

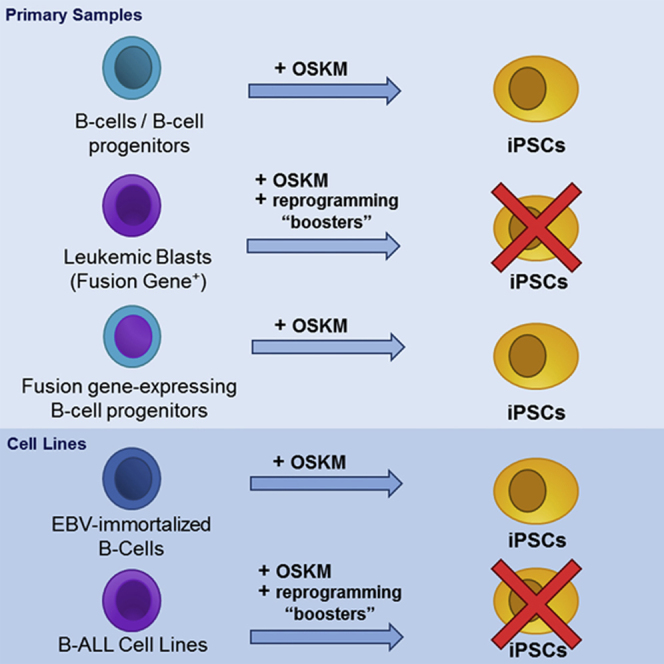
Summary
Induced pluripotent stem cells (iPSCs) are a powerful tool for disease modeling. They are routinely generated from healthy donors and patients from multiple cell types at different developmental stages. However, reprogramming leukemias is an extremely inefficient process. Few studies generated iPSCs from primary chronic myeloid leukemias, but iPSC generation from acute myeloid or lymphoid leukemias (ALL) has not been achieved. We attempted to generate iPSCs from different subtypes of B-ALL to address the developmental impact of leukemic fusion genes. OKSM(L)-expressing mono/polycistronic-, retroviral/lentiviral/episomal-, and Sendai virus vector-based reprogramming strategies failed to render iPSCs in vitro and in vivo. Addition of transcriptomic-epigenetic reprogramming “boosters” also failed to generate iPSCs from B cell blasts and B-ALL lines, and when iPSCs emerged they lacked leukemic fusion genes, demonstrating non-leukemic myeloid origin. Conversely, MLL-AF4-overexpressing hematopoietic stem cells/B progenitors were successfully reprogrammed, indicating that B cell origin and leukemic fusion gene were not reprogramming barriers. Global transcriptome/DNA methylome profiling suggested a developmental/differentiation refractoriness of MLL-rearranged B-ALL to reprogramming into pluripotency.
Key words: iPSC, cancer reprogramming, MLL-AF4, B-ALL, Sendai virus, transcriptome, DNA methylome
Graphical Abstract
Highlights
-
•
Neither primary B-ALL blasts nor leukemic B cell lines can be reprogrammed to iPSCs
-
•
Global transcriptome and DNA methylome suggest a developmental refractoriness
Despite the interest in generating iPSCs from human primary acute leukemias for disease modeling, reprogramming leukemias is an extremely inefficient process. In this article, Menéndez, Bueno, and colleagues show that many reprogramming strategies reported to date are not sufficient to generate B-ALL-derived iPSCs. Global transcriptome/DNA methylome profiling suggested a developmental/differentiation refractoriness of B-ALL to reprogramming into pluripotency.
Introduction
Leukemia is generally studied once the full transformation events have already occurred and, therefore, the mechanisms by which leukemia-specific mutations transform to a pre-leukemic state followed by rapid transition to overt leukemia are not amenable to analysis with patient samples (Ramos-Mejia et al., 2012c). Therefore, it is imperative to develop effective disease models to study the developmental impact of leukemia-specific genetic aberrations on human stem cell fate. Induced pluripotent stem cells (iPSCs) are a powerful tool for modeling different aspects of human disease that cannot otherwise be addressed by patient sample analyses or animal models (Menendez et al., 2006, Wu and Hochedlinger, 2011). Because leukemia manifests as a developmental cell blockage, the generation and differentiation of leukemia-specific iPSCs offers a promising strategy to study the earliest events leading to the specification of both normal and abnormal hematopoietic tissue, thus illuminating molecular mechanisms underlying the pathogenesis of human leukemia.
iPSCs are routinely generated from tissues obtained from healthy donors and patients and cell types at different developmental stages. Reprogramming human primary cancer cells, however, remains challenging. Despite significant interest in generating iPSCs from leukemia cells (Curry et al., 2015, Ramos-Mejia et al., 2012c, Yilmazer et al., 2015), only a few reports have demonstrated successful reprogramming and, unfortunately, only seven of these studies reprogrammed human primary leukemias (the remaining studies used cell lines) (Bedel et al., 2013, Carette et al., 2010, Gandre-Babbe et al., 2013, Hu, 2014, Kumano et al., 2012, Yamamoto et al., 2015, Ye et al., 2009) (Table S1). Intriguingly, iPSCs from hematological primary cancer cells have exclusively been generated from chronic leukemias of myeloid origin, including Philadelphia+ chronic myeloid leukemia (CML), primary myelofibrosis (PMF), JAK2-V617F+ polycythemia vera (PV), and juvenile myelomonocytic leukemia (JMML) (Table S1). iPSCs from acute myeloid leukemia (AML) or acute lymphoid leukemia (ALL) have not been reported so far, whereas iPSCs have been generated from normal myeloid and T cells (Bueno et al., 2016) and, very recently, from CD19+ B cells from human cord blood (CB), peripheral blood (PB), and fetal liver (FL) using non-integrative tetracistronic OCT4/KLF4/SOX2/MYC (OSKM)-expressing Sendai virus (SeV) (Bueno et al., 2016, Munoz-Lopez et al., 2016).
Here, we attempted to reprogram highly fluorescence-activated cell sorting (FACS)-purified (100% purity) leukemia blasts from three subtypes of B-ALL, t(4;11)/MLL-AF4+, t(1;11)+MLL-EPS15+, and t(12;21)/ETV6-RUNX1 B-ALL, to establish novel iPSC-based disease models to address the developmental impact of these leukemia-specific fusion genes on human stem cell fate. Our data demonstrate that despite multiple technical and biological reprogramming strategies, neither primary blasts nor B-ALL cell lines could be reprogrammed to pluripotency. Functional assays coupled with global transcriptome and DNA methylome profiling suggest a developmental/differentiation refractoriness of MLL-rearranged human B-ALL to reprogramming to pluripotency.
Results
Reprogramming B Cell Leukemic Blasts Results in Generation of iPSCs from Contaminating Normal Myeloid Cells
iPSCs from primary leukemic cells harboring specific genetic mutations offer an unprecedented opportunity to understand how cancer-specific mutations impair tissue homeostasis by deregulating cell differentiation and proliferation. We attempted to reprogram blasts from t(4;11)/MLL-AF4+, t(1;11)+MLL-EPS15+, and t(12;21)/ETV6-RUNX1+ B-ALL. FACS-purified leukemic blasts (>99%, Figures 1A and 1B) were infected (or transfected) with different combinations of monocistronic or polycistronic retroviral, lentiviral, and SeV vectors (or episomal vectors) expressing either OKSM or OKSL reprogramming factors (Table 1). No iPSC clones were generated when reprogramming factors were expressed via episomal vectors or viral retro-/lentivectors for any of the cytogenetically different leukemias tested (n = 7, Table 1). iPSC clones were exclusively generated when OKSM-expressing SeV vectors were employed (Figures 1C and 1D; Table 1). However, all of the resultant clones were negative for the corresponding fusion gene at the genomic (fluorescent in situ hybridization [FISH] and PCR) and RNA (RT-PCR) level (Figures 1E and 1F; Table 1).
Figure 1.

Reprogramming Highly Purified B Cell Leukemic Blasts Results in Generation of iPSCs from Contaminating Normal Myeloid Cells
(A) Representative flow cytometry of high purity (>99%) FACS-sorted B cell leukemia blasts.
(B) Representative FISH showing that leukemia blasts homogeneously harbor the leukemia-specific chromosomal rearrangements 11q23 (MLL) or ETV6-RUNX1 (white arrows). Scale bars, 5 μm.
(C) Scheme of the polycistronic SeV-OKSM-mir302 vector used for reprogramming.
(D) Scheme of the strategy used to reprogram leukemia blasts.
(E) Resulting iPSC colonies do not harbor either MLL or ETV6-RUNX1 rearrangements. Scale bars, 5 μm.
(F) Genomic PCR confirming the absence of both MLL and ETV6-RUNX1 in resulting iPSC colonies.
(G) Representative experiment (n = 1) showing that factors such as sodium salicylate (NaS), decitabine, and iDot1L enhance the reprogramming of contaminating cells lacking either MLL or ETV6-RUNX1 rearrangements (n = 3).
Table 1.
Summary of the Reprogramming Conditions Used in This Study and Their Outcome
| Reprogramming Factors | Additional Factors | B-ALL t(4;11) | B-ALL t(1;11) | B-ALL t(12;21) | Sort Purity (No. of Sorts) | Clones | iFISH for MLL Locus | PCR for MLL Fusion |
|---|---|---|---|---|---|---|---|---|
| Lentiviral OKSM polycistronic | None | √ | √ | √ | ≥99% (single) | 0 | NA | NA |
| ascorbic acid | √ | ND | ND | ≥99% (single) | 0 | NA | NA | |
| sodium butyrate | √ | ND | ND | ≥99% (single) | 0 | NA | NA | |
| valproic acid | √ | ND | ND | ≥99% (single) | 0 | NA | NA | |
| LiCl | √ | ND | ND | ≥99% (single) | 0 | NA | NA | |
| pLVX-mir302 | √ | √ | √ | ≥99% (single) | 0 | NA | NA | |
| Episomal OKSM | none | √ | √ | √ | ≥99% (single) | 0 | NA | NA |
| ascorbic acid | √ | ND | ND | ≥99% (single) | 0 | NA | NA | |
| sodium butyrate | √ | ND | ND | ≥99% (single) | 0 | NA | NA | |
| valproic acid | √ | ND | ND | ≥99% (single) | 0 | NA | NA | |
| LiCl | √ | ND | ND | ≥99% (single) | 0 | NA | NA | |
| pLVX-mir302 | √ | √ | √ | ≥99% (single) | 0 | NA | NA | |
| Episomal OKSL | none | √ | √ | √ | ≥99% (single) | 0 | NA | NA |
| pLVX-mir302 | √ | √ | √ | ≥99% (single) | 0 | NA | NA | |
| Retroviral OKSM | none | √ | √ | √ | ≥99% (single) | 0 | NA | NA |
| SeV-OKSM | none | √ | √ | √ | ≥99% (single) | yes∗ | negative | negative |
| SeVdp-OKSM polycistronic | none | √ | √ | √ | ≥99% (single) | yes∗ | negative | negative |
| sodium salicylate | √ | √ | √ | ≥99% (single) | yes∗ | negative | negative | |
| decitabine | √ | ND | ND | ≥99% (single) | yes∗ | negative | negative | |
| iDot1L (SGC0946) | √ | ND | ND | ≥99% (single) | yes∗ | negative | negative | |
| iDot1L (epz004777) | √ | ND | ND | ≥99% (single) | yes∗ | negative | negative | |
| iMenin-MLL (iML2) | √ | ND | ND | ≥99% (single) | yes∗ | negative | negative | |
| ectopic c/EBPα | √ | ND | ND | ≥99% (single) | yes∗ | negative | ND | |
| iPTEN (bVp(HO)pic) | √ | ND | ND | ≥99% (single) | yes∗ | negative | ND | |
| c/EBPα + iPTEN | √ | ND | ND | ≥99% (single) | yes∗ | negative | ND | |
| decitabine | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| trichostatin A | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| valproic acid | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| sodium butyrate | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| iSUV39H1 (Chaetocin) | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| iEZH2 (GSK126) | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| iEZH2 (DZNep) | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| iBRD4 (JQ1) | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| iCDK-P-TEFb (flavopiridol) | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| ascorbic acid | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| octyl-α-ketoglutarate | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| shRing1a MOI = 10 | √ | ND | ND | ∼100% (double) | 0 | NA | NA | |
| shMacroH2A MOI = 10 | √ | ND | ND | ∼100% (double) | 0 | NA | NA |
OKSM, Oct4, Klf4, Sox2, Myc; OKSL, Oct4, Klf4, Sox2, Lin28; SeVdp, Sendai vector-defective persistent; ND, not done; NA, not applicable; yes∗, the number of clones varies between 5 and 50.
Several molecules that promote or enhance reprogramming, so-called reprogramming “boosters,” have been reported (Esteban et al., 2010, Goyal et al., 2013, Onder et al., 2012, Soria-Valles et al., 2015, Zhang and Wu, 2013). SeV-OKSM-mediated reprogramming experiments were performed (mainly with MLL-AF4+ B-ALL blasts) using many reprogramming epigenetic/transcriptomic factors described to improve reprogramming (Hu, 2014, Lin and Wu, 2015). Although some of these factors enhanced the reprogramming efficiency (Figure 1G), FISH and RT-PCR assays revealed the absence of the fusion gene in resulting clones, indicating that residual non-leukemic myeloid cells were reprogrammed to pluripotency (Table 1). Furthermore, healthy adult B cells are known to be difficult to reprogram. We therefore attempted to reprogram MLL-AF4+ B cell blasts using SeV-OKSM in combination with (1) compounds which specifically target MLL fusion-driven signaling such as the Dot1L inhibitor and an inhibitor of Menin-MLL interaction (He et al., 2016) (2) the lymphoid “path breaker” cEBPα (Bueno et al., 2016, Di Stefano et al., 2014), or (3) phosphatase and tensin homolog (PTEN) inhibitors that constitutively activate the phosphoinositol 3-kinase (PI3K) pathway resulting in increased iPSC generation (Liao et al., 2013) and rescue of B cell receptor (BCR)-defective B cells (Srinivasan et al., 2009). Although these conditions rendered iPSC clones, they were consistently negative for the MLL fusions (Table 1; Figures S1A and S1B). Importantly, OKSM-SeV-mediated reprogramming, with or without additional reprogramming boosters, of MLL-AF4+ leukemic blasts after double FACS sorting (virtually 100% purity) rendered no iPSC colonies, indicating that the limited number of iPSC clones lacking the MLL fusion were derived from easy-to-reprogram residual/contaminating non-leukemic myeloid cells (Table 1).
Immortalized B Cell Lines and Xenograft-Expanded Proliferating MLL-AF4+ Leukemic Blasts Are Refractory to Reprogramming to Pluripotency
Because human acute leukemias do not proliferate in vitro, we hypothesized that successful iPSC generation from leukemic blasts would rely on our ability to induce their proliferation. As a first approach, we used human leukemic B cell lines derived from B-ALL patients with MLL rearrangements (SEM and THP1) or with ETV6-RUNX1 (REH), rather than non-proliferating primary blasts. To this end, B cell lines were infected with OKSM-, OKSL-, or OKSML-expressing SeV vectors alone or in combination with epigenetic and transcriptomic reprogramming boosters; however, no iPSC clones could be generated (Figure S2A and Table 2). Moreover, immortalized B cell lines primed with decitabine and trichostatin A (TSA) prior to OKSM-SeV infection and then exposed to the aforementioned additional chemical inducers, also failed to generate iPSC clones (Figure S2B and Table 2). In another approach, SEM cells were stably knocked down for genes reported to act as barriers to induced pluripotency prior to OKSM-SeV infection (Gaspar-Maia et al., 2013, Menendez et al., 2010, Nashun et al., 2015, Pasque et al., 2011). Intriguingly, small hairpin RNA (shRNA)-mediated knockdown of the tumor suppressor p53, the master B cell transcription factor Pax5, the Polycomb protein RING1a, and the histone variant macroH2A1 failed to facilitate the generation of iPSCs (Table 2; Figures S1C and S1D). Combination of p53 knockdown with 7 days’ treatment with demethylating agents (5-azacytidine, decitabine) before and after OKSM infection also failed to generate iPSCs. The knockdown of macroH2A1 was shown to reactivate a reporter gene on the inactive X chromosome only when combined with decitabine and TSA (Hernandez-Munoz et al., 2005). As reactivation of the inactive X is a hallmark of reprogramming (Ohhata and Wutz, 2013), we tested the same and other triple combinations but found that SEM cells remained resistant to OKSM-induced reprogramming (Table 2).
Table 2.
Summary of the Conditions Used to Reprogram the Leukemic B Cell Lines SEM, THP1, and REH
| Reprogramming Factors | Additional Factors | SEM t(4;11) | THP1 t(9;11) | REH t(12;21) | Clones | iFISH | Genomic PCR |
|---|---|---|---|---|---|---|---|
| SeVdp-OSKM polycistronic | none | √ | √ | √ | no | NA | NA |
| shp53 (MOI = 10) | √ | √ | √ | no | NA | NA | |
| shPax5 (MOI = 10) | √ | √ | √ | no | NA | NA | |
| cEBPα (MOI = 10) | √ | √ | √ | no | NA | NA | |
| ShRING1a (MOI = 10) | √ | ND | ND | no | NA | NA | |
| shMacroH2A1 (MOI = 10) | √ | ND | ND | no | NA | NA | |
| iPTEN (bVp(HO)pic) | √ | √ | √ | no | NA | NA | |
| decitabine | √ | ND | ND | no | NA | NA | |
| iDot1L (SGC0946) | √ | ND | ND | no | NA | NA | |
| iDot1L (epz004777) | √ | ND | ND | no | NA | NA | |
| iMenin-Dot1L (iML2) | √ | ND | ND | no | NA | NA | |
| trichostatin A | √ | ND | ND | no | NA | NA | |
| valproic acid | √ | ND | ND | no | NA | NA | |
| sodium butyrate | √ | ND | ND | no | NA | NA | |
| sodium salicylate | √ | ND | ND | no | NA | NA | |
| iSUV39H1 (Chaetocin) | √ | ND | ND | no | NA | NA | |
| iEZH2 (GSK126) | √ | ND | ND | no | NA | NA | |
| iEZH2 (DZNep) | √ | ND | ND | no | NA | NA | |
| iBRD4 (JQ1) | √ | ND | ND | no | NA | NA | |
| iCDK/iP-TEFb (Flavopiridol) | √ | ND | ND | no | NA | NA | |
| ascorbic acid | √ | ND | ND | no | NA | NA | |
| octyl-α-ketoglutarate | √ | ND | ND | no | NA | NA | |
| SeVdp-OSKL polycistronic | none | √ | √ | √ | no | NA | NA |
| SeVdp-OSKLN polycistronic | none | √ | √ | √ | no | NA | NA |
| SeVdp-OSKM polycistronic + decitabine 0.1 μM + trichostatin A 2 μM | none | √ | √ | √ | no | NA | NA |
| shRING1a MOI = 10 | √ | ND | ND | no | NA | NA | |
| shMacroH2A1 MOI = 10 | √ | ND | ND | no | NA | NA | |
| iDot1L (SGC09469) | √ | ND | ND | no | NA | NA | |
| iDot1L (epz004777) | √ | ND | ND | no | NA | NA | |
| iMenin-MLL (iML2) | √ | ND | ND | no | NA | NA | |
| valproic acid | √ | ND | ND | no | NA | NA | |
| sodium butyrate | √ | ND | ND | no | NA | NA | |
| sodium salicylate | √ | ND | ND | no | NA | NA | |
| iSUV39H1 (Chaetocin) | √ | ND | ND | no | NA | NA | |
| iEZH2 (GSK126) | √ | ND | ND | no | NA | NA | |
| iEZH2 (DZNep) | √ | ND | ND | no | NA | NA | |
| iBRD4 (JQ1) | √ | ND | ND | no | NA | NA | |
| iCDK/iP-TEFb (flavopiridol) | √ | ND | ND | no | NA | NA | |
| ascorbic acid | √ | ND | ND | no | NA | NA | |
| octyl-α-ketoglutarate | √ | ND | ND | no | NA | NA |
ND, not done; NA, not analyzed.
We next induced primary blasts to proliferate through xenograft expansion. Two approaches were followed: (1) in vivo expansion of OKSM-SeV-infected primary B cell blasts or (2) OKSM-SeV infection of in vivo expanded primary B cell blasts. In the second scenario, engrafted mice were treated with iDoT1L, decitabine, or left untreated, to (epi)-genetically prime the blasts prior to OKSM-SeV-infection (Figure 2A). Although these strategies generated some iPSC clones after in vivo expansion of primary blasts in xenografted mice, all iPSCs analyzed lacked the MLL fusion gene by FISH and PCR, and were of mouse origin (Figures 2A and 2B). Together these results show that in vivo expanded leukemic blasts consistently failed to be reprogrammed.
Figure 2.

Reprogramming of Xenograft-Expanded Proliferating Highly Purified MLL-AF4+ Leukemic Blasts Results in Generation of iPSCs from Contaminating Mouse Cells
(A) Schematic depicting the in vivo strategies used to reprogram proliferating xenograft-expanded MLLr leukemic blasts. MLL-AF4+ blasts were either first OKSM-infected and then xenografted for proliferation (“expand infected blasts”) or xenografted first for proliferation (and treated in vivo with decitabine or iDot1L), then OKSM infected ex vivo (“infect expanded blasts”).
(B) Summary of the outcome of reprogramming in vivo xenografted MLL-AF4+ blasts (n = 2 independent experiments).
MLL-AF4 Expression by Itself Is Not a Reprogramming Barrier
Our results show that neither primary MLL-AF4+ blasts nor proliferating leukemic B cell lines can be reprogrammed. However, and in line with previous work (Munoz-Lopez et al., 2016), Epstein-Barr virus (EBV)-immortalized healthy B cells as well as healthy pro-B and pre-B cells could be successfully reprogrammed (Figure S2C), suggesting that the leukemia-initiating event (e.g., MLL fusion genes) may represent a reprogramming barrier. To test this idea, we lentivirally transduced both CB-CD34+ hematopoietic stem/progenitor cells (HSPCs) and CD34+CD19+ B cell progenitors with MLL-AF4-GFP, and after several days infected MLL-AF4-expressing CD34+ and CD34+CD19+ cells with OKSM-SeV (Bueno et al., 2015). MLL-AF4 expression did not impair the generation of iPSCs, and the reprogramming efficiency was similar to that of GFP-transduced CD34+ HSPCs (Figure 3A) and CD34+CD19+ B cell progenitors (Figure S3A). Resulting iPSC clones displayed human embryonic stem cell (hESC)-like morphology and expressed MLL-AF4-GFP (Figures 3B and S3B). Further characterization revealed that MLL-AF4 was present in the majority of the iPSC clones and was always expressed (Figures 3C, 3D, and S3B) after ten passages. In addition, MLL-AF4-expressing iPSC clones were OKSM transgene independent (Figure 3E), diploid (Figure 3F), positive for alkaline phosphatase (Figure 3G), and expressed the pluripotency factors NANOG, OCT4, SOX2, REX1, DNMT3β, and CRIPTO (Figure 3H) and the surface markers TRA-1-60, SSEA3, and SSEA4 (Figure 3I). Importantly, iPSCs derived from MLL-AF4-expressing CD34+CD19+ B cell progenitors carried complete VDJH immunoglobulin gene monoclonal rearrangements, confirming the B lineage identity (Figure S3C). Collectively, these results suggest that MLL-AF4 expression does not seem to represent a reprogramming barrier in either CD34+ cells or CD34+CD19+ B cell progenitors, and is compatible with pluripotency.
Figure 3.

MLL-AF4 Expression Does Not Constitute a Reprogramming Barrier on Its Own
(A) Representative TRA-1-60 staining of iPSC colonies generated from CB-CD34+ HSPCs ectopically expressing GFP alone (empty vector; EV) or MLL-AF4 (n = 3 independent experiments). No iPSC colonies were obtained from SEM, THP1, or REH cell lines (n = 3 independent experiments).
(B) Phase-contrast and fluorescence images of iPSC colonies generated from EV- and MLL-AF4-expressing CB-CD34+ cells. Scale bar, 100 μm.
(C) Genomic PCR revealing that ∼85% of the iPSCs harbor MLL-AF4 provirus.
(D) RT-PCR revealing that all iPSC clones carrying MLL-AF4 provirus express MLL-AF4 transcript.
(E) Representative qRT-PCR demonstrating SeV elimination after ten passages.
(F) Representative diploid karyotype of iPSCs (p15) derived from MLL-AF4-expressing CD34+ cells.
(G) Representative morphology and alkaline phosphatase staining of iPSCs derived from MLL-AF4-expressing CD34+ cells.
(H) qRT-PCR for the pluripotency transcription factors OCT4, SOX2, NANOG, REX1, CRIPTO, and DNMT3β in MLL-AF4+ iPSCs.
(I) Representative flow cytometry expression of the pluripotency-associated surface markers TRA-1-60, SSEA-3, and SSEA-4 by MLL-AF4+ iPSCs.
Global Transcriptome and DNA Methylome Analyses Suggest a Developmental Refractoriness of MLL-Rearranged B-ALL to Reprogramming to Pluripotency
To identify patterns of gene expression that might provide a molecular explanation for the refractoriness of leukemic blasts to reprogramming, we compared gene expression profiles of FACS-purified MLL-AF4+ blasts from infant B-ALL (n = 3) with hematopoietic stem cells (HSCs) (n = 2), B cell hematopoietic progenitor cells (HPCs) (n = 2), and myeloid HPCs (n = 2) from healthy CB. A heatmap representation of hierarchical clustering of genes differentially expressed (2-fold regulated; p < 0.01) in MLL-AF4+ blasts versus healthy HSPCs is shown in Figure 4A. A total of 87 genes were differentially expressed in MLL-AF4+ blasts (Figures 4B and 4C). To gain insight into the biological functions affected by differentially expressed genes, we performed gene ontology (GO) analysis comparing MLL-AF4+ blasts with normal HSPCs (Figure 4D). Among the top significant GO biological processes enriched in MLL-AF4+ blasts, we found “cell differentiation,” “cell morphogenesis,” “developmental process,” and “cell proliferation” (Figure 4C), suggesting that the intrinsic developmental (differentiation) blockage and proliferative defects of leukemic blasts, rather than leukemia-specific genetic alterations, may constitute a reprogramming barrier.
Figure 4.

Gene Expression Profiling Comparing MLL-AF4+ B Cell Blasts with HSCs, Myeloid HPCs, and B Cell HPCs
(A) Heatmap depicting the genes differentially expressed (2-fold up- or downregulated; p < 0.01) in MLL-AF4+ B cell blasts versus normal HSCs and HPCs. The left color bar categorizes the gene expression level in a log2 scale.
(B) Venn diagrams showing the number of transcripts differentially expressed between MLL-AF4+ blasts and HSCs, B cell HPCs, and myeloid HPCs.
(C) Identification of the 87 genes shared by normal HSC, B cell HPCs, and myeloid HPCs but differentially expressed in MLL-AF4+ blasts. Red and blue identify upregulated and downregulated genes, respectively.
(D) Statistically significant GO biological functions identified using GOrilla software of the genes differentially expressed in MLL-AF4+ blasts versus normal HSCs/HPCs ranked p value. −log p value, black bars (left y axis); enrichment score, filled red circle with red line (right y axis).
Similarly, to identify potential DNA methylation changes explaining the refractoriness of leukemic blasts to reprogramming, we performed global DNA methylation (LINE-1) profiling on FACS-purified MLL-AF4+ blasts from infant B-ALL (n = 3), B cell HPCs (n = 2), and MLL-AF4-expressing CD34+ HSPCs. Although no major quantitative changes in global DNA methylation were revealed by bisulfite pyrosequencing (Figure 5A), DNA methylation 450K BeadChip arrays identified ∼6,700 CpG sites differentially methylated (dmCpGs; false discovery rate <0.05) between MLL-AF4+ blasts and both B cell HPCs and MLL-AF4-expressing HSPCs (Figures 5B and 5C). Specifically, 1,691 dmCpGs were hypomethylated and 5,012 CpGs hypermethylated in MLL-AF4+ leukemic blasts (Figure 5C). GO analysis of hypermethylated dmCpGs revealed “cell differentiation,” “cell morphogenesis,” and “developmental process” as significant biological processes enriched in MLL-AF4+ blasts (Figure 5D). GO analysis of hypomethylated dmCpGs identified RAS/JAK-STAT/MAPK activities (through which BCR-mediated signaling regulates B cell activation and differentiation) (Marshall et al., 2000) as significant biological processes enriched in MLL-AF4+ blasts (Figure 5D). Thus, in line with the transcriptome data, these results suggest that the intrinsic differentiation blockage and proliferative status of leukemic blasts constitute a bona fide reprogramming barrier.
Figure 5.

DNA Methylation Differences Observed in MLL-AF4+ B Cell Blasts Versus CD34+ Cells Expressing MLL-AF4 and CD34+CD19+ B Cell HPCs
(A) Global DNA methylation analysis by pyrosequencing of LINE-1 elements in MLL-AF4+ blasts and normal CD34+CD19+ B cell HPCs (n = 3 independent experiments). Error bars indicate SD.
(B) Unsupervised hierarchical clustering and heatmap showing the CpG sites with differential DNA methylation between MLL-AF4+ blasts versus normal CD34+CD19+ B cell HPCs and CD34+ cells expressing ectopic MLL-AF4. Average methylation values are displayed from 0 (blue) to 1 (yellow).
(C) Venn diagrams showing the number of CpG sites differentially hypomethylated (left) or hypermethylated (right) in MLL-AF4+ blasts versus normal CD34+CD19+ B cell HPCs and CD34+ ectopically expressing MLL-AF4.
(D) Selection of GO terms from the top 50 statistically significant biological functions, ranked by p value (x axis), of genes differentially hypomethylated (left) or hypermethylated (right) in MLL-AF4+ blasts versus normal CD34+CD19+ B cell HPCs and CD34+ ectopically expressing MLL-AF4. The y axis indicates the relative risk (±95% confidence intervals) as a measure of effect size. The relative risk is the ratio of the proportion of genes belonging to a given GO term in a selected subset of genes to the same proportion in the remaining, background genes.
Discussion
iPSCs reprogrammed from cancer cells have the potential to illuminate molecular mechanisms underlying the pathogenesis of cancer (Barrett et al., 2014, Curry et al., 2015, Ramos-Mejia et al., 2012c, Yilmazer et al., 2015). However, reprogramming human primary cancer cells remains challenging, and only a few reports have demonstrated successful reprogramming of malignant cells. Moreover, iPSCs from primary leukemic cells have exclusively been generated from chronic hematological malignances, including Philadelphia+ CML, PMF, JAK2-V617F+ PV, and JMML (Bedel et al., 2013, Carette et al., 2010, Gandre-Babbe et al., 2013, Hu, 2014, Kumano et al., 2012, Yamamoto et al., 2015, Ye et al., 2009) (Table S1). No iPSCs reprogrammed from acute leukemias have been reported, but iPSCs have been successfully generated from normal myeloid, T cells, and B cells using non-integrative tetracistronic OKSM-expressing SeV (Bueno et al., 2016, Munoz-Lopez et al., 2016). Here, we attempted to establish an iPSC-based disease model to address the developmental impact of leukemia-specific fusion genes on human stem cell fate. We provide insights into the difficulty of reprogramming primary leukemia blasts from cytogenetically different subtypes of B-ALL, including t(4;11)/MLL-AF4+, t(1;11)+MLL-EPS15+, and t(12;21)/ETV6-RUNX1 B-ALL. Our data demonstrate that neither primary blasts nor B-ALL cell lines could be reprogrammed to pluripotency. The few iPSC clones that were generated consistently lacked the leukemic fusion gene, indicating that only residual/contaminating non-leukemic myeloid cells, which are less demanding to reprogram than lymphoid cells, were reprogrammed (Bueno et al., 2016).
The question remains open as to whether biological or technical reprogramming barriers underlie the inability of B-ALL leukemic blasts to be reprogrammed. Technically, multiple reprogramming strategies were attempted, but neither transient nor stable expression of reprogramming factors using a variety of monocistronic and polycistronic vectors rendered leukemic iPSCs. The choice of reprogramming factors (OKSM, OKSL, or OKSML) also had little impact on the reprogramming outcome, suggesting that the methods used for transgene delivery and c-myc dependency are not causal mechanisms responsible for the lack of success. From a biological standpoint, cell identity is a reflection of cell-type-specific gene expression and epigenetic signatures. A variety of transcription factors, tumor suppressors, microRNAs, and chromatin-remodeling enzymes, as well as chemical regulators of histone and DNA modifications, have been extensively reported to provide a permissive environment for cell-fate change during cellular reprogramming (Nashun et al., 2015). We combined different reprogramming strategies with several reprogramming “boosters” acting on transcription factor expression and chromatin structure; yet, iPSCs could not be generated. Recently, the differentiation blockage of BCR-ABL1+ B cell ALL cells was overcome by forcing cells to reprogram to the myeloid lineage through exposure to myeloid differentiation-promoting cytokines in vitro or by transient expression of the myeloid transcription factor C/EBPα (McClellan et al., 2015). Similarly, mouse and human healthy B cells were efficiently reprogrammed upon C/EBPα-mediated myeloid priming (Bueno et al., 2016, Di Stefano et al., 2014). Unfortunately, neither C/EBPα expression (Tables 1 and 2) nor exposure to myeloid differentiation-promoting cytokines (interleukin-3 [IL-3], IL-6, FLT3, granulocyte macrophage colony-stimulating factor, and macrophage colony-stimulating factor; data not shown) sensitized MLL-AF4+ blasts to undergo myeloid priming and subsequent reprogramming.
Active cell proliferation is key for transcription factor-induced cell-fate change during cellular reprogramming. Because acute leukemias barely or do not proliferate in vitro, we endeavored to reprogram leukemic B cell lines as well as MLL-rearranged primary blasts induced to proliferate through xenograft expansion. Again, these approaches consistently failed to reprogram in vivo expanded leukemic blasts; only some murine iPSCs were generated as a result of traces of OKSM-SeV carry-over after in vivo transplantation. However, EBV-immortalized healthy B cells as well as healthy pro-B and pre-B precursors could be successfully reprogrammed (Munoz-Lopez et al., 2016), suggesting that the leukemia-initiating genetic event might represent a reprogramming barrier. However, functional data proved that MLL-AF4 expression in both CB-derived CD34+ HSPCs and CD34+CD19+ B cell progenitors was compatible with pluripotency and did not impede reprogramming, indicating that the leukemia driver genetic event does not itself represent a barrier.
Cell-type-specific gene expression profiles and specific chromatin signatures establish epigenetic barriers to transcription factor-mediated reprogramming. Consequently, global transcriptome and DNA methylome analyses were performed to gain insight into the refractoriness of leukemic blasts to reprogramming. Our data suggest a developmental refractoriness of MLL-rearranged B-ALL to reprogramming to pluripotency, reflecting that the intrinsic differentiation blockage of leukemic blasts constitutes a bona fide reprogramming barrier. In addition, analysis of hypomethylated dmCpGs identified RAS/JAK-STAT/MAPK signaling to be likely involved in refractoriness to reprogram MLL-AF4+ blasts (Figure 5D). These pathways are master signaling effectors of BCR-mediated signaling regulation of B cell activation and differentiation (Marshall et al., 2000). However, chemical activation of the PI3K-AKT-Ras pathway (through PTEN inhibition), which can rescue BCR-defective B cells (Srinivasan et al., 2009), failed to reprogram primary B-ALL blasts/cell lines. In line with the transcriptome data, we suggest that the intrinsic differentiation blockage and proliferative status of leukemic blasts constitutes a bona fide reprogramming barrier.
Recent studies on the 3D chromosome regulatory landscape of human pluripotent stem cells have revealed the importance of chromosome structure and topologically associating domains in maintaining pluripotency (Ji et al., 2016). In addition, an elegant study has suggested that some large genetic rearrangements (e.g., ring chromosomes) are not compatible with the pluripotent state, likely due to the inability of establishing the characteristic chromosomal topology of iPSCs (Bershteyn et al., 2014). Whether genetically complex leukemic chromosomal translocations impede leukemic blast reprogramming cannot be ruled out. In addition, these genetic rearrangements may simply be lethal in a pluripotent cell context. Preliminary data from our laboratory suggest that after generating a t(4;11)/MLL-AF4+ translocation in human iPSCs using the CRISPR/Cas9 system (Torres et al., 2014), t(4;11)-carrying clones are lost after a few passages, suggesting that large genetic rearrangements may not be compatible with pluripotency. Further work is needed to identify and overcome the biological barriers impeding reprogramming of acute leukemias and primary cancer cells, which may reveal information on the links between pluripotency and oncogenic transformation that would be instrumental for the development of new therapies.
Experimental Procedures
Primary Leukemic Samples, Normal HSC/HPCs, EBV-Immortalized B Cells, and Leukemic Cell Lines
Human primary B cell leukemic samples, and CB- and FL-derived HSPCs were obtained in accordance with procedures approved by the Clinic Hospital of Barcelona or the Erasmus University Medical Center in Rotterdam. Informed consent was obtained in accordance with the Declaration of Helsinki. B cell leukemias used corresponded to the following cytogenetic subtypes: t(4;11)/MLL-AF4+ (n = 4), t(1;11)/MLL-EPS15+ (n = 2), and t(12; 21)/ETV6-RUNX1+ (n = 2). Primary leukemias with >80% of blasts were processed as reported previously (Bueno et al., 2014, Menendez et al., 2009). In brief, after Ficoll-based gradient centrifugation, mononuclear cells were stained with anti-human CD45 (555482, Beckton Dickinson), CD34 (130-081-001, Miltenyi), CD19 (130-091-328, Miltenyi), and CD10 (555376, Becton Dickinson). Live (7AAD−) blast cells (CD45dimCD19+) were highly purified using a FACSAria-III sorter (Becton Dickinson, Figure 1A). A one- or two-round sorting strategy was used as indicated with resulting purities >99% and practically 100%, respectively (Figure 1A and Table 1). CB-derived CD34+ HSPCs and FL-derived CD34+CD19+ B cell HPCs were processed as described previously (Bueno et al., 2015, Munoz-Lopez et al., 2016, Prieto et al., 2016, Ramos-Mejia et al., 2012d). Where indicated, CD34+ cells were lentivirally transduced with MLL-AF4-GFP as reported (Montes et al., 2014) and MLL-AF4-GFP-expressing CD34+ cells were FACS enriched for iPSC generation and DNA methylome studies (see below). SEM (MLL-AF4+), THP1 (MLL-AF9+), and REH (ETV6-RUNX1+) B cell lines were grown in DMEM + 10% fetal bovine serum (FBS) + 1% P/Sas as described by Bueno et al. (2008). A lymphoblastoid B cell line was obtained from the Spanish National DNA Bank (Salamanca). This cell line was established by immortalizing PB B cells of a healthy female donor with EBV. Culture conditions were RPMI + GlutaMAX + 15% FBS + 1% penicillin-streptomycin (Munoz-Lopez et al., 2016).
Vectors, iPSC Generation, and Characterization
Human primary leukemias were infected with OKSM(L)-expressing vectors immediately after FACS sorting. The following vectors were used: (1) OKSM polycistronic lentivector, (2) OKSM or OKSL non-integrative monocistronic episomal vectors, (3) OKSM monocistronic retroviral vectors, (4) OKSM monocistronic SeV vector, and (5) OKSM polycistronic SeV vectors (Bueno et al., 2016, Munoz-Lopez et al., 2016) (Table 1). Primary HSPCs, EBV-immortalized B lymphocytes, and in vivo expanded blasts were infected with OKSM polycistronic SeV vector immediately after FACS sorting. Wild-type and shRNA-knockdown (KD) SEM, THP1, and REH cell lines were transduced with OKSM, OKSL, or OKSML polycistronic SeV vector after 3 days of priming with reprogramming factors (Figure S2A and Table 2) or decitabine + TSA (Figure S2B and Table 2). KD B cell lines were generated using shRNA-expressing pLKO-based lentiviral vectors for P53, PAX5, RING1a, and macroH2A1 as described previously (Ramos-Mejia et al., 2014, Real et al., 2012). cEBPα was ectopically expressed using a cEBPα-expressing SeV vector previously generated in our laboratory (Bueno et al., 2016, Di Stefano et al., 2014). SeV vectors, lenti-/retrovectors, and shRNA-expressing pLKO lentivectors were used at MOI of 3–4, 5–10, and 10, respectively (Figure 1 and Table 2). OKSM-infected human primary cells (5 × 105 per experiment) and SEM/THP1/REH cell lines were stimulated/maintained with 10%–20% FBS supplemented with either the hematopoietic cytokines IL-7, IL-3, SCF, FLT3L, and TPO (Bueno et al., 2016, Munoz-Lopez et al., 2016) or reprogramming factors. Cells were plated onto irradiated mouse embryonic fibroblasts (MEFs) for 3 days without a medium change and were thereafter maintained on MEF-conditioned medium supplemented with 8 ng/mL basic fibroblast growth factor (bFGF) (Miltenyi Biotec) with medium changes every other day until iPSC colonies emerged (Bueno et al., 2016, Munoz-Lopez et al., 2016). Identification of the first emerging iPSC colonies (∼6–8 days after OKSM infection) and passage onto fresh feeders was done mechanically as described. Depending on the reprogramming conditions, iPSC colonies were established by days 18–30, and were immunostained for TRA-1-60 to determine the reprogramming efficiency. iPSC clones were further maintained on irradiated MEFs in hESC medium supplemented with 8 ng/mL bFGF. hESC medium was changed daily, and the colonies were passaged mechanically weekly (Montes et al., 2009, Ramos-Mejia et al., 2012b).
Established iPSCs were characterized as previously described in detail (Bueno et al., 2016, Munoz-Lopez et al., 2016) upon confirmation of their bona fide morphology and transgene independency. Each iPSC clone was analyzed beforehand for the presence of the chromosomal rearrangement by FISH (see below), genomic PCR, and RT-PCR, using standard procedures and primers shown in Table S2. SeV elimination was determined by qRT-PCR. Expression of pluripotency markers was performed by alkaline phosphatase and TRA-1-60 immunostaining, by qRT-PCR for OCT4, NANOG, SOX2, REX1, CRIPTO, and DNMT3B, and by flow cytometry using anti-human SSEA-3 (560237), SSEA-4 (561156), and TRA-1-60 (563188) antibodies (Beckton Dickinson). Primers and flow cytometry antibodies used are shown in Table S2. Normal karyotype was confirmed by conventional G banding (Catalina et al., 2008).
Expansion of Leukemic Blasts in NSG Mice and iPSC Generation
Human acute leukemias barely or do not proliferate in vitro. In an attempt to enhance the efficiency of iPSC generation from leukemic blasts, primary leukemias were intratibia-transplanted in NSG mice and allowed to proliferate in vivo. MLL-AF4+ blasts were (1) OKSM infected, in vivo expanded, and then retrieved and plated for iPSC emergence (“expand infected blasts”) or (2) in vivo expanded, retrieved by FACS sorting, OKSM infected, and then plated for iPSC emergence (“infect expanded blasts”) (Figure 2A). In this second scenario, once mice achieved 5%–10% PB engraftment with MLL-AF4+ blasts, they were divided into three groups: (1) untreated, (2) treated with a Dot1L inhibitor (iDoT1L), or (3) treated with decitabine (both at 2.5 mg/kg, subcutaneously, for 3 days). Three days later, mice were killed and blasts were purified from bone marrow and PB for OKSM-SeV infection (Figure 2A).
Chemicals
Information about the compounds used for enhancing reprogramming, their concentration, biological function, and suppliers is detailed in Table S3.
FISH Studies
FISH was performed on leukemic blasts at disease presentation, and on patient-derived iPSCs as described (Bueno et al., 2009, Menendez et al., 2009) using commercially available probes (Vysis). ETV6-RUNX1 and MLL-AF4 fusions were detected using locus-specific LSI Dual Color Translocation probes. MLL-EPS15 and MLL-AF4 rearrangements were analyzed using the LSI MLL Dual Color Break Apart Rearrangement Probe. At least 500 nuclei were analyzed. The slides were analyzed under a fluorescence microscope equipped with appropriate filters using ISIS software (Metasystems).
Gene Expression Profiling
Profiling was performed on FACS-purified (purity >98%) MLL-AF4+ blasts from infant B-ALL (n = 3), CB-derived CD34+CD38−CD19−CD33− HSCs (n = 2), CD34+CD19+CD33− B cell HPCs (n = 2), and CD34+CD33+CD19− myeloid HPCs (n = 2). Technical duplicates were performed for each independent sample. Microarray data have been deposited in the NCBI Gene Expression Omnibus (GSE79450). Microarrays, statistical analysis, hierarchical clustering, and GO analysis were performed as detailed elsewhere (Irizarry et al., 2003, Tusher et al., 2001).
Bisulfite Pyrosequencing, Human Methylation 450 BeadChip Array, and Data Analysis
Differences in global DNA methylation were compared between MLL-AF4+ blasts and healthy CD34+CD19+ B cell HPCs and in B cell lines before and after decitabine treatment by bisulfite pyrosequencing of LINE-1 elements using the EZ DNA Methylation-Gold kit (Zymo Research). PCR amplification of modified DNA was performed using a set of primers reported previously (Bollati et al., 2007, Ramos-Mejia et al., 2012a, Ramos-Mejia et al., 2012b). Microarray-based DNA methylation profiling was performed with the Illumina Infinium HumanMethylation450 BeadChip as described elsewhere (Bibikova et al., 2011, Heinz et al., 2010).
Further detailed information is provided in Supplemental Experimental Procedures.
Author Contributions
A.M.-L. and C.B. conceived the study, designed and performed experiments, analyzed data, and wrote the manuscript. D.R.-M., C.P., V.R.-M., A.A.-D., I.V., M.B., A.P., X.C.-V., A.G., A.F., M.L., I.G., N.R-X., R.T, S.R.-P., M.F., A.F.F., and M.B. performed experiments and analyzed data. R.W.S., G.C., and M.N. provided key biological samples and reagents and helped with the experimental design. P.M. conceived the study, designed experiments, analyzed data, and wrote the manuscript. All authors read and approved the manuscript.
Acknowledgments
This work was supported by the European Research Council to P.M. (ERC-2014-CoG-646903), the ISCIII/FEDER (E-Rare-2 Call PI12/03112 to P.M. and PI14/01191 to C.B.), MINECO (SAF2013-43065 to P.M.), and the Spanish Association Against Cancer (AECC) to P.M. and C.B. C.B. is supported by a Miguel Servet II contract (CPII13/00011). D.R.M. and A.M.-L. are supported by PFIS (FI11/0511) and FPI (BES-2014-067844) scholarships, respectively. P.M. also acknowledges financial support from the Obra Social La Caixa, Fundaciò Josep Carreras and The Generalitat de Catalunya (SGR330). C.B. and P.M. are investigators of TERCEL.
Published: September 22, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, three figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.08.013.
Contributor Information
Clara Bueno, Email: cbueno@carrerasresearch.org.
Pablo Menendez, Email: pmenendez@carrerasresearch.org.
Accession Numbers
Microarray data have been deposited in NCBI Gene Expression Omnibus (GEO: GSE79450).
Supplemental Information
References
- Barrett R., Ornelas L., Yeager N., Mandefro B., Sahabian A., Lenaeus L., Targan S.R., Svendsen C.N., Sareen D. Reliable generation of induced pluripotent stem cells from human lymphoblastoid cell lines. Stem Cells Transl. Med. 2014;3:1429–1434. doi: 10.5966/sctm.2014-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedel A., Pasquet J.M., Lippert E., Taillepierre M., Lagarde V., Dabernat S., Dubus P., Charaf L., Beliveau F., de Verneuil H. Variable behavior of iPSCs derived from CML patients for response to TKI and hematopoietic differentiation. PLoS One. 2013;8:e71596. doi: 10.1371/journal.pone.0071596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershteyn M., Hayashi Y., Desachy G., Hsiao E.C., Sami S., Tsang K.M., Weiss L.A., Kriegstein A.R., Yamanaka S., Wynshaw-Boris A. Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature. 2014;507:99–103. doi: 10.1038/nature12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M., Barnes B., Tsan C., Ho V., Klotzle B., Le J.M., Delano D., Zhang L., Schroth G.P., Gunderson K.L. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Bollati V., Baccarelli A., Hou L., Bonzini M., Fustinoni S., Cavallo D., Byun H.M., Jiang J., Marinelli B., Pesatori A.C. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- Bueno C., Montes R., Martin L., Prat I., Hernandez M.C., Orfao A., Menendez P. NG2 antigen is expressed in CD34+ HPCs and plasmacytoid dendritic cell precursors: is NG2 expression in leukemia dependent on the target cell where leukemogenesis is triggered? Leukemia. 2008;22:1475–1478. doi: 10.1038/leu.2008.134. [DOI] [PubMed] [Google Scholar]
- Bueno C., Catalina P., Melen G.J., Montes R., Sanchez L., Ligero G., Garcia-Perez J.L., Menendez P. Etoposide induces MLL rearrangements and other chromosomal abnormalities in human embryonic stem cells. Carcinogenesis. 2009;30:1628–1637. doi: 10.1093/carcin/bgp169. [DOI] [PubMed] [Google Scholar]
- Bueno C., Roldan M., Anguita E., Romero-Moya D., Martin-Antonio B., Rosu-Myles M., del Canizo C., Campos F., Garcia R., Gomez-Casares M. Bone marrow mesenchymal stem cells from patients with aplastic anemia maintain functional and immune properties and do not contribute to the pathogenesis of the disease. Haematologica. 2014;99:1168–1175. doi: 10.3324/haematol.2014.103580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno C., van Roon E.H., Muñoz-López A., Sanjuan-Pla A., Juan M., Navarro A., Stam R.W., Menendez P. Immunophenotypic analysis and quantification of B-1 and B-2 B cells during human fetal hematopoietic development. Leukemia. 2015;30:1603–1606. doi: 10.1038/leu.2015.362. [DOI] [PubMed] [Google Scholar]
- Bueno C., Sardina J.L., Di Stefano B., Romero-Moya D., Munoz-Lopez A., Ariza L., Chillon M.C., Balanzategui A., Castano J., Herreros A. Reprogramming human B cells into induced pluripotent stem cells and its enhancement by C/EBPalpha. Leukemia. 2016;30:674–682. doi: 10.1038/leu.2015.294. [DOI] [PubMed] [Google Scholar]
- Carette J.E., Pruszak J., Varadarajan M., Blomen V.A., Gokhale S., Camargo F.D., Wernig M., Jaenisch R., Brummelkamp T.R. Generation of iPSCs from cultured human malignant cells. Blood. 2010;115:4039–4042. doi: 10.1182/blood-2009-07-231845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalina P., Montes R., Ligero G., Sanchez L., de la Cueva T., Bueno C., Leone P.E., Menendez P. Human ESCs predisposition to karyotypic instability: is a matter of culture adaptation or differential vulnerability among hESC lines due to inherent properties? Mol. Cancer. 2008;7:76. doi: 10.1186/1476-4598-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry E.L., Moad M., Robson C.N., Heer R. Using induced pluripotent stem cells as a tool for modelling carcinogenesis. World J. Stem Cells. 2015;7:461–469. doi: 10.4252/wjsc.v7.i2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano B., Sardina J.L., van Oevelen C., Collombet S., Kallin E.M., Vicent G.P., Lu J., Thieffry D., Beato M., Graf T. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature. 2014;506:235–239. doi: 10.1038/nature12885. [DOI] [PubMed] [Google Scholar]
- Esteban M.A., Wang T., Qin B., Yang J., Qin D., Cai J., Li W., Weng Z., Chen J., Ni S. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell. 2010;6:71–79. doi: 10.1016/j.stem.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Gandre-Babbe S., Paluru P., Aribeana C., Chou S.T., Bresolin S., Lu L., Sullivan S.K., Tasian S.K., Weng J., Favre H. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood. 2013;121:4925–4929. doi: 10.1182/blood-2013-01-478412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar-Maia A., Qadeer Z.A., Hasson D., Ratnakumar K., Leu N.A., Leroy G., Liu S., Costanzi C., Valle-Garcia D., Schaniel C. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 2013;4:1565. doi: 10.1038/ncomms2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A., Chavez S.L., Reijo Pera R.A. Generation of human induced pluripotent stem cells using epigenetic regulators reveals a germ cell-like identity in partially reprogrammed colonies. PLoS One. 2013;8:e82838. doi: 10.1371/journal.pone.0082838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S., Malik B., Borkin D., Miao H., Shukla S., Kempinska K., Purohit T., Wang J., Chen L., Parkin B. Menin-MLL inhibitors block oncogenic transformation by MLL-fusion proteins in a fusion partner-independent manner. Leukemia. 2016;30:508–513. doi: 10.1038/leu.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Munoz I., Lund A.H., van der Stoop P., Boutsma E., Muijrers I., Verhoeven E., Nusinow D.A., Panning B., Marahrens Y., van Lohuizen M. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc. Natl. Acad. Sci. USA. 2005;102:7635–7640. doi: 10.1073/pnas.0408918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K. All roads lead to induced pluripotent stem cells: the technologies of iPSC generation. Stem Cells Dev. 2014;23:1285–1300. doi: 10.1089/scd.2013.0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X., Dadon D.B., Powell B.E., Fan Z.P., Borges-Rivera D., Shachar S., Weintraub A.S., Hnisz D., Pegoraro G., Lee T.I. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016;18:262–275. doi: 10.1016/j.stem.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumano K., Arai S., Hosoi M., Taoka K., Takayama N., Otsu M., Nagae G., Ueda K., Nakazaki K., Kamikubo Y. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood. 2012;119:6234–6242. doi: 10.1182/blood-2011-07-367441. [DOI] [PubMed] [Google Scholar]
- Liao J., Marumoto T., Yamaguchi S., Okano S., Takeda N., Sakamoto C., Kawano H., Nii T., Miyamato S., Nagai Y. Inhibition of PTEN tumor suppressor promotes the generation of induced pluripotent stem cells. Mol. Ther. 2013;21:1242–1250. doi: 10.1038/mt.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T., Wu S. Reprogramming with small molecules instead of exogenous transcription factors. Stem Cells Int. 2015;2015:794632. doi: 10.1155/2015/794632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall A.J., Niiro H., Yun T.J., Clark E.A. Regulation of B-cell activation and differentiation by the phosphatidylinositol 3-kinase and phospholipase Cgamma pathway. Immunol. Rev. 2000;176:30–46. doi: 10.1034/j.1600-065x.2000.00611.x. [DOI] [PubMed] [Google Scholar]
- McClellan J.S., Dove C., Gentles A.J., Ryan C.E., Majeti R. Reprogramming of primary human Philadelphia chromosome-positive B cell acute lymphoblastic leukemia cells into nonleukemic macrophages. Proc. Natl. Acad. Sci. USA. 2015;112:4074–4079. doi: 10.1073/pnas.1413383112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez P., Bueno C., Wang L. Human embryonic stem cells: a journey beyond cell replacement therapies. Cytotherapy. 2006;8:530–541. doi: 10.1080/14653240601026654. [DOI] [PubMed] [Google Scholar]
- Menendez P., Catalina P., Rodriguez R., Melen G.J., Bueno C., Arriero M., Garcia-Sanchez F., Lassaletta A., Garcia-Sanz R., Garcia-Castro J. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J. Exp. Med. 2009;206:3131–3141. doi: 10.1084/jem.20091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez S., Camus S., Izpisua Belmonte J.C. p53: guardian of reprogramming. Cell Cycle. 2010;9:3887–3891. doi: 10.4161/cc.9.19.13301. [DOI] [PubMed] [Google Scholar]
- Montes R., Ligero G., Sanchez L., Catalina P., de la Cueva T., Nieto A., Melen G.J., Rubio R., Garcia-Castro J., Bueno C., Menendez P. Feeder-free maintenance of hESCs in mesenchymal stem cell-conditioned media: distinct requirements for TGF-beta and IGF-II. Cell Res. 2009;19:698–709. doi: 10.1038/cr.2009.35. [DOI] [PubMed] [Google Scholar]
- Montes R., Ayllon V., Prieto C., Bursen A., Prelle C., Romero-Moya D., Real P.J., Navarro-Montero O., Chillon C., Marschalek R. Ligand-independent FLT3 activation does not cooperate with MLL-AF4 to immortalize/transform cord blood CD34+ cells. Leukemia. 2014;28:666–674. doi: 10.1038/leu.2013.346. [DOI] [PubMed] [Google Scholar]
- Munoz-Lopez A., van Roon E.H., Romero-Moya D., Lopez-Millan B., Stam R.W., Colomer D., Nakanishi M., Bueno C., Menendez P. Cellular ontogeny and hierarchy influence the reprogramming efficiency of human B cells into induced pluripotent stem cells. Stem Cells. 2016;34:581–587. doi: 10.1002/stem.2303. [DOI] [PubMed] [Google Scholar]
- Nashun B., Hill P.W., Hajkova P. Reprogramming of cell fate: epigenetic memory and the erasure of memories past. EMBO J. 2015;34:1296–1308. doi: 10.15252/embj.201490649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohhata T., Wutz A. Reactivation of the inactive X chromosome in development and reprogramming. Cell Mol. Life Sci. 2013;70:2443–2461. doi: 10.1007/s00018-012-1174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder T.T., Kara N., Cherry A., Sinha A.U., Zhu N., Bernt K.M., Cahan P., Marcarci B.O., Unternaehrer J., Gupta P.B. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasque V., Gillich A., Garrett N., Gurdon J.B. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J. 2011;30:2373–2387. doi: 10.1038/emboj.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto C., Stam R.W., Agraz-Doblas A., Ballerini P., Camos M., Castano J., Marschalek R., Bursen A., Varela I., Bueno C., Menendez P. Activated KRAS cooperates with MLLAF4 to promote extramedullary engraftment and migration of cord blood CD34+ HSPC but is insufficient to initiate leukemia. Cancer Res. 2016;76:2478–2489. doi: 10.1158/0008-5472.CAN-15-2769. [DOI] [PubMed] [Google Scholar]
- Ramos-Mejia V., Bueno C., Roldan M., Sanchez L., Ligero G., Menendez P., Martin M. The adaptation of human embryonic stem cells to different feeder-free culture conditions is accompanied by a mitochondrial response. Stem Cells Dev. 2012;21:1145–1155. doi: 10.1089/scd.2011.0248. [DOI] [PubMed] [Google Scholar]
- Ramos-Mejia V., Fernandez A.F., Ayllon V., Real P.J., Bueno C., Anderson P., Martin F., Fraga M.F., Menendez P. Maintenance of human embryonic stem cells in mesenchymal stem cell-conditioned media augments hematopoietic specification. Stem Cells Dev. 2012;21:1549–1558. doi: 10.1089/scd.2011.0400. [DOI] [PubMed] [Google Scholar]
- Ramos-Mejia V., Fraga M.F., Menendez P. iPSCs from cancer cells: challenges and opportunities. Trends Mol. Med. 2012;18:245–247. doi: 10.1016/j.molmed.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Ramos-Mejia V., Montes R., Bueno C., Ayllon V., Real P.J., Rodriguez R., Menendez P. Residual expression of the reprogramming factors prevents differentiation of iPSC generated from human fibroblasts and cord blood CD34+ progenitors. PLoS One. 2012;7:e35824. doi: 10.1371/journal.pone.0035824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Mejia V., Navarro-Montero O., Ayllon V., Bueno C., Romero T., Real P.J., Menendez P. HOXA9 promotes hematopoietic commitment of human embryonic stem cells. Blood. 2014;124:3065–3075. doi: 10.1182/blood-2014-03-558825. [DOI] [PubMed] [Google Scholar]
- Real P.J., Ligero G., Ayllon V., Ramos-Mejia V., Bueno C., Gutierrez-Aranda I., Navarro-Montero O., Lako M., Menendez P. SCL/TAL1 regulates hematopoietic specification from human embryonic stem cells. Mol. Ther. 2012;20:1443–1453. doi: 10.1038/mt.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria-Valles C., Osorio F.G., Gutierrez-Fernandez A., De Los Angeles A., Bueno C., Menendez P., Martin-Subero J.I., Daley G.Q., Freije J.M., Lopez-Otin C. NF-kappaB activation impairs somatic cell reprogramming in ageing. Nat. Cell Biol. 2015;17:1004–1013. doi: 10.1038/ncb3207. [DOI] [PubMed] [Google Scholar]
- Srinivasan L., Sasaki Y., Calado D.P., Zhang B., Paik J.H., DePinho R.A., Kutok J.L., Kearney J.F., Otipoby K.L., Rajewsky K. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R., Martin M.C., Garcia A., Cigudosa J.C., Ramirez J.C., Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 2014;5:3964. doi: 10.1038/ncomms4964. [DOI] [PubMed] [Google Scholar]
- Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.M., Hochedlinger K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat. Cell Biol. 2011;13:497–505. doi: 10.1038/ncb0511-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S., Otsu M., Matsuzaka E., Konishi C., Takagi H., Hanada S., Mochizuki S., Nakauchi H., Imai K., Tsuji K., Ebihara Y. Screening of drugs to treat 8p11 myeloproliferative syndrome using patient-derived induced pluripotent stem cells with fusion gene CEP110-FGFR1. PLoS One. 2015;10:e0120841. doi: 10.1371/journal.pone.0120841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z., Zhan H., Mali P., Dowey S., Williams D.M., Jang Y.Y., Dang C.V., Spivak J.L., Moliterno A.R., Cheng L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmazer A., de Lazaro I., Taheri H. Reprogramming cancer cells: a novel approach for cancer therapy or a tool for disease-modeling? Cancer Lett. 2015;369:1–8. doi: 10.1016/j.canlet.2015.06.027. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Wu W.S. Sodium butyrate promotes generation of human induced pluripotent stem cells through induction of the miR302/367 cluster. Stem Cells Dev. 2013;22:2268–2277. doi: 10.1089/scd.2012.0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.