

Abstract
The synthesis, processing, and joining of Okazaki fragments during DNA replication is complex, requiring the sequential action of a large number of proteins. Proliferating cell nuclear antigen, a DNA sliding clamp, interacts with and coordinates the activity of several DNA replication proteins, including the enzymes flap endonuclease 1 (FEN-1) and DNA ligase I that complete the processing and joining of Okazaki fragments, respectively. Although it is evident that maintaining the appropriate relative stoichiometry of FEN-1 and DNA ligase I, which compete for binding to proliferating cell nuclear antigen, is critical to prevent genomic instability, little is known about how the steady state levels of DNA replication proteins are regulated, in particular the proteolytic mechanisms involved in their turnover. Because DNA ligase I has been reported to be ubiquitylated, we used a proteomic approach to map ubiquitylation sites and screen for DNA ligase I-associated E3 ubiquitin ligases. We identified three ubiquitylated lysine residues and showed that DNA ligase I interacts with and is targeted for ubiquitylation by DCAF7, a specificity factor for the Cul4-DDB1 complex. Notably, knockdown of DCAF7 reduced the degradation of DNA ligase I in response to inhibition of proliferation and replacement of ubiquitylated lysine residues reduced the in vitro ubiquitylation of DNA ligase I by Cul4-DDB1 and DCAF7. In contrast, a different E3 ubiquitin ligase regulates FEN-1 turnover. Thus, although the expression of many of the genes encoding DNA replication proteins is coordinately regulated, our studies reveal that different mechanisms are involved in the turnover of these proteins.
Keywords: cell cycle, DNA replication, proliferating cell nuclear antigen (PCNA), proteasome, protein turnover, proteolysis, ubiquitin, ubiquitin ligase, DNA ligase I, ubiquitylation
Introduction
A large number of single-strand interruptions are generated during DNA replication because of the discontinuous nature of lagging strand DNA synthesis. These unlinked Okazaki fragments are joined together by a DNA ligase to generate an intact strand. In humans, there are three genes that encode DNA ligases, LIG1, LIG3, and LIG4 (1–3). There is compelling cell biology, biochemical, and molecular genetic evidence indicating that the DNA ligase encoded by the mammalian LIG1 gene, DNA ligase I (LigI),4 plays the predominant role in DNA replication. The expression of the LIG1 gene is increased when quiescent cells are induced to proliferate (4). In addition, LigI physically and functionally interacts with two key DNA replication protein complexes, proliferating cell nuclear antigen (PCNA) and replication factor C (RFC), and co-localizes with these factors in replication foci during S phase (5–8). Finally, the human ligI cell line 46BR.1G1 has a defect in joining Okazaki fragments that is complemented by the expression of wild-type LigI (6, 9).
The 46BR.1G1 cell line was established from a patient with growth retardation, sunlight sensitivity, and severe immunodeficiency (10). This individual was a compound heterozygote with one allele (Glu-566 to Lys) encoding an inactive polypeptide and the other (Arg-771 to Trp) encoding a version of LigI with about 20-fold less activity than wild-type LigI (9). Notably, knock-in mice expressing a mouse version of LigI equivalent to the Arg-771 to Trp version of human LigI exhibited genome instability and cancer predisposition in addition to a defect in joining Okazaki fragments, (11). Thus, the LIG1 gene is a member of the caretaker class of tumor suppressor genes with LigI deficiency resulting in an increased incidence of cancer.
Interestingly, elevated levels of LigI also cause genomic instability with the overexpression of LigI causing trinucleotide repeat instability in human cells (12). Similar studies in Saccharomyces cerevisiae showed that the overexpression of the yeast LigI homolog, Cdc9, not only resulted in increased instability of trinucleotide repeat sequences but also increased mitotic recombination with this effect being dependent on the interaction of Cdc9 with PCNA, not Cdc9 catalytic activity (13). Furthermore, we and others have shown that LigI is frequently overexpressed in tumor biopsies and cancer cell lines (14, 15). Together, these observations suggest that the overexpression of LigI may contribute to the genomic instability that drives tumor progression and the acquisition of resistance to chemotherapeutics.
The PCNA-interacting protein, flap endonuclease 1 (FEN-1), that processes the 5′ end of Okazaki fragments during the replication of the lagging strand is also overexpressed in a variety of cancers (16). Similar to LigI, both reduced FEN-1 activity and increased steady state levels of FEN-1 result in genomic instability in human cells (17, 18). Although these studies indicate that maintaining the appropriate relative levels of DNA replication proteins, in particular PCNA-interacting proteins involved in Okazaki fragment synthesis, processing, and ligation, is critical for genomic stability, relatively little is know about the post-translational mechanisms that regulate the steady state levels of DNA replication proteins. Although proteomic studies have shown that LigI is ubiquitylated (19–21), neither the proteins responsible for LigI ubiquitylation nor the role(s) of ubiquitylation in regulating the cellular functions of LigI have been described. In this study, we show that the steady state levels of LigI are markedly reduced in response to inhibition of cell proliferation and that LigI is also degraded by the proteasome in a ubiquitin-dependent manner. Interestingly, LigI is targeted for degradation by a different E3 ubiquitin ligase activity than the one implicated in the ubiquitylation and degradation of FEN-1 (18). Specifically, we have shown that LigI is targeted for degradation by the Cul4-DDB1 complex with specificity conferred by the conferred by the DDB1- and Cul4-associated factor 7 (DCAF7) that is also known as HAN11 (22).
Results
Periodic Expression of LigI during the Cell Cycle
Previously, it was shown that the steady state levels of LigI mRNA were markedly reduced in non-proliferating cells when compared with dividing cells and the levels of LigI mRNA increased when cells were stimulated to proliferate (4). In accordance with these observations, the steady state levels of LigI mRNA increased when confluent MCF10A cells derived from normal breast epithelium were plated at a lower density, reaching a maximum after about 15 h (Fig. 1a). Interestingly, the LigI mRNA levels declined and then peaked again at 27 h as the cell population progressed through the first cell cycle and into the next one, indicating that the LIG1 gene is transcribed periodically during the cell cycle as well as in response to proliferation (Fig. 1a). The peak of LigI mRNA preceded the peak of S phase cells by about 6 h (Fig. 1a). Changes in the steady state levels of LigI and FEN-1 proteins followed a similar pattern. The protein levels began to rise as cells entered the first S phase and continued to rise until the next peak of S phase cells before declining (Fig. 1b). In contrast, the levels of PCNA, which plays a critical role in coordinating lagging strand DNA synthesis (23, 24), remained relatively constant under these conditions (Fig. 1b). To determine the contribution of proteolytic degradation to the steady state levels of LigI, FEN-1, and PCNA, asynchronous MCF10A cells were treated with cycloheximide to inhibit protein synthesis. Although the steady state levels of PCNA remained relatively constant under these conditions, LigI and FEN-1 had half-lives of about 9 and 12 h, respectively (Fig. 2, a and b).
FIGURE 1.
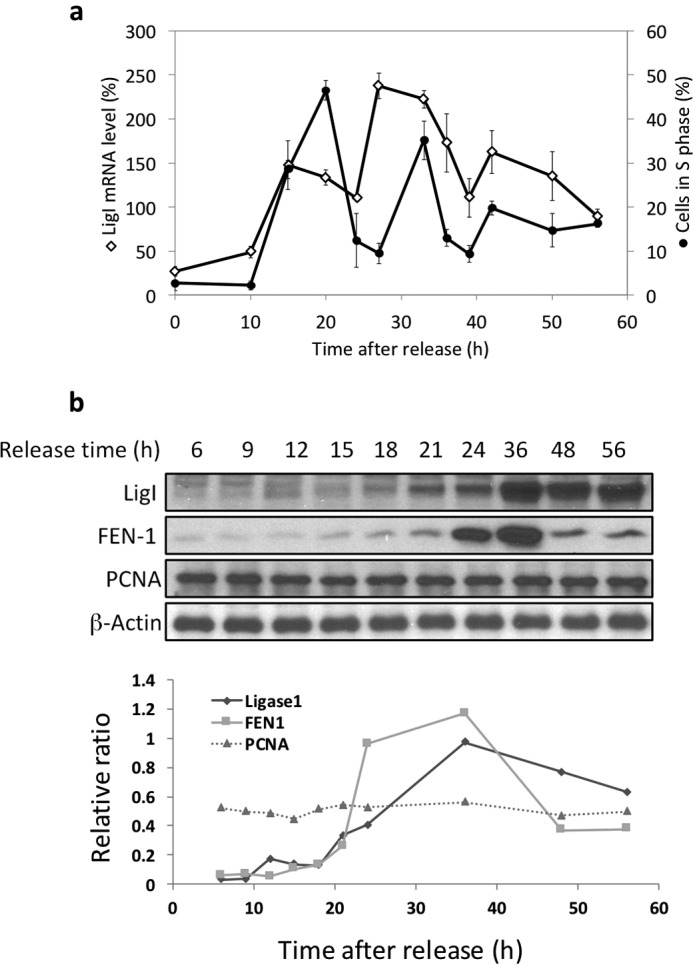
Expression of the LIG1 gene and LigI, FEN-1, and PCNA proteins during the cell cycle. Confluent MCF10A cells were trypsinized, plated at a lower density, and then incubated for the indicated times prior to harvesting. a, the percentage of the cell population in S phase was measured by flow cytometry, and the steady state levels of LigI mRNA were measured by quantitative RT-PCR as described under “Experimental Procedures.” LigI mRNA levels are expressed as a percentage of the LigI mRNA level in asynchronously proliferating cells. The data shown graphically represent the average ± S.D. of three independent experiments. b, upper panel, LigI, FEN-1, PCNA, and β-actin proteins were detected in total cell lysates (20 μg) from the synchronized cell populations by immunoblotting as indicated. Lower panel, the film was digitized and then quantitated by band densitometry using the ImageJ software (National Institutes of Health). The results shown graphically for LigI, FEN-1, and PCNA are expressed as ratio of β-actin levels.
FIGURE 2.
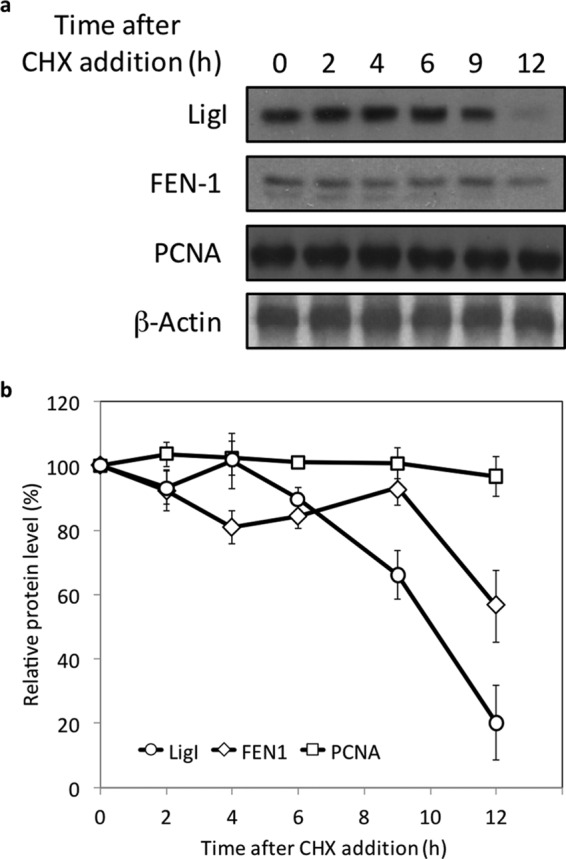
Half-lives of LigI and FEN-1. a, asynchronous MCF10A cells were treated with 100 μg/ml cycloheximide (CHX) for the indicated times prior to lysis. LigI, FEN-1, PCNA, and β-actin proteins were detected in total cell lysates (20 μg) by immunoblotting. b, the relative levels of LigI, FEN-1, and PCNA were determined using the ImageJ software (National Institutes of Health) and expressed as the percentage of the signal in untreated cells. The data shown graphically represent the average ± S.D. of three independent experiments.
Degradation of LigI by the Proteasome in Response to Inhibition of Cell Proliferation and DNA Damage
To confirm that growth inhibition results in a reduction in the level of LigI, we examined the effect of serum starvation and contact inhibition on the steady state levels of LigI mRNA and protein in MCF10A cells. As expected, the steady state levels of both LigI mRNA and protein were reduced after serum starvation for 24 h (Fig. 3a) and contact inhibition for 24 h (Fig. 3b). Under these conditions, the levels of FEN-1 and PCNA did not change (data not shown). In the non-dividing cells, the reduction of LigI protein levels (about 70–80%) was greater than the reduction of LigI mRNA (45–65%), suggesting that proteolytic degradation of LigI protein may be increased and/or translation of LigI mRNA may be decreased in non-dividing cells. Notably, levels of LigI protein but not mRNA were maintained in the growth-inhibited cells in the presence of the proteasome inhibitors, MG132 (Fig. 3a, lane 3) and epoxomicin (Fig. 3b, lane 3). In accordance with a published study (25), we observed that treatment with the DNA-damaging agent, doxorubicin, also caused a reduction in LigI protein levels (Fig. 3a, lane 4). Once again the reduction in LigI protein but not mRNA was prevented by co-incubation with the proteasome inhibitor MG132 (Fig. 3a, lane 5). The effect of doxorubicin on LigI levels is likely to be due to cell cycle arrest mediated by DNA damage-activated cell cycle checkpoints (25).
FIGURE 3.
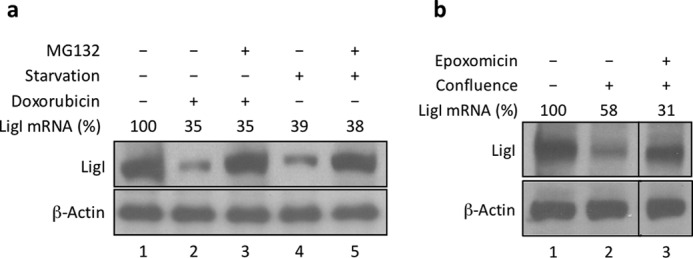
Degradation of LigI in human breast epithelial cells following inhibition of cell proliferation and DNA damage. a, asynchronously proliferating MCF10A cells were either untreated (lane 1), serum-starved for 24 h (lanes 4 and 5), or incubated with doxorubicin (0.1 μm) for 48 h (lanes 2 and 3) in the absence or presence of the proteasome inhibitor MG132 (20 μm) for the last 8 h as indicated. b, lane 1, asynchronous, proliferating MCF10A cells. Lanes 2 and 3, confluent MCF10A cells incubated in the absence (lane 2) or presence (lane 3) of epoxomicin (0.2 μm) for 8 h. The steady state levels of LigI mRNA were measured by quantitative RT-PCR as described under “Experimental Procedures.” LigI and β-actin were detected by immunoblotting.
Serum starvation of fibroblast cell lines from normal individuals that had been immortalized either with telomerase (HCA-Ltrt, Fig. 4a, left panel) or with SV40 T antigen (GM00847, Fig. 4a, right panel) also resulted in a reduction in the steady state levels of LigI that was prevented by a proteasome inhibitor. Because these results indicate that the proteasome plays an important role in determining the steady state levels of LigI, we asked whether LigI was ubiquitylated in GM00847 cells that were transiently transfected with a plasmid expressing HA-tagged ubiquitin. Only low levels of LigI ubiquitylation were detected in LigI immunoprecipitates from extracts of asynchronous cell populations even in the presence of a proteasome inhibitor (Fig. 4b, lanes 2 and 3). As expected, there were higher levels of LigI ubiquitylation in LigI immunoprecipitates from extracts of serum-starved cells, and these levels were further increased by the inclusion of the proteasome inhibitor epoxomicin during serum starvation (Fig. 4b, lanes 4 and 5).
FIGURE 4.
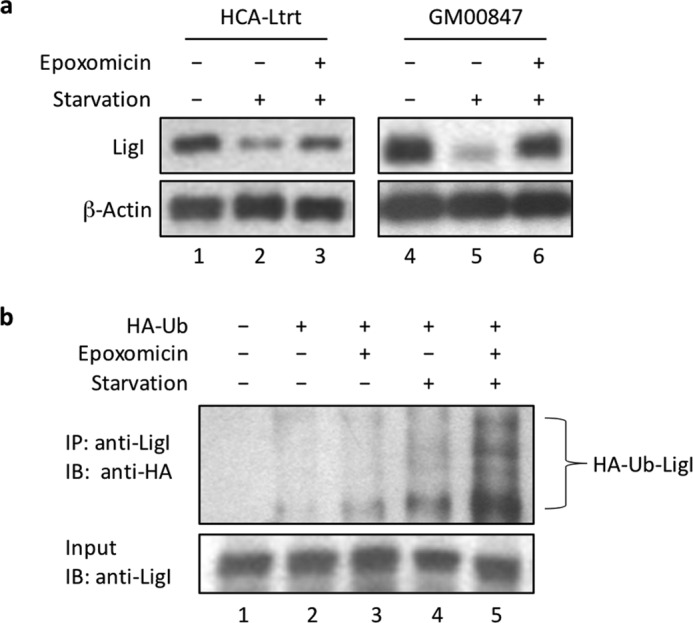
Ubiquitylation and degradation of LigI in growth-arrested immortalized human fibroblasts. a, asynchronous, proliferating immortalized fibroblasts were either untreated (lanes 1 and 4), serum-starved for 24 h (lanes 2 and 5), or serum-starved for 24 h in the presence of epoxomicin (0.2 μm 8 h, lanes 3 and 6). Left panel, telomerase-immortalized fibroblasts, HCA-Ltrt. Right panel, SV40-immortalized fibroblasts, GM00847. LigI and β-actin were detected by immunoblotting. b, LigI immunoprecipitates (IP) from GM00847 cells expressing HA-tagged ubiquitin (Ha-Ub) were probed for ubiquitylated LigI by immunoblotting (IB). Lane 1, untreated GM00847 cells. GM00847 cells transiently transfected with a plasmid expressing HA-tagged ubiquitin were: untreated (lane 2); incubated with 0.2 μm epoxomicin for 8 h (lane 3); serum-starved for 8 h (lane 4); serum-starved and incubated with 0.2 μm epoxomicin for 8 h (lane 5). Lower panel, LigI in the cell lysates was detected by immunoblotting. Upper panel, LigI with covalently linked HA-tagged ubiquitin was detected in the LigI immunoprecipitates by immunoblotting with anti-HA antibody.
Identification of LigI Ubiquitylation Sites and Association with the Cul4-DDB1 E3 Ligase
To map the sites of ubiquitylation, anti-FLAG immunoprecipitates from extracts of 293T cells that stably express FLAG-tagged human LigI were analyzed by mass spectrometry for peptides with the di-Gly modification that is characteristic of ubiquitylation peptides (19–21). The peptide coverage for LigI in these experiments was greater than 90%. Lys-376 (Fig. 5a) was definitively identified as a major site of ubiquitylation site by mass spectrometry, and Lys-79 (Fig. 5b) and Lys-192 (Fig. 5c) were identified as possible ubiquitylation sites.
FIGURE 5.

Identification of ubiquitylated lysine residues in LigI, and the effect of replacing ubiquitylated lysine residues with arginine in LigI steady state levels in serum-starved cells. Anti-FLAG immunoprecipitates from nuclear extracts of labeled 293T cells expressing FLAG-tagged LigI mixed with nuclear extracts of unlabeled parental 293T cells were digested with trypsin and tryptic peptides identified by mass spectrometry as described under “Experimental Procedures.” a–c, MS/MS spectra of peptides bearing ubiquitylation of Lys-376 (a), Lys-79 (b), and Lys-192 (c) in LigI. K#, ubiquitylation site. R′, [13C615N4]arginine. Red peaks correspond to b ions (masses of fragments generated from the peptide N terminus), and blue peaks correspond to y ions (masses of fragments generated from the peptide C terminus). d, asynchronously proliferating GM00087 cells expressing the FLAG-tagged K79R,K192R,K226R,K376R version of LigI were serum-starved for the indicated times. FLAG-tagged and endogenous versions of LigI were detected by immunoblotting with the LigI antibody.
To examine the role of these lysine residues, Lys-79, Lys-192, and Lys-376 and another lysine residue, Lys-226, identified as a LigI ubiquitylation site in other studies (19, 20), we constructed FLAG-tagged versions of LigI in which one or more of the lysine residues was replaced with an arginine, and then we isolated stable derivatives of SV40-immortalized GM00847 cells that stably express the FLAG-tagged mutant versions of LigI at levels about two times higher than endogenous LigI. Although none of the single amino acid substitutions significantly impacted protein stability, a mutant version of LigI, in which all four of the lysine residues were replaced with arginine (K79R,K192R,K226R,K376R), was more resistant to serum starvation-induced degradation (∼50% after 24 h) when compared with endogenous wild-type LigI (∼25% after 24 h), indicating that ubiquitylation of these lysine residues contributes to LigI degradation by the proteasome (Fig. 5d). Because the levels of the mutant version of LigI are reduced in response to serum starvation, it is likely that additional lysine residues within LigI are also ubiquitylated.
FLAG immunoprecipitates were also examined by stable isotope labeling by amino acids in cell culture (SILAC)-based mass spectrometry for associated E3 ubiquitin ligases that may be involved in the ubiquitylation of LigI. When compared with parental 293T cells, higher levels of the Cul4 and DDB1 subunits of the core Cul4-DDB1 complex and DCAF7, a specificity factor for Cul4-DDB1 complex, were detected in FLAG immunoprecipitates from derivatives of 293T cells expressing FLAG-LigI (Table 1), suggesting that LigI associates with the Cul4-DDB1 complex. The co-immunoprecipitation of DDB1 and DCAF7 with FLAG-tagged LigI was confirmed by immunoblotting (Fig. 6a). In reciprocal experiments, FLAG-tagged LigI and the DDB1 subunit of the Cul4-DDB1 complex were co-immunoprecipitated by a Cul4 antibody. Notably, the amount FLAG-tagged LigI co-immunoprecipitated with Cul4 increased in extracts from serum-starved cells (Fig. 6b, compare lanes 4 and 5), and as expected, this was further increased by co-incubation with a proteasome inhibitor (Fig. 6b, lane 3). The increased association of LigI with the Cul4-DDB1 E3 ligase in serum-starved cells suggests that the capacity or rate of proteasome-mediated degradation may be limiting under these conditions.
TABLE 1.
Identification of CUL family proteins, DDB1 and DCAF7 interacting with LigI
| UniProt accession | Gene name | Protein name | HEK293-FLAG-hLIG1/HEK293a |
|||
|---|---|---|---|---|---|---|
| Ratio | S.D. | CV | No. of peptides | |||
| Q13616 | CUL1 | Cullin-1 | 1.97 | 0.12 | 5.9 | 7 |
| Q13617 | CUL2 | Cullin-2 | 2.15 | 0.52 | 24.2 | 2 |
| Q13618 | CUL3 | Cullin-3 | 2.57 | 0.18 | 6.8 | 3 |
| Q13619 | CUL4A | Cullin-4A | 2.72 | 0.28 | 10.3 | 6 |
| Q13620 | CUL4B | Cullin-4B | 2.3 | 0.14 | 6.2 | 5 |
| Q93034 | CUL5 | Cullin-5 | 2.84 | 1 | ||
| Q86VP6 | CAND1 | Cullin-associated NEDD8-dissociated protein 1 | 2.73 | 0.39 | 14.3 | 23 |
| Q16531 | DBB1 | DNA damage-binding protein 1 | 1.44 | 0.03 | 1.8 | 4 |
| P61962 | DCAF7 | DDB1- and CUL4-associated factor 7 | 1.17 | 0.01 | 0.8 | 2 |
a The ratio of selected LigI-associated proteins in HEK293-FLAG-hLIG1 cells versus parental HEK293 cells was derived from the ratios of their individual identified peptides (No. of peptides). The S.D. and coefficient of variation (CV) for these peptide ratios are shown.
FIGURE 6.
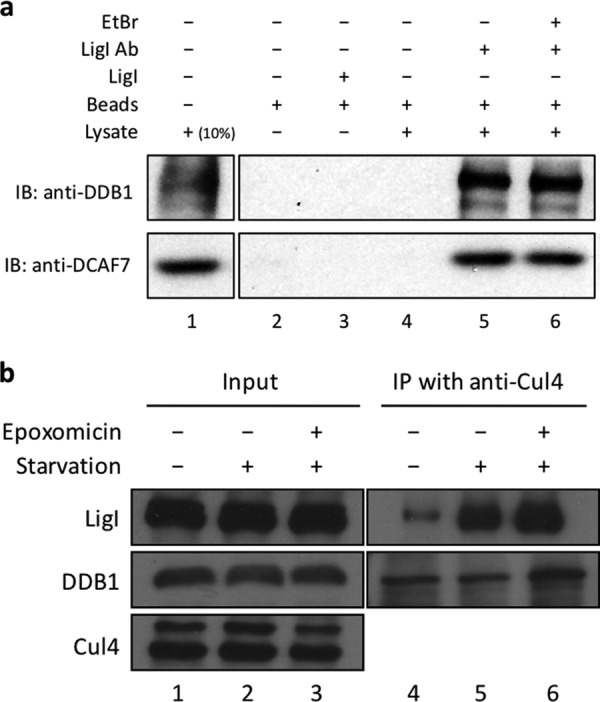
Association of LigI with the Cul4-DDB1-DCAF7 complex. a, lane 1, 293T cell lysate, 5 μg (10% input). Lane 2, protein A/G beads only. Lane 3, protein A/G beads plus LigI. Lanes 4–6, 293T cell lysate incubated with: protein A/G beads (lane 4); LigI antibody (LigI Ab) followed by protein A/G beads (lane 5); LigI antibody plus 10 μg/ml ethidium bromide followed by protein A/G beads (lane 6). DDB1 and DCAF7 proteins were detected by immunoblotting (IB). b, input lysates (5 μg, 10% of input) and Cul4 immunoprecipitates (IP) from GM00847 cells that were: untreated (lanes 1 and 4); serum-starved for 8 h (lanes 2 and 5); serum-starved for 8 h in the presence of 0.2 μm epoxomicin for 8 h (lanes 3 and 6). Cul4, DDB1, and LigI proteins were detected by immunoblotting.
DCAF7 Interacts with and Targets LigI for Degradation
To determine whether DCAF7 is the specificity factor that targets LigI for ubiquitylation by the Cul4-DDB1 E3 ligase and subsequent degradation by the proteasome, we initially carried out pulldown experiments with purified proteins. As shown in Fig. 7a, DCAF7 bound specifically to nickel beads liganded by His-tagged LigI. Next we asked whether depletion of DCAF7 alters LigI for degradation in vivo. GM00847 cells were stably transfected with either a DCAF7-specific shRNA or a modified control shRNA construct that differs in two nucleotides from the DCAF7 shRNA. Knockdown of DCAF7 levels by about 70% reduced the degradation of LigI in response to serum starvation (Fig. 7b).
FIGURE 7.
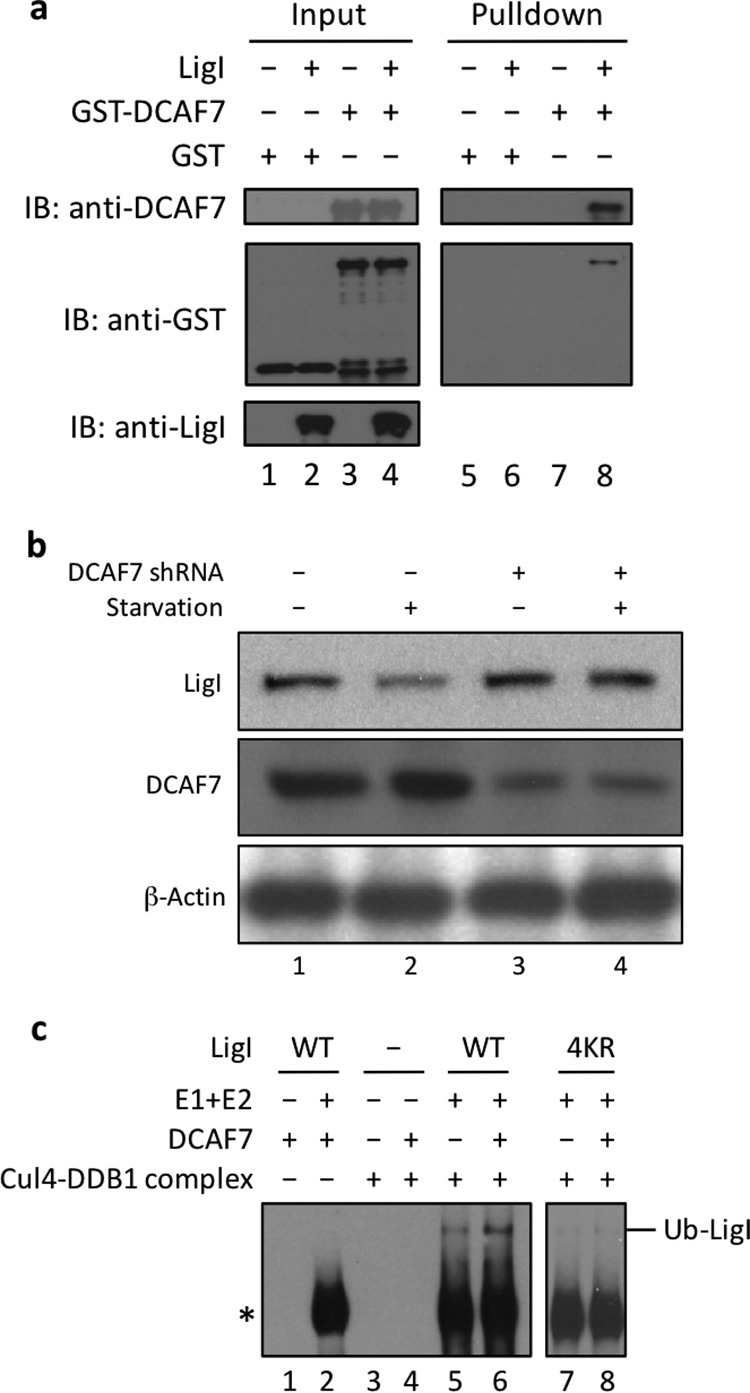
DCAF7 interacts with and targets LigI for ubiquitylation and degradation. a, pulldown assay with nickel beads. Purified GST (lanes 1 and 5); GST and His-tagged LigI (lanes 2 and 6); GST-DCAF7 (lanes 3 and 7); and GST-DCAF7 and His-tagged LigI (lanes 4 and 8) were incubated with nickel beads. The input lanes (lanes 1–4) contain 0.2 μg of the GST and 0.2 μg His-tagged LigI as indicated (10% of total input). Proteins retained on the beads were eluted with SDS-PAGE sample buffer and then separated by SDS-PAGE. Proteins were detected by immunoblotting (IB) with antibodies against GST, DCAF7, and LigI. b, GM00847 cells that were stably transfected with either a control shRNA (lanes 1 and 2) or a DCAF7 shRNA (lanes 3 and 4) were serum-starved or not as indicated. LigI, DCAF7, and β-actin were detected in whole cell lysates (20 μg) by immunoblotting. c, purified wild-type LigI (lanes 1, 2, 5, and 6, 1 μm) and the K79R,K192R,K226R,K376R (4KR) version (lanes 7 and 8, 1 μm) were incubated with biotinylated ubiquitin (Ub) provided by the ENZO Ubiquitinylation kit, a Cul4 immunoprecipitate from GM00847 cells, a purified E1 enzyme, and a mixture of E2 enzymes and purified DCAF7 (1 μm) as indicated. Ubiquitylated proteins were detected by immunoblotting with the Pierce Far-Western Blot Kit for Biotinylated Proteins. The ubiquitylated band indicated by the asterisk is generated by the E1 and E2 enzymes in the absence of LigI.
Finally, we asked whether the Cul4 complex immunoprecipitated from GM00847 cells could ubiquitylate LigI in vitro in the presence of human ubiquitin-activating enzyme E1, a mixture of ubiquitin-conjugating E2 enzymes, and biotinylated ubiquitin. Low levels of LigI ubiquitylation were detected in assays with the Cul4 immunoprecipitate that presumably contains low levels of DCAF7 (Fig. 7c, lane 5). The addition of recombinant DCAF7 enhanced LigI ubiquitylation but only in the presence of the Cul4 immunoprecipitate (Fig. 7c, compare lanes 2, 5, and 6). In accordance with our results showing that the 4KR version (K79R,K192R,K226R,K376R) of LigI was more resistant to degradation in vivo (Fig. 5d), ubiquitylation of this mutant version by immunoprecipitated Cul4, either in the absence or in the presence of DCAF7, was severely reduced (Fig. 7c, compare lanes 5 and 6 with lanes 7 and 8).
Discussion
Although there is compelling evidence showing that alterations in the relative stoichiometry of proteins involved in DNA replication result in genomic instability (13, 18), the proteolytic mechanisms that regulate the steady state levels of DNA replication proteins are relatively unexplored. Because proteomic studies have shown that LigI is ubiquitylated (19–21), we examined the role of ubiquitin-dependent proteolysis in determining the steady state levels of LigI. In this study, we have shown that LigI is ubiquitylated by a member of the cullin-RING finger E3 ligase family, Cul4-DDB1, and then degraded by the proteasome. Cullin-RING finger E3 ligases, which are composed of one of seven cullins plus an adaptor and substrate receptor, are the largest family of ubiquitin ligases in eukaryotes (26). The complexes formed by cullins, Cul4A and Cul4B, with DDB1 have been implicated in several different aspects of genome maintenance, including the repair of DNA damage caused by UV light (27–29), replication licensing (30–32), DNA damage- and stress-activated signaling (33, 34), replication-associated nucleosome assembly (35), and DNA polymerase δ (Polδ) subunit structure (36, 37). Furthermore, mutations in CUL4B have been identified in humans with mental retardation, macrocephaly, and peripheral neuropathy with cell lines from these patients exhibiting defects in the repair of camptothecin-induced DNA single-strand breaks (38).
The substrate specificity of Cul4-DDB1 complexes is determined by substrate receptor proteins that interact with DDB1 (39). Here we have demonstrated that the Cul4-DDB1 substrate receptor DCAF7 directly interacts with LigI and targets it for ubiquitylation by the Cul4-DDB1 E3 ligase complex in vitro. Previous studies have shown that DCAF7, also known as HAN11 and WDR68, interacts with and negatively regulates the signaling activity of several protein kinases, including DYRK1A, DYRK1B, HIPK2, and MEKK1 (22, 40, 41). In these instances, it is not known whether DCAF7 is acting as a scaffolding protein for the kinases or is targeting them for degradation. Notably, depletion of cellular DCAF7 increases the stability of LigI, confirming that DCAF7 plays a key role in determining the steady state levels of LigI.
When cells are induced to proliferate, the expression of both the FEN-1 and LIG1 genes is increased (4, 42). Furthermore, our studies revealed periodic changes in the expression of the LIG1 gene in synchronized, proliferating human cells, indicating that transcription of the LIG1 gene is also cell cycle-regulated. Thus, the regulation of human LIG1 gene expression is more similar to that of the functionally homologous CDC9 gene of S. cerevisiae than the Schizosaccharomyces pombe CDC17 gene (43). Because the level of LigI protein correlates with the mRNA levels, it appears that, in cycling cells, changes in the steady state levels of LigI are primarily determined by transcription rates rather than by proteolysis. The similar changes in the steady state levels of LigI and FEN-1 proteins as cells enter and progress through S phase suggest that the expression of the FEN-1 and LIG1 genes is coordinately regulated in a cell cycle-dependent manner.
Previously, it has been demonstrated that FEN-1 is ubiquitylated by the UBE1-UBE2-PRP19 E3 ligase during the G2/M phase of the cell cycle and that this ubiquitylation is regulated by sequential post-translational modifications of FEN-1 (18). Our studies identifying the Cul4-DDB1 E3 ligase as the major enzyme directing the proteolytic degradation of LigI reveal that two distinct mechanisms are utilized to maintain the relative steady state levels of these PCNA-interacting enzymes that catalyze the last two steps of Okazaki fragment processing and joining. Although it appears that degradation of LigI is enhanced in non-dividing cells, further studies are needed to determine whether the ubiquitylation of LigI by the Cul4-DDB1-DCAF7 complex varies during the cell cycle and in response to inhibition of proliferation and whether cell cycle-dependent phosphorylation of LigI regulates its ubiquitylation (44, 45)
The efficient and accurate replication of DNA requires a high degree of coordination among a large number of proteins. This is particularly evident for lagging strand DNA synthesis where protein-protein interactions with PCNA coordinate the sequential actions of the enzymes that synthesize, process, and join Okazaki fragments (46). This mechanism, which involves transient interactions with overlapping binding sites on the PCNA trimer, is prone to disruption by changes in the relative stoichiometry of the PCNA-interacting proteins (12, 13, 18). Although the expression of many of the genes encoding DNA replication proteins are coordinately regulated, our studies reveal that different mechanisms control the turnover of these proteins. Although this increases the complexity of the network of pathways that regulate the relative stoichiometry of DNA replication proteins, it appears to be designed to integrate DNA replication with other aspects of the cell cycle because FEN-1 degradation is linked to the degradation of cyclin B (18). It is possible that alterations in the proteolytic mechanisms that target LigI and FEN-1 may underlie the frequently elevated steady state levels of these proteins observed in cancers. This may constitute an unexplored source of genomic instability in cancer.
Experimental Procedures
Cell Lines and Culture Conditions
The human MCF10A cell line established from normal breast epithelium (ATCC) was cultured in DMEM/F12 supplemented with 5% horse serum, 20 ng/ml epidermal growth factor, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 10 μg/ml insulin, and 10 μg/ml penicillin and streptomycin at 37 °C. SV40- (GM00847, ATCC) and telomerase-immortalized (HCA-Ltrt, a gift from Dr. Murnane) human fibroblasts (47) and human embryonic kidney 293T cells (ATCC) were cultured at 37 °C in DMEM supplemented with 10% fetal bovine serum, 50 units/ml penicillin, and 50 μg/ml streptomycin. To synchronize cell populations, cultures were grown until confluent. After 48 h, the confluent cells were trypsinized and then plated at a lower density. The synchronization of the cell population and its distribution among the cell cycle stages were determined by flow cytometry in the University of New Mexico Comprehensive Cancer Center Flow Cytometry Shared Resource. To investigate the effects of serum starvation, the culture medium was removed from proliferating cells and replaced with medium lacking serum. Incubation was continued in the serum-free medium for 24 h unless specified. For experiments with doxorubicin hydrochloride (Sigma), cells were incubated either in the absence or in the presence of 0.1 μm doxorubicin for 48 h. Where indicated, a proteasome inhibitor, either epoxomicin (0.2 μm, Millipore) or MG132 (20 μm, Sigma), was added to the culture medium for the final 8 h of the incubation without serum or with doxorubicin.
Quantification of Gene Expression by Real-time PCR
Total RNA was purified from synchronized MCF10A cells using an RNeasy kit (Qiagen) and reverse-transcribed using the TaqMan Reverse Transcription Reagent kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. The expression of the LIG1 gene was measured with a quantitative real-time PCR assay as reported previously (48). Expression levels were calculated as a ratio of the LigI mRNA level relative to the mRNA level for GAPDH in the same cDNA sample. Primer sequences used here are: LIGI-1F, 5′-GAA TTC TGA CGC CAA CAT GCA-3′, and LIGI-1R, 5′-CCG TCT CTC TGC TGC TAT TGG A-3′; GAPDH F, 5′-TCT GGT AAA GTG GAT ATT GTT G-3′, and GAPDH R, 5′-GAT GGT GAT GGG ATT TCC-3′.
Immunoblotting
Cell extracts were prepared as described previously, and their protein concentration was determined using the method of Bradford (50). Proteins in whole cell extracts (20 μg) were detected by immunoblotting after separation by SDS-PAGE using the following primary antibodies: anti-FLAG M2 (1:10,000, Sigma); anti-FEN-1 (1:1000, Novus) and anti-GST (1:10,000, Novus); anti-Cul4 and anti-DDB1 (1:1000, GeneTex); anti-PCNA (1:10,000, Santa Cruz Biotechnology); anti-HA antibody (1:2000, Bethyl Laboratories, Inc.); anti-DCAF7 (1:1000, Abcam); and anti-actin (1:10,000, Abcam). A rabbit polyclonal antibody generated against purified human LigI (1:5000) was used as described previously (6, 7, 51).
Determination of Protein Half-life
Half-life experiments employing cycloheximide, an inhibitor of protein synthesis, were performed as described previously (19). The levels of FEN-1, LigI, and PCNA were detected by immunoblotting and quantitated using the ImageJ software (National Institutes of Health).
Detection of LigI Ubiquitylation in Vivo
GM00847 cells were transiently transfected with a plasmid encoding HA-tagged ubiquitin using Lipofectamine (Invitrogen) according to the manufacturer's instructions. After 24 h, the culture medium was replaced with medium lacking serum and incubation was continued for 24 h. Where indicated, a proteasome inhibitor, either epoxomicin (0.2 μm, Millipore) or MG132 (20 μm, Sigma), was added to the culture medium for the final 8 h of the 24-h incubation. Cells were lysed in immunoprecipitation lysis buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 1 mm EDTA, 1 mm DTT, 5% glycerol, 1% Triton X-100, containing a protease inhibitor cocktail (Roche Applied Science) and phosphatase inhibitor cocktails 2 and 3 (Sigma) according to the manufacturers' recommendations). Clarified lysates (20 mg) were incubated with anti-LigI antibody (1:500) for 1 h at 4 °C prior to overnight the incubation with 20 μl of protein A beads (GE Healthcare). After washing five times with immunoprecipitation lysis buffer, bound proteins were eluted from the beads with SDS-PAGE sample buffer and then separated by SDS-PAGE. Ubiquitylated proteins were detected by immunoblotting with anti-HA antibody.
Identification of LigI Ubiquitylation Sites
To identify ubiquitylated lysine residues in LigI, we selected a derivative of the 293T cell line that stably expresses FLAG-tagged wild-type LigI at levels about two times higher than endogenous LigI. FLAG-tagged LigI was immunoprecipitated from clarified extracts (1 mg) of the 293T cell line (1 × 108 cells) using anti-FLAG M2 affinity beads (Sigma) as described above. Proteins retained on the beads were reduced, alkylated, and then digested with trypsin. The resulting peptides were analyzed by a nano HPLC-coupled LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific) as described previously (52). Raw data were searched against the human database with a SEQUEST algorithm built in BioWorks (version 3.3.1 SP1, Thermo Fisher Scientific). A variable modification of lysine with a mass shift of +114.04 Da was set to identify the ubiquitylation sites. Other modifications included a fixed modification of carbamidomethyl cysteine and a variable modification of oxidized methionine.
Identification of LigI-associated Proteins by Mass Spectrometry
293T cells that stably express FLAG-tagged LIG1 were metabolically labeled with l-[13C215N6]lysine and l-[13C415N6]arginine (53). Nuclear extracts were prepared from labeled 293T cells expressing FLAG-tagged hLigI and unlabeled parental 293T cells as described previously (54). Equal amounts of the labeled and unlabeled nuclear extracts were mixed and then incubated with anti-FLAG M2 affinity beads (Sigma) as described above. Proteins were eluted from the beads with 3×FLAG peptide (Sigma) and then digested with trypsin using the filter-aided sample preparation (FASP) procedure (55). The resulting peptides were desalted on a Sep-Pak C18 cartridge (Waters) and dried using a SpeedVac (Thermo Fisher Scientific). After further fractionation using stop and go extraction (STAGE) tips made in-house with a strong anion exchange disk and desalting with a STAGE tip containing three layers of C18 membrane disks (3M) (56, 57), peptide solutions were dried and stored at −80 °C prior to analysis by nano HPLC-coupled LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific). A database search was carried out as described previously to identify the proteins from which the tryptic peptides were derived (52). The ratios of peptides from labeled and non-labeled cells to parental cells were calculated using the IsoQuant software (58).
Generation of Cell Lines That Stably Express Tagged Versions of LigI
A pVP-FLAG5 plasmid encoding an N-terminal FLAG-tagged version of LigI (6, 7, 51) was mutated by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The mutations altered the coding sequence such that lysine residues 79, 192, 226, and 376 were replaced with arginine residues either singly or all together. After verification of the nucleotide sequence by DNA sequencing, N-terminally FLAG-tagged wild-type LigI (WT) and FLAG-tagged mutant LigI cDNAs were subcloned into the mammalian expression vector pRC/RSV. After selection for G418 resistance, single colonies were isolated and evaluated for the level of tagged LigI expression relative to endogenous LigI using FLAG and LigI antibodies. Cell lines that stably express FLAG-tagged mutant versions of LigI at levels about two times higher than endogenous LigI were selected for analysis.
Immunoprecipitation
A total of 4 × 108 cells were harvested, and lysed in 1 ml of 10 mm Tris-HCl, pH 7.4, 100 mm NaCl, 2.5 mm MgCl2, and 0.5% Triton X-100 containing protease and phosphatase inhibitors. After centrifugation, the supernatant was incubated at 4 °C overnight with a 100-μl 50% suspension of protein A/G-Sepharose (Amersham Biosciences) coupled with 2 μl of anti-FLAG M2 antibody. The beads were washed extensively with the buffer described above prior to the elution of bound proteins with SDS sample buffer. After separation by SDS-PAGE, eluted proteins were identified by immunoblotting.
Proteins
Plasmids encoding either GST or GST-DCAF7 were transformed into Escherichia coli BL21(DE3) cells. GST and GST-DCAF7 proteins were purified from cell extracts by glutathione-Sepharose 4B affinity column chromatography (GE Healthcare). Wild-type and mutant LigI cDNAs were subcloned into the bacterial expression vector pET28a. After expression in E. coli BL21(DE3) cells, the N-terminally His-tagged versions of hLigI were purified by HisTrap and HiTrap Q (GE Healthcare Life Sciences) column chromatography. Purified proteins fractions were separated by SDS-PAGE prior to Coomassie blue staining.
Pulldown Assay
Purified His-tagged LigI (1 μg) was incubated with either GST (2 μg) or GST-DCAF7 (2 μg) in 300 μl of buffer containing 50 mm NaH2PO4 (pH 7.4), 250 mm NaCl, and 10 mm imidazole for 1 h on ice, followed by the addition of 15 μl of Ni-nitrilotriacetic acid-agarose beads (Qiagen) with rotation for 1 h at 4 °C. The beads were collected by centrifugation and then washed five times with 500 μl of 50 mm NaH2PO4 (pH 7.4), 250 mm NaCl, and 30 mm imidazole. After resuspension in 2× SDS-PAGE sample buffer, the beads were incubated at 100 °C for 5 min. Eluted proteins were separated by SDS-PAGE prior to detection both by staining with Coomassie Blue and by immunoblotting with GST and LigI antibodies.
Generation of Cell Lines That Stably Express DCAF7 shRNA
GM00847 fibroblast cells were transfected with plasmids encoding DCAF7 shRNA or a scrambled shRNA (22) using the Lipofectamine transfection reagent (Invitrogen) according to the manufacturer's directions. After selection for resistance to puromycin, single colonies were isolated. The steady state levels of DCAF7 and hLigI proteins in these clones were determined by immunoblotting with antibodies against hLigI and DCAF7.
In Vitro Ubiquitylation Assay
hLigI ubiquitylation in vitro was performed using the ubiquitylation kit (Enzo Life Science) according to the manufacturer's directions. Briefly, purified His-tagged versions of wild-type and mutant versions of LigI (1 μm) were incubated as indicated with E1 and E2 enzymes, biotinylated ubiquitin, 0.75 μl of ubiquitin aldehyde, ATP, Cul4-DDB1 complex immunoprecipitated from GM00847 cells, and purified DCAF7 (1 μm) in a final 15-μl volume at 30 °C for 1 h. Ubiquitylated LigI protein was detected by immunoblotting using the Pierce Far-Western Blot Kit for Biotinylated Protein (Thermo Fisher) according to the manufacturer's directions.
Statistical Analysis
Data represent the average ± S.D. from at least from three independent experiments. Statistical comparisons were performed with Student's two-tailed paired t test and analysis of variance. Values of p < 0.01 were considered statistically significant.
Author Contributions
A. E. T. and A. Y. conceived and coordinated the study and wrote the paper. Z. P. performed and analyzed the cell biology and biochemical studies. Z. L. carried out and analyzed the mass spectrometry experiments. Y. M. purified proteins, provided technical expertise, and contributed to the preparation of the figures. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Dr. John Murnane for the HCA-Ltrt cell line, Dr. Michael Lienhard Schmitz for the DCAF7 plasmids, and Dr. Annahita Sallmyr for assistance with figure preparation.
This work was supported by the University of New Mexico Comprehensive Cancer Center (Grant P30 CA118100) and National Institute of Health Grant R01 GM057479 (to A. E. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LigI
- DNA ligase I
- hLigI
- human LigI
- Cul4
- cullin4
- DDB1
- DNA damage-specific DNA-binding protein 1
- PCNA
- proliferating cell nuclear antigen
- FEN-1
- flap endonuclease 1.
References
- 1. Barnes D. E., Johnston L. H., Kodama K., Tomkinson A. E., Lasko D. D., and Lindahl T. (1990) Human DNA ligase I cDNA: cloning and functional expression in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 87, 6679–6683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen J., Tomkinson A. E., Ramos W., Mackey Z. B., Danehower S., Walter C. A., Schultz R. A., Besterman J. M., and Husain I. (1995) Mammalian DNA ligase III: molecular cloning, chromosomal localization, and expression in spermatocytes undergoing meiotic recombination. Mol. Cell. Biol. 15, 5412–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wei Y. F., Robins P., Carter K., Caldecott K., Papin D. J. C., Yu G.-L., Wang R.-P., Shell B. K., Nash R. A., Schär P., Barnes D. E., Haseltine W. A., and Lindahl T. (1995) Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and genetic recombination. Mol. Cell. Biol. 15, 3206–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Petrini J. H., Huwiler K. G., and Weaver D. T. (1991) A wild-type DNA ligase I gene is expressed in Bloom's syndrome cells. Proc. Natl. Acad. Sci. U.S.A. 88, 7615–7619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Levin D. S., Bai W., Yao N., O'Donnell M., and Tomkinson A. E. (1997) An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment synthesis and joining. Proc. Natl. Acad. Sci. U.S.A. 94, 12863–12868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levin D. S., McKenna A. E., Motycka T. A., Matsumoto Y., and Tomkinson A. E. (2000) Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr. Biol. 10, 919–922 [DOI] [PubMed] [Google Scholar]
- 7. Levin D. S., Vijayakumar S., Liu X., Bermudez V. P., Hurwitz J., and Tomkinson A. E. (2004) A conserved interaction between the replicative clamp loader and DNA ligase in eukaryotes: implications for Okazaki fragment joining. J. Biol. Chem. 279, 55196–55201 [DOI] [PubMed] [Google Scholar]
- 8. Montecucco A., Rossi R., Levin D. S., Gary R., Park M. S., Motycka T. A., Ciarrocchi G., Villa A., Biamonti G., and Tomkinson A. E. (1998) DNA ligase I is recruited to sites of DNA replication by an interaction with proliferating cell nuclear antigen: identification of a common targeting mechanism for the assembly of replication factories. EMBO J. 17, 3786–3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barnes D. E., Tomkinson A. E., Lehmann A. R., Webster A. D., and Lindahl T. (1992) Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell 69, 495–503 [DOI] [PubMed] [Google Scholar]
- 10. Webster A. D., Barnes D. E., Arlett C. F., Lehmann A. R., and Lindahl T. (1992) Growth retardation and immunodeficiency in a patient with mutations in the DNA ligase I gene. Lancet 339, 1508–1509 [DOI] [PubMed] [Google Scholar]
- 11. Harrison C., Ketchen A. M., Redhead N. J., O'Sullivan M. J., and Melton D. W. (2002) Replication failure, genome instability, and increased cancer susceptibility in mice with a point mutation in the DNA ligase I gene. Cancer Res. 62, 4065–4074 [PubMed] [Google Scholar]
- 12. López Castel A., Tomkinson A. E., and Pearson C. E. (2009) CTG/CAG repeat instability is modulated by the levels of human DNA ligase I and its interaction with proliferating cell nuclear antigen: a distinction between replication and slipped-DNA repair. J. Biol. Chem. 284, 26631–26645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Subramanian J., Vijayakumar S., Tomkinson A. E., and Arnheim N. (2005) Genetic instability induced by overexpression of DNA ligase I in budding yeast. Genetics 171, 427–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X., Zhong S., Zhu X., Dziegielewska B., Ellenberger T., Wilson G. M., MacKerell A. D. Jr., and Tomkinson A. E. (2008) Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 68, 3169–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun D., Urrabaz R., Nguyen M., Marty J., Stringer S., Cruz E., Medina-Gundrum L., and Weitman S. (2001) Elevated expression of DNA ligase I in human cancers. Clin Cancer Res. 7, 4143–4148 [PubMed] [Google Scholar]
- 16. Singh P., Yang M., Dai H., Yu D., Huang Q., Tan W., Kernstine K. H., Lin D., and Shen B. (2008) Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol. Cancer Res. 6, 1710–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng L., Dai H., Zhou M., Li M., Singh P., Qiu J., Tsark W., Huang Q., Kernstine K., Zhang X., Lin D., and Shen B. (2007) Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat. Med. 13, 812–819 [DOI] [PubMed] [Google Scholar]
- 18. Guo Z., Kanjanapangka J., Liu N., Liu S., Liu C., Wu Z., Wang Y., Loh T., Kowolik C., Jamsen J., Zhou M., Truong K., Chen Y., Zheng L., and Shen B. (2012) Sequential posttranslational modifications program FEN1 degradation during cell-cycle progression. Mol. Cell 47, 444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., and Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., and Choudhary C. (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell Proteomics 10, M111.013284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan F., Lu L., Cai Y., Wang J., Xie Y., Wang L., Gong Y., Xu B. E., Wu J., Luo Y., Qiang B., Yuan J., Sun X., and Peng X. (2008) Proteomic analysis of ubiquitinated proteins in normal hepatocyte cell line Chang liver cells. Proteomics 8, 2885–2896 [DOI] [PubMed] [Google Scholar]
- 22. Ritterhoff S., Farah C. M., Grabitzki J., Lochnit G., Skurat A. V., and Schmitz M. L. (2010) The WD40-repeat protein Han11 functions as a scaffold protein to control HIPK2 and MEKK1 kinase functions. EMBO J. 29, 3750–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waga S., and Stillman B. (1998) The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 67, 721–751 [DOI] [PubMed] [Google Scholar]
- 24. Waga S., and Stillman B. (1994) Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature 369, 207–212 [DOI] [PubMed] [Google Scholar]
- 25. Gajewski E., Gaur S., Akman S. A., Matsumoto L., van Balgooy J. N., and Doroshow J. H. (2007) Oxidative DNA base damage in MCF-10A breast epithelial cells at clinically achievable concentrations of doxorubicin. Biochem. Pharmacol. 73, 1947–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petroski M. D., and Deshaies R. J. (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 27. Groisman R., Kuraoka I., Chevallier O., Gaye N., Magnaldo T., Tanaka K., Kisselev A. F., Harel-Bellan A., and Nakatani Y. (2006) CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 20, 1429–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groisman R., Polanowska J., Kuraoka I., Sawada J., Saijo M., Drapkin R., Kisselev A. F., Tanaka K., and Nakatani Y. (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113, 357–367 [DOI] [PubMed] [Google Scholar]
- 29. Sugasawa K., Okuda Y., Saijo M., Nishi R., Matsuda N., Chu G., Mori T., Iwai S., Tanaka K., Tanaka K., and Hanaoka F. (2005) UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121, 387–400 [DOI] [PubMed] [Google Scholar]
- 30. Jin J., Arias E. E., Chen J., Harper J. W., and Walter J. C. (2006) A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 [DOI] [PubMed] [Google Scholar]
- 31. Kim Y., Starostina N. G., and Kipreos E. T. (2008) The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 22, 2507–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., and Dutta A. (2008) PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 22, 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leung-Pineda V., Huh J., and Piwnica-Worms H. (2009) DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 69, 2630–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Katiyar S., Liu E., Knutzen C. A., Lang E. S., Lombardo C. R., Sankar S., Toth J. I., Petroski M. D., Ronai Z., and Chiang G. G. (2009) REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep. 10, 866–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han J., Zhang H., Zhang H., Wang Z., Zhou H., and Zhang Z. (2013) A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 155, 817–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Terai K., Shibata E., Abbas T., and Dutta A. (2013) Degradation of p12 subunit by CRL4Cdt2 E3 ligase inhibits fork progression after DNA damage. J. Biol. Chem. 288, 30509–30514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang S., Zhao H., Darzynkiewicz Z., Zhou P., Zhang Z., Lee E. Y., and Lee M. Y. (2013) A novel function of CRL4Cdt2: regulation of the subunit structure of DNA polymerase δ in response to DNA damage and during the S phase. J. Biol. Chem. 288, 29550–29561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kerzendorfer C., Whibley A., Carpenter G., Outwin E., Chiang S. C., Turner G., Schwartz C., El-Khamisy S., Raymond F. L., and O'Driscoll M. (2010) Mutations in Cullin 4B result in a human syndrome associated with increased camptothecin-induced topoisomerase I-dependent DNA breaks. Hum. Mol. Genet. 19, 1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee J., and Zhou P. (2007) DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 26, 775–780 [DOI] [PubMed] [Google Scholar]
- 40. Bouwmeester T., Bauch A., Ruffner H., Angrand P. O., Bergamini G., Croughton K., Cruciat C., Eberhard D., Gagneur J., Ghidelli S., Hopf C., Huhse B., Mangano R., Michon A. M., Schirle M., et al. (2004) A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell Biol. 6, 97–105 [DOI] [PubMed] [Google Scholar]
- 41. Skurat A. V., and Dietrich A. D. (2004) Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J. Biol. Chem. 279, 2490–2498 [DOI] [PubMed] [Google Scholar]
- 42. Kim I. S., Lee M. Y., Lee I. H., Shin S. L., and Lee S. Y. (2000) Gene expression of flap endonuclease-1 during cell proliferation and differentiation. Biochim. Biophys. Acta 1496, 333–340 [DOI] [PubMed] [Google Scholar]
- 43. White J. H., Barker D. G., Nurse P., and Johnston L. H. (1986) Periodic transcription as a means of regulating gene expression during the cell cycle: contrasting modes of expression of DNA ligase genes in budding and fission yeast. EMBO J. 5, 1705–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ferrari G., Rossi R., Arosio D., Vindigni A., Biamonti G., and Montecucco A. (2003) Cell cycle-dependent phosphorylation of human DNA ligase I at the cyclin-dependent kinase sites. J. Biol. Chem. 278, 37761–37767 [DOI] [PubMed] [Google Scholar]
- 45. Rossi R., Villa A., Negri C., Scovassi I., Ciarrocchi G., Biamonti G., and Montecucco A. (1999) The replication factory targeting sequence/PCNA-binding site is required in G1 to control the phosphorylation status of DNA ligase I. EMBO J. 18, 5745–5754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Howes T. R., and Tomkinson A. E. (2012) DNA ligase I, the replicative DNA ligase. Subcell. Biochem 62, 327–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pirzio L. M., Freulet-Marrière M. A., Bai Y., Fouladi B., Murnane J. P., Sabatier L., and Desmaze C. (2004) Human fibroblasts expressing hTERT show remarkable karyotype stability even after exposure to ionizing radiation. Cytogenet. Genome Res. 104, 87–94 [DOI] [PubMed] [Google Scholar]
- 48. Liang L., Deng L., Nguyen S. C., Zhao X., Maulion C. D., Shao C., and Tischfield J. A. (2008) Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 36, 3297–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 50. Goetz J. D., Motycka T. A., Han M., Jasin M., and Tomkinson A. E. (2005) Reduced repair of DNA double-strand breaks by homologous recombination in a DNA ligase I-deficient human cell line. DNA Repair (Amst.) 4, 649–654 [DOI] [PubMed] [Google Scholar]
- 51. Vijayakumar S., Dziegielewska B., Levin D. S., Song W., Yin J., Yang A., Matsumoto Y., Bermudez V. P., Hurwitz J., and Tomkinson A. E. (2009) Phosphorylation of human DNA ligase I regulates its interaction with replication factor C and its participation in DNA replication and DNA repair. Mol. Cell. Biol. 29, 2042–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liao Z., Thomas S. N., Wan Y., Lin H. H., Ann D. K., and Yang A. J. (2013) An Internal standard-assisted synthesis and degradation proteomic approach reveals the potential linkage between VPS4B depletion and activation of fatty acid β-oxidation in breast cancer cells. Int. J. Proteomics 2013, 291415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 54. Boisvert F. M., Lam Y. W., Lamont D., and Lamond A. I. (2010) A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell. Proteomics 9, 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Winiewski J. R., Zougman A., Nagaraj N., and Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 56. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 57. Winiewski J. R., Zougman A., and Mann M. (2009) Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8, 5674–5678 [DOI] [PubMed] [Google Scholar]
- 58. Liao Z., Wan Y., Thomas S. N., and Yang A. J. (2012) IsoQuant: a software tool for stable isotope labeling by amino acids in cell culture-based mass spectrometry quantitation. Anal. Chem. 84, 4535–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]