011), do not induce stringent response (Table 4), and produce a similar amount of FtsZ as the pbp1a+ parent strain (data not shown). Likewise, mltG(Y488D) mutants grow normally (Fig. 7), exhibit orderly division planes when stained with fluorescent D-amino acids, and do not contain an elevated amount of FtsZ compared to the parent strain (data not shown). Moreover, the inviability of the mltG(Y488D) Δpbp1a Δpbp2b and mltG(Y488D) Δpbp1a ΔrodA mutants is hard to understand by simple FtsZ overexpression.
MltGSpn is the structural and functional homologue of MltGEco
Several observations strongly support the hypothesis that MltGSpn is the structural homologue of MltGEco and functions as an endo-LT. The predicted structure of the YceG domain, including a putative LysM-like PG binding domain, of MltGSpn is nearly identical to the determined structures of the YceG domains of MltGEco and MltGLmo (Fig. 5, S6, and S7). Moreover, critical conserved catalytic (e.g., E428Spn) and regulatory (e.g., Y488Spn) aa’s overlap completely in the predicted and determined structures of MltGSpn, MltGEco, and MltGLmo (Fig. 5 and S7). Consistent with this conservation, the conserved Glu residue required for catalysis by LTs is required for function of MltGSpn(E428) and MltGEco(E218) (Table 2) (Yunck et al., 2015). As noted in Results, the specific activity of the purified soluble MltGEco(YceG) domain is low, and robust kinetic characterization is not yet possible (Yunck et al., 2015). We could not detect LT activity of the purified soluble MltGSpn(YceG) domain.
However, an MltGSpn-Eco chimera containing the cytoplasmic and TM domains of MltGSpn and the YceG endo-LT domain of MltGEco is functional in S. pneumoniae (Table 2; Fig. 2C), directly implicating MltGSpn as an endo-LT. The morphology of cells expressing the MltGSpn-Eco chimera is more similar to the wild-type strain and less spherical than those of the mltG(Y488D) mutant (Fig. 7; data not shown), suggesting that the MltGSpn-Eco chimera is more active than MltG(Y488D). Transformation of the pbp1a+ mltG(Y488D) or pbp1a+ mltGSpn-Eco mutant with Δpbp2b amplicon gave >500 normal-sized or >500 tiny colonies, respectively (Table 2, rows 10 and 17), further supporting that MltGSpn-Eco is more active than MltG(Y488D). Consistent with this interpretation, a mltGSpn-Eco Δpbp1a Δpbp2b mutant is impaired for growth, but viable, whereas a mltG(Y488D) Δpbp1a Δpbp2b is inviable (Table 2, lines 9 and 18). Together, these results indicate that the MltGSpn-Eco chimera protein is nearly fully functional in S. pneumoniae. In fact, strains in which the entire mltGEco reading frame replaces the chromosomal mltGSpn reading frame have phenotypic properties similar to strains expressing the MltGSpn-Eco chimera (Fig 2C; Table 2, lines 17–20). This result reiterates that MltGSpn functions as an endo-LT and that the cytoplasmic DUF_1346 domain of MltGSpn, which is absent in MltGEco, is not required for endo-LT activity (Table 2, lines 15 and 16; Results).
Longer PG glycan strands were detected for an E. coli ΔmltG mutant compared to its wild-type parent strain (Yunck et al., 2015). This quantitative assay of glycan chain length is currently difficult to perform for S. pneumoniae PG, which contains longer glycan strands than E. coli PG (Bui et al., 2012, Wheeler et al., 2011). In addition, the relative amount of muropeptides with LT-cleaved anhydro ends decreased in an E. coli ΔmltG mutant compared to the parent strain (Yunck et al., 2015). However, the distribution of muropeptides with anhydro ends has not been reported for wild-type S. pneumoniae strains (Bui et al., 2012). We expect that the percentage of muropeptides with anhydro ends will be considerably lower for pneumococcal PG than for E. coli PG, because besides MltG, E. coli produces a periplasmic soluble LT (sLT) and several outer-membrane attached mLTs that play roles in PG turnover and recycling (see (Cho et al., 2014)). In addition, E. coli sLT functions in PG quality control and in pathways that enhance sensitivity to killing by β-lactam antibiotics (Cho et al., 2014). In contrast, pneumococcal PG undergoes minimal turnover and no known recycling in growing cells (Boersma et al., 2015). Besides the MltG endo-LT reported here, S. pneumoniae produces only one other putative mLT (Pmp23) (Pagliero et al., 2008) and no homologues to sLTs. ΔmltG and Δpmp23 mutations were not synthetically lethal in the Δpbp1a background (Table S4; Fig. S14B), suggesting separate functions. Further work is needed to determine the effects of the mutations reported here on pneumococcal PG composition and whether MltGSpn or MltGEco endo-LT activity is autoinhibited and activated by interactions with other proteins.
Pneumococcal MltG functions in peripheral PG synthesis
The complex synthetic-viable genetic relationships summarized above (see Fig. 4) suggest that pneumococcal MltG is involved in peripheral PG synthesis in S. pneumoniae. This hypothesis is supported by two additional pieces of evidence. First, depletion of MltG in unencapsulated strains, absence of MltG in poorly growing encapsulated ΔmltG D39 mutants, and reduction of MltG activity in unencapsulated mltG(Y488D) mutants increase the sphericity of cells (Fig. 6, 7, and 8), indicative of a defect in peripheral PG synthesis (Berg et al., 2013, Land & Winkler, 2011, Tsui et al., 2014). Second, MltG localized differently from FtsZ in later stages of cell division in a pattern that matches the localization of MreC, PBP2b, and PBP1a (Fig. 9 and 10), which mediate peripheral PG synthesis in S. pneumoniae (Fig. S1B) (Land et al., 2013, Tsui et al., 2014). In addition, MltG localizes the same way as MreC and other peripheral PG synthesis proteins relative to regions of PG pentapeptide accumulation (Fig. S13). Bacterial two-hybrid assays suggest that E. coli MltG interacts with PBP1b (Yunck et al., 2015), which may function in both septal and sidewall (peripheral) PG synthesis in E. coli, as indicated by its synthetic-lethal relationship with PBP1a (Paradis-Bleau et al., 2010). Further work is needed to understand the timing of protein associations of MltG in these two differently shaped bacteria.
The single mltG(Y488D) mutation has the unusual capacity to eliminate the requirement for peripheral PG synthesis in unencapsulated S. pneumoniae strains. A corollary of this conclusion is that the mltG(Y488D) allele can be used to distinguish whether other PG synthesis proteins likely play primary roles in peripheral PG synthesis. For example, the StkP protein kinase localizes in a ring with peripheral PG synthesis proteins adjacent to PBP2x, which separates to the centers of septa in later stages of pneumococcal cell division (Land et al., 2013). Previous work showed that StkP interacts with PBP2x, suggesting a role for StkP in regulating PBP2x TP activity at some stage of septal PG synthesis (Morlot et al., 2013). Consistent with this conclusion, the mltG(Y488D) mutation does not suppress the poor growth of ΔstkP mutants (data not shown). Likewise, the mltG(Y488D) mutation did not suppress the requirement for the PcsB PG hydrolase or the GpsB regulatory protein (data not shown), consistent with their primary roles in septal PG remodeling (Sham et al., 2011) and septal ring closure (Land et al., 2013), respectively, instead of in peripheral PG synthesis. Conversely, the pbp2b or rodA genes can be disrupted in strain CNRZ368 of Streptococcus thermophilus (Thibessard et al., 2002), suggesting that this partially sequenced strain may contain an mltG mutation with comparable suppression properties as pneumococcal mltG(Y488D).
Reduced activity of MltG(Y488D) mutant protein
Y488 is highly conserved in MltG homologues (Fig. 5, S6, and S7). The mutant MltG(Y488D) and wild-type MltG proteins are expressed at similar levels (Fig. S2A), and the mltG(Y488D) allele makes unencapsulated S. pneumoniae cells more spherical (Fig. 7C). These phenotypes and the fact that mltG(Y488D) was selected along with mltG null mutations (Table 1; Fig. 1) suggest that MltG(Y488D) has decreased endo-LT activity compared to wild-type MltG+. In support of this conclusion, gain-of-function mutations that increase MltG activity or alter its function would be exceedingly rare in the limited selection performed. In addition, overexpression of MltG in a mltG+//PZn-mltG+ merodiploid stain grown in the presence of zinc (+manganese) does not cause spherical cell formation or suppression of a Δpbp2b mutation (data not shown). Tellingly, MltG(Y488D) expressed in a mltG(Y488D)//PZn-mltG+ merodiploid stain suppresses the requirement for PBP2b in the absence of zinc, but loses this ability when MltG+ amount is increased by the addition of zinc (+manganese). Conversely, reduced expression of MltG+ in a ΔmltG//PZn-mltG+ merodiploid strain grown in limiting zinc (+manganese) that just allows growth (Fig. S10) is transformable by a Δpbp2b amplicon. Finally, mltGSpn-Eco, mltGEco, and mltG(Y488D) mutants show different degrees of some shared phenotypes (Table 2; Results), again consistent with reduced endo-LT activity. At this point, we cannot distinguish whether decreased activity of MltG(Y488D), MltGSpn-Eco, and MltGEco involves reduced catalysis or changes in protein interactions or regulation.
Phenotypes of ΔmltG mutants in encapsulated pneumococcal strains
The mltG(Y488D) mutation has not occurred spontaneously in clinical isolates of different serotypes of S. pneumoniae, which are encapsulated (Chewapreecha, et al., 2014). However, unlike in unencapsulated derivatives, mltG can be deleted in encapsulated serotype 2 strain D39, resulting in slow, biphasic growth and the formation of spherical cells (aspect ratio ≈1.1; Fig. 8 and S12). Consistent with this result, previous Tn-Seq analyses showed that disruption of mltG decreased growth of serotype 4 strain TIGR4 in several culture media and caused severe attenuation of colonization and invasive pneumonia in a murine model of infection (van Opijnen & Camilli, 2012). The capsule of strain D39 is covalently attached to PG glycan strands (Gisch et al., 2015, Standish & Morona, 2015), and the mechanisms underlying the damping of cell division and PG biosynthesis mutant phenotypes by capsule remains poorly understood in S. pneumoniae (Barendt et al., 2009, Barendt et al., 2011).
Phenotypes caused by combinations of mltG and other mutations suggest a model involving a shared function of PBP2b and RodA
The synthetic viable genetic relationships in this paper could arise by two mechanisms. In one mechanism, the requirement for essential regulatory proteins becomes unnecessary when the protein being regulated is eliminated. In the second mechanism, the absence of an enzyme from a consecutive pathway leads to the accumulation of a toxic intermediate that is not made when an enzyme that catalyzes an earlier step in the pathway is also absent. We can use these two mechanisms along with redundancy of class A PBP TP and TG activities to model the synthetic-viable and suppression phenotypes in this paper (Fig. 12). In wild-type cells, the primary pathway for peripheral PG synthesis is initiated when the TG activity of PBP1a builds a new glycan strand from external Lipid II precursors. The genetic relationships in this paper are not easily reconciled with RodA acting exclusively as a peripheral-PG Lipid II flippase; instead, they suggest that RodA is additionally necessary for PBP2b TP function, possibly by direct interaction, as suggested by biochemical studies (Philippe et al., 2014). Functional interaction between RodA and PBP2b is suggested by the observations that Δpbp1a mutations do not suppress Δpbp2b or ΔrodA mutations, but suppress ΔmltG, ΔmreCD, or ΔrodZ mutations (Fig. 4). Likewise, mltG(Y488D) Δpbp1a Δpbp2b and mltG(Y488D) Δpbp1a ΔrodA mutants are not viable, consistent with a shared function of PBP2b and RodA.
Fig. 12.

Model for the steps in peripheral PG synthesis of Streptococcus pneumoniae in wild-type and Δpbp1a mutant strains. The model proposes that new anchored-glycan PG strand is synthesized primarily by PBP1a (TG) activity, that un-crosslinked strands with anhydro ends are released by the MltG endo-LT, and that released strands are primarily crosslinked into growing peripheral PG by a PBP2b TP activity, which depends on a PBP2b:RodA interaction. Accumulation of the new anchored-glycan strands and released-strands with an anhydro end are assumed to be toxic in mutants. In this model, MreCD and RodZ control PBP1a activity and/or localization, although they may also regulate MltG activity and/or localization (see Discussion). In Δpbp1a mutants, TG activity is provided by other Class A PBPs and MltG still cleaves anchored-glycan strands, along with PG hydrolases induced in amount by activation of the WalRK two-component regulatory system. In a Δpbp1a ΔmltG Δpbp2b mutant an alternative pathway is used that replaces MltG with WalRK-induced PG hydrolases. Evidence is presented that MltG(Y488D) has partial endo-LT activity, and the mltG(Y488D) mutation also induces WalRK TCS regulon expression. The model can account for the phenotypes of mutants presented in this paper. See Discussion for additional details.
MreCD and RodZ likely play the different functions of regulating the activity and/or localization of PBP1a and/or MltG, such that loss of either regulator leads to aberrant PBP1a and/or MltG function that is harmful to cells. There is a precedent for MreC regulation of the spatial organization of a mLT (MltA) in Caulobacter crescentus (Divakaruni et al, 2007). We cannot yet distinguish whether MreCD and/or RodZ regulates PBP1a or MltG activity or both activities or the order of regulation. For the sake of argument, here we assume that MreCD and RodZ separately negatively regulate PBP1a (Fig. 12). In this model, newly synthesized, non-crosslinked glycan strand anchored to C55-PP (undecaprenol pyrophosphate) is released by MltG endo-LT activity, and the released strands with anhydro ends are crosslinked into the growing peripheral PG mainly by the TP activity of PBP2b complexed with RodA and secondarily by the TP activity of PBP1a (Fig. 12). Accumulation of either anchored-glycan strands or released glycan-strands with anhydro ends becomes toxic in a ΔmltG or Δpbp2b mutant, respectively, contributing to lack of growth (Fig. 12). Consequently, Δpbp1a mutations suppress ΔmltG mutations, and mltG null mutations suppress Δpbp2b mutations and accumulate pbp1a suppressor mutations.
In Δpbp1a mutants, anchored glycan strand is synthesized by the TG activity of PBP2a and/or PBP1b (Fig. 12). MltG cleavage activity is augmented by PG hydrolases activated by the WalRK TCS (discussed below), such that Δpbp1a ΔmltG mutants are viable, and MreCD and RodZ are not needed to regulate PBP1a activity or localization. However, Δpbp1a Δpbp2b and Δpbp1a ΔrodA mutants are inviable, because the toxic released-strand intermediate accumulates in the absence of the PBP2b:RodA or PBP1a TP activities. On the other hand, in a Δpbp1a ΔmltG Δpbp2b mutant, peripheral PG synthesis is carried out by an alternate pathway that is independent of PBP1a, MltG, and PBP2b (Fig. 12). In this alternate pathway, TG activity of PBP2a and/or PBP1b builds the glycan chain, PG hydrolases induced in amount by activation of the WalRK TCS release the glycan chain, and TP activity of PBP2a and/or PBP1b crosslinks the released strand into the growing PG.
In pbp1a+ mltG(Y488D) mutants, the components of the normal peripheral PG synthesis machine can be eliminated entirely. New anchored-glycan strand is synthesized by PBP1a TG, even when PBP1a is misregulated in the absence of MreCD and RodZ. Cleavage and release of new anchored-glycan strand is carried out by the reduced activity of MltG(Y488D) augmented by PG hydrolases induced by the WalRK TCS. Crosslinking of released-strand is catalyzed by the PBP2b:RodA and/or PBP1a TP activity. This redundancy accounts for the inviability of mltG(Y488D) Δpbp1a Δpbp2b or mltG(Y488D) Δpbp1a ΔrodA mutants that lack both PBP1a and PBP2b:RodA TP activities. Thus, although this model is speculative at this stage, it can account for all of the phenotypes caused by combinations of mutations in this study. Hypotheses about mechanisms that are consistent with these phenotypes will be tested in future experiments.
Activation of the WalRK TCS by Δpbp1a, mltG(Y488D), or Δpbp1b mutations
Changes in gene expression are often overlooked in interpretations of phenotypes caused by PG biosynthesis mutations. The finding that certain mutations, such as Δpbp1a and mltG(Y488D), induce expression of additional PG hydrolases was important for formulating an alternative pathway of peripheral PG synthesis (Fig. 12). We found that the Δpbp1a or mltG(Y488D) mutation strongly induces the expression of the WalRK TCS regulon (Table 4; Fig. S15) and that growth of ΔmltG Δpbp1a and mltG(Y488D) mutants is dependent on the WalK histidine kinase (Fig. 11). In addition, WalRK regulon expression is induced in Δpbp1b, but not in Δpbp2a, mutants (data not shown). The function of class A PBP1b is unknown and is not synthetically lethal with class A PBP1a or PBP2a (Land & Winkler, 2011, Massidda et al., 2013). PBP2a and PBP1a have a synthetic lethal relationship, but Δpbp1a and Δpbp2a single mutants do not show equivalent phenotypes (Land & Winkler, 2011). The conserved WalR response regulator is essential in Firmacutes, and the WalRK TCS induces regulon transcription in response to cell wall stresses (Dubrac et al., 2008, Winkler & Hoch, 2008). The pneumococcal WalK response regulator differs from its homologues in other low-GC Gram-positive species, such as B. subtilis, in several ways. Pneumococcal WalK is not essential for growth and contains a single transmembrane domain and no extracellular sensing domain (Ng & Winkler, 2004, Winkler & Hoch, 2008). S, pneumoniae lacks the ancillary WalHI (YycHI) regulatory proteins found in B. subtilis, and WalKSpn is not strongly localized to division septa, like WalKBsu (Wayne et al., 2010).
General cell-wall stress caused by treatment of S. pneumoniae with antibiotics that block early steps (e.g., cycloserine) or late steps (e.g. penicillin G and vancomycin) in PG synthesis activates both the WalRK and CiaRH TCSs (Krystyna Kazmierczak, unpublished results). The CiaRH TCS has been linked to antibiotic resistance, avoidance of autolysis, and repression of competence development (Halfmann et al., 2011, Schnorpfeil et al., 2013). In contrast, the Δpbp1a mutation activates the WalRK TCS specifically and does not induce expression of the CiaRH TCS regulon (Table 4). Previously, we showed that the default activity of WalK is as a WalR~P phosphatase in exponentially growing and non-stressed cells (Gutu et al., 2010, Wayne et al., 2012). WalK phosphatase activity depends on a cytoplasmic PAS domain, suggesting that the switch of WalK from a phosphatase to a kinase involves binding a ligand by the PAS domain (Gutu et al., 2010, Wayne et al., 2012). Recently, a cytoplasmic C55-PP-bound intermediate of wall teichoic acid biosynthesis was shown to bind the PAS domain and negatively regulate the kinase activity of the PhoR histidine kinase of B. subtilis (Botella et al., 2014). These two sets of results suggest an attractive hypothesis for activation of the pneumococcal WalRK TCS by the Δpbp1a, mltG(Y488D), and Δpbp1b mutations (Table 4; Fig. 11 and S15). These mutations all limit the incorporation of external Lipid II precursor into new anchored-PG strands (Fig. 12). We speculate that this blockage backs up the intracellular PG biosynthesis pathway, and accumulated internal Lipid II, Lipid I, or another intermediate binds to the PAS domain of WalK, switching on its autokinase activity that phosphorylates WalR to WalR~P.
PBP1a sets pneumococcal cell width during septal PG synthesis
Finally, this work points to the critical role played by class A PBP1a in setting the width of pneumococcal cells. The absence of PBP1a invariably results in a narrowing of the diameter of pneumococcal cells in a variety of genetic backgrounds, including in divIVA mutants (data not shown), which form chains of large, rounded cells (Boersma et al., 2015, Massidda, et al., 2013). Δpbp1a mutants are shorter, narrower, and characterized by a greater aspect ratio and significantly smaller volume than pbp1a+ cells (Fig. S11) (Land & Winkler, 2011). Decreases in cell length and width are consistent with PBP1a providing TG activity to both peripheral and septal PG synthesis (Fig. S1) (see (Land et al., 2013, Land & Winkler, 2011, Tsui et al., 2014)). However, a role for PBP1a in setting cell width through septal PG synthesis is suggested by comparing the dimensions of mltG(Y488D) and mltG(Y488D) Δpbp1a mutant cells (Fig. 7). The mltG(Y488D) cells have the same width, but a shorter length, compared to the parent mltG+ cells, resulting in increased sphericity (Fig. 7), consistent with the localization of MltG with other components of the peripheral PG synthesis machine (Fig. 9,10, and S13). Cellular spherical shape is further increased in the mltG(Y488D) Δpbp2b mutant (Fig. 7). In contrast, mltG(Y488D) Δpbp1a cells have reduced widths, but similar lengths, compared to mltG(Y488D) cells and similar aspect ratios compared to parent cells (Fig. 7B and 7C). Similarly, ΔmltG Δpbp2b Δpbp1a mutant cells, which lack peripheral PG synthesis, have the same aspect ratio as parent cells (Fig. S11). Together, these results indicate that PBP1a sets cell width in mutants defective in or lacking peripheral PG synthesis, implying that cell width is set by septal PG synthesis.
EXPERIMENTAL PROCEDURES
Bacterial strains and growth conditions
Unencapsulated pneumococcal strains used in this study were derived from strains IU1824 (D39 Δcps rpsL1) or IU1945 (D39 Δcps), which are derivatives of encapsulated serotype 2 D39 strain IU1690 (Table S1) (Lanie et al., 2007). Strains containing antibiotic markers were constructed by transforming linear DNA amplicons synthesized by overlapping fusion PCR into competent pneumococcal cells as described previously (Tsui et al., 2010). Strains containing markerless alleles of pbp1a, mltG, or ftsZ in native chromosomal loci (see Table S1) were generated using allele replacement with the Pc-[kan-rpsL+] (Janus cassette) (Sung et al., 2001). Primers used for the generation of amplicons are listed in Table S2. All constructs were confirmed by DNA sequencing of chromosomal regions corresponding to the amplicon region used for transformation. Bacteria were grown on plates containing trypticase soy agar II (modified; Becton-Dickinson) and 5% (vol/vol) defibrinated sheep blood (TSAII-BA). Plates were incubated at 37°C in an atmosphere of 5% CO2. For antibiotic selections, TSAII BA plates contained 250 μg kanamycin mL−1, 150 μg spectinomycin mL−1, 0.3 μg erythromycin mL−1, 0.25 μg tetracycline mL−1, 250 μg streptomycin mL−1, and/or 2.5 μg chloramphenicol mL−1. Transformants were single-colony-isolated on TSAII-BA plates containing antibiotics twice before growth in antibiotic-containing BHI broth for storage. Strains were cultured statically in Becton-Dickinson brain heart infusion (BHI) broth at 37°C in an atmosphere of 5% CO2, and growth was monitored by OD620 using a Spectronic 20 spectrophotometer fitted for measurement of capped tubes (outer diameter, 16 mm). Bacteria were inoculated into BHI broth from frozen cultures or colonies, serially diluted into the same medium, and propagated overnight for 11 to 15 h. For most growth experiments, overnight cultures that were still in exponential phase (OD620 = 0.1 to 0.4) were diluted back to OD620 ≈ 0.003 to start final cultures, which lacked antibiotics.
Transformation assays with ΔmltG, Δpbp2b, ΔrodA, ΔmreCD, and ΔrodZ amplicons
ΔmltG::Pc-erm, Δpbp2b<>aad9, ΔrodA::Pc-erm, ΔrodA::Pc-[kan-rpsL+], ΔmreCD<>Pc-erm, ΔmreCD<>aad9, ΔrodZ<>aad9, ΔrodZ::Pc-erm, ΔrodZ::Pc-[kan-rpsL+] amplicons, and positive control Δpbp1b::Pc-erm amplicon with ~ 1 kb flanking DNA sequences were obtained from PCR reactions using primers and templates listed in Table S2. 100 μL of recipient strains grown to OD620 ≈0.03 from ice stock were added to 900 μL of transformation mix (10% heat-treated horse serum, 0.18% (wt/vol) glucose, 100 ng CSP−1 (competence stimulatory peptide, type 1) mL−1 in BHI). The mixture was incubated for 10 min at 37°C in an atmosphere of 5% CO2. 30 or 100 ng of purified PCR amplicon (for unencapsulated and encapsulated strains, respectively) was added to the transformation mixture, which was incubated for 1 h at 37°C in an atmosphere of 5% CO2. An aliquot or the entire final transformation mixture was mixed with 3 mL of soft agar containing 72 μL of 0.1mg erythromycin mL−1, 36 μL of 0.1 mg spectinomycin−1, or 120 μL of 50 mg kanamycin mL−1 and plated onto TSAII-BA plates. Plates were examined at 20 to 22 h after incubation at 37°C in an atmosphere of 5% CO2. The numbers of colonies indicated are adjusted to the numbers obtained with 1 mL of transformation mixture. Transformation using a control amplicon with the wild-type unencapsulated strains IU1945 and IU1824 and the encapsulated strain IU1690 yielded >500 colonies and ≈300 colonies, respectively.
Mapping of Δpbp2b suppressor mutations by whole-genome DNA sequencing
Strains IU7476, IU7477, IU7567, IU7570 and IU7765 containing suppressor mutations that allowed the growth of a Δpbp2b deletion mutant were isolated as described in Results (Table S1). Overnight cultures still in exponential phase were diluted into 5 ml of BHI broth to an OD620 ≈0.01 and grown to an OD620 ≈ 0.3 to 0.4. Cells were collected by centrifugation (10,000 xg for 10 min). Genomic DNA was purified from collected cells using a MasterPure Gram-positive DNA purification kit (Epicenter Biotechnologies) according to the manufacturer’s protocol. DNA library construction, Illumina MiSeq DNA sequencing, and bioinformatic analyses are described in procedures in Supplemental Information.
Boc-FL labeling of PBPs in Δpbp2b strains
Labeling of pneumococcal PBPs with fluorescent bocillin (Boc-FL) (Molecular Probes) to determine the Boc-FL labeling profiles of suppressed Δpbp2b mutants were carried out as previously described (Kocaoglu et al., 2015, Tsui et al., 2014, Zhao et al., 1999).
Cell length and width measurements
Cell lengths and widths of strains growing in BHI broth were measured from phase-contrast images by using Nikon NIS-Element AR software as described before (Barendt et al., 2009, Tsui et al., 2014). For strains that do not show chaining morphology, separated stage 1 cells or daughter cells of stage 4 cells were measured. For strains that form short chains of cells, measurements included cells in chains whose widths at constriction sites were < 50% of cell widths. Fifty or more cells from two independent experiments were measured for each strain. P values were obtained by one-way ANOVA analysis (GraphPad Prism, nonparametric Kruskal-Wallis test).
2D immunofluorescence microscopy (IFM), 3D-SIM (structured illumination microscopy) IFM, and double IFM-fluorescent-vancomycin (FL-V) labeling
Localization of FLAG-tagged MreC and Myc- or HA-tagged MltG by IFM was performed on exponentially growing cells as described before (Tsui et al., 2014) on strain IU7580 (mreC-L-FLAG3 mltG-HA) or strain IU7582 (mreC-L-FLAG3 mltG-Myc). Double IFM-FL-V labeling was performed on strain IU7399 (mltG-HA) as described previously (Tsui et al., 2014). Image analyses of 2D IFM images using a graphical user interface (GUI) organized by MATLAB (The Mathworks) scripts were performed as described previously (Land et al., 2013, Tsui et al., 2014).
RNA extraction, library construction and RNA-Seq analysis
Starter cultures were used to inoculate 30 mL cultures of BHI broth in 50 mL conical tubes. RNA was extracted from 23 ml of exponentially growing culture (OD620 ≈ 0.15) using a FastRNA Pro Blue Kit (MP Bio) according to the manufacturer’s guidelines. Briefly, cells were collected by centrifugation at 14, 500 ×g for 5 min at 4°C. Cells were resuspended in 1 mL RNApro (MP Bio) and processed twice in a Fast-Prep 24 homogenizer (MP Bio) for 40 s at setting 6.0. Chloroform and 100% ethanol were used to extract and precipitate the RNA from the resulting lysate, and the miRNeasy minikit (Qiagen) with on-column DNase I treatments was used to purify RNA. Procedures for cDNA library construction, single-end, 50 bp-sequencing performed on a HiSeq 2000 sequencer (Illumina), and bioinformatic analyses are described in Supplemental Information. RNA-Seq data were deposited in the GEO database under accession number GSE77021.
MltG structural modeling
The aa sequence of MltGSpn was entered into the Phyre2 server (Kelley & Sternberg, 2009), which was run in intensive mode. The resulting PDB threading model of MltGSpn(YceG) from aa 266 to 547 was visualized and aligned using PyMOL (The PyMOL Molecular Graphics System, Version 1.7.4.3 Schrödinger, LLC 2) with homologous YceG domains whose structures are in the PDB database (Table S3). RMSDs were determined using the PyMOL PDB model of MltGSpn(YceG) compared to the homologues listed in Table S3. Z-scores were determined using the DALI server (Table S3) (Holm & Rosenstrom, 2010).
Supplementary Material
ABBREVIATED SUMMARY.
An endo-lytic transglycosylase, named MltGSpn, is reported as a new component of the peripheral (side-wall-like) peptidoglycan (PG) synthesis machine in the ovococcus pathogen, Streptococcus pneumoniae. “Synthetic viable” combinations of null mutations and a single MltG(Y488D) change suppress the requirement for the essential PBP2b, MreCD, RodZ, and RodA proteins and suggest a genetic interaction between PBP2b and RodA. A model is proposed in which MltG releases newly synthesized glycan chains for crosslinking in peripheral PG synthesis.
Acknowledgments
We thank Jean-Pierre Claverys for supplying pUC57-gfp(Sp), Jan-Willem Veening for supplying pJWV25 and pKB01_mKate2, Orietta Massidda for supplying anti-FltZ antibody, Stephen Vella for assistance with IFM experiments, Krystyna Kazmierczak for conversations and unpublished results, Kevin Bruce for assistance with RNA preparation for RNA-Seq analyses, and James Ford, Kurt Zimmer, and Doug Rusch for assistance with Illumina DNA sequencing and RNA-Seq analyses. This work was supported by grants 1RO1GM113172 and 1R01GM114315 (to M.E.W.), by grant 1R01AI083365 (to T.G.B.), and by predoctoral Quantitative and Chemical Biology (QCB) institutional training grant T32 GM109825 (to B.E.R.).
References
- Barendt SM, Land AD, Sham LT, Ng WL, Tsui HC, Arnold RJ, Winkler ME. Influences of capsule on cell shape and chain formation of wild-type and pcsB mutants of serotype 2 Streptococcus pneumoniae. J Bacteriol. 2009;191:3024–3040. doi: 10.1128/JB.01505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barendt SM, Sham LT, Winkler ME. Characterization of mutants deficient in the L,D-carboxypeptidase (DacB) and WalRK (VicRK) regulon, involved in peptidoglycan maturation of Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol. 2011;193:2290–2300. doi: 10.1128/JB.01555-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 2009;28:193–204. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KH, Stamsas GA, Straume D, Havarstein LS. Effects of low PBP2b levels on cell morphology and peptidoglycan composition in Streptococcus pneumoniae R6. J Bacteriol. 2013;195:4342–4354. doi: 10.1128/JB.00184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KH, Straume D, Havarstein LS. The function of the transmembrane and cytoplasmic domains of pneumococcal penicillin-binding proteins 2x and 2b extends beyond that of simple anchoring devices. Microbiology. 2014;160:1585–1598. doi: 10.1099/mic.0.078535-0. [DOI] [PubMed] [Google Scholar]
- Boersma MJ, Kuru E, Rittichier JT, VanNieuwenhze MS, Brun YV, Winkler ME. Minimal peptidoglycan (PG) turnover in wild-type and PG hydrolase and cell division mutants of Streptococcus pneumoniae D39 growing planktonically and in host-relevant biofilms. J Bacteriol. 2015;197:3472–3485. doi: 10.1128/JB.00541-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botella E, Devine SK, Hubner S, Salzberg LI, Gale RT, Brown ED, Link H, Sauer U, Codee JD, Noone D, Devine KM. PhoR autokinase activity is controlled by an intermediate in wall teichoic acid metabolism that is sensed by the intracellular PAS domain during the PhoPR-mediated phosphate limitation response of Bacillus subtilis. Molec Microbiol. 2014;94:1242–1259. doi: 10.1111/mmi.12833. [DOI] [PubMed] [Google Scholar]
- Bui NK, Eberhardt A, Vollmer D, Kern T, Bougault C, Tomasz A, Simorre JP, Vollmer W. Isolation and analysis of cell wall components from Streptococcus pneumoniae. Analyt Biochem. 2012;421:657–666. doi: 10.1016/j.ab.2011.11.026. [DOI] [PubMed] [Google Scholar]
- Cadby IT, Lovering AL. Molecular surveillance of the subtle septum: discovering a new mode of peptidoglycan synthesis in streptococci. Molec Microbiol. 2014;94:1–4. doi: 10.1111/mmi.12756. [DOI] [PubMed] [Google Scholar]
- CDC. Antibiotic resistance threats in the United States, 2013. 2013 http://www.cdc.gov/drugresistance/threat-report-2013/
- Chao Y, Marks LR, Pettigrew MM, Hakansson AP. Streptococcus pneumoniae biofilm formation and dispersion during colonization and disease. Front Cell Infect Microbiol. 2014;4:194. doi: 10.3389/fcimb.2014.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chewapreecha C, Marttinen P, Croucher NJ, Salter SJ, Harris SR, Mather AE, Hanage WP, Gloldblatt D, Nosten FH, Turner C, Turner P, Bentley SD, Parkhill J. Comphrensive identification of single nucleotide polymorphisms associated with beta-lactam resistance within pneumococcal mosaic genes. PloS Genet. 2014;10:e1004547. doi: 10.1371/journal.pgen.1004547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Uehara T, Bernhardt TG. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell. 2014;159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalia AB, Weiser JN. Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell Host Microbe. 2011;10:486–496. doi: 10.1016/j.chom.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel RA, Errington J. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell. 2003;113:767–776. doi: 10.1016/s0092-8674(03)00421-5. [DOI] [PubMed] [Google Scholar]
- den Blaauwen T, Andreu JM, Monasterio O. Bacterial cell division proteins as antibiotic targets. Bioorg Chem. 2014;55(2):7–38. doi: 10.1016/j.bioorg.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Divakaruni AV, Baida C, White CL, Gober JW. The cell shape proteins MreB and MreC control cell morphogenesis by positioning cell wall synthetic complexes. Molec Microbiol. 2007;66:174–188. doi: 10.1111/j.1365-2958.2007.05910.x. [DOI] [PubMed] [Google Scholar]
- Donkor ES. Understanding the pneumococcus: transmission and evolution. Front Cell Infection Microbiol. 2013;3:7. doi: 10.3389/fcimb.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrac S, Bisicchia P, Devine KM, Msadek T. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Molec Microbiol. 2008;70:1307–1322. doi: 10.1111/j.1365-2958.2008.06483.x. [DOI] [PubMed] [Google Scholar]
- Eberhardt A, Hoyland CN, Vollmer D, Bisle S, Cleverley RM, Johnsborg O, Havarstein LS, Lewis RJ, Vollmer W. Attachment of capsular polysaccharide to the cell wall in Streptococcus pneumoniae. Microb Drug Resis. 2012;18:240–255. doi: 10.1089/mdr.2011.0232. [DOI] [PubMed] [Google Scholar]
- Egan AJ, Biboy J, van’t Veer I, Breukink E, Vollmer W. Activities and regulation of peptidoglycan synthases. Phil Trans Royal Socf London Series B, Biological sciences. 2015;370:1679. doi: 10.1098/rstb.2015.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan AJ, Vollmer W. The physiology of bacterial cell division. Ann NY Acad Sci. 2013;1277:8–28. doi: 10.1111/j.1749-6632.2012.06818.x. [DOI] [PubMed] [Google Scholar]
- Eldholm V, Johnsborg O, Straume D, Ohnstad HS, Berg KH, Hermoso JA, Havarstein LS. Pneumococcal CbpD is a murein hydrolase that requires a dual cell envelope binding specificity to kill target cells during fratricide. Molec Microbiol. 2010;76:905–917. doi: 10.1111/j.1365-2958.2010.07143.x. [DOI] [PubMed] [Google Scholar]
- Ferreira DM, Gordon SP. Mechanisms causing inflammatory response to Streptococcus pneumoniae. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 383–400. [Google Scholar]
- Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, Freton C, Combet C, Guiral S, Soufi B, Macek B, Kuru E, VanNieuwenhze MS, Brun YV, Di Guilmi AM, Claverys JP, Galinier A, Grangeasse C. Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet. 2014;10:e1004275. doi: 10.1371/journal.pgen.1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisch N, Peters K, Zahringer U, Vollmer W. The pneumococcal cell wall. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 145–167. [Google Scholar]
- Gratz N, Loh LN, Tuomanen EI. Pneumococcal invasion: development of bacteremia and meningitis. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 433–451. [Google Scholar]
- Gutu AD, Wayne KJ, Sham LT, Winkler ME. Kinetic characterization of the WalRKSpn (VicRK) two-component system of Streptococcus pneumoniae: dependence of WalKSpn (VicK) phosphatase activity on its PAS domain. J Bacteriol. 2010;192:2346–2358. doi: 10.1128/JB.01690-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson AP, Marks LR, Roche-Hakansson H. Pneumococcal genetic transformation during colonization and biofilm formation. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 129–142. [Google Scholar]
- Hakenbeck R. Discovery of beta-lactam-resistant variants in diverse pneumococcal populations. Genome Med. 2014;6:72. doi: 10.1186/s13073-014-0072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakenbeck R, Bruckner R, Denapaite D, Maurer P. Molecular mechanisms of beta-lactam resistance in Streptococcus pneumoniae. Fut Microbiol. 2012;7:395–410. doi: 10.2217/fmb.12.2. [DOI] [PubMed] [Google Scholar]
- Halfmann A, Schnorpfeil A, Muller M, Marx P, Gunzler U, Hakenbeck R, Bruckner R. Activity of the two-component regulatory system CiaRH in Streptococcus pneumoniae R6. J Molec Microbiol Biotech. 2011;20:96–104. doi: 10.1159/000324893. [DOI] [PubMed] [Google Scholar]
- Henriques-Normark B, Tuomanen EI. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harbor perspectives in medicine. 2013;3:a010215. doi: 10.1101/cshperspect.a010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nuc Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins J, Matsushima P, Mullen DL, Tang J, Zhao G, Meier TI, Nicas TI, Jaskunas SR. Gene disruption studies of penicillin-binding proteins 1a, 1b, and 2a in Streptococcus pneumoniae. J Bacteriol. 1999;181:6552–6555. doi: 10.1128/jb.181.20.6552-6555.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nature Prot. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kocaoglu O, Carlson EE. Profiling of beta-lactam selectivity for penicillin-binding proteins in Escherichia coli strain DC2. Antimicrob Agents Chemother. 2015;59:2785–2790. doi: 10.1128/AAC.04552-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocaoglu O, Tsui HC, Winkler ME, Carlson EE. Profiling of beta-lactam selectivity for penicillin-binding proteins in Streptococcus pneumoniae D39. Antimicrob Agents Chemother. 2015;59:3548–3555. doi: 10.1128/AAC.05142-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse T, Bork-Jensen J, Gerdes K. The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Molec Microbiol. 2005;55:78–89. doi: 10.1111/j.1365-2958.2004.04367.x. [DOI] [PubMed] [Google Scholar]
- Land AD, Tsui HC, Kocaoglu O, Vella SA, Shaw SL, Keen SK, Sham LT, Carlson EE, Winkler ME. Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Molec Microbiol. 2013;90:939–955. doi: 10.1111/mmi.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land AD, Winkler ME. The requirement for pneumococcal MreC and MreD is relieved by inactivation of the gene encoding PBP1a. J Bacteriol. 2011;193:4166–4179. doi: 10.1128/JB.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. Genome sequence of Avery’s virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol. 2007;189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering AL, Safadi SS, Strynadka NC. Structural perspective of peptidoglycan biosynthesis and assembly. Ann Rev Biochem. 2012;81:451–478. doi: 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]
- Mao F, Dam P, Chou J, Olman V, Xu Y. DOOR: a database for prokaryotic operons. Nuc Acids Res. 2009;37:D459–463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massidda O, Novakova L, Vollmer W. From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ Microbiol. 2013;15:3133–3157. doi: 10.1111/1462-2920.12189. [DOI] [PubMed] [Google Scholar]
- Mellroth P, Daniels R, Eberhardt A, Ronnlund D, Blom H, Widengren J, Normark S, Henriques-Normark B. LytA, major autolysin of Streptococcus pneumoniae, requires access to nascent peptidoglycan. J Biol Chem. 2012;287:11018–11029. doi: 10.1074/jbc.M111.318584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlot C, Bayle L, Jacq M, Fleurie A, Tourcier G, Galisson F, Vernet T, Grangeasse C, Di Guilmi AM. Interaction of penicillin-binding protein 2x and Ser/Thr protein kinase StkP, two key players in Streptococcus pneumoniae R6 morphogenesis. Molec Microbiol. 2013;90:88–102. doi: 10.1111/mmi.12348. [DOI] [PubMed] [Google Scholar]
- Ng WL, Kazmierczak KM, Winkler ME. Defective cell wall synthesis in Streptococcus pneumoniae R6 depleted for the essential PcsB putative murein hydrolase or the VicR (YycF) response regulator. Molec Microbiol. 2004;53:1161–1175. doi: 10.1111/j.1365-2958.2004.04196.x. [DOI] [PubMed] [Google Scholar]
- Ng WL, Robertson GT, Kazmierczak KM, Zhao J, Gilmour R, Winkler ME. Constitutive expression of PcsB suppresses the requirement for the essential VicR (YycF) response regulator in Streptococcus pneumoniae R6. Molec Microbiol. 2003;50:1647–1663. doi: 10.1046/j.1365-2958.2003.03806.x. [DOI] [PubMed] [Google Scholar]
- Ng WL, Tsui HC, Winkler ME. Regulation of the pspA virulence factor and essential pcsB murein biosynthetic genes by the phosphorylated VicR (YycF) response regulator in Streptococcus pneumoniae. J Bacteriol. 2005;187:7444–7459. doi: 10.1128/JB.187.21.7444-7459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng WL, Winkler ME. Singular structures and operon organizations of essential two-component systems in species of Streptococcus. Microbiology. 2004;150:3096–3098. doi: 10.1099/mic.0.27550-0. [DOI] [PubMed] [Google Scholar]
- Oliver MB, Swords WE. Pneumococcal biofilms and bacterial perisistence during otitis media. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 293–308. [Google Scholar]
- Pagliero E, Dublet B, Frehel C, Dideberg O, Vernet T, DiGuilmi AM. The inactivation of a new peptidoglycan hydrolase Pmp23 leads to abnormal septum formation in Streptococcus pneumoniae. Open Microbiol. 2008;2:107–114. doi: 10.2174/1874285800802010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J, Kern I, Lurz R, Hakenbeck R. Mutational analysis of the Streptococcus pneumoniae bimodular class A penicillin-binding proteins. J Bacteriol. 1999;181:3852–3856. doi: 10.1128/jb.181.12.3852-3856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis-Bleau, Markovski CM, Uehara T, Lupoli TJ, Walker S, Kahne DE, Bernhardt TG. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell. 2010;143:110–1120. doi: 10.1016/j.cell.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Dorado I, Gonzalez A, Morales M, Sanles R, Striker W, Vollmer W, Mobashery S, Garcia JL, Martinez-Ripoll M, Garcia P, Hermoso JA. Insights into pneumococcal fratricide from the crystal structures of the modular killing factor LytC. Nature Structl & Molec Biol. 2010;17:576–581. doi: 10.1038/nsmb.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters K, Schweizer I, Beilharz K, Stahlmann C, Veening JW, Hakenbeck R, Denapaite D. Streptococcus pneumoniae PBP2x mid-cell localization requires the C-terminal PASTA domains and is essential for cell shape maintenance. Molec Microbiol. 2014;92:733–755. doi: 10.1111/mmi.12588. [DOI] [PubMed] [Google Scholar]
- Philippe J, Gallet B, Morlot C, Denapaite D, Hakenbeck R, Chen Y, Vernet T, Zapun A. Mechanism of beta-lactam action in Streptococcus pneumoniae: the piperacillin paradox. Antimicrob Agents and Chemother. 2015;59:609–621. doi: 10.1128/AAC.04283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe J, Vernet T, Zapun A. The elongation of ovococci. Microbl Drug Resist. 2014;20:215–221. doi: 10.1089/mdr.2014.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho MG, Errington J. Recruitment of penicillin-binding protein PBP2 to the division site of Staphylococcus aureus is dependent on its transpeptidation substrates. Molec Microbiol. 2005;55:799–807. doi: 10.1111/j.1365-2958.2004.04420.x. [DOI] [PubMed] [Google Scholar]
- Pinho MG, Kjos M, Veening JW. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol. 2013;11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- Potluri LP, Kannan S, Young KD. ZipA is required for FtsZ-dependent preseptal peptidoglycan synthesis prior to invagination during cell division. J Bacteriol. 2012;194:5334–5342. doi: 10.1128/JB.00859-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JL, Dalia AB, Weiser JN. Increased chain length promotes pneumococcal adherence and colonization. Infect Immun. 2012;80:3454–3459. doi: 10.1128/IAI.00587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneewind O, Missiakas D. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochimica et biophysica acta. 2013;1843:1687–1697. doi: 10.1016/j.bbamcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorpfeil A, Kranz M, Kovacs M, Kirsch C, Gartmann J, Brunner I, Bittmann S, Bruckner R. Target evaluation of the non-coding csRNAs reveals a link of the two-component regulatory system CiaRH to competence control in Streptococcus pneumoniae R6. Molec Microbiol. 2013;89:334–349. doi: 10.1111/mmi.12277. [DOI] [PubMed] [Google Scholar]
- Sham LT, Barendt SM, Kopecky KE, Winkler ME. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proceed Nat Acad Sci USA. 2011;108:E1061–1069. doi: 10.1073/pnas.1108323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham LT, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014;345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham LT, Tsui HC, Land AD, Barendt SM, Winkler ME. Recent advances in pneumococcal peptidoglycan biosynthesis suggest new vaccine and antimicrobial targets. Curr Opinion Microbiol. 2012;15:194–203. doi: 10.1016/j.mib.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Diavatopoulos DA. Nasopharyngeal colonization with Streptococcus pneumoniae. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 279–291. [Google Scholar]
- Standish AJ, Morona R. Capsule, structure, synthesis, and regulation. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 169–179. [Google Scholar]
- Sung CK, Li H, Claverys JP, Morrison DA. An rpsL cassette, Janus, for gene replacement through negative selection in Streptococcus pneumoniae. App Environ Microbiol. 2001;67:5190–5196. doi: 10.1128/AEM.67.11.5190-5196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo ACK, Roper DI. Core steps of membrane-bound peptidoglycan biosynthesis: recent advance, insight and opportunities. Antibiotics. 2015;4:495–520. doi: 10.3390/antibiotics4040495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibessard A, Fernandez A, Gintz B, Leblond-Bourget N, Decaris B. Effects of rodA and pbp2b disruption on cell morphology and oxidative stress response of Streptococcus thermophilus CNRZ368. J Bacteriol. 2002;184:2821–2826. doi: 10.1128/JB.184.10.2821-2826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorova K, Maurer P, Rieger M, Becker T, Bui NK, Gray J, Vollmer W, Hakenbeck R. Transfer of penicillin resistance from Streptococcus oralis to Streptococcus pneumoniae identifies murE as resistance determinant. Molec Microbiol. 2015;97:866–880. doi: 10.1111/mmi.13070. [DOI] [PubMed] [Google Scholar]
- Tomasz A, Fischer W. The cell wall of Streptococcus pneumonaie. In: Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, Rood JI, editors. Gram-Postive Pathogens. Washington, D. C: ASM Press; 2006. pp. 230–240. [Google Scholar]
- Tsui HC, Boersma MJ, Vella SA, Kocaoglu O, Kuru E, Peceny JK, Carlson EE, VanNieuwenhze MS, Brun YV, Shaw SL, Winkler ME. Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of Streptococcus pneumoniae D39. Molec Microbiol. 2014;94:21–40. doi: 10.1111/mmi.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Mukherjee D, Ray VA, Sham LT, Feig AL, Winkler ME. Identification and characterization of noncoding small RNAs in Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol. 2010;192:264–279. doi: 10.1128/JB.01204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RD, Vollmer W, Foster SJ. Different walls for rods and balls: the diversity of peptidoglycan. Molec Microbiol. 2014;91:862–874. doi: 10.1111/mmi.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol. 2012;10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ploeg R, Verheul J, Vischer NO, Alexeeva S, Hoogendoorn E, Postma M, Banzhaf M, Vollmer W, den Blaauwen T. Colocalization and interaction between elongasome and divisome during a preparative cell division phase in Escherichia coli. Molecr Microbiol. 2013;87:1074–1087. doi: 10.1111/mmi.12150. [DOI] [PubMed] [Google Scholar]
- van Opijnen T, Camilli A. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res. 2012;22:2541–2551. doi: 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernatter J, Pirofski LA. Current concepts in host-microbe interaction leading to pneumococcal pneumonia. Curr Opinion Infect Dis. 2013;26:277–283. doi: 10.1097/QCO.0b013e3283608419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinella D, Joseleau-Petit D, Thevenet D, Bouloc P, D’Ari R. Penicillin-binding protein 2 inactivation in Escherichia coli results in cell division inhibition, which is relieved by FtsZ overexpression. J Bacteriol. 1993;175:6704–6710. doi: 10.1128/jb.175.20.6704-6710.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne KJ, Li S, Kazmierczak KM, Tsui HC, Winkler ME. Involvement of WalK (VicK) phosphatase activity in setting WalR (VicR) response regulator phosphorylation level and limiting cross-talk in Streptococcus pneumoniae D39 cells. Molec Microbiol. 2012;86:645–660. doi: 10.1111/mmi.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne KJ, Sham LT, Tsui HC, Gutu AD, Barendt SM, Keen SK, Winkler ME. Localization and cellular amounts of the WalRKJ (VicRKX) two-component regulatory system proteins in serotype 2 Streptococcus pneumoniae. J Bacteriol. 2010;192:4388–4394. doi: 10.1128/JB.00578-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser JN. The battle with the host over microbial size. Curr Opin Microbiol. 2013;16:59–62. doi: 10.1016/j.mib.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler R, Mesnage S, Boneca IG, Hobbs JK, Foster SJ. Super-resolution microscopy reveals cell wall dynamics and peptidoglycan architecture in ovococcal bacteria. Molec Microbiol. 2011;82:1096–1109. doi: 10.1111/j.1365-2958.2011.07871.x. [DOI] [PubMed] [Google Scholar]
- Wilson M. Microbial inhabitants of humans. Cambridge University Press; Cambridge, United Kingdom: 2005. [Google Scholar]
- Winkler ME, Hoch JA. Essentiality, bypass, and targeting of the YycFG (VicRK) two-component regulatory system in gram-positive bacteria. J Bacteriol. 2008;190:2645–2648. doi: 10.1128/JB.01682-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunck R, Cho H, Bernhardt TG. Identification of MltG as a potential terminase for peptidoglycan polymerization in bacteria. Molec Microbiol. 2015 doi: 10.1111/mmi.13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev. 2008a;32:361–385. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- Zapun A, Vernet T, Pinho MG. The different shapes of cocci. FEMS Microbiol Rev. 2008b;32:345–360. doi: 10.1111/j.1574-6976.2007.00098.x. [DOI] [PubMed] [Google Scholar]
- Zhao G, Meier TI, Kahl SD, Gee KR, Blaszczak LC. Bocillin-FL, a sensitive and commercially available reagent for detection of penicillin-binding proteins. Antimicrobl Agents Chemother. 1999;43:1124–1128. doi: 10.1128/aac.43.5.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.