

Abstract
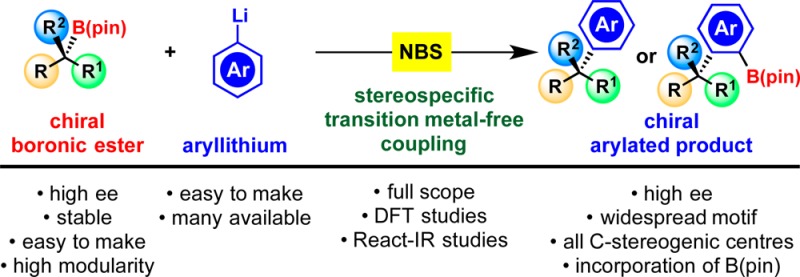
The stereospecific cross-coupling of secondary boronic esters with sp2 electrophiles (Suzuki–Miyaura reaction) is a long-standing problem in synthesis, but progress has been achieved in specific cases using palladium catalysis. However, related couplings with tertiary boronic esters are not currently achievable. To address this general problem, we have focused on an alternative method exploiting the reactivity of a boronate complex formed between an aryl lithium and a boronic ester. We reasoned that subsequent addition of an oxidant or an electrophile would remove an electron from the aromatic ring or react in a Friedel–Crafts-type manner, respectively, generating a cationic species, which would trigger 1,2-migration of the boron substituent, creating the new C–C bond. Elimination (preceded by further oxidation in the former case) would result in rearomatization giving the coupled product stereospecifically. Initial work was examined with 2-furyllithium. Although the oxidants tested were unsuccessful, electrophiles, particularly NBS, enabled the coupling reaction to occur in good yield with a broad range of secondary and tertiary boronic esters, bearing different steric demands and functional groups (esters, azides, nitriles, alcohols, and ethers). The reaction also worked well with other electron-rich heteroaromatics and 6-membered ring aromatics provided they had donor groups in the meta position. Conditions were also found under which the B(pin)- moiety could be retained in the product, ortho to the boron substituent. This protocol, which created a new C(sp2)–C(sp3) and an adjacent C–B bond, was again applicable to a range of secondary and tertiary boronic esters. In all cases, the coupling reaction occurred with complete stereospecificity. Computational studies verified the competing processes involved and were in close agreement with the experimental observations.
1. . Introduction
The cross-coupling of organoboron derivatives with C-sp2 electrophiles, the Suzuki–Miyaura reaction (Scheme 1), is one of the most broadly used reactions with applications that span materials, pharmaceuticals, and agrochemicals.1,2 Its mild reaction conditions, functional group tolerance, broad scope, and scalability are features that have contributed to its extensive use in both academic and industrial settings.2 There is currently a strong impetus to extend this reaction to sp3–sp2 cross-coupling because it offers new strategic disconnections for the rapid assembly of chiral (3D) molecules.3,4 However, such a coupling reaction has proved challenging.5,6 This is because the desirable features of the high configurational stability of the C–B bond that enables boronic esters to be isolated and purified also renders them relatively unreactive, and further activation is required in order to engage them in the key transmetalation step of the catalytic cycle.7,8 Furthermore, competing β-hydride elimination can also occur from the aliphatic organo-Pd(II) species, which can have an impact on both yield and stereoselectivity.
Scheme 1. Suzuki–Miyaura Cross-Coupling of Aliphatic Organoborons.

Despite these inherent difficulties, examples of Pd-catalyzed sp2–sp3 cross-coupling of chiral organoborons have been reported (Scheme 2).5,6,9−20 Pioneering work by Crudden showed that benzylic pinacol boronic esters and dibenzylic neopentyl boronic esters could be coupled with high stereospecificity with retention of configuration with aryl iodides.21−23 Liao found that the stereospecific coupling of dibenzylic organoborons could be rendered invertive using the potassium trifluoroborate salt in place of the neopentyl boronic ester, which occurred with retention.24 Molander and Hall employed internal activating groups to promote the coupling of chiral potassium trifluoroborates with inversion of configuration.25−28 Suginome developed a unique stereospecific cross-coupling of benzylic α-amino boronic esters that depending on the addition of a Bronsted or a Lewis acid occurred with inversion or retention of configuration.29−32 More recently, Biscoe reported the first stereoinvertive cross-coupling of unactivated secondary potassium trifluoroborates.33
Scheme 2. Suzuki–Miyaura Stereospecific Cross-Coupling of Aliphatic Secondary Organoborons.

Single-electron processes have also been used for the construction of sp2–sp3 bonds, and Molander successfully explored the application of dual photoredox and Ni0 catalysis for the coupling of secondary benzylic trifluoroborates and aryl bromides (Scheme 3).34,35 Despite the moderate enantioselectivity achieved in the single example reported (52% yield, 75:25 e.r.), this exciting approach opens new avenues for exploration.
Scheme 3. Molander’s Stereoselective Cross-Coupling of Aliphatic Organoborons.

None of the transition-metal-based processes have been applied to tertiary boronic esters, and their application to secondary boronic esters are rather substrate-specific, limiting the applicability of the methodology. This is because the transition-metal-catalyzed processes are especially sensitive to steric effects. Indeed, the assembly of all-carbon quaternary stereogenic centers by Suzuki–Miyaura cross-coupling has not been reported.5,6
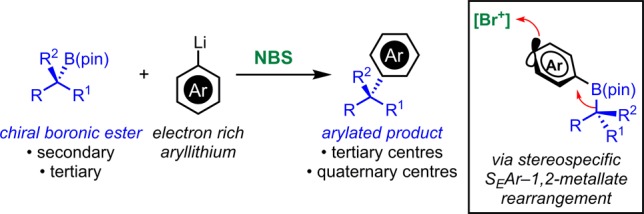
We have recently developed an alternative nontransition-metal-mediated strategy for the stereospecific cross-coupling of secondary and tertiary boronic esters with complete retention of configuration (Scheme 4).36 Our protocol relies on the formation of a chiral boronate complex that upon addition of N-bromosuccinimide (NBS) undergoes a reaction cascade comprising SEAr, 1,2-metallate rearrangement, and elimination. This chemistry was inspired by reactions developed more than 40 years ago on simple and symmetrical boranes.37−43 In this paper we provide a full account of this work, describing its genesis, mechanistic and DFT studies, as well as illustrating the full scope of the coupling reaction.
Scheme 4. Planned Coupling Protocol.
2. Design Plan
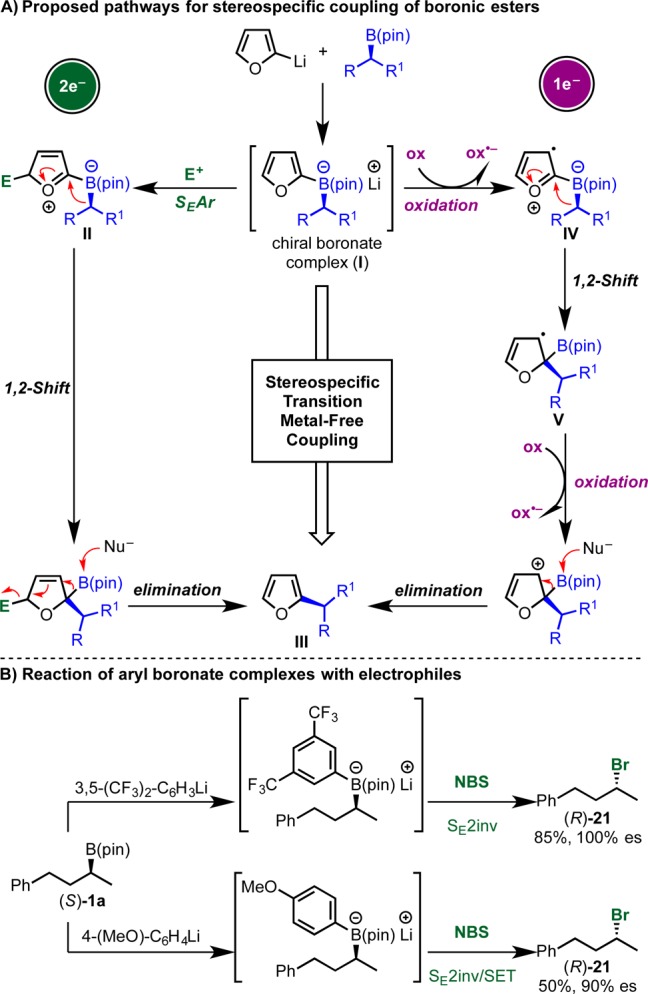
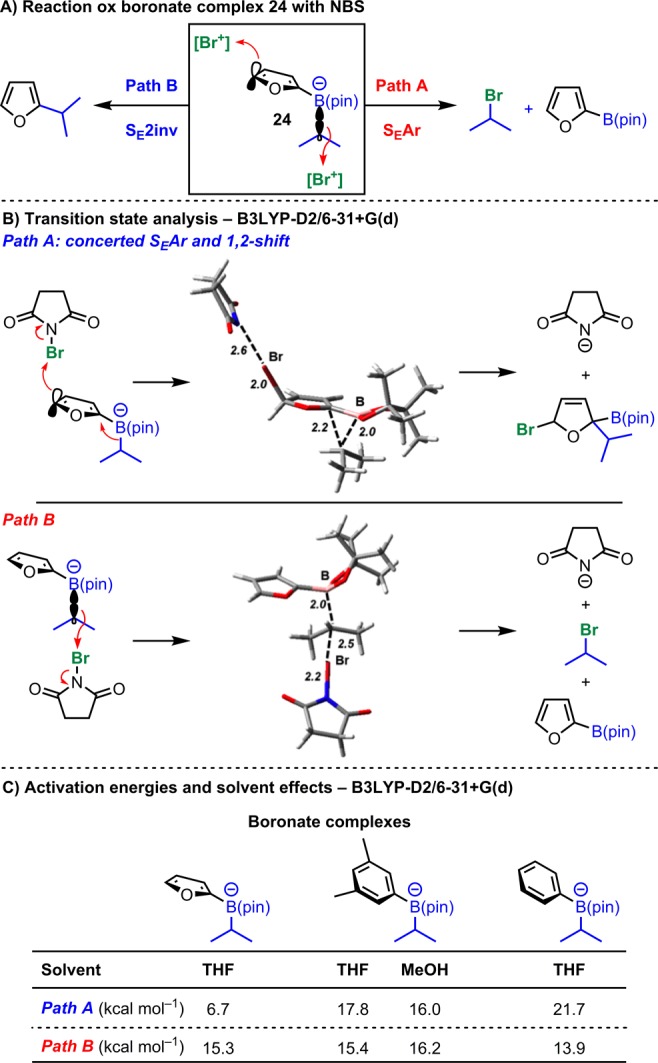
At the outset, we reasoned that a possible way to achieve a transition-metal-free stereospecific cross-coupling of boronic esters would be to exploit the inherent nucleophilicity of aryl-boronate complex I (Scheme 5A, left). These species are very easy to prepare by the addition of aryllithiums (e.g., 2-lithiofuran) to boronic esters and are both chemically and configurationally stable. To trigger a stereospecific 1,2-metallate rearrangement, a positive charge needs to be generated at the aromatic B-bearing carbon. Inspired by the work of Suzuki, Negishi, and Levy on the coupling of symmetrical boranes,37−45 we reasoned that addition of a suitable electrophile would generate cation II by SEAr. This was expected to trigger a 1,2-migration and following elimination would give aryl-coupled product III stereospecifically. However, while feasible, this approach was not without concern because our previous studies on the reactivity of related aryl boronates toward electrophilic reagents (e.g., NBS, I2,46 iminium ions,47 and Selectfluor48) showed that they efficiently serve as chiral nucleophiles and undergo electrophilic substitution with inversion of stereochemistry (SE2inv) at the sp3 carbon (Scheme 5B).49 We therefore considered an alternative mode of activation. In particular, we envisaged that treatment of I with an appropriate oxidant would deliver the radical cation IV by SET oxidation (Scheme 5A, right).50,51 This was expected to trigger a 1,2-migration and following a second oxidation (from the radical intermediate V) and elimination would deliver arylated product III again with retention of stereochemistry.
Scheme 5. Proposed Pathways for Coupling (A) and Potential Competing Reactions (B).
3. Synthesis of Boronic Esters
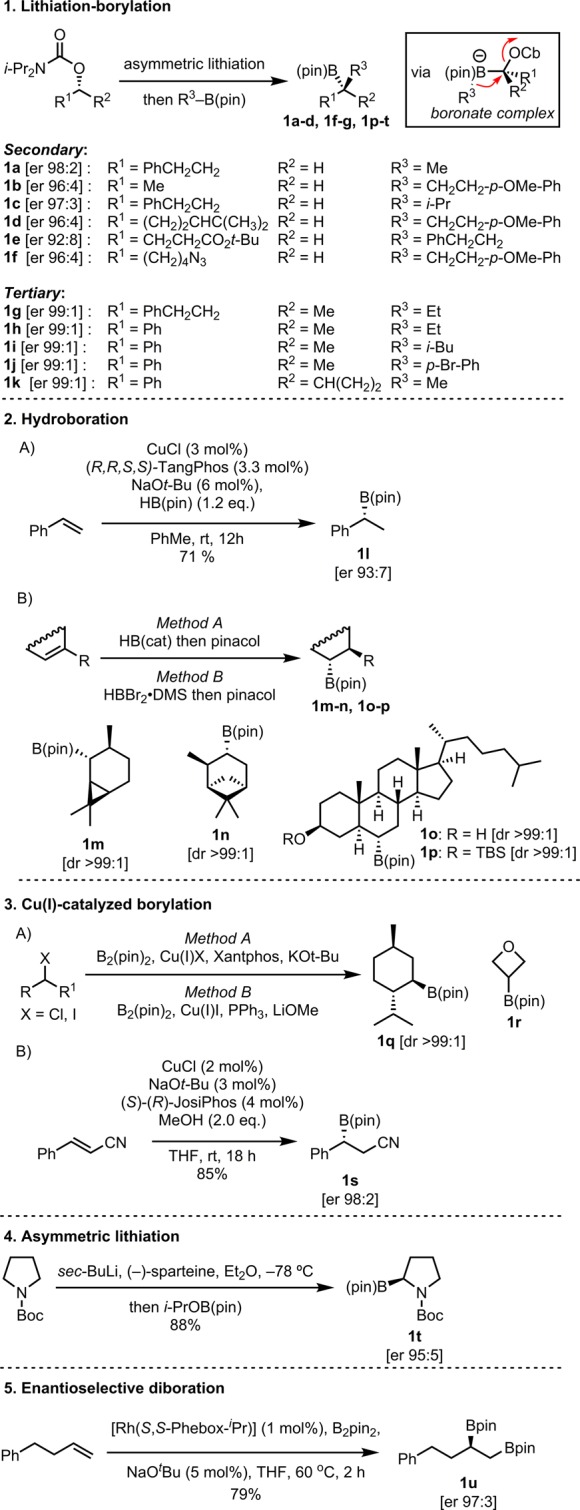
All of the required boronic esters were prepared using five general protocols as described in Scheme 6 and the Supporting Information: (1) Secondary and tertiary boronic esters 1a–k were prepared by lithiation–borylation methodology developed by us.52−55 (2) Benzylic boronate 1l was prepared by enantioselective hydroboration as reported by Yun,56 whereas carene-, pinene-, and cholesterol-based substrates (1m–n and 1o–p) were prepared by diastereoselective hydroboration as described by Renaud57 and in situ transesterification with pinacol. (3A) Menthyl and oxetane boronic ester 1q and 1r were obtained by Cu(I)-catalyzed borylation as reported by Ito58 and Marder.59 (3B) Nitrile 1s was synthesized according to Yun’s β-boration protocol.60 (4) 2-B(pin)-N-Boc-pyrrolidine 1t was prepared by asymmetric lithiation of N-Boc-pyrrolidine and quenching with i-PrOB(pin) as described by Whiting.61 (5) 1,2-Bis-boronic ester 1u was prepared by asymmetric Morken–Nishiyama diboration.62,63
Scheme 6. Synthesis of Secondary and Tertiary Boronic Esters.
4. Results and Discussion
4.1. Oxidative Couplings
We began our studies with the addition of 2-lithiofuran to secondary boronic ester 1a and explored a variety of oxidants covering a wide range of electrochemical potential (Scheme 7A). Out of all the oxidants tested, DDQ (2,3-dichloro-5,6-dicyano-1,4-benozquinone) was found to be uniquely effective, resulting in complete reaction after just 5 min at −78 °C (entry 1). Furthermore, the process was completely stereospecific (100% es). At this point a range of enantioenriched secondary and tertiary boronic esters were tested under the optimized reaction conditions (Scheme 7B). In general, secondary dialkyl boronic esters 1a, 1c, and 1e reacted well, and corresponding products 2a, 2c, and 2e were obtained in high yields and complete stereospecificity. This initial screening showed that the process tolerated increased steric hindrance on the boronic ester and ester functional groups. The methodology was also applied to hindered cyclic terpene-derived boronic esters 1m–n giving desired products 2m–n in high yields and complete diastereospecificity. However, secondary benzylic, α-amino, and tertiary boronic esters 1l, 1t, and 1g failed. Furthermore, boronate complex 22 obtained by addition of p-MeO-phenyllithium to 1a also failed to deliver the desired product. This was particularly surprising because (i) boronic ester 1a efficiently underwent intramolecular arylation with 2-lithiofuran (2a) and (ii) the p-MeO-phenyl group can be readily oxidized by DDQ.64
Scheme 7. Reaction Optimization and Substrate Scope with DDQ.
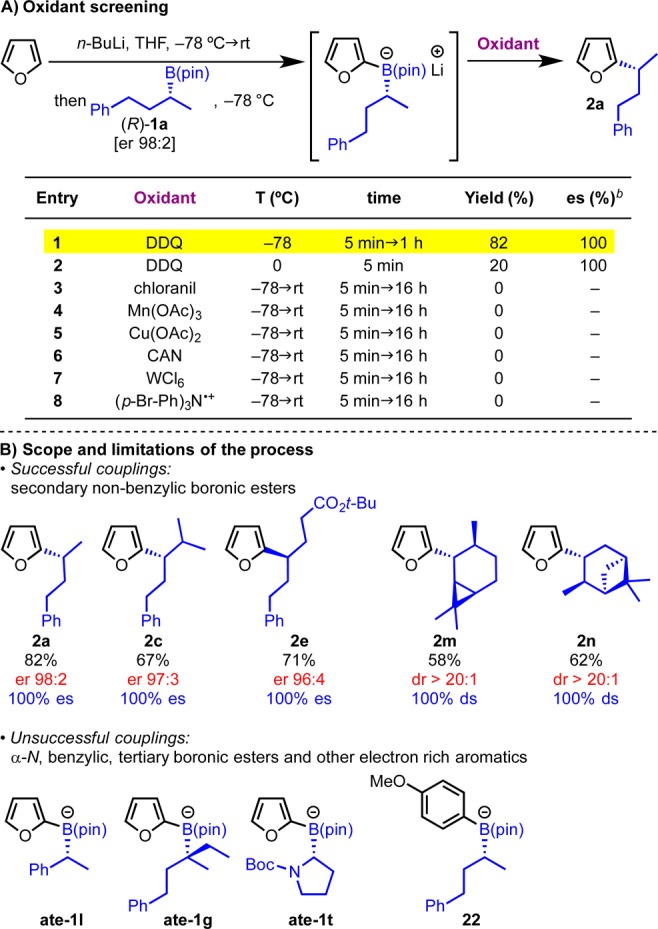
Reaction conditions: furan (1.2 equiv), n-BuLi (1.2 equiv) in THF (0.3 M) at −78 °C → rt for 1 h, then 1a (1.0 equiv) in THF (0.3 M) at −78 °C, then oxidant (1.5 equiv) in THF (0.3 M).
Determined by chiral HPLC analysis.
4.1.1. DFT Calculations and Proposed Mechanism of Oxidative Couplings
The surprising difference in behavior of these boronate complexes prompted us to try to understand the cause of the unsuccessful couplings (Scheme 8). Analysis of the different failed reactions revealed that 2-B(pin)-furan [or p-MeO-C6H4–B(pin)] was the major product, and using boronic ester 1g, we were able to isolate olefin 23 (as a mixture of regioisomers; Scheme 8A). The formation of these products suggested to us that preferential SET oxidation of the sp3-C–B bond over the aromatic unit was occurring which would generate the 2-B(pin)-furan, the radical anion of DDQ, and a stable tertiary (or secondary benzylic) radical which could ultimately give olefins 23. This proposed mechanism implied a surprising chemoselectivity in the DDQ oxidation step whereby secondary (nonbenzylic) boronate complexes react at the furyl unit (Scheme 8B, path A), whereas the sp3-C–B bond is oxidized in the cases of secondary benzylic and tertiary substrates (path B). To analyze this further we performed computational studies. DFT calculations based on model i-Pr boronate 24 showed the HOMO and hence the site for SET in species such as boronate complex I to be mostly localized on the sp3-C–B bond. DFT geometry optimization of the radical produced by SET led to spontaneous C–B bond breaking with formation of a radical. While it does provide an explanation for the unsuccessful couplings, this predicted behavior is inconsistent with (i) the observed stereospecific coupling with the dialkyl secondary boronic esters and (ii) the retention of the cyclopropane unit in example 2m. This led us to consider a different mechanism that did not involve SET. In fact, DDQ is known to be a strong electrophile with a Mayr reactivity parameter of −3.59, comparable to a tropylium ion.65 Thus, a mechanism similar to the one delineated for the electrophilic quench in Scheme 5A might be operative. As described in Scheme 8C, SEAr from the boronate complex ate-1a with DDQ would give intermediate 25, which upon stereospecific 1,2-rearrangement and elimination would account for the successful formation of 2a with retention of stereochemistry. DFT studies showed that DDQ could indeed react via this pathway after initial formation of the encounter complex 26·DDQ. The low calculated barriers for this mechanism are consistent with the fact that reaction with DDQ is complete in minutes at −78 °C, and it is proposed that in the successful arylation cases this pathway is followed (see Supporting Information for more details).
Scheme 8. Experimental Observations and Proposed Mechanism for Unsuccessful Couplings.

While this mechanism would fully explain the successful couplings reported in Scheme 8B, a unified mechanistic picture would still imply a remarkable ability of DDQ to serve as an electrophile or a one-electron oxidant depending on the furyl-boronate substitution pattern: secondary nonbenzylic substrates would undergo SEAr (path C), whereas secondary benzylic and tertiary would undergo sp3-C–B bond SET oxidation (path D). We believe that in the latter cases path D operates owing to the greater ease of removal of an electron from the benzylic sp3-C–B bond leading to a more stable benzylic or tertiary radical, and we performed further calculations using model isopropyl and benzylic boronate complexes 24 and ate-1l. We estimated the free energy of activation ΔG* for one-electron oxidation of the boronate in encounter complex 26·DDQ with Marcus theory,66,67 using DFT (B3LYP) calculations to obtain the parameters needed (see the Supporting Information for details of this Marcus theory modeling and other computations). This yielded a ΔG* of 4.7 kcal mol–1 for R = Me and 2.5 kcal mol−1 for R = Ph (Scheme 8E). Also, regular B3LYP-D2 calculations were used to predict free energies of activation ΔG* for SEAr starting from 26·DDQ, which were 3.7 kcal mol–1 in both cases. Hence, as shown in Scheme 8D, reaction through path C in encounter complex 26·DDQ is favored by 1.0 kcal mol–1 for the isopropyl case (consistent with the successful coupling with secondary alkyl substrates), whereas reaction through path D is favored by 1.2 kcal mol–1 for the benzylic substrate (accounting for the lack of coupling in such cases). The uncertainties in the Marcus theory calculations mean that this agreement with experiment is in part fortuitous, but the conclusions that both SEAr and SET are facile and that the latter proceeds more readily from 26·DDQ are more robust.
4.2. Electrophile-Mediated Couplings
Our initial study on the DDQ-mediated arylation of chiral boronic esters revealed that the electrophile-promoted pathway was feasible, so we decided to evaluate this strategy further. As described in Scheme 9, we subjected boronate complex ate-1a (obtained by addition of 2-lithiofuran to boronic ester 1a) to a range of electrophiles and identified NBS to be highly effective at both −78 and 0 °C (entries 3 and 4). Under these reaction conditions, product 2a was obtained in 92% yield and complete stereospecificity with retention of configuration.
Scheme 9. Optimization of Reaction Conditions for Electrophile-Promoted Couplings.
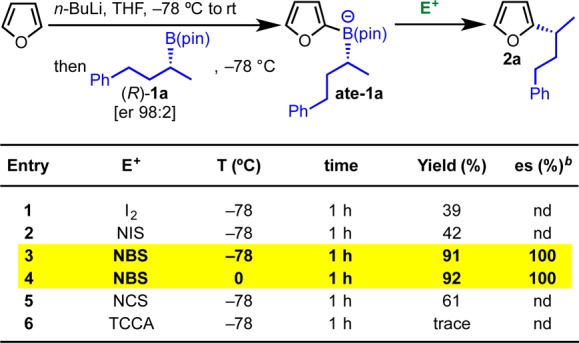
Reaction conditions: furan (1.2 equiv), n-BuLi (1.2 equiv) in THF (0.3 M) at −78 °C → rt for 1 h, then 1a (1.0 equiv) in THF (0.3 M) at −78 °C, then electrophile (1.2 equiv) in THF (0.3 M).
Determined by chiral HPLC analysis. TCCA = trichloroisocyanuric acid.
4.2.1. Couplings between Chiral Secondary Boronic Esters with 5-Membered Ring Heterocycles
We then decided to evaluate the scope of this transition-metal-free cross-coupling strategy. As described in Scheme 10, a broad range of boronic esters was evaluated with electron-rich 5-membered ring aryllithiums. In general, secondary dialkyl substituted boronic esters reacted very well with 2-furyllithium, giving the desired products in high yields and complete es. Pleasingly, secondary benzylic and pyrrolidinyl boronic esters 1l and 1t also reacted well and afforded the desired products in high yields and with complete retention of stereochemistry (2l–2t). Furthermore, we found that the stereospecific coupling was compatible with the following functional groups: ester 2e, azide 2f, nitrile 2s, trisubstituted double bond 2d, and unprotected hydroxyl group 2o. Oxetanyl organoboronic ester 1r also coupled effectively under our standard conditions, providing the first example of an oxetanyl organoboron being engaged in a C–C coupling reaction.68 This moiety is gaining in popularity as it is emerging as a privileged pharmacophore in medicinal chemistry. In particular, Carreira has shown that 3,3-disubstituted oxetanes are effective polar replacements for carbonyl and gem-dimethyl groups in drug-like molecules.69
Scheme 10. Scope of Electrophile-Mediated Coupling of Secondary Alkylboronic Esters with 5-Membered Heteroaromatics.

Reaction conditions: see Scheme 9.
NBS added at 0 °C.
NBS added at −78 °C.
NBS added at −100 °C.
NIS was used instead of NBS.
Finally, we evaluated the use of other electron-rich heteroaromatics as coupling partners and found that the reaction scope was quite broad. Thus, the coupling reaction was applicable to the following lithiated heterocycles: C-3 furan 3e, C-2 benzofuran 4e, C-2 N-methyl pyrrole 5b, C-2 N-methylindole 6a and 6q, C-2 thiophene 7a and 7q, and C-2 benzothiophene 8b. In some cases, N-iodosuccinimide (NIS) was found to be superior to NBS because the latter caused the bromination of the electron-rich aromatic ring of the reaction product. However, attempts to carry out the coupling with indoles bearing other groups on nitrogen failed, as did coupling of C-3 N-methylindole. Moreover, reaction with 2-lithio-5-methylfuran resulted in low yield. The power of this approach for the simple modification of natural products was showcased by the successful coupling of cholesterol-B(pin) 1o with 2-lithiofuran and 2-lithio-N-methyl-indole (2o and 6p). Furthermore, by using 1,2-bis-boronic ester 1u, a stereospecific diarylation process was developed affording 2u in 58% yield and complete es.
4.2.2. Couplings between Chiral Secondary Boronic Esters with 6-Membered Ring Aromatics
Having evaluated the ability of chiral secondary boronic esters to undergo the stereospecific NBS-coupling with electron-rich heteroaromatics, we moved on to the more challenging 6-membered ring electron-rich aromatics. Because their corresponding boronate complexes are competent sp3-nucleophiles (SE2inv, see Scheme 5B),46−49 the reactions conditions had to be modified to promote reaction on the aromatic ring (Scheme 11A).
Scheme 11. Optimization (A) and Scope (B) for Coupling of Secondary Boronic Esters to 6-Membered Electron-Rich Aromatics.

Reaction conditions: for lithiation conditions, see Supporting Information; boronic ester (1.0 equiv) in THF (0.3 M) at −78 °C, then solvent switched to MeOH, then NBS (1.2 equiv) in MeOH (0.2 M), −78 °C, 1 h.
Optimization perfomed with GC-MS; isolated yields given in parentheses.
NIS used instead of NBS.
Reaction performed at −15 °C.
DBDMH (1,3-dibromo-5,5-dimethylhydantoin) used instead of NBS.
We started our investigation using 3,5-(MeO)2-C6H3–Li, taking advantage of the synergistic effect of all three donor groups of the boronate complex promoting bromination on the aromatic ring at the para position.70 This would be followed by stereospecific 1,2-metallate rearrangement and elimination. As shown in Scheme 11A, when the reaction was performed using the standard conditions a mixture of the desired product 9a, the B(pin)-incorporated product 9aa (see section 4.3), and the alkyl bromide 21 was obtained in a 81:6:13 ratio as determined by crude GC analysis (entry 1). Compound 9aa arises from an incomplete nucleophilic elimination step, so we repeated the reaction adding NBS in MeOH, in order to promote the elimination pathway (MeOH could act as the nucleophile). Pleasingly, this simple modification of the reaction conditions completely suppressed the formation of both 9aa and 21 (entry 2). SEAr can be modulated by the reaction media; thus, we now believe the increased polarity of the mixed solvent system to be the reason for the improved selectivity.71 Under these reaction conditions, 9a was isolated in 83% yield. When the reaction was tested with a single methoxy group in the aromatic unit (using 3-MeO-C6H4–Li), product 11a was not observed under standard reaction conditions (entry 3), but following addition of NBS in MeOH, a mixture of 11a, 11aa, and 21 in 69:29:3 ratio was obtained (entry 4). We therefore reasoned that in order to maximize the beneficial effect of MeOH a complete solvent exchange was required. Thus, after formation of the boronate complex in THF at −78 °C, the solvent was removed under high vacuum at 0 °C. MeOH was then added, and the mixture was cooled to −78 °C prior to NBS addition. Under these conditions, desired product 11a was obtained with complete selectivity and isolated in 72% yield (entry 5).
As shown in Scheme 11B, a broad range of electron-rich benzene derivatives was evaluated. The following lithiated benzene derivatives coupled successfully with both secondary dialkyl as well as benzylic boronic esters furnishing products in high yield and complete stereospecificity: 3,5-dimethoxy (9a, 9q, and 9l), 3-N,N-dimethylamino (10a and 10q), and 3-methoxy (11a and 11l). Disubstituted electron-rich aromatics (e.g., 2,3- or 3,4-dimethoxy (12b and 13b)) aryllithiums also worked, but lower yields were observed in these cases as a result of competing ipso-substitution (giving the aryl bromide and starting boronic ester) and competing reaction at the sp3 carbon (giving bromide 21). Pleasingly, Br substitution (e.g., at C-5) was tolerated giving 14b in good yield and complete es. The coupling was successful with the weakly nucleophilic naphthyl (both at C-1 and C-2) and the phenanthryl moiety giving products 15a, 16b, and 17b. Furthermore, the coupling was also successful with weakly donating aromatics (e.g., 3,5-dimethylphenyl and 2-lithio-6-methoxypyridine), giving products 18b and 20b, respectively.
However, not all aromatics worked due to competing processes. If bromination occurs on the aromatic ring, then the desired arylation takes place, but the competing reaction can also occur at the sp3 carbon of the boronate complex. Substitution on the aromatic ring, which pushes electron density onto boron (i.e., o- and p-donor groups), favors the latter process. This explains why 2- or 4-methoxyphenyllithium were not effective, whereas 3-methoxyphenyllithium, giving 11a, was successful. Without a meta donor group (e.g., phenyllithium), bromination at the sp3 carbon dominated and no arylation was observed. With a weak meta donor group (e.g., 3-methylphenyllithium), a ∼3:1 mixture of 2-bromo-4-phenylbutane and the desired coupled product was obtained showing the lower limit of the aromatic group that can be employed.
We have previously found that benzylic boronate complexes are better nucleophiles at the sp3 carbon of the boronate than their nonbenzylic counterparts. This competing undesired reaction could account for the lower yields of the desired arylation obtained in the benzylic versus the nonbenzylic substrates (e.g., compare 9a/9l, 11a/11l, and 18b/18l). Cholesterol B(pin) 1o reacted well with the electron-rich aryllithiums, giving products 9o and 10p in high yield and complete diastereospecificity.
4.2.3. Couplings of Chiral Tertiary Boronic Esters
Having evaluated the use of secondary boronic esters in the NBS-mediated stereospecific cross-couplings methodology, we moved on to the more challenging tertiary boronic esters, a class of substrates for which transition-metal-based methodologies are currently not available.5,6 Our approach goes through a distinct mechanistic pathway, so we were keen to explore if our methodology had the potential to solve this unmet synthetic challenge. As shown in Scheme 12, a range of tertiary alkyl and benzylic boronic esters with both 5- and 6-membered ring aryllithiums were found to work well. Trialkyl, benzylic, and dibenzylic boronic esters 1g–k were coupled with 2-furyllithium in high yields and complete es, furnishing products 2g–k. The chemistry was also extended to 2-lithiobenzofuran giving 4g and 4h. Electron-rich 6-membered aryllithiums were evaluated next. In the case of 3,5-dimethoxy-phenyllithium, both alkyl and benzylic substrates were successful, and desired products 9g and 9h were obtained in high yield and complete es. The coupling was successful with other electron-rich aromatics such as 3-methoxy-phenyllithium (11h), 1- and 2-naphthyllithium (15g and 16g), 6-methoxy 2-lithio-pyridine (20g), and 3,5-dimethyl-phenyllithium (18g). A remarkable example is the successful coupling with the weakly electron-rich aromatic, 3-methyl-phenyllithium, which gave product 19g in 46% yield and complete es. We rationalized the success of this example with the increased steric congestion offered by the three alkyl groups that slows down the rate of sp3 halogenation, enabling reaction at the aromatic ring to compete more favorably.
Scheme 12. Coupling of Tertiary Boronic Esters.
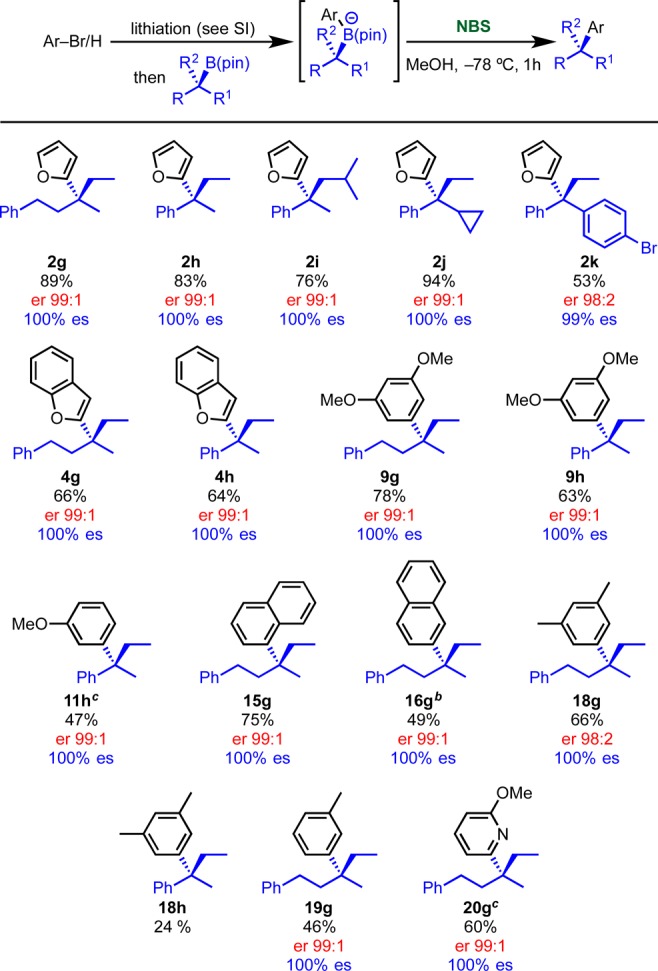
Reaction conditions: for lithiation conditions, see Supporting Information; boronic ester (1.0 equiv) in THF (0.3 M) at −78 °C, then solvent switched to MeOH, then NBS (1.2 equiv) in MeOH (0.2 M), −78 °C, 1 h.
Reaction performed at −15 °C.
DBDMH (1,3-dibromo-5,5-dimethylhydantoin) used instead of NBS.
4.3. Stereospecific Arylation-B(pin) Incorporations
During the optimization of the NBS-mediated coupling of secondary boronic esters and 6-membered ring aryllithiums, we observed the unexpected formation of products where the B(pin) group was incorporated into the aromatic ring (Scheme 11A, see entries 1 and 4). This observation set the stage for the development of a unique ortho-borylation-coupling reaction. Mechanistically, we believe this product arises from an incomplete nucleophilic B(pin)-elimination reaction from intermediate 27 (Scheme 13A). This would lead to a nucleophilic 1,2-Wagner–Meerwein shift of the B(pin) moiety forming the more stable carbocation, 28. Final deprotonation would furnish B(pin)-incorporated product 18aa. On the basis of this mechanism, we speculated that while a polar solvent (e.g., MeOH) was required to facilitate the SEAr we needed to reduce its ability to serve as a nucleophile in order to favor the 1,2-Wagner–Meerwein shift.71 We therefore tested sterically hindered alcohol i-PrOH in place of MeOH and the ratio of 18a/18aa/21 was improved to 5:88:7 as determined by GC-MS. Furthermore, by using a 1:1 i-PrOH–CH3CN solvent mixture, we were able to selectively obtain product 18aa and isolate it in 90% yield. With the optimized reaction conditions, we evaluated the scope of this novel coupling reaction. As described in Scheme 13B, a variety of aryllithiums reacted well providing the desired products in good yields and complete es (9ba, 11ba, 15ba, and 18aa). We were pleased to see that tertiary groups were compatible as well and delivered remarkably high yields furnishing very sterically congested quaternary scaffolds (9ga and 18ga). It was also possible to perform this reaction on heterocyclic substrate such as benzothiophene (8ba). We believe that this particular result is a very attractive tool for introducing neighboring functionalities in a single transformation. Moreover, the B(pin) moiety serves as a useful handle for further functionalization either by transition metal catalysis or the protocol described herein.
Scheme 13. Method for Retaining the Boronic Ester in the Coupling Reaction.

GC conversion; isolated yield given in parentheses.
Reaction conditions: for lithiation conditions, see Supporting Information; boronic ester (1.0 equiv) in THF (0.3 M) at −78 °C, then solvent switched to CH3CN–i-PrOH, then NBS (1.2 equiv) in MeCN (0.2 M), 0 °C, 1 h.
4.4. Trends in Reactivity
The successful outcome of the coupling reaction is dependent on a number of factors, but the extent of competing reactions at the sp3 carbon versus those at the aromatic ring appears to dominate in many cases. Considering these two factors only, trends in reactivity can be readily understood (Scheme 14). For tertiary and secondary nonbenzylic boronates, as the aromatic becomes less electron-rich, the yield for the coupled product declines because reaction of NBS at the less electron-rich aromatic becomes increasingly less favored. Comparing benzylic secondary 2l, 9l, 11l, and 18l) and tertiary (2h, 9h, 11h, and 18h) products, the same trend is observed. In addition, the increased resistance to reactions at the sp3 carbon of the tertiary alkyl substrates leads to increased yields of coupled products compared to the corresponding secondary substrates (cf. 19g and 19b).
Scheme 14. Main Competing Processes Which Determine the Outcome of the Coupling Reaction Illustrate Trends in Reactivity.
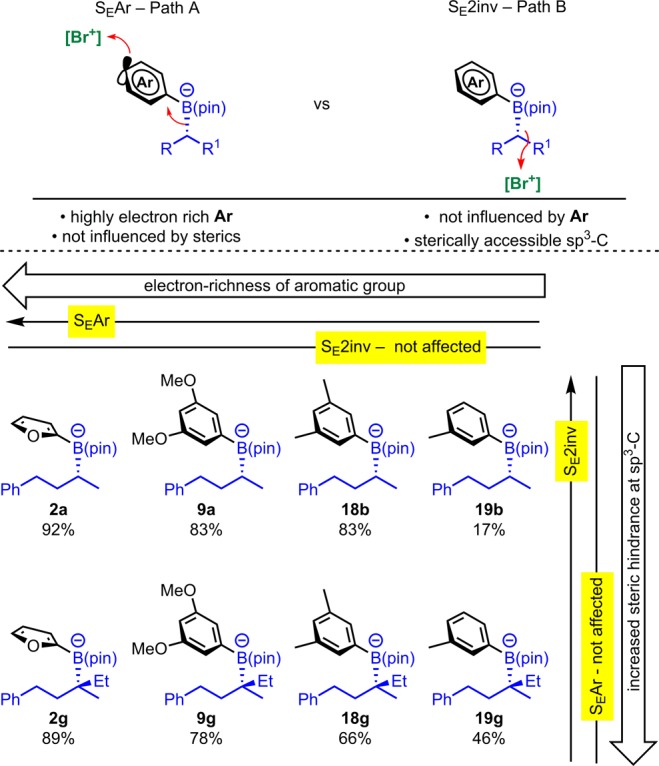
Yields for arylation process.
4.5. DFT and React-IR Studies
Additional DFT calculations were performed for the electrophilic reactions with NBS. Using the same model isopropyl boronate anionic complex, 24, two mechanisms were considered: One involves electrophilic addition at the aromatic ring (path A, SEAr), whereas the other involves electrophilic attack at the sp3-C–B bond (path B, SE2inv) (Scheme 15A). In both cases, transition states were located (Scheme 15B) involving near-linear N–Br–C arrangements corresponding to nucleophilic substitution at bromine. For path B, the transition state (TS) leads directly to succinimide anion and the bromoalkyl species with release of boronate ester. For path A, electrophilic attack of the bromine triggered migration of the alkyl group in the same reaction step, without further barrier. The migration is already well under way at the TS. The discrete cationic intermediate, II (Scheme 5A), is never actually formed but is nevertheless a useful species to consider when formulating the overall process. This TS is significantly more stable one with the furyl model boronate complex, consistent with the observed high yields for alkylated furan. Additional calculations were performed using 3,5-dimethylphenyl and phenyl substrates. The TS for path B has almost the same free energy in all three cases, but the barrier height for path A increases sharply for these less nucleophilic aryl groups (Scheme 15C). In the case of phenyl, the barrier is now much higher than for oxidation of the C–B bond, consistent with experiment. For 3,5-dimethylphenyl, SE2inv (path B) is predicted to be strongly favored in THF as observed. However, in MeOH the two barriers are almost the same, although experimentally path A is favored. The TS for path A is much more polar than the reactants, so the solvation free energy for the TS is large compared to that for reactants. This helps to explain why more polar solvents favor path A over path B. Also, considering the uncertainties in continuum models of solvation free energy, one must bear in mind that the calculated free energy for this TS is likely somewhat inaccurate. In previous work,72 we have shown based on calibration with very accurate coupled cluster calculations that B3LYP with dispersion corrections as used here yields reasonably accurate results for similar boronate chemistry. However, it should also be noted that conformational complexity, treatment of the lithium counterion, and modeling of solvation introduces further sources of error.
Scheme 15. DFT Calculations of the Main Competing Processes Illustrating Trends in Reactivity.
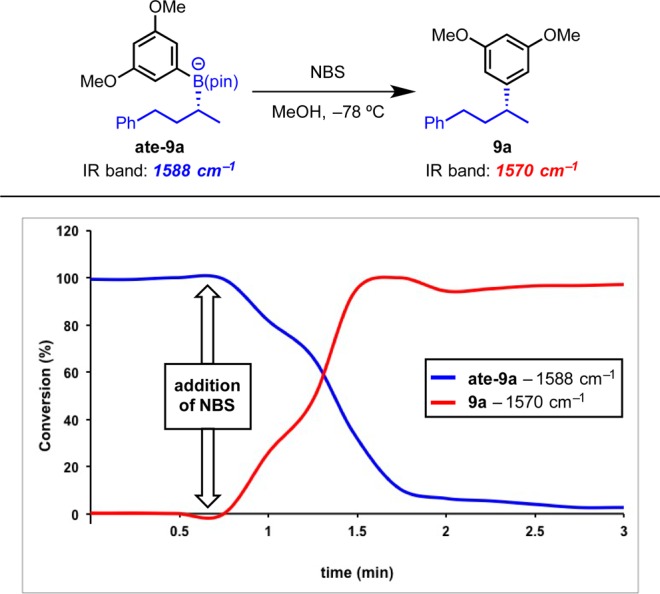
The boronate reaction with NBS could be followed by React-IR.73 The boronate complex was prepared and cooled to −78 °C and the mixture monitored by React-IR.74 Addition of NBS in MeOH over 60 s resulted in rapid consumption of ate complex ate-9a, essentially instantaneously, indicating that the reaction was extremely rapid (Scheme 16). The low free energy barrier calculated for the reaction with the furan boronate complex suggests that reaction with NBS should be very rapid at −78 °C, and indeed, reaction was observed to occur in less than a minute for the dimethoxyphenyl boronate, which should have a similar reactivity.
Scheme 16. React-IR Studies of Reaction at −78 °C.
5. Conclusions
We set out to explore two different pathways by which boronate complexes derived from an aliphatic secondary or tertiary boronic ester and 2-furyllithium could react with either an oxidant or an electrophile to give a coupled product stereospecifically. Of the oxidants tested, DDQ was uniquely effective, but it only worked with a limited range of secondary boronic esters: Benzylic and tertiary boronic esters were ineffective. From analysis of the products obtained from the unsuccessful reactions and from DFT studies, we concluded that in those cases DDQ oxidized the aliphatic C–B bond leading to a relatively stable (benzylic or tertiary) radical. In the cases of nonbenzylic aliphatic secondary boronic esters which did react successfully, DDQ acted as an electrophile and not as an oxidant, triggering 1,2-migration and subsequent elimination. NBS was a better electrophile than DDQ because it was less prone to oxidize the aliphatic C–B bond. This reagent was applicable to a broad range of secondary and tertiary boronic esters with very different steric demands and tolerated a range of functional groups (esters, azides, nitriles, alcohols, and ethers). It was also applicable to a range of electron-rich heteroaromatics including pyrrole, indole, thiophene, benzofuran, and benzothiophene. It was important that the NBS-mediated bromination occurred at the ortho or para position of the aromatic ring in relation to the boronate; if the electronics favored reaction at the ipso position, then competing ipso bromination occurred returning the boronic ester (e.g., 2-lithioindole was successful but the 3-lithioindole failed) (Scheme 17).
Scheme 17. Reactions with 5-Membered Ring Aromatics.
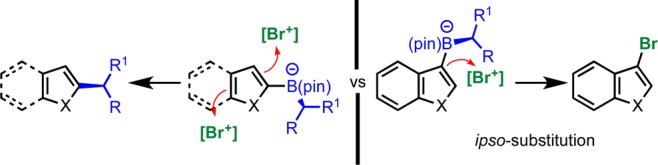
The coupling reaction was also applicable to 6-membered ring aromatics provided they had donor groups in the meta position (Scheme 18). The meta donor group together with the (electron-rich) boronate worked synergistically, promoting bromination on the aromatic ring. If the donor group was para to the boronate, then it promoted bromination at the sp3 carbon instead and no coupled product was obtained. This competing reaction was increasingly observed when weaker meta donors on the 6-membered ring were employed (e.g., using 3-methylaryllithium). The coupling reactions with 6-membered ring aromatics required different conditions; it was found that solvent exchange from THF to MeOH led to improved yields of the desired coupling product. Using these conditions, a range of 6-membered ring aromatics, including 1- and 2-naphthalenes, were successfully coupled to secondary and tertiary boronic esters. Solvent choice played a major role in the outcome of the coupling reaction. Using MeCN-iPrOH, we found that the B(pin)- moiety was retained in the product, ortho to the boron substituent. We believe this arises from a 1,2-Wagner–Meerwein shift of the B(pin) group, which relieves steric hindrance, followed by loss of a proton. Again this protocol, which created a new C(sp2)–C(sp3) and an adjacent C–B bond, was applicable to a range of secondary and tertiary boronic esters. All of the coupling reactions occurred with complete stereospecificity.
Scheme 18. Reactions with 6-Membered Ring Aromatics.

DFT calculations supported the mechanistic scenario put forward, and the very low barriers calculated for addition of NBS to electron-rich aromatics were commensurate with the almost instantaneous reaction observed by React-IR at −78 °C. Interestingly, no cationic intermediates are involved during the process; the 1,2-migration occurs as the NBS reacts with the aromatic ring.
The methodology described provides a new way of coupling secondary and tertiary boronic esters with electron-rich aromatics. There are a growing number of practical methods for making aliphatic secondary and tertiary boronic esters with very high enantioselectivity, so this new stereospecific method should find wide application. Even in the cases where racemic or achiral substrates are used, the absence of costly or difficult-to-remove transition metals, which are often avoided at late stage processes of drug manufacture, are further attractive features of the methodology.
Acknowledgments
We thank EPSRC (EP/I038071/1) and the European Research Council (FP7/2007-2013, ERC grant no. 246785) for financial support. A.B. thanks the Marie Curie Fellowship program (EC FP7 No 329578).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b03963.
Author Present Address
A.B.: Department of Chemistry, University of Hull, Cottingham Road, Hull, HU6 7RX, United Kingdom.
Author Present Address
J.N.H.: Department of Chemistry, Katholieke Universiteit Leuven, Celestijnenlaan 200F, B-3001 Heverlee, Belgium.
Author Present Address
D.L.: School of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, United Kingdom.
The authors declare no competing financial interest.
Supplementary Material
References
- Suzuki A. Angew. Chem., Int. Ed. 2011, 50, 6722. 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]
- Slagt V. F.; de Vries A. H. M.; de Vries J. G.; Kellogg R. M. Org. Process Res. Dev. 2010, 14, 30. 10.1021/op900221v. [DOI] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. J. Med. Chem. 2009, 52, 6752. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Lovering F. MedChemComm 2013, 4, 515. 10.1039/c2md20347b. [DOI] [Google Scholar]
- Leonori D.; Aggarwal V. K. Angew. Chem., Int. Ed. 2015, 54, 1082. 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]
- Wang C.-Y.; Derosa J.; Biscoe M. R. Chem. Sci. 2015, 6, 5105. 10.1039/C5SC01710F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift E.; Jarvo E. R. Tetrahedron 2013, 69, 5799. 10.1016/j.tet.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennox A. J. J.; Lloyd-Jones G. C. Angew. Chem. 2013, 125, 7506. 10.1002/ange.201301737. [DOI] [Google Scholar]
- Ridgway B. H.; Woerpel K. A. J. Org. Chem. 1998, 63, 458. 10.1021/jo970803d. [DOI] [PubMed] [Google Scholar]
- Matos K.; Soderquist J. J. Org. Chem. 1998, 63, 461. 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]
- Hildebrand J.; Marsden S. Synlett 1996, 1996, 893. 10.1055/s-1996-5608. [DOI] [Google Scholar]
- Charette A.; DeFreitas-Gil R. Tetrahedron Lett. 1997, 38, 2809. 10.1016/S0040-4039(97)00478-4. [DOI] [Google Scholar]
- Fang G.-H.; Yan Z.-J.; Deng M.-Z. Org. Lett. 2004, 6, 357. 10.1021/ol036184e. [DOI] [PubMed] [Google Scholar]
- Soderquist J.; Huertas R.; Leon-Colon G. Tetrahedron Lett. 2000, 41, 4251. 10.1016/S0040-4039(00)00605-5. [DOI] [Google Scholar]
- Zhou S.-M.; Deng M.-Z.; Xia L.-J.; Tang M.-H. Angew. Chem., Int. Ed. 1998, 37, 2845. 10.1002/(SICI)1521-3773(19981102)37:20<2845::AID-ANIE2845>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Luithle J.; Pietruszka J. J. Org. Chem. 2000, 65, 9194. 10.1021/jo0056601. [DOI] [PubMed] [Google Scholar]
- Rubina M.; Rubin M.; Gevorgyan V. J. Am. Chem. Soc. 2003, 125, 7198. 10.1021/ja034210y. [DOI] [PubMed] [Google Scholar]
- Littke A.; Dai C.; Fu G. C. J. Am. Chem. Soc. 2000, 122, 4020. 10.1021/ja0002058. [DOI] [Google Scholar]
- Sun C.; Potter B.; Morken J. P. J. Am. Chem. Soc. 2014, 136, 6534. 10.1021/ja500029w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs J. R.; Zhang L.; Morken J. P. J. Am. Chem. Soc. 2014, 136, 16140. 10.1021/ja510081r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasspoole B.; Oderinde M.; Moore B.; Antoft-Finch A.; Crudden C. Synthesis 2013, 45, 1759. 10.1055/s-0033-1338875. [DOI] [Google Scholar]
- Imao D.; Glasspoole B.; Laberge V.; Crudden C. J. Am. Chem. Soc. 2009, 131, 5024. 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- Matthew S. C.; Glasspoole B.; Eisenberger P.; Crudden C. J. Am. Chem. Soc. 2014, 136, 5828. 10.1021/ja412159g. [DOI] [PubMed] [Google Scholar]
- Lou Y.; Cao P.; Jia T.; Zhang Y.; Wang M.; Liao J. Angew. Chem., Int. Ed. 2015, 54, 12134. 10.1002/anie.201505926. [DOI] [PubMed] [Google Scholar]
- Molander G. A. J. Org. Chem. 2015, 80, 7837. 10.1021/acs.joc.5b00981. [DOI] [PubMed] [Google Scholar]
- Sandrock D.; Jean-Gerard L.; Chen C.; Dreher S.; Molander G. A. J. Am. Chem. Soc. 2010, 132, 17108. 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander G. A.; Wisniewski S. J. Am. Chem. Soc. 2012, 134, 16856. 10.1021/ja307861n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; McDonald R.; Hall D. Nat. Chem. 2011, 3, 894. 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]
- Ohmura T.; Awano T.; Suginome M. J. Am. Chem. Soc. 2010, 132, 13191. 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]
- Awano T.; Ohmura T.; Suginome M. J. Am. Chem. Soc. 2011, 133, 20738. 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]
- Dreher S.; Dormer P.; Sandrock D.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257. 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.; Jeon H.; Yun J. Angew. Chem., Int. Ed. 2013, 52, 3989. 10.1002/anie.201208610. [DOI] [PubMed] [Google Scholar]
- Li L.; Zhao S.; Joshi-Pangu A.; Diane M.; Biscoe M. R. J. Am. Chem. Soc. 2014, 136, 14027. 10.1021/ja508815w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonet A.; Odachowski M.; Leonori D.; Essafi S.; Aggarwal V. K. Nat. Chem. 2014, 6, 584. 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]
- Levy A. B. J. Org. Chem. 1978, 43, 4684. 10.1021/jo00418a044. [DOI] [Google Scholar]
- Ishikura M.; Kato M. A. Tetrahedron 2002, 58, 9827. 10.1016/S0040-4020(02)01293-0. [DOI] [Google Scholar]
- Akimoto I.; Suzuki A. Synthesis 1979, 1979, 146. 10.1055/s-1979-28596. [DOI] [Google Scholar]
- Pelter A.; Williamson H.; Davies G. M. Tetrahedron Lett. 1984, 25, 453. 10.1016/S0040-4039(00)99909-X. [DOI] [Google Scholar]
- Marinelli E. R.; Levy A. B. Tetrahedron Lett. 1979, 20, 2313. 10.1016/S0040-4039(01)93960-7. [DOI] [Google Scholar]
- Kagan J.; Arora S. K. Tetrahedron Lett. 1983, 24, 4043. 10.1016/S0040-4039(00)88257-X. [DOI] [Google Scholar]
- Davies G. M.; Davies P. S.; Paget W. E.; Wardleworth J. M. Tetrahedron Lett. 1976, 17, 795. 10.1016/S0040-4039(00)77954-8. [DOI] [Google Scholar]
- Negishi E.-I.; Abramovitch A.; Merrill R. E. J. Chem. Soc., Chem. Commun. 1975, 138. 10.1039/c39750000138. [DOI] [Google Scholar]
- Suzuki A.; Miyaura N.; Itoh M. Tetrahedron 1971, 27, 2775. 10.1016/S0040-4020(01)98068-8. [DOI] [Google Scholar]
- Larouche-Gauthier R.; Elford T. G.; Aggarwal V. K. J. Am. Chem. Soc. 2011, 133, 16794. 10.1021/ja2077813. [DOI] [PubMed] [Google Scholar]
- Mohiti M.; Rampalakos C.; Feeney K.; Leonori D.; Aggarwal V. K. Chem. Sci. 2014, 5, 602. 10.1039/C3SC52409D. [DOI] [Google Scholar]
- Sandford C.; Rasappan R.; Aggarwal V. K. J. Am. Chem. Soc. 2015, 137, 10100. 10.1021/jacs.5b05848. [DOI] [PubMed] [Google Scholar]
- Feeney K.; Berionni G.; Mayr H.; Aggarwal V. K. Org. Lett. 2015, 17, 2614. 10.1021/acs.orglett.5b00918. [DOI] [PubMed] [Google Scholar]
- Amaya T.; Tsukamura Y.; Hirao T. Adv. Synth. Catal. 2009, 351, 1025. 10.1002/adsc.200800781. [DOI] [Google Scholar]
- Mizuno H.; Sakurai H.; Amaya T.; Hirao T. Chem. Commun. 2006, 5042. 10.1039/b610731a. [DOI] [PubMed] [Google Scholar]
- Leonori D.; Aggarwal V. K. Acc. Chem. Res. 2014, 47, 3174. 10.1021/ar5002473. [DOI] [PubMed] [Google Scholar]
- Stymiest J. L.; Dutheuìl G.; Mahmood A.; Aggarwal V. K. Angew. Chem., Int. Ed. 2007, 46, 7491. 10.1002/anie.200702146. [DOI] [PubMed] [Google Scholar]
- Pulis A. P.; Blair D. J.; Torres E.; Aggarwal V. K. J. Am. Chem. Soc. 2013, 135, 16054. 10.1021/ja409100y. [DOI] [PubMed] [Google Scholar]
- Stymiest J. L.; Bagutski V.; French R. M.; Aggarwal V. K. Nature 2008, 456, 778. 10.1038/nature07592. [DOI] [PubMed] [Google Scholar]
- Noh D.; Chea H.; Ju J.; Yun J. Angew. Chem., Int. Ed. 2009, 48, 6062. 10.1002/anie.200902015. [DOI] [PubMed] [Google Scholar]
- Pozzi D.; Scanlan E. M.; Renaud P. J. Am. Chem. Soc. 2005, 127, 14204. 10.1021/ja055691j. [DOI] [PubMed] [Google Scholar]
- Ito H.; Kubota K. Org. Lett. 2012, 14, 890. 10.1021/ol203413w. [DOI] [PubMed] [Google Scholar]
- Yang C.-T.; Zhang Z.-Q.; Tajuddin H.; Wu C.-C.; Liang J.; Liu J.-H.; Fu Y.; Czyzewska M.; Steel P. G.; Marder T. B.; Liu L. Angew. Chem., Int. Ed. 2012, 51, 528. 10.1002/anie.201106299. [DOI] [PubMed] [Google Scholar]
- Lee J.-E.; Yun J. Angew. Chem., Int. Ed. 2008, 47, 145. 10.1002/anie.200703699. [DOI] [PubMed] [Google Scholar]
- Batsanov A. S.; Grosjean C.; Schutz T.; Whiting A. J. Org. Chem. 2007, 72, 6276. 10.1021/jo0708792. [DOI] [PubMed] [Google Scholar]
- Toribatake K.; Nishiyama H. Angew. Chem., Int. Ed. 2013, 52, 11011. 10.1002/anie.201305181. [DOI] [PubMed] [Google Scholar]
- Coombs J. R.; Haeffner F.; Kliman L. T.; Morken J. P. J. Am. Chem. Soc. 2013, 135, 11222. 10.1021/ja4041016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horita K.; Yoshioka T.; Tanaka T.; Oikawa Y.; Yonemitsu O. Tetrahedron 1986, 42, 3021. 10.1016/S0040-4020(01)90593-9. [DOI] [Google Scholar]
- Guo X.; Mayr H. J. Am. Chem. Soc. 2013, 135, 12377. 10.1021/ja405890d. [DOI] [PubMed] [Google Scholar]
- Saveant J. M. J. Am. Chem. Soc. 1987, 109, 6788. 10.1021/ja00256a037. [DOI] [Google Scholar]
- Marcus R. A. Angew. Chem., Int. Ed. Engl. 1993, 32, 1111. 10.1002/anie.199311113. [DOI] [Google Scholar]
- Although an oxetanyl trifluoroborate has been reported, it has not been used in cross-coupling. SeePresset M.; Fleury-Bregeot N.; Oehlrich D.; Rombouts F.; Molander G. A. J. Org. Chem. 2013, 78, 4615. 10.1021/jo4005519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhard J. A.; Wuitschik G.; Rogers-Evans M.; Muller K.; Carreira E. M. Angew. Chem., Int. Ed. 2010, 49, 9052. 10.1002/anie.200907155. [DOI] [PubMed] [Google Scholar]
- Berionni G.; Maji B.; Knochel P.; Mayr H. Chem. Sci. 2012, 3, 878. 10.1039/C2SC00883A. [DOI] [Google Scholar]
- Reichardt C.; Welton T.. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; John Wiley and Sons: Weinheim, Germany, 2011. [Google Scholar]
- Essafi S.; Tomasi S.; Aggarwal V. K.; Harvey J. N. J. Org. Chem. 2014, 79, 12148. 10.1021/jo502020e. [DOI] [PubMed] [Google Scholar]
- Dimethoxy boronate ate-9a was used in this experiment as it had clear IR bands for the boronate complex and product whilst for furan the bands were in a more crowded region of the spectrum and so were difficult to identify.
- Llaveria J.; Leonori D.; Aggarwal V. K. J. Am. Chem. Soc. 2015, 137, 10958. 10.1021/jacs.5b07842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.