

Abstract
Pacemaker activity of the sino‐atrial node generates the heart rate. Disease of the sinus node and impairment of atrioventricular conduction induce an excessively low ventricular rate (bradycardia), which cannot meet the needs of the organism. Bradycardia accounts for about half of the total workload of clinical cardiologists. The ‘sick sinus’ syndrome (SSS) is characterized by sinus bradycardia and periods of intermittent atrial fibrillation. Several genetic or acquired risk factors or pathologies can lead to SSS. Implantation of an electronic pacemaker constitutes the only available therapy for SSS. The incidence of SSS is forecast to double over the next 50 years, with ageing of the general population thus urging the development of complementary or alternative therapeutic strategies. In recent years an increasing number of mutations affecting ion channels involved in sino‐atrial automaticity have been reported to underlie inheritable SSS. L‐type Cav1.3 channels play a major role in the generation and regulation of sino‐atrial pacemaker activity and atrioventricular conduction. Mutation in the CACNA1D gene encoding Cav1.3 channels induces loss‐of‐function in channel activity and underlies the sino‐atrial node dysfunction and deafness syndrome (SANDD). Mice lacking Cav1.3 channels (Cav1.3−/−) fairly recapitulate SSS and constitute a precious model to test new therapeutic approaches to handle this disease. Work in our laboratory shows that targeting G protein‐gated K+ (I KACh) channels effectively rescues SSS of Cav1.3−/− mice. This new concept of ‘compensatory’ ion channel targeting shines new light on the principles underlying the pacemaker mechanism and may open the way to new therapies for SSS.
Keywords: cardiac electrophysiology, Cav1.3 channels, Girk4 channels, HCN4 channels, pacemaker activity, sick sinus syndrome, sino‐atrial node
Abbreviations
- AVN
atrioventricular node
- [Ca2+]i
intracellular Ca2+
- HRV
heart rate variability
- RyR
ryanodine receptors
- SAN
sino‐atrial node
- SANDD
sinus node dysfunction and deafness syndrome
- SSS
sick sinus syndrome
Introduction: cardiac pacemaker activity and sinus node dysfunction
Pacemaker activity of the sino‐atrial node (SAN) underlies heart automaticity and constantly regulates heart rate. The cardiac conduction system ensures a proper spatial and timely spread of the SAN impulse to the ventricles. SAN automaticity is due to a specialized population of myocytes referred to as ‘pacemaker’ cells (Mangoni & Nargeot, 2008). These cells are differentiated by the activation of a specialized genetic differentiation programme, which is distinct from that of the working myocardium and display peculiar morphology (Christoffels et al. 2010). The electric hallmark of pacemaker cells is the presence of the diastolic depolarization phase of the action potential, which leads the membrane voltage from the maximum diastolic potential to the threshold of the following action potential (DiFrancesco et al. 1986). This specific genetic programme of SAN differentiation induces the expression of a set of ion channels that differ qualitatively and quantitatively from those of the other cardiac chambers (Marionneau et al. 2005; Chandler et al. 2009). In comparison to the myocytes of the working myocardium, pacemaker cells are characterized also by the presence of local intracellular Ca2+ () release from ryanodine receptors (RyRs) during the diastolic depolarization at discrete points of the subsarcolemmal space (Huser et al. 2000; Rigg et al. 2000; Bogdanov et al. 2001). During spontaneous activity, local release precedes the cell‐wide [Ca2+]i transient evoked in correspondence to the action potential upstroke phase. The cardiac pacemaker mechanism has been the subject of an intensive research effort during the last 20 years (Mangoni & Nargeot, 2008). Research in the field is centred on three main issues: which specific genetic pathways control pacemaker differentiation; how membrane ion channels and RyR‐dependent release generate spontaneous activity; and the identification of downstream effectors of membrane receptors regulating pacemaker activity. The picture emerging from this intensive and strenuous work is that an intricate and still only partially understood functional association of membrane ion channels and dynamics generates SAN pacemaker activity (for recent reviews see: Mangoni & Nargeot, 2008; DiFrancesco, 2010; Lakatta et al. 2010; Monfredi et al. 2013; Capel & Terrar, 2015). Several classes of ion channels driving inward current contribute to the diastolic depolarization, including those for the hyperpolarization‐activated current I f (DiFrancesco, 2010), L‐ and T‐type voltage‐gated Ca2+ channels (Mesirca et al. 2015), and those for the sustained inward current I st (Mitsuiye et al. 2000). In addition, voltage‐independent channels and Ca2+‐activated conductances have been proposed to contribute to pacemaking. These currents are generally referred to as ‘background’ conductances. These mechanisms have not been fully understood yet; however, chloride channels and channels belonging to TRP gene families contribute to background currents (see for review Capel & Terrar, 2015). Finally, the Na+/Ca2+ exchanger contributes to pacemaking (Ju & Allen, 1998) via its functional interaction with RyRs (Lakatta et al. 2010) and possibly by acting as a ‘background’ conductance (Kang & Hilgemann, 2004).
Beside the great interest in identifying the molecular pathways involved in pacemaking as a fundamental physiological function, this research field has important translational aspects for clinical cardiology. Diseases of heart automaticity and/or impulse conduction are relatively common conditions. SAN or atrioventricular failures result in low ventricular rate, which is inappropriate to meet the physiological demand (Sanders et al. 2014). This condition, commonly referred to as ‘bradycardia’, is a primary cause of electronic pacemaker implantations. In addition, SAN disease is frequently associated with atrial tachyarrhythmias, including atrial fibrillation and flutter. The ‘sick sinus’ syndrome (SSS) is a typical example of tachycardia–bradycardia syndromes in which patients alternate from periods of low SAN rate and consequent bradycardia to atrial fibrillation (Semelka et al. 2013). The aetiology of SSS disease can be complex. Indeed, a broad spectrum of acquired or genetic causes can induce SAN failure or block of atrioventricular conduction. Among acquired causes, ageing, endocrine dysfunction, inflammation, infection and autoimmune disease, as well as improper medication intake or intoxication, are important risk factors (Semelka et al. 2013; Monfredi & Boyett, 2015). Irrespective of the underlying causes, symptomatic SAN disease and conduction block are handled by electronic pacemaker implantation, which constitutes the only currently available therapy. Because of the constantly increasing incidence of SAN disease, the number of electronic pacemaker implantations has been forecast to double over the next 50 years, thus making pathologies of heart automaticity a potential future societal problem (Jensen et al. 2014).
Pacemaker activity and models of sinus dysfunction
During the last few years the description of mutations in families showing SAN disease has significantly improved knowledge of the aetiological causes of this disease. These mutations affect a wide range of different gene families, including genes encoding cytoskeletal and structural proteins, membrane ion channels and RyRs (Monfredi & Boyett, 2015). While inherited channelopathies do not account for the majority of SAN diseases, they nevertheless constitute a precious model system to investigate the pacemaker mechanism and to test potential therapeutic strategies. Our group has been the first to introduce the use of mouse SAN pacemaker cells in the field (Mangoni, 2001; Mangoni & Nargeot, 2001), showing the feasibility of this approach and the close similarity between the expression pattern of ionic currents and channels between the mouse SAN and that of other animal models used so far (Marionneau et al. 2005). We also showed that this approach was extendable also to atrioventricular node (AVN) (Mangoni et al. 2006 b) and Purkinje fibres (Miquerol et al. 2004). These works opened the way to investigate the pacemaker mechanism using genetically modified mice and to associate gene activity to a specific function in automaticity (Mangoni et al. 2006 a). Many laboratories in Europe and North America have since used mouse models with altered pacemaker activity.
While research conducted on transgenic mice undoubtedly shed a new light in the cardiac pacemaker mechanism, it also demonstrated that these models fairly recapitulate SAN and impulse conduction disease observed in humans (see for review Monfredi & Boyett, 2015). This symposium article will focus on models of congenital SAN diseases due to mutations in I f‐mediating hyperpolarization‐activated f‐channels (HCN) and voltage‐gated L‐type Cav1.3 Ca2+ channels.
Cardiac f‐channels underlie the hyperpolarization‐activated current (I f) in the SAN, as well as in the AVN and Purkinje fibres (DiFrancesco, 2010). The sympathetic and parasympathetic branches of the autonomic nervous system regulate the open probability of f‐channels in opposite ways through the intracellular concentration of cAMP (DiFrancesco & Tortora, 1991), which directly binds to the channel C‐terminus (Ludwig et al. 1998). f‐Channels play a major role in SAN pacemaker activity (DiFrancesco, 2006), impulse conduction (Baruscotti et al. 2011; Mesirca et al. 2014) and heart rate determination in mice and humans (Milanesi et al. 2006; Alig et al. 2009; D'Souza et al. 2014; Baruscotti et al. 2016). The relevance of I f in cardiac pacemaking is underscored by the development of the heart rate reducing drug ivabradine, which is now prescribed in the clinical practice for ischaemic heart disease (Bucchi et al. 2007). Importantly, several forms of bradycardia (DiFrancesco, 2015) and tachycardia (Baruscotti et al. 2016) induced by mutations affecting the predominant SAN f‐channel isoforms HCN4 have been described during the last years.
While I f is an important inward current, it is not the only ionic current to be activated during the diastolic depolarization phase. L‐type Cav1.3 channels are also activated during the diastolic depolarization phase of SAN cells (Mangoni et al. 2003; Torrente et al. 2016). Cav1.3 channels present peculiar steady‐state activation and inactivation properties in comparison to the classic cardiac excitation–contraction coupling L‐type Cav1.2 isoform. In cardiac cells and neurons, Cav1.3 channel activation and inactivation lie about half way between those of low voltage‐activated T‐type and high voltage‐activated Cav1.2 L‐type channels. Because of these biophysical properties, Cav1.3 channels play also an important role in regulation of firing in neuroendocrine cells and in some central neurons (see for recent review Vandael et al. 2015) and in the control of dendritic Ca2+ oscillations in dopaminergic neurons (Guzman et al. 2009). In SAN cells, the Cav1.3‐mediated I Ca,L activation threshold is close to –55 mV under adrenergic activation (Mangoni et al. 2003). Even under basal conditions, Cav1.3‐mediated I Ca,L half‐activation and ‐inactivation voltages lie 20 mV negative to that of Cav1.2‐mediated I Ca,L, thus distinguishing the function of Cav1.3 channels as ‘pacemaker’ channels from that of Cav1.2 channels contributing to the SAN action potential upstroke (Mangoni et al. 2006 a). Similarly to Cav1.2, the SAN Cav1.3‐mediated I Ca,L is positively regulated by the cAMP‐dependent signalling pathway (Mangoni et al. 2003). However, new studies are needed to elucidate the other potential cellular factors that regulate Cav1.3 channels in SAN cells.
Global gene knockout of Cav1.3 channels induces a 60–70% reduction in the density of SAN I Ca,L (Mangoni, 2001; Mangoni et al. 2003) and a positive shift in the current half‐activation voltage (Mangoni, 2001; Zhang et al. 2002; Mangoni et al. 2003). Consequently, Cav1.3−/− mice show bradycardia and atrioventricular block (Platzer et al. 2000; Marger et al. 2011; Mesirca et al. 2016). In addition, Cav1.3 channels contribute to pacemaking not only by supplying inward current in the full range of voltages of the diastolic depolarization, but also by regulating the activity of RyRs (Torrente et al. 2016). Previous work has shown that reduction in Cav1.3‐mediated I Ca,L contributes to SAN disease due to loss‐of‐function of ankyrin‐B (Hund & Mohler, 2008). However, only recently it has been possible to show directly that loss‐of‐function of Cav1.3 channel gating induces SAN bradycardia and congenital deafness in humans (Baig et al. 2011). It has also been proposed that reduction in Cav1.3‐mediated I Ca,L is responsible for bradycardia observed in patients affected by chronic iron overload (Rose et al. 2011). Collectively, these works indicate that a significant fraction of SSS and atrioventricular conduction diseases could be ascribed to functional alteration in ion channels contributing to the diastolic depolarization and that mouse models of human channelopathies can provide a valuable tool to test potential molecular and pharmacological rescuing strategies.
The role of the autonomic nervous system input in establishing the hallmarks of sino‐atrial and atrioventricular dysfunctions
During the last 10 years, different groups have made an intensive effort to dissect the consequences for pacemaker activity and heart rate following knockout of the predominant SAN f‐channel isoform HCN4. Global or cardiac specific knockout of hcn4 in mice is embryonically lethal (Stieber et al. 2003). Conditional hcn4 knockout in adult mice gave apparently contrasting results, with phenotypes ranging from SAN pauses (Herrmann et al. 2007) to SAN bradycardia and quickly developing lethal heart block (Baruscotti et al. 2011). The effects on cardiac pacemaker activity of conditional and time‐controlled expression of dominant negative HCN4 subunits lacking either cAMP‐dependent regulation of the channel gating (Schulze‐Bahr et al. 2003; Alig et al. 2009) or mutated pore sequence silencing channel conductance (Mesirca et al. 2014) have been recently described. Incidentally, these mouse lines express mutated HCN4 channels similar to those found in two familial congenital SAN dysfunctions (Schweizer et al. 2010; Schweizer et al. 2014). In particular, our group showed that genetic silencing of I f in mice via conditional time‐ and cardiac‐specific expression of non‐conducting HCN4 mutant channels (HCN4‐AYA) induces complex arrhythmia with SAN dysfunction, atrioventricular block and high incidence of ventricular tachycardia (Mesirca et al. 2014). While not being lethal, this phenotype shares qualitative similarities with that reported by Baruscotti et al. (2011). This mouse model also recapitulates familial SAN dysfunction induced by a mutation in the same pore motif of the HCN4 channel subunit (Fig. 1), which reduces (but does not eliminate) f‐channel conductance (Schweizer et al. 2010, 2014). Expression of HCN4‐AYA channels induces complete silencing of I f in the SAN, the AVN and Purkinje fibres. The basal spontaneous activity of automatic cells is slowed, with SAN cells being the most affected cell type in comparison to the other cells of the conduction system. Slowing of spontaneous SAN activity by I f silencing augments the Ca2+ content of the sarcoplasmic reticulum, impairs local diastolic release and delays also the generation of spontaneous [Ca2+]i transients. This observation suggests that the activity of f‐channels is an important factor in coupling membrane voltage to RyR‐dependent release (Lakatta et al. 2010). I f loss‐of‐function does not affect the relative responsiveness of heart rate to adrenergic activation, and cannot ameliorate SAN dysfunction (SAN pauses) and atrioventricular blocks, nor does it improve ventricular tachycardia (Mesirca et al. 2014). However, pharmacological inhibition of the autonomic nervous system by combined injection of atropine and propranolol ‘rescues’ atrioventricular conduction of HCN4‐AYA‐expressing hearts. This observation is reminiscent of the effects of the autonomic nervous system in mice conditionally expressing HCN4 channels lacking cAMP‐dependent regulation. Indeed, while these mice have lower basal heart rate than their control counterparts, inhibition of the autonomic nervous system abolishes the difference in heart rate of these strains (Alig et al. 2009). These observations showed, for the first time, that the autonomic nervous system input in some way regulates the functional role of f‐channels in pacemaker activity in vivo and that this input is responsible for the manifestations of SAN dysfunction and atrioventricular conduction block. Similarly, pharmacological inhibition of the autonomic nervous system is able to reduce SAN dysfunction and to rescue atrioventricular dysfunction of Cav1.3−/− mice, showing that the role of the autonomic nervous system in modulating the symptoms of automaticity failure is not specific to f‐channel loss‐of‐function (Mesirca et al. 2016).
Figure 1. Examples of two congenital dysfunctions of heart automaticity in humans, related to loss‐of‐function in Cav1.3 channels (SANDD; A and B), or familial bradycrdia due to a mutation in the pore sequence of the f‐channel HCN4 (C–F) .
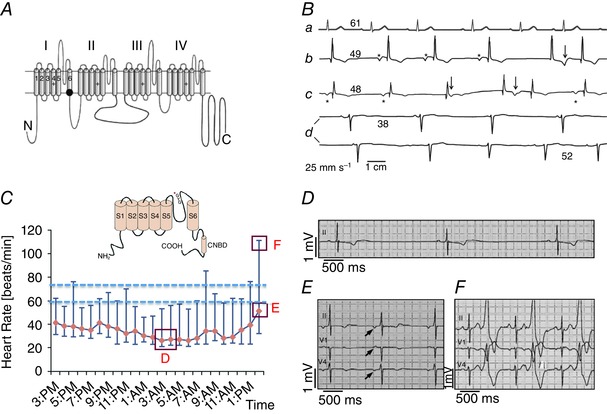
A, schematic transmembrane topology of the α1 subunit of the Cav1.3 Ca2+ channel. The black dot indicates the position of the SANDD mutation. B, sample Holter ECG recordings from individuals with normal heart rate (a), or from SANDD affected subjects, who were homozygous for the mutation (b–d). The number above each ECG baseline indicates the averaged heart rate. Asterisks mark P waves. Arrows identify ECG time points suggesting P waves coincide with T waves. Adapted from Baig et al. (2011), with permission. C, daily Holter heart rate profile of a patient with familial SAN dysfunction due to HCN4 pore mutation. Data points indicate hourly averaged data between the maximum and minimum heart rate of a patients carrying the glycine–tyrosine–glycine (GYG) mutation in the pore motif of HCN4. The inset shows a schematic transmembrane topology of the f‐channel HCN4 subunit. The asterisk indicates the position of the mutation within the motif of the channel pore. Dashed lines indicate the expected basal heart rate interval for normal subjects. Red boxes mark time points corresponding to ECG samples shown in panels D–F. D, a sample recording of SAN bradycardia. E and F, ectopic atrial rhythms and ventricular bigeminisms, respectively. Adapted from Schweizer et al. (2014), with permission.
The ‘compensatory’ channel‐targeting concept
The G protein‐activated K+ current (I KACh) is involved in the negative chronotropic effect of acetylcholine (ACh) on heart rate (Giles & Tsien, 1975; Giles & Noble, 1976; Noma & Trautwein, 1978; DiFrancesco et al. 1989). A gene family comprising four channel subunit isoforms named GIRK1–4 encodes I KACh (Wickman & Clapham, 1995). Only GIRK1 and GIRK4 subunits are expressed in the heart, but mice lacking GIRK4 channels (Girk4−/−) are devoid of I KACh since GIRK1 subunits cannot be targeted to the membrane in these mice (Wickman et al. 1998). A previous study showed that in rabbit SAN I KACh is activated at concentrations of ACh higher than those regulating f‐channels (DiFrancesco et al. 1989). However, I KACh also exhibits a background conductance due to spontaneous channel opening (Ito et al. 1994). Inactivation of this background conductance induces a detectable elevation of the basal pacemaker activity of Girk4−/− SAN cells (Mesirca et al. 2013). In addition, there is solid evidence that I KACh exerts a tonic regulation on heart rate in mice (Wickman et al. 1998; Mesirca et al. 2013). Indeed, Girk4−/− mice show a significant reduction of heart rate variability (HRV) even in the high frequency domain (Wickman et al. 1998; Mesirca et al. 2013), as well as a delayed recovery of resting heart rate following physical exercise (Mesirca et al. 2013). We thus reasoned that abolition of atrioventricular blocks observed in HCN4‐AYA mice after administration of atropine and propranolol could be explained by I KACh suppression by atropine, via blockade of muscarinic receptors. We validated this working hypothesis by crossing HCN4‐AYA with Girk4−/− mice (HCN4‐AYA/Girk4−/−, Fig. 2). These animals are devoid of SAN dysfunction and atrioventricular blocks, as well as of the other arrhythmias associated with genetic silencing of f‐channels, including atrial and ventricular (Fig. 2) tachycardias (Mesirca et al. 2014). In contrast to Girk4−/− mice, the recovery of resting heart rate following exercise in HCN4‐AYA/Girk4−/− mice was similar to that of control animals (Mesirca et al. 2014). Taken together, these observations suggested that genetic ablation of GIRK4 (I KACh) channels was restoring equilibrium between ionic currents stimulating (I f) or braking (I KACh) cardiac pacemaker activity. Inactivation of GIRK4 channels was thus acting as a ‘compensatory’ mechanism allowing the SAN to control atrial rate and to suppress ventricular tachycardias by normalizing atrioventricular conduction.
Figure 2. Inactivation of GIRK4 channels rescues the complex cardiac arrhythmia observed in mice with silenced If (HCN4‐AYA) .
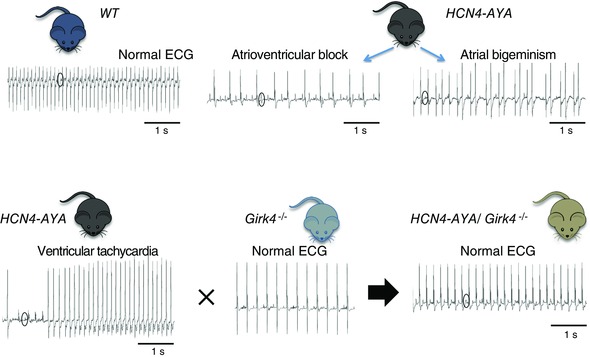
Wild‐type (WT) mice exhibit fast heart rate and a normal ECG profile (top left). In contrast, HCN4‐AYA double transgenic mice show low SAN rate, 2nd degree atrioventricular blocks and signs of atrial arrhythmia (atrial bigeminism, top centre and right). HCN4‐AYA mice show also ventricular tachycardia (bottom left). Crossing HCN4‐AYA mice with Girk4−/− mice produces HCN4‐AYA/Girk4−/− mice with normal ECG (Mesirca et al. 2014). Circles indicate P waves in ECG recordings.
Cav1.3−/− mice constitute a faithful model of tachy–brady syndromes, since they present with both bradycardia (Platzer et al. 2000; Zhang et al. 2002; Mesirca et al. 2016) and atrial tachyarrhythmia (Zhang et al. 2005; Mesirca et al. 2016). In addition, the similarity between SSS observed in Cav1.3−/− mice and SAN dysfunction associated with SANDD (Figs 1 A and 3) indicates that this mouse strain can be relevant for testing new therapeutic approaches. Similarly to HCN4‐AYA mice, genetic ablation of I KACh in Cav1.3−/− mice produces Cav1.3−/−/Girk4−/− animals presenting normal heart rate and rhythm (Mesirca et al. 2016). Taken together, the phenotype of HCN4‐AYA/Girk4−/− and Cav1.3−/−/Girk4−/ − demonstrates that it is possible to rescue SAN dysfunction and improve heart rate of genetically modified mice models of congenital channelopathy of heart automaticity. Importantly, pharmacological block of I KACh by the bee venom petide tertiapin mimics rescuing of Cav1.3−/− heart rate by genetic inactivation of GIRK4 channels, which demonstrates that improvement of pacemaker activity is not the result of remodelling phenomena (Fig. 3).
Figure 3. Either genetic ablation (top) or pharmacological targeting (bottom) of Girk4‐mediated IKACh channels is able to prevent sick sinus syndrome and atrioventricular block in SANDD mimicking Cav1.3−/− mice (Mesirca et al. 2016 ) .
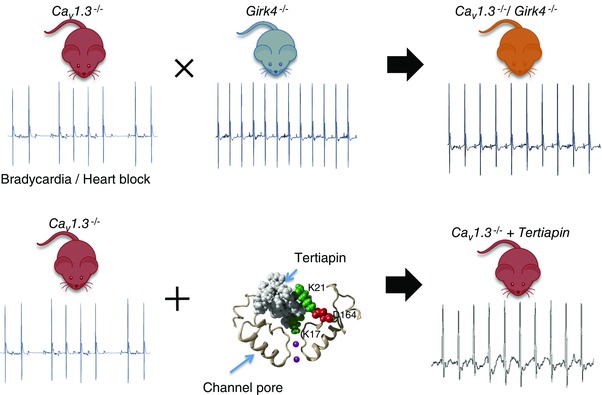
Telemetric ECG recordings of Cav1.3−/− mice show SAN bradycardia and 2nd degree atrioventricular block (top left). In contrast, Girk4−/− mice present with normal SAN rate and no atrioventricular blocks (top centre). Crossing these mouse lines produces viable Cav1.3−/−/Girk4−/− animals with normal heart rate and rhythm (top right). When Cav1.3−/− mice undergo an intraperitoneal injection of the GIRK pore blocker tertiapin‐Q (tertiapin, bottom centre) normalization of heart rate is observed (bottom right). The central panel shows a close up view of a structural model of tertiapin bound to the GIRK pore. K17 and D164 indicate residues important for tertiapin activity.
Prevention of atrial tachycardias typical of Cav1.3−/− hearts by inactivation of GIRK4 channels can be due to normalization of the atrial action potential duration and/or to improvement of SAN rate and rhythm (Mesirca et al. 2016). Investigation of the mechanism underlying rescuing of heart automaticity and impulse conduction in Cav1.3−/‐ mice by I KACh ablation shows that targeting GIRK4 channels reduces the cholinergic negative chronotropic response in rhythmogenic centres. Since cholinergic I KACh activation generates an outward current it can be expected that loss of f‐ or Cav1.3 channels will reduce the depolarization reserve of SAN pacemaker cells. In this context, analysis of the net current flowing during the SAN diastolic depolarization indicates that in Cav1.3−/− pacemaker cells ACh switches the net diastolic current from inward to outward, predicting arrest of automaticity. In contrast, the net diastolic current is maintained in the inward direction in Cav1.3−/−/Girk4−/− SAN cells even under ACh, thus predicting regular pacemaking (Mesirca et al. 2016).
The concept of ‘compensatory’ ion channel targeting is summarized and idealized in Fig. 4. Here the behaviour of spontaneous activity is compared with heart rate and rhythm of wild‐type mice, as well as with mice having Cav1.3 loss‐of‐function and with Cav1.3−/−/Girk4−/− mice showing ‘compensated’ pacemaker activity and heart rate. SAN automaticity depends on the equilibrium between inward and outward ionic currents, which regulate pacemaker activity in opposite ways. Normal SAN rate and atrioventricular conduction are possible when inward and outward currents balance in such a way that the direction of the sum of membrane currents during the diastolic depolarization is inward. Under this condition, the SAN will generate constant pacemaker activity at a rate determined by the inputs of the sympathetic and parasympathetic branches of the autonomic nervous system. Similarly the AVN will conduct the SAN impulse to the ventricles in a 1:1 ratio because the depolarization reserve of conduction cells will allow reaching the action potential threshold. This balance is impaired following Cav1.3 loss‐of‐function, and during opening of I KACh channels the net current will switch in the outward direction. This will eventually cause failure in the generation of SAN (or AVN) action potentials (asterisk in Fig. 4) and consequent low SAN rate with pauses and atrioventricular blocks (Mesirca et al. 2016). Targeting I KACh channels in Cav1.3‐deficient cells will restore the balance between inward and outward currents preventing SAN bradycardia and SAN pauses, and allowing constant 1:1 conduction through AVN. While Fig. 4 shows an example of Cav1.3 loss‐of‐function, results indicate that the concept could be extended to loss or decreased function of other channels involved in pacemaking. Indeed, as discussed above, genetic ablation of I KACh in HCN4‐AYA mice effectively rescues the complex arrhythmic profile observed in these mice (Mesirca et al. 2014). This additional evidence is suggestive of a high degree of functional redundancy between inward ionic currents contributing in the pacemaker mechanisms, so that the loss of one current can be compensated by ablation of I KACh. Interestingly, I KACh ablation seems able to compensate also for partial loss of RyR‐dependent Ca2+ release in SAN cells, provided that Cav1.3 channels are still present (Mesirca et al. 2016).
Figure 4. Summary of the principle of re‐establishment of the equilibrium among pacemaker mechanisms by ‘compensatory’ targeting of an ion channel in the mouse heart .
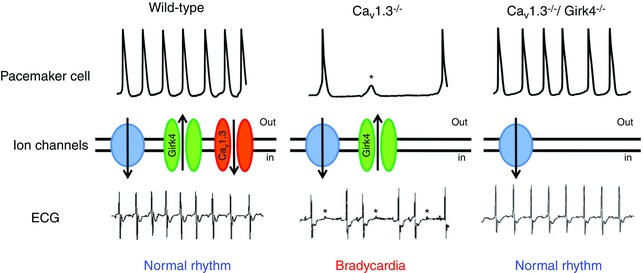
Top, samples of consecutive spontaneous action potentials recorded in isolated SAN pacemaker cells from wild‐type (left), Cav1.3−/− (centre) and Cav1.3−/−/Girk4−/− (right) mice. Middle, ion channels in the plasmalemma of SAN pacemaker cells. Green identifies I KACh (GIRK4) channels, orange shows L‐type Cav1.3 channels and the blue circle represents the idealized sum of other inward currents involved in the generation of pacemaker activity. Bottom, samples of ECGs of wild‐type mice with normal heart rate (left), Cav1.3−/− mice showing slow SAN rate and atrioventricular blocks leading to bradycardia (centre), or Cav1.3−/−/Girk4−/− mice with ‘compensated’ heart rate (left). Asterisks indicate failure of action potential generation (top line) or atrioventricular blocks (bottom line). Adapted after permission from Mesirca et al. (2016).
Conclusions
Failure in the generation or regulation of the cardiac impulse is a common clinical condition which, with the ageing of the population, is becoming a societal issue. The aetiological and mechanistic bases underlying congenital diseases in SAN automaticity and atrioventricular conduction have remained undefined for many years. However, the discovery of mutations in ion channels involved in pacemaker activity sheds new light on these pathologies, showing that some of them were indeed channelopathies (Dobrzynski et al. 2007; Monfredi & Boyett, 2015).
The development and functional exploration of mouse lines that recapitulate SSS and associated arrhythmias allows new therapeutic strategies to be tested with the aim of offering alternative or complementary therapies to the implantation of electronic pacemakers. ‘Compensatory’ I KACh targeting will possibly constitute a new research avenue to manage bradycardia and SSS (Mesirca et al. 2016). A corollary consideration emerging from this work is that, at least in the case of channelopathies of cardiac pacemaker activity, while I KACh constitutes a physiological mechanism regulating normal heart rate under physiological conditions, its activity becomes highly detrimental to impulse generation and conduction under SSS conditions. This unwanted action eventually causes the symptoms of SSS and associated arrhythmias in mice. Thus, compensatory I KACh targeting aims chiefly to re‐establishment an intrinsic equilibrium in pacemaker mechanism that is lost during the onset and progression of SAN disease. Future works will define the clinical applicability of this approach and whether, beside I KACh, other ion channels could be targeted to improve and normalize heart rate in cardiac disease.
Additional information
Competing interests
The authors declare no conflicts of interest.
Author contributions
All authors have contributed to writing the paper. All authors approved the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors, qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors’ work is supported by the Agence Nationale pour la Recherche (ANR, Paris) grant ANR‐15‐CE14‐0004. P.M was supported by the CavNet a Research Training Network (RTN) funded through the European Union Research Programme (6FP) MRTN‐CT‐2006‐035367. The group is a member of the Laboratory of Excellence ‘Ion Channel Science and Therapeutics’ (ICST) supported by a grant from ANR (ANR‐11‐LABX‐0015).
Acknowledgements
We thank Prof. Philippe Chevalier (CHU, Lyon, France) for helpful discussion on SSS. We are indebted to the staff of the RAM animal facility of Montpellier for managing mouse lines.
Biography
Matteo Mangoni is Research Director in the Department of Physiology of the Institute of Functional Genomics in Montpellier (France). He obtained his doctoral degree in Physiology at the University of Milan (Italy) with Dario DiFrancesco. His team works on the mechanisms underlying cardiac automaticity using a wide collection of genetically modified mouse strains. Pietro Mesirca is Research Associate in the same department. He obtained his doctoral degree in Physics at the University of Bologna (Italy). His work has contributed to the understanding of the antagonistic roles of G protein‐activated K+ (I KACh) and other critical inward currents underlying cardiac automaticity such as I f and Cav1.3‐mediated I Ca,L. Isabelle Bidaud is Senior Research Engineer in the same Department. She obtained her doctoral degree in Molecular Biology at the University of Paris VI. She characterized the genetic and pharmacological factors able to regulate heart rate in mice recapitulating disease of cardiac automaticity.

Contributor Information
Pietro Mesirca, Email: pietro.mesirca@igf.cnrs.fr.
Matteo E. Mangoni, Email: matteo.mangoni@igf.cnrs.fr
References
- Alig J, Marger L, Mesirca P, Ehmke H, Mangoni ME & Isbrandt D (2009). Control of heart rate by cAMP sensitivity of HCN channels. Proc Natl Acad Sci USA 106, 12189–12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, Ali A, Ahmad I, Sinnegger‐Brauns MJ, Brandt N, Engel J, Mangoni ME, Farooq M, Khan HU, Nurnberg P, Striessnig J & Bolz HJ (2011). Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci 14, 77–84. [DOI] [PubMed] [Google Scholar]
- Baruscotti M, Bucchi A, Milanesi R, Paina M, Barbuti A, Gnecchi‐Ruscone T, Bianco E, Vitali‐Serdoz L, Cappato R & DiFrancesco D (2016). A gain‐of‐function mutation in the cardiac pacemaker HCN4 channel increasing cAMP sensitivity is associated with familial Inappropriate Sinus Tachycardia. Eur Heart J DOI:10.1093/eurheartj/ehv582. [DOI] [PubMed] [Google Scholar]
- Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, Gnecchi‐Rusconi T, Montano N, Casali KR, Micheloni S, Barbuti A & Difrancesco D (2011). Deep bradycardia and heart block caused by inducible cardiac‐specific knockout of the pacemaker channel gene Hcn4. Proc Natl Acad Sci USA 108, 1705–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov KY, Vinogradova TM & Lakatta EG (2001). Sinoatrial nodal cell ryanodine receptor and Na+‐Ca2+ exchanger: molecular partners in pacemaker regulation. Circ Res 88, 1254–1258. [DOI] [PubMed] [Google Scholar]
- Bucchi A, Barbuti A, Baruscotti M & DiFrancesco D (2007). Heart rate reduction via selective ‘funny’ channel blockers. Curr Opin Pharmacol 7, 208–213. [DOI] [PubMed] [Google Scholar]
- Capel RA & Terrar DA (2015). The importance of Ca2+‐dependent mechanisms for the initiation of the heartbeat. Front Physiol 6, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P, Difrancesco D, Baruscotti M, Longhi R, Anderson RH, Billeter R, Sharma V, Sigg DC, Boyett MR & Dobrzynski H (2009). Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. Circulation 119, 1562–1575. [DOI] [PubMed] [Google Scholar]
- Christoffels VM, Smits GJ, Kispert A & Moorman AF (2010). Development of the pacemaker tissues of the heart. Circ Res 106, 240–254. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D (2006). Serious workings of the funny current. Prog Biophys Mol Biol 90, 13–25. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D (2010). The role of the funny current in pacemaker activity. Circ Res 106, 434–446. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D (2015). HCN4, sinus bradycardia and atrial fibrillation. Arrhythm Electrophysiol Rev 4, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D, Ducouret P & Robinson RB (1989). Muscarinic modulation of cardiac rate at low acetylcholine concentrations. Science 243, 669–671. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Ferroni A, Mazzanti M & Tromba C (1986). Properties of the hyperpolarizing‐activated current (i f) in cells isolated from the rabbit sino‐atrial node. J Physiol 377, 61–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D & Tortora P (1991). Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351, 145–147. [DOI] [PubMed] [Google Scholar]
- Dobrzynski H, Boyett MR & Anderson RH (2007). New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115, 1921–1932. [DOI] [PubMed] [Google Scholar]
- D'Souza A, Bucchi A, Johnsen AB, Logantha SJ, Monfredi O, Yanni J, Prehar S, Hart G, Cartwright E, Wisloff U, Dobryznski H, DiFrancesco D, Morris GM & Boyett MR (2014). Exercise training reduces resting heart rate via downregulation of the funny channel HCN4. Nat Commun 5, 3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles W & Noble SJ (1976). Changes in membrane currents in bullfrog atrium produced by acetylcholine. J Physiol 261, 103–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles W & Tsien RW (1975). Effects of acetylcholine on membrane currents in frog artrial muscle. J Physiol 246, 64P–66P. [PubMed] [Google Scholar]
- Guzman JN, Sanchez‐Padilla J, Chan CS & Surmeier DJ (2009). Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29, 11011–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann S, Stieber J, Stockl G, Hofmann F & Ludwig A (2007). HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J 26, 4423–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hund TJ & Mohler PJ (2008). Ankyrin‐based targeting pathway regulates human sinoatrial node automaticity. Channels (Austin) 2, 404–406. [DOI] [PubMed] [Google Scholar]
- Huser J, Blatter LA & Lipsius SL (2000). Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J Physiol 524, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Ono K & Noma A (1994). Background conductance attributable to spontaneous opening of muscarinic K+ channels in rabbit sino‐atrial node cells. J Physiol 476, 55–68. [PMC free article] [PubMed] [Google Scholar]
- Jensen PN, Gronroos NN, Chen LY, Folsom AR, deFilippi C, Heckbert SR & Alonso A (2014). Incidence of and risk factors for sick sinus syndrome in the general population. J Am Coll Cardiol 64, 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YK & Allen DG (1998). Intracellular calcium and Na+–Ca2+ exchange current in isolated toad pacemaker cells. J Physiol 508, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TM & Hilgemann DW (2004). Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature 427, 544–548. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, Maltsev VA & Vinogradova TM (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res 106, 659–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F & Biel M (1998). A family of hyperpolarization‐activated mammalian cation channels. Nature 393, 587–591. [DOI] [PubMed] [Google Scholar]
- Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J & Nargeot J (2003). Functional role of L‐type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA 100, 5543–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangoni ME, Couette B, Marger L, Bourinet E, Striessnig J & Nargeot J (2006. a). Voltage‐dependent calcium channels and cardiac pacemaker activity: From ionic currents to genes. Prog Biophys Mol Biol 90, 38–63. [DOI] [PubMed] [Google Scholar]
- Mangoni ME & Nargeot J (2001). Properties of the hyperpolarization‐activated current (If) in isolated mouse sino‐atrial cells. Cardiovasc Res 52, 51–64. [DOI] [PubMed] [Google Scholar]
- Mangoni ME & Nargeot J (2008). Genesis and regulation of the heart automaticity. Physiol Rev 88, 919–982. [DOI] [PubMed] [Google Scholar]
- Mangoni ME, Striessnig J, Platzer J & Nargeot J (2001). Pacemaker currents in mouse pacemaker cells. Circulation 104, R1047. [Google Scholar]
- Mangoni ME, Traboulsie A, Leoni AL, Couette B, Marger L, Le Quang K, Kupfer E, Cohen‐Solal A, Vilar J, Shin HS, Escande D, Charpentier F, Nargeot J & Lory P (2006. b). Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/α1G T‐type calcium channels. Circ Res 98, 1422–1430. [DOI] [PubMed] [Google Scholar]
- Marger L, Mesirca P, Alig J, Torrente A, Dubel S, Engeland B, Kanani S, Fontanaud P, Striessnig J, Shin HS, Isbrandt D, Ehmke H, Nargeot J & Mangoni ME (2011). Functional roles of Cav1.3, Cav3.1 and HCN channels in automaticity of mouse atrioventricular cells: insights into the atrioventricular pacemaker mechanism. Channels (Austin) 5, 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, Lei M, Escande D & Demolombe S (2005). Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol 562, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesirca P, Alig J, Torrente AG, Muller JC, Marger L, Rollin A, Marquilly C, Vincent A, Dubel S, Bidaud I, Fernandez A, Seniuk A, Engeland B, Singh J, Miquerol L, Ehmke H, Eschenhagen T, Nargeot J, Wickman K, Isbrandt D & Mangoni ME (2014). Cardiac arrhythmia induced by genetic silencing of ‘funny’ (f) channels is rescued by GIRK4 inactivation. Nat Commun 5, 4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesirca P, Bidaud I, Briec F, Evain S, Torrente AG, Le Quang K, Leoni AL, Baudot M, Marger L, Chung You Chong A, Nargeot J, Striessnig J, Wickman K, Charpentier F & Mangoni ME (2016). G protein‐gated IKACh channels as therapeutic targets for treatment of sick sinus syndrome and heart block. Proc Natl Acad Sci USA 113, E932–E941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesirca P, Marger L, Toyoda F, Rizzetto R, Audoubert M, Dubel S, Torrente AG, Difrancesco ML, Muller JC, Leoni AL, Couette B, Nargeot J, Clapham DE, Wickman K & Mangoni ME (2013). The G‐protein‐gated K+ channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J Gen Physiol 142, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesirca P, Torrente AG & Mangoni ME (2015). Functional role of voltage gated Ca2+ channels in heart automaticity. Front Physiol 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanesi R, Baruscotti M, Gnecchi‐Ruscone T & DiFrancesco D (2006). Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med 354, 151–157. [DOI] [PubMed] [Google Scholar]
- Miquerol L, Meysen S, Mangoni M, Bois P, van Rijen HV, Abran P, Jongsma H, Nargeot J & Gros D (2004). Architectural and functional asymmetry of the His‐Purkinje system of the murine heart. Cardiovasc Res 63, 77–86. [DOI] [PubMed] [Google Scholar]
- Mitsuiye T, Shinagawa Y & Noma A (2000). Sustained inward current during pacemaker depolarization in mammalian sinoatrial node cells. Circ Res 87, 88–91. [DOI] [PubMed] [Google Scholar]
- Monfredi O & Boyett MR (2015). Sick sinus syndrome and atrial fibrillation in older persons – A view from the sinoatrial nodal myocyte. J Mol Cell Cardiol 83, 88–100. [DOI] [PubMed] [Google Scholar]
- Monfredi O, Maltsev VA & Lakatta EG (2013). Modern concepts concerning the origin of the heartbeat. Physiology (Bethesda) 28, 74–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A & Trautwein W (1978). Relaxation of the ACh‐induced potassium current in the rabbit sinoatrial node cell. Pflugers Arch 377, 193–200. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott‐Fischer A, Stephan K, Bova S, Chen H, Zheng H & Striessnig J (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L‐type Ca2+ channels. Cell 102, 89–97. [DOI] [PubMed] [Google Scholar]
- Rigg L, Heath BM, Cui Y & Terrar DA (2000). Localisation and functional significance of ryanodine receptors during β‐adrenoceptor stimulation in the guinea‐pig sino‐atrial node. Cardiovasc Res 48, 254–264. [DOI] [PubMed] [Google Scholar]
- Rose RA, Sellan M, Simpson JA, Izaddoustdar F, Cifelli C, Panama BK, Davis M, Zhao D, Markhani M, Murphy GG, Striessnig J, Liu PP, Heximer SP & Backx PH (2011). Iron overload decreases CaV1.3‐dependent L‐type Ca2+ currents leading to bradycardia, altered electrical conduction, and atrial fibrillation. Circ Arrhythm Electrophysiol 4, 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders P, Lau DH & Kalman JK (2014). Sinus node abnormalities In Cardiac Electrophysiology: From Cell to Bedside, 6th edn, ed. Zipes DP. & Jalife J, pp. 691–696. Elsevier Saunders, Philadelphia. [Google Scholar]
- Schulze‐Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O & Isbrandt D (2003). Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest 111, 1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer PA, Duhme N, Thomas D, Becker R, Zehelein J, Draguhn A, Bruehl C, Katus HA & Koenen M (2010). cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ Arrhythm Electrophysiol 3, 542–552. [DOI] [PubMed] [Google Scholar]
- Schweizer PA, Schroter J, Greiner S, Haas J, Yampolsky P, Mereles D, Buss SJ, Seyler C, Bruehl C, Draguhn A, Koenen M, Meder B, Katus HA & Thomas D (2014). The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J Am Coll Cardiol 64, 757–767. [DOI] [PubMed] [Google Scholar]
- Semelka M, Gera J & Usman S (2013). Sick sinus syndrome: a review. Am Fam Physician 87, 691–696. [PubMed] [Google Scholar]
- Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M, Hofmann F & Ludwig A (2003). The hyperpolarization‐activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci USA 100, 15235–15240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente AG, Mesirca P, Neco P, Rizzetto R, Dubel S, Barrere C, Sinegger‐Brauns M, Striessnig J, Richard S, Nargeot J, Gomez AM & Mangoni ME (2016). L‐type Cav1.3 channels regulate ryanodine receptor‐dependent Ca2+ release during sino‐atrial node pacemaker activity. Cardiovasc Res 109, 451–461. [DOI] [PubMed] [Google Scholar]
- Vandael DH, Marcantoni A & Carbone E (2015). Cav1.3 channels as key regulators of neuron‐like firings and catecholamine release in chromaffin cells. Curr Mol Pharmacol 8, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickman K, Nemec J, Gendler SJ & Clapham DE (1998). Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20, 103–114. [DOI] [PubMed] [Google Scholar]
- Wickman KD & Clapham DE (1995). G‐protein regulation of ion channels. Curr Opin Neurobiol 5, 278–285. [DOI] [PubMed] [Google Scholar]
- Zhang Z, He Y, Tuteja D, Xu D, Timofeyev V, Zhang Q, Glatter KA, Xu Y, Shin HS, Low R & Chiamvimonvat N (2005). Functional roles of Cav1.3(α1D) calcium channels in atria: insights gained from gene‐targeted null mutant mice. Circulation 112, 1936–1944. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS & Chiamvimonvat N (2002). Functional roles of Cav1.3 (α1D) calcium channel in sinoatrial nodes: insight gained using gene‐targeted null mutant mice. Circ Res 90, 981–987. [DOI] [PubMed] [Google Scholar]