

Abstract
Site selectivity of protein N-linked glycosylation is dependent on many factors, including accessibility of the modification site, amino acid composition of the glycosylation consensus sequence, and cellular localization of target proteins. Previous studies have shown that the bacterial oligosaccharyltransferase, PglB, of Campylobacter jejuni favors acceptor proteins with consensus sequences ((D/E)X1NX2(S/T), where X1,2 ≠ proline) in flexible, solvent-exposed motifs; however, several native glycoproteins are known to harbor consensus sequences within structured regions of the acceptor protein, suggesting that unfolding or partial unfolding is required for efficient N-linked glycosylation in the native environment. To derive insight into these observations, we generated structural homology models of the N-linked glycoproteome of C. jejuni. This evaluation highlights the potential diversity of secondary structural conformations of previously identified N-linked glycosylation sequons. Detailed assessment of PglB activity with a structurally characterized acceptor protein, PEB3, demonstrated that this natively folded substrate protein is not efficiently glycosylated in vitro, whereas structural destabilization increases glycosylation efficiency. Furthermore, in vivo glycosylation studies in both glyco-competent Escherichia coli and the native system, C. jejuni, revealed that efficient glycosylation of glycoproteins, AcrA and PEB3, depends on translocation to the periplasmic space via the general secretory pathway. Our studies provide quantitative evidence that many acceptor proteins are likely to be N-linked-glycosylated before complete folding and suggest that PglB activity is coupled to general secretion-mediated translocation to the periplasm. This work extends our understanding of the molecular mechanisms underlying N-linked glycosylation in bacteria.
Keywords: N-linked glycosylation, oligosaccharyltransferase, protein folding, protein translocation, substrate specificity
Introduction
N-Linked protein glycosylation is a ubiquitous post-translational modification that influences protein stability, folding, and host-cell interactions (1–3). In eukaryotes this phenomenon occurs at the rough endoplasmic reticulum (RER)2 membrane. Here, glycans are assembled onto a polyprenol diphosphate-linked substrate on the cytoplasmic face of the RER membrane and subsequently flipped to the RER luminal face, where the glycans are further extended before transfer to an asparagine residue within a glycosylation consensus sequence by the oligosaccharyltransferase (OTase) (4, 5). In mammalian cells N-linked glycosylation is mediated by hetero-oligomeric OTase complexes that include either the STT3A or STT3B isoform of the catalytic subunit, which have distinct functions (6). The mammalian STT3A is found in a complex that associates with the general secretion (Sec) pathway, coordinating co-translational glycosylation of nascent proteins entering the RER. This macromolecular association is mediated by direct interactions between subunits of the Sec61 translocation machinery and the OTase subunits (7–9). The STT3B isoform is part of a distinct OTase complex in the RER that is not associated with the Sec translocon and rather glycosylates sites missed by STT3A in a post-translational and/or post-translocational manner (10, 11).
Less is known about the structural determinants of the more recently discovered N-linked protein glycosylation pathway of δ- and ϵ-proteobacteria (12). To date, the best characterized bacterial N-linked glycosylation system is the Pgl (protein glycosylation) pathway of Campylobacter jejuni (13, 14). C. jejuni is part of the natural gut flora of poultry and cattle; however, it is a human pathogen and one of the leading causes of bacterial gastroenteritis worldwide (15). Studies have shown that protein N-linked glycosylation plays a role in pathogenicity, as this pathway is important for colonization of the gastrointestinal tract of chickens and mice (16, 17).
The glycosylation process in C. jejuni begins at the cytoplasmic face of the inner membrane, where the glycan, a branched heptasaccharide, is assembled by stepwise addition of monosaccharides onto an undecaprenol diphosphate (Und-PP) moiety and then flipped to the periplasmic face of the inner membrane. In the periplasm, the heptasaccharide is transferred en bloc to an amide nitrogen of an asparagine located within a consensus sequence ((D/E)X1NX2(S/T), X1,2 ≠ proline) on an acceptor protein by a single integral membrane OTase, PglB, that is homologous to the catalytic subunit of eukaryotic OTases (STT3) (13, 18) (Fig. 1). The enzymes that biosynthesize and transfer this heptasaccharide are chromosomally encoded within a gene locus (pgl locus). Notably, heterologous glycosylation of selected target proteins can be achieved by expression of the pgl locus in Escherichia coli (glycocompetent E. coli) (19).
FIGURE 1.
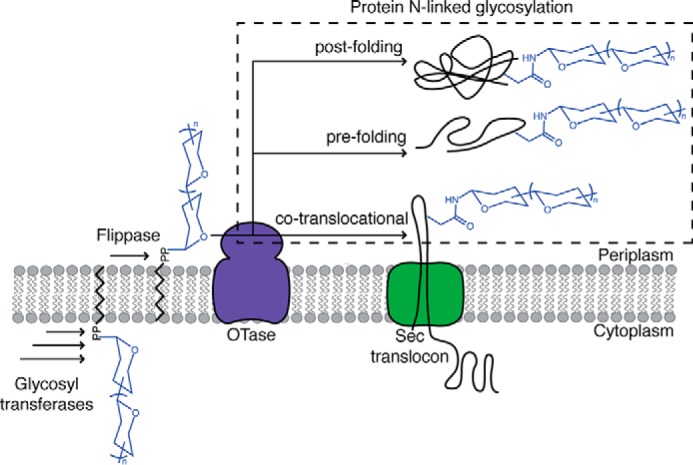
Schematic overview of bacterial N-linked glycosylation and proposed conformation states of substrate proteins during modification. Glycosyltransferases assemble a polyprenol diphosphate-linked glycan in a stepwise manner at the cytoplasmic membrane. The glycosyl donor is subsequently translocated by a flippase to the periplasmic face of the membrane. The OTase transfers the glycan onto an asparagine residue of a substrate protein. Three conformational states of substrate proteins during N-glycosylation are proposed: 1) before folding is complete, co-translocationally; 2) before folding is complete, post-translocationally; 3) after folding is complete, post-translocationally.
To date, >60 proteins from C. jejuni have been demonstrated to be N-glycosylated, and many of these have predicted Sec signal peptides (20). Previous studies have demonstrated that PglB is capable of glycosylating both folded and unfolded proteins in vitro and in glycocompetent E. coli. For example, AcrA (Cj0367c), a component of the AcrABC (Cj0365–7c) multidrug efflux system of C. jejuni, is a native glycoprotein that can be N-glycosylated in vitro or in glycocompetent E. coli when it is exported to the periplasm through the Sec pathway, which transports proteins in an unfolded conformation, or though the twin arginine translocation (TAT) pathway, which transports fully folded proteins (21, 22). Furthermore, Fisher et al. (23) demonstrated that in glycocompetent E. coli, proteins carrying engineered glycosylation tags are glycosylated through diverse export pathways (Sec, TAT, or SRP (signal recognition particle)), although variations in glycosylation efficiencies were observed. These studies show that N-linked glycosylation is compatible with diverse secretory pathways; however, the native spatial and temporal aspects of N-linked glycosylation in the context of bacterial cells remain unclear. Specifically, it is not known whether N-glycosylation and protein translocation are coupled in bacteria, as they are in eukaryotes.
In this work we used complementary approaches to determine how the structural context of N-linked glycosylation substrates affects glycosylation efficiency. In silico evaluation of N-linked glycosylation sequons within C. jejuni glycoproteins was performed using structural homology modeling. This analysis showed that N-linked glycosylation sites are predicted to adopt diverse secondary structures. Next, a detailed investigation was performed on a C. jejuni glycoprotein, PEB3 (Cj0289c), whose x-ray crystallographic structure was previously determined (24, 25). Using in vitro site-directed mutagenesis, we introduced N-linked glycosylation sequons into selected positions in the sequence of PEB3, and examined the relative glycosylation efficiency at each of these sites. We found that these new glycosylation sites were susceptible to increased levels of modification compared with the wild type protein, which was likely due to higher accessibility of the glycosylation site and partial destabilization of the protein. We also investigated whether the mode of protein substrate translocation of glycosylation substrates into the periplasm had an effect on glycosylation efficiencies. Using a glycocompetent E. coli strain, we found that Sec-translocation of AcrA and PEB3 is important for efficient glycosylation. Moreover, we show for the first time that in the native system, C. jejuni, efficient glycosylation requires the delivery of protein substrates by the Sec pathway. In light of these findings and the observation that a subset of native glycoproteins contains sequons in structured regions, we postulate that bacterial protein N-linked glycosylation of selected protein substrates is likely coupled to protein translocation, as it is in eukaryotes.
Results
In Silico Analysis of Glycosylation Sites
Previously, a survey of eukaryotic N-linked glycoproteins structures (x-ray crystallographic data) revealed that N-glycosylation sequons adopt diverse secondary structure conformations (26). N-Glycosylation at these sites is possible because glycosylation occurs before folding in eukaryotes (27, 28). Although >60 glycoproteins have been identified from C. jejuni and Campylobacter lari, only four x-ray crystallographic structures (CjaA, PEB3, JlpA, and PglB) and one NMR structure of truncated AcrA have been determined (24, 25, 29–32). Analysis of the four known x-ray crystal structures shows that the bacterial glycosylation consensus sequence also adopts various conformational states (Fig. 2A). These four protein structures include five solvent-exposed glycosylation sites: one in an α-helix, one in a structured turn between a β-strand and α-helix, and three in non-structured loops. Due to the limited number of x-ray crystallographic structures of known C. jejuni glycoproteins, we generated protein models of additional protein substrates to obtain a broader understanding of the conformations of glycosylation sequons in C. jejuni. Using Phyre2 (Protein homology/analogy recognition engine 2) we generated three-dimensional structural models of experimentally determined C. jejuni glycoproteins (33). A total of 35 high-confidence models containing 53 glycosylation sequons were obtained, and the backbone dihedral angles of each residue within the experimentally determined glycosylation sequon were categorized as α-helix, β-strand, structured turn (β, γ), or non-structured loop (Fig. 2B and supplemental Table S1). In this study we define a non-structured loop as a region that has more than five residues that are not appropriately positioned to form backbone hydrogen-bonding interactions. All residues within glycosylation sequons were predicted to be surface-exposed. Interestingly, >50% of the glycosylation sites were found in predicted α-helices, β-strands, or turns, whereas the remaining were found in larger non-structured loops. These analyses highlight the potential structural diversity of bacterial glycosylation sites. In the context of a folded protein, these conformations could interfere with N-glycosylation activity by PglB. Thus, we hypothesized that native glycosylation sites of C. jejuni proteins located in structurally defined regions are poor substrates of PglB and that partial unfolding is required for glycosylation to occur at these sites.
FIGURE 2.

In silico analysis of diverse secondary structures of N-glycosylation sequons. A, schematic representation of x-ray crystallographic structures of CjaA, PEB3, JlpA, and PglB (PDB codes 1XT8, 2HXW, 3UAU and 3RCE, respectively). The glycosylation consensus sequence ((D/E)X1NX2(S/T), X1,2≠P) is colored in magenta. B, in silico analysis of 53 glycosylation consensus sequence secondary structures. Each residue was classified as α-helix, β-strand, structured turn, or non-structured loop and is represented as an average distribution for each site. Structures were predicted using Phyre2.
In Vitro, PEB3 Is a Poor Glycosylation Substrate
To evaluate whether PglB shows a preference for folded or unfolded protein targets, glycosylation efficiencies were evaluated using a purified target substrate. PEB3, a C. jejuni homodimeric surface-associated protein with immunogenic properties and predicted transporter functions (25, 34), was selected for these studies because it is a reported soluble glycoprotein, and the x-ray crystallographic structure was previously determined, which shows the native glycosylation site in a structurally defined region of the protein (24, 25) (Fig. 2A). Relative levels of N-glycosylated PEB3 appear to be strain-dependent. In C. jejuni 11168, PEB3 is reported to be ∼50% N-glycosylated (35). In this study we used C. jejuni 81-176, wherein 80–100% of PEB3 is N-glycosylated. Based on x-ray crystallographic analysis, the glycosylation site of PEB3 is located in a structured exposed loop between a β-strand and α-helix (24, 25). Glycosylation efficiencies of purified PEB3, compared with a peptide representing the glycosylation sequon, were assessed using an in vitro radioactivity-based glycosylation assay with a readily accepted tritiated disaccharide donor (Und-PP-diNAcBac-[3H]GalNAc) (36). Purified PglB and glycan donor were incubated with purified PEB3 wild type, a non-glycosylatable mutant of PEB3 (N90Q), or a peptide containing the glycosylation consensus sequence. Glycosylated products were separated from glycan donor using Ni-NTA chromatography and analyzed by liquid scintillation counting to determine the percent of disaccharide incorporated onto the protein. N-Glycosylation of the peptide (∼87%) far exceeded that of wild type PEB3 (∼1%) (Fig. 3A). Although PEB3 wild type exhibited very low levels of N-glycosylation, the extent was reproducibly higher than background levels of the assay, as determined using PEB3 N90Q. These data show that although PEB3 is an N-glycosylation substrate in vivo, it is not robustly modified in this in vitro glycosylation assay, suggesting that partial destabilization or unfolding is required for efficient glycosylation in the cell.
FIGURE 3.

In vitro analysis of PEB3, PEB3 variants, and peptide substrate. A, the percentage of radiolabeled disaccharide transferred to protein (green bars) or peptide substrate (blue bar) was measured by liquid scintillation counting. Results are representative of one data set from triplicate experiments. Background activity is indicated by dashed line. B, schematic representation of PEB3 and the position where the glycosylation consensus sequence is substituted for each variant. The native consensus sequence is colored in magenta, repositioned consensus sequences are colored in blue, and the positions of the glycosylated asparagine residues are represented as sticks. First derivative curves of averaged thermal denaturation data collected from three replicates (C), and calculated Tm values of each PEB3 variant with standard deviation were determined from triplicate experimental results (D).
In Vitro, Partial Destabilization of PEB3 Increases Glycosylation Efficiency
To investigate the structural determinants of protein substrates contributing to N-glycosylation, we determined the glycosylation efficiencies of PEB3 variants with repositioned glycosylation sites. The x-ray structure of PEB3 guided the selection of five sites for integration of the glycosylation consensus sequence (Fig. 3B, Table 1). To avoid intrinsic variation of the local sequence, which has been shown to effect glycosylation efficiencies in eukaryotes (37) and prokaryotes (38), we integrated the same, native PEB3 sequence at each selected site (DFNVS). Two sites are located in predicted flexible loops (A179N and I199N), and the other three are found in a structured helix-turn-helix (Q152N), a partially buried turn between an α-helix and β-strand (D68N), and a buried β-strand (G72N). For simplicity and to ensure incorporation of a single glycan, consensus sequences were introduced by site-directed mutagenesis in a PEB3 mutant lacking the native glycosylated asparagine residue (N90Q). Purification by Ni-NTA chromatography followed by size-exclusion chromatography confirmed that all PEB3 variants were soluble and monodisperse (supplemental Fig. S1).
TABLE 1.
Structural features of PEB3 glycosylation site variants. Determined using PEB3 x-ray crystallographic structure (PDB code 2HXW)
| Variant | Local structure | Solvent exposure |
|---|---|---|
| D68N | Turn | Partially buried |
| G72N | β-Strand | Buried |
| Q152N | Turn | Surface |
| A179N | Loop | Surface |
| I199N | Loop | Surface |
In vitro end-point glycosylation assays with Und-PP-diNAcBac-[3H]GalNAc were performed with the panel of PEB3 variants. Compared with wild type, an increase in the level of the N-glycoprotein was observed in four of the five variants, with the most efficient incorporation into D68N, Q152N, and A179N, whereas only background levels of glycosylation were observed for the fully buried site at G72N (Fig. 3A). Despite the enhanced levels of N-glycosylation for several PEB3 variants (5–15%), they did not achieve N-glycosylation levels comparable to the peptide (87%) under identical experimental conditions.
The observed increase in glycosylation at these sites may be attributed to increased flexibility due to destabilization of the native fold. To determine the extent of destabilization, average melting temperature (Tm) values of PEB3 variants were measured by thermofluor analysis using SYPRO Orange (39). The average Tm values of all variants were lower (4–5 °C) than the wild type, suggesting that these variants were structurally less stable (Fig. 3, C and D).
Together these data confirm that buried glycosylation sites are not targeted by PglB in vitro. The results also suggest that conformational changes to the native glycosylation sites may be necessary to allow for efficient modification.
Efficient N-Linked Glycosylation Is Dependent on Sec Translocation in Glycocompetent E. coli
Although N-linked glycosylation of PEB3 in C. jejuni has been experimentally validated (35), our data indicate that it is a very poor substrate under in vitro conditions. Several factors may contribute to this observation. First, the native glycosylation consensus sequence is located between two defined elements of secondary structure, affording rigidity that may decrease the accessibility of the glycosylation site to PglB. Second, the folded state (local or global) of PEB3 during glycosylation in vivo, which is currently unknown, may impose constraints that preclude efficient glycosylation. Based on the signal sequence prediction software (PRED-TAT), PEB3 is predicted to be delivered to the periplasm via the Sec export pathway wherein proteins are translocated to the periplasm in an unfolded conformation and fold during or after translocation (40). We postulated that the poor in vitro N-glycosylation levels of wild type PEB3 is due to its folded state and that N-linked glycosylation occurs before or during folding, either co- or post-translocationally.
To better understand the dynamics of in vivo glycosylation, we evaluated the glycosylation efficiency of PEB3 when it was translocated via the Sec pathway or through an alternative translocation pathway, the TAT pathway, which transports fully folded proteins and oligomeric protein complexes across the inner membrane into the periplasm (41, 42). This experiment helps to evaluate the importance of environmental factors that differ between the in vitro glycosylation assay conditions and the native periplasmic space. This approach was previously used by Kowarik et al. to demonstrate that glycosylation of AcrA, which has two glycosylation sites, can occur on folded or unfolded proteins in glycocompetent E. coli (21). Currently, an x-ray crystallographic structure of full-length AcrA has not yet been determined; however, homology modeling suggests that one glycosylation site is in an α-helix, and the other is in a flexible loop (18). For the current study we used densitometry analysis to compare relative glycosylation efficiencies of two proteins, PEB3 and AcrA, when they are transported through the Sec and TAT pathways in glycocompetent E. coli. A soluble periplasmic variant of AcrA, which is a native lipoprotein of C. jejuni, was previously produced in E. coli by substituting the native Sec signal sequence of AcrA, including the lipidation site, with the Sec signal sequence of pelB (21). The same strategy was employed for the current experiments. The Sec signal sequence of pelB and TAT signal sequence of ycbk were substituted for the native signal sequence of AcrA, and a C-terminal hexahistidine (His6) tag was added. Both signal sequences are known to direct translocation through the designated machinery (43, 44). PEB3 contains a predicted native Sec signal sequence; thus, it was kept intact for Sec-mediated translocation and was substituted with the ycbk signal sequence for rerouting through that TAT pathway. Each construct was co-expressed in E. coli W3110 together with the pgl locus. Target proteins purified from the periplasmic fraction were analyzed by SDS-PAGE and immunoblotting with anti-His antibody, for detection of both non-glycosylated and glycosylated protein, and anti-N-glycan antibody (R1), which recognizes the C. jejuni heptasaccharide, for detection of glycosylated protein (45). Both glycosylation sites on AcrA were modified as noted by observation of two higher molecular weight bands that reacted with anti-N-glycan antibodies corresponding to mono- and di-glycosylated AcrA (Fig. 4A and supplemental Fig. S2A). Previous studies have confirmed these shifts by mass spectrometry (21). Analysis by densitometry indicated a modest, but significant increase in total di-glycosylated Sec-translocated AcrA (19 ± 3%) compared with TAT-translocated AcrA (11 ± 2%) (Fig. 4B). Of note, the slight difference in molecular weights between non-glycosylated Sec- and TAT-translocated AcrA are due to a linker that was included when constructing the plasmid containing pelB.
FIGURE 4.
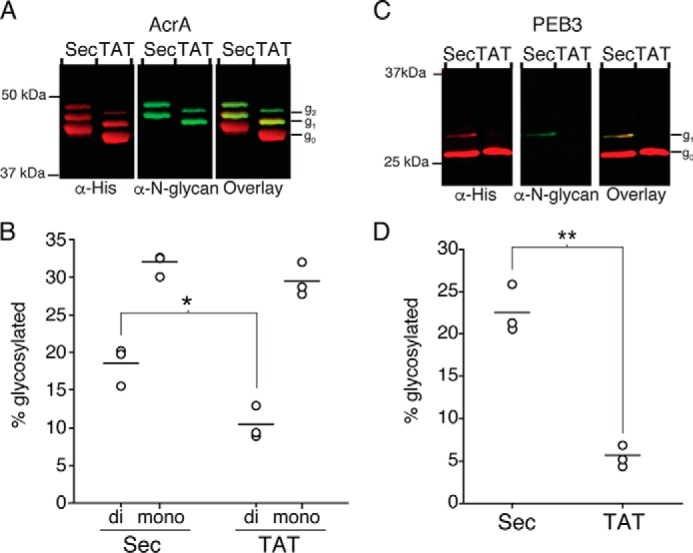
Glycosylation of protein substrates in glycocompetent E. coli. Representative Western blot analysis of periplasmically-localized AcrA (A) and PEB3 (C) translocated via the Sec or TAT pathway in E. coli (n = 3). Total AcrA or PEB3 was detected with anti-His antibody, and N-linked glycosylated proteins were detected with anti-N-glycan antibody (R1). Non-glycosylated (g0), mono-glycosylated (g1), and di-glycosylated (g2) species are indicated. Average glycosylation of AcrA (B) and PEB3 (D) from three biological replicates was determined by densitometry analysis of non-glycosylated and glycosylated protein bands (*, p < 0.02; **, p < 0.002).
More strikingly, glycosylation efficiencies of PEB3 were significantly higher for Sec-translocated PEB3 (23 ± 3%), compared with TAT translocated PEB3 (6 ± 1%) (Fig. 4, C and D, and supplemental Fig. S2B). These results indicate that glycosylation in glycocompetent E. coli is partially dependent on the Sec translocation pathway.
Consistent with previous studies in glycocompetent E. coli (19, 46), total glycosylation levels of AcrA (50 ± 2%) and PEB3 (23 ± 3%) are low. Several known factors contribute to inefficient glycosylation in glycocompetent E. coli. For example, WecA, a priming glycosyltransferase, can interfere with Und-PP-heptasaccharide production by transferring GlcNAc-P from UDP-GlcNAc onto the Und-P carrier, thereby providing a non-native substrate for the Pgl pathway, rather than the native Und-PP-diNAcBac (19, 47). In addition, in E. coli, PglB can compete with WaaL, an O-antigen ligase, for substrates by incorporating O-antigen subunits onto acceptor proteins (48). Efforts to improve glycosylation efficiencies by targeting these enzymes as well as controlling carbon flux in E. coli have resulted in only modest increases in N-glycoprotein yields (49, 50). In addition to these limitations, we propose that interactions between PglB and its natively partnered translocon may be lost upon transfer to E. coli, thus uncoupling protein translocation from N-glycosylation in the periplasm. To address this, we examined the contribution of Sec translocation to N-glycosylation in the native host, C. jejuni.
Efficient N-Linked Glycosylation Is Dependent on Sec Translocation in C. jejuni
To direct translocation through the Sec or TAT pathways of C. jejuni, expression vectors encoding acrA and peb3 with the Sec or TAT signal sequences were generated. For Sec-mediated translocation, the pelB signal sequence from E. coli was used for acrA, and the native signal sequence was used for peb3. For TAT-mediated translocation, acrA and peb3 were modified with the TAT signal sequence from C. jejuni torA (cj0264c), which was previously determined to be a TAT-transported protein (51). Analysis of periplasmic purified AcrA demonstrated overall higher levels of N-linked glycosylation for both Sec- and TAT-translocated protein (91 ± 7% and 81 ± 5%, respectively) compared with the levels observed in glycocompetent E. coli (Fig. 5, A and B, and supplemental Fig. S2C). Although there was no significant difference in total glycosylation, we observed a statistically significant increase in di-glycosylated AcrA for Sec-dependent translocation, consistent with the glycocompetent E. coli results (Sec, 74 ± 10%; TAT, 35 ± 2%).
FIGURE 5.
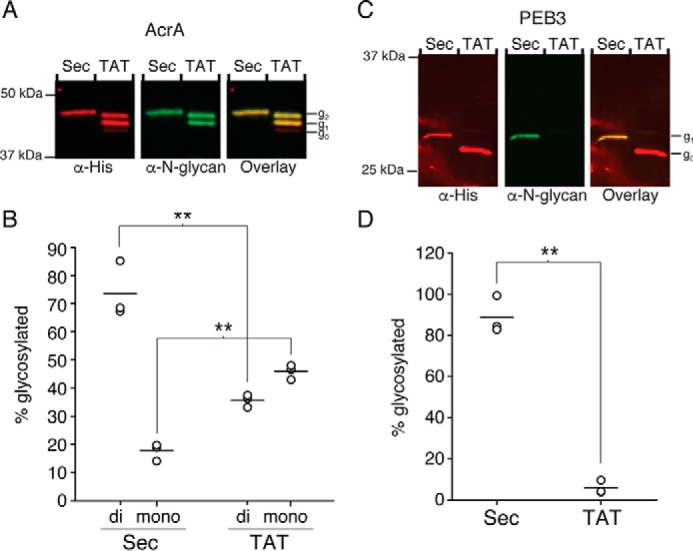
Glycosylation of protein substrates in C. jejuni. Representative Western blot results of periplasmically-localized AcrA (A) and PEB3 (C) translocated via the Sec or TAT pathway in C. jejuni (n = 3) are shown. Total AcrA or PEB3 were detected with anti-His antibody, and N-linked glycosylated proteins were detected with anti-N-glycan antibody (R1). Non-glycosylated (g0), mono-glycosylated (g1), and di-glycosylated (g2) species are indicated. Average glycosylation of AcrA (B) and PEB3 (D) from three biological replicates was determined by densitometry analysis of non-glycosylated and glycosylated protein bands (**, p < 0.002).
N-Linked glycosylation in C. jejuni was markedly higher for Sec translocated PEB3 (89 ± 9%) than for TAT-translocated PEB3 (6 ± 3%) (Fig. 5, C and D, supplemental Fig. S2D). These results in C. jejuni are consistent with those in glycocompetent E. coli, although the relative levels of glycosylation differ between the two systems.
These data support the hypothesis that PglB specificity depends on the substrate protein and the conformation of the glycosylation sequon that is presented as proteins are delivered to the periplasm. To further validate these findings and to determine whether the correlation between in vitro and in vivo glycosylation efficiencies are consistent, we assessed glycosylation in C. jejuni of the aforementioned PEB3 variant (A179N) that was shown to be N-linked-glycosylated more efficiently compared with wild type PEB3 in vitro (Fig. 3A). When PEB3 A179N was expressed with the native Sec signal sequence or TAT signal sequence in C. jejuni, the protein was fully glycosylated for both conditions, confirming the hypothesis that the structural context influences glycosylation efficiency (Fig. 6 and supplemental Fig. S2E). Together with the results from native PEB3, these findings suggest that coupling with the Sec translocation pathway will be useful for efficient glycosylation of proteins wherein glycosylation sites in the native target are predicted to be structurally defined.
FIGURE 6.
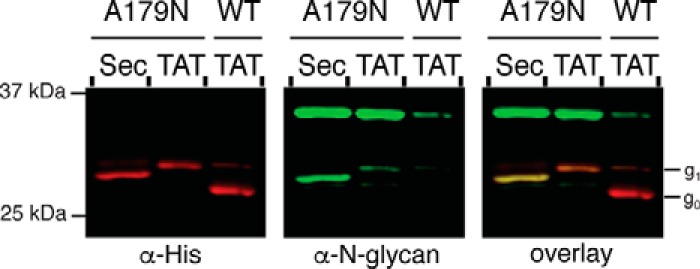
Glycosylation of PEB3 variant A179N in C. jejuni. Representative Western blot results of Sec or TAT translocated PEB3 variant A179N (n = 3). For comparison, TAT-translocated wild type PEB3 is shown. Total PEB3 was detected with anti-His antibody, and N-linked glycosylated proteins were detected with anti-N-glycan antibody (R1). Non-glycosylated (g0) and mono-glycosylated (g1) species are indicated.
Discussion
Through the analysis of native N-glycosylation sites of C. jejuni glycoproteins in vitro and in vivo, we show that PglB substrate selectivity is dependent on the structure and accessibility of the target asparagine residue. In the cell, Sec-dependent translocation of substrate proteins promotes efficient N-glycosylation, in contrast to protein substrates translocated in a TAT-dependent manner. These findings underscore the complexity of this post-translational modification system and suggest that protein N-linked glycosylation occurs before complete folding of substrate proteins in bacteria, reminiscent of eukaryotic N-linked glycosylation (Fig. 1).
The previously determined x-ray structure of PglB from C. lari (56% sequence identity to C. jejuni) shows that the co-crystallized hexapeptide, which contains the glycosylation consensus peptide (DQNATF), forms a 180° chain reversal in the active site of PglB (31). Moreover, this binding site does not appear to be large enough to accommodate fully folded proteins, consistent with the findings in our study. Together, our data are in agreement with a scenario in which N-glycosylation activity by PglB is in competition with substrate protein folding and diffusion away from the active site. Although direct evidence is yet to be shown, there are several possible underlying mechanisms by which PglB could overcome these competing factors in C. jejuni. Our data support the hypothesis that N-linked glycosylation is coupled with Sec-mediated protein translocation through interactions between Sec subunits and PglB. In eukaryotes, direct interactions between OTase subunits and Sec61 subunits position the OTase for glycosylation of nascent proteins entering the RER, thus reducing diffusion effects and favoring glycosylation before folding can be completed (7–9, 52). Identification of direct or indirect interactions between PglB and the Sec translocon would support the coupling model in bacteria.
The discovery of bacterial glycosylation systems has ignited interest in their use for the production of vaccines and therapeutic proteins (53). Nevertheless, this study and previous studies have observed suboptimal N-glycosylation levels of C. jejuni substrate proteins when expressed in glycocompetent E. coli. Efforts to engineer E. coli for glycosylation with bacterial or eukaryotic-like glycans have been successful; however, establishing robust systems remains a challenge (50, 54, 55). Based on the model that protein N-linked glycosylation and translocation are coupled, we hypothesize that PglB interactions with the Sec translocon would be lost in the heterologous system, contributing further to inefficient glycosylation. Indeed, there is low sequence identity between SecYEG subunits of C. jejuni and E. coli (41%, 31%, 29%, respectively). Deciphering the mechanistic basis of glycosylation in C. jejuni could provide valuable information for the optimization of glycosylation in a heterologous host.
In addition to potential interactions between PglB and the Sec translocon, one should also consider accessory proteins or chaperones that could maintain protein substrates in an optimal conformation for modification by PglB. With the exception of some lower eukaryotes, in particular parasites Leishmania major and Trypanosoma brucei, which contain a single subunit OTase (56–58), eukaryotic OTases are hetero-oligomeric complexes comprising the catalytic subunit (STT3) and up to seven additional subunits, some of which have been shown to have distinct roles in stabilizing the complex, interacting with the Sec61 translocon or mediating glycosylation before complete folding of substrate proteins (59, 60). For example, subunits of the yeast OTase complex, Ost3p/Ost6p, and the mammalian STT3B OTase complex, MagT1/TUSC3, exhibit oxidoreductase activity, which is thought to delay oxidative protein folding through mixed disulfide bond formation (61–63). Although the enzymes required to synthesize and transfer the C. jejuni heptasaccharide are encoded adjacent to each other on the chromosome, there may be other nonessential components elsewhere on the chromosome with analogous functions to the eukaryotic OTase subunits. One candidate protein is PpiD (peptidyl prolyl isomerase), which was recently identified in C. jejuni (64). The PpiD E. coli homolog is anchored in the inner membrane and interacts with SecYEG, modulating folding of newly exported proteins (65, 66). It is conceivable that unidentified accessory proteins of PglB or periplasmic chaperones could play a role in the N-glycosylation process.
In conclusion, we have provided strong evidence that bacterial N-glycosylation is a temporally and spatially coordinated process. Further identification of the molecular dynamics underlying this pathway will enhance our understanding of functional macromolecular complexes and potentially enhance the bioengineering application of this system.
Experimental Procedures
In Silico Analysis of N-Linked Glycosylation Consensus Sequences
Predicted secondary and tertiary structures of experimentally identified C. jejuni glycoproteins were determined using Phyre2 (Protein Homology/analogY Recognition Engine V 2.0) (33). Predicted structures with > 80% confidence were used for the analysis. The secondary structure of each amino acid in a glycosylation site was classified as an α-helix, β-strand, turn, or non-structured loop. A non-structured loop is defined as a region that has more than five residues that are not appropriately positioned to form hydrogen bonding interactions between backbone-amide bond partners.
Bacterial Strains and Growth Conditions
Strains and plasmids used in this study are listed in supplemental Table S2. E. coli strains were grown in lysogeny broth (LB) broth supplemented with 30 μg/ml chloramphenicol, 100 μg/ml carbenicillin, or 30 μg/ml kanamycin as required. C. jejuni 81-176 cells were grown in Mueller Hinton (MH) broth under microaerophilic conditions (BD Biosciences CampyPak or pre-mixed gas: 5% O2, 10% CO2, and 85% N2) supplemented with 10 μg/ml trimethoprim and 15 μg/ml chloramphenicol when required (67). For expression in C. jejuni, pCE11128 derivatives were transformed into DH5α E. coli cells carrying a conjugation helper plasmid, RK212.2 (68–70). C. jejuni 81-176 wild type recipients were grown on MH agar supplemented with 10 μg/ml trimethoprim overnight. Donor and recipient were mixed 1:1 on MH agar with no antibiotic and incubated for 6 h under microaerophilic conditions at 37 °C. Harvested cells were resuspended in MH broth and plated onto agar plates containing 10 μg/ml trimethoprim and 15 μg/ml chloramphenicol. Plates were incubated at 37 °C for 1–2 days under microaerophilic conditions. Colonies were restreaked and grown in liquid culture. To confirm success of conjugation and expression of target proteins, cell lysates were prepared for Western blot analysis from liquid cultures, and His6-tagged proteins were probed with primary mouse IgG2b anti-His monoclonal antibody (1:5000 dilution, LifeTein LLC, South Plainfield, NJ; catalog #LT 04256 Lot #411005100202) and secondary antibody IRDye® 680LT goat anti-mouse (Li-COR Biosciences, Lincoln; NE catalog #926-68020 lot #C50721–02) and visualized with a Li-COR Odyssey IR-Scanner.
Plasmid Construction
Primers used to generate plasmids for this study are listed in supplemental Table S2. For cytoplasmic expression of PEB3 wild type and variants, PCR-amplified peb3 (residues 21–251) from C. jejuni 81-176 genomic DNA was inserted into pET24a at restriction sites NdeI and XhoI. Glycosylation consensus sequences were substituted using site-directed mutagenesis (QuikChange, Agilent). For expression of Sec-translocated PEB3 in glycocompetent E. coli, full-length peb3 was PCR-amplified and inserted into pET22b at restriction sites NdeI and XhoI. This construct was used as a template for PCR amplification, digestion with BspH1 and EcoRI, and insertion into pBAD-mNectarine (71) digested with NcoI and EcoRI. For Sec-translocated AcrA (pBAD::pelB::acrA) the acrA gene encoding residues 23–347 was PCR-amplified, digested with BamHI and XhoI, and inserted into pET22b. This was used as a template for PCR amplification, digestion with BspH1 and EcoRI, and insertion into pBAD-mNectarine digested with NcoI and EcoRI. For TAT-translocated substrates, the pelB signal sequence of pET22b was replaced with either ycbK or torAcj signal sequences by PCR amplification, digestion with NdeI and BamHI, and insertion into pET22b. peb3 or acrA lacking their native signal sequences were inserted at BamHI and XhoI restriction sites of the modified pET22b plasmids and subcloned into pBAD-mNectarine. The C. jejuni expression vector, pCE11128, was modified to contain a ribosome-binding site (RBS) before the BamHI site followed by a multiple cloning site and a C-terminal His6 tag after the XhoI site using synthesized oligonucleotides to generate pCE11128H::rbs (Sigma). pET22b containing peb3 or acrA derivatives were digested with BglII and XhoI. The insert was subcloned into pCE11128H::rbs digested with BamHI and XhoI. All constructs were confirmed by sequencing.
Expression and Purification of Recombinant PEB3
C-terminal His6-tagged PEB3 wild type and PEB3 variants were overexpressed in E. coli BL21 cells. Specifically, overnight cultures were sub-inoculated at 1:1000 into 1 liter of LB with the required antibiotics. Cell cultures were grown with shaking at 37 °C to A600 = 0.6 and subsequently induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside for 16 h at 16 °C. Cells were harvested in a Beckman Allegra 6 Series swing bucket centrifuge at 3200 × g at 4 °C for 15 min. Pellets were washed with 50 ml of Buffer 1 (50 mm HEPES, pH 7.5, 100 mm NaCl) and resuspended in Buffer 1 supplemented with 10 mm imidazole and EDTA-free protease inhibitor mixture. The resuspension was kept on ice and sonicated 2 times, with a 10-min recovery period after 40 s of 1-s pulses. Lysed cells were centrifuged in a Beckman ultracentrifuge at 9000 × g for 40 min at 4 °C. Clarified lysates were incubated with Ni-NTA resin for 30 min at 4 °C with gentle agitation. The bead/lysate mixture was applied to a gravity flow column and washed with 10 column volumes (CV) of buffer 1 with 20 mm followed by 45 mm imidazole. Protein was eluted in 4 × 1 CV fractions with buffer 1 with 300 mm imidazole. Samples of each fraction were mixed 1:1 with SDS loading dye, boiled, and analyzed by SDS-PAGE. A 3-ml aliquot of fraction 1 of the eluted sample was injected onto a Superdex 200 16/60 HiLoad prep grade column. Fractions were collected, analyzed by SDS-PAGE, and concentrated to 100–400 μm using Amicon® Ultra Centrifugal Filters molecular weight cut off 10,000 kDa and stored at −80°C.
Thermal Stability Assays
Melting temperatures (Tm) of PEB3 variants were determined using SYPRO Orange thermal melt assays (39). SYPRO Orange (Sigma) is an environmentally sensitive fluorescent dye that undergoes dequenching when it binds to proteins with exposed hydrophobic regions. A solution containing 20 μm of PEB3 in 50 mm HEPES, pH 7.5, 100 mm NaCl, and 10× SYPRO Orange (5000× stock) was subjected to temperature increasing from 20 °C to 99 °C at 0.06 °C/s in a LightCycler 480 instrument (Roche Applied Science), and fluorescence was monitored and recorded (excitation and emission wavelengths of 465 and 610 nm, respectively). Tm values were determined using the negative first derivative of the fluorescence curve (change in fluorescence/change in temperature). The reported melting temperatures are the mean ± S.D. for three replicates.
In Vitro Glycosylation Assay
Und-PP-diNAcBac-[3H]GalNAc (36) and purified PglB were prepared as previously reported (72). Peptide substrate, Ac-HHHHHHYGSGSDFNVS-CONH2 (>96% purity) was purchased from Boston Open Labs (Cambridge, MA). Dried Und-PP-diNAcBac-[3H]GalNAc (specific activity = 15 nCi/nmol) was resuspended in DMSO (5% final). Samples including 20 μm Und-PP-diNAcBac-[3H]GalNAc, 20 μm PEB3 variant or peptide substrate, 140 mm sucrose, 1.2% Triton X-100 (v/v), 140 μm HEPES, pH 7.5, 10 mm MnCl2, and 20 nm concentrations of purified PglB were incubated at room temperature for 4 h with shaking. The reaction was stopped by washing the mixture over Ni-NTA resin in Millipore Ultrafree-MC GV centrifugal filters. The resin was washed with 2 CV of buffer 1 with 20 mm imidazole followed by 45 mm imidazole. Protein was eluted in 4 × 1 CV fractions of buffer 1 with 300 mm imidazole. For each wash the flow-through was collected and measured for glycan incorporation by scintillation counting on a LS6500 Beckman Scintillation Counter. Experiments were performed in triplicate. The percent glycosylated protein was calculated by dividing the total counts of the 300 mm imidazole-eluted samples over the total counts for all flow-through samples.
E. coli- and C. jejuni-based Glycosylation Assay
E. coli strains harboring plasmids encoding target proteins and pACYC::pgl or pACYC::pgl::ΔpglB were sub-inoculated at 1:1000 in 250 ml of LB broth with required antibiotics. Cell cultures were grown with shaking at 37 °C to A600 = 0.6 and subsequently induced with 0.1 mm isopropyl β-d-1-thiogalactopyranoside and 0.08% arabinose for 3–4 h. Cell cultures were centrifuged at 3000 × g at 4 °C for 15 min. The periplasmic fraction was extracted by osmotic shock. Cell pellets were gently resuspended in 1.5 ml of cold spheroplasting buffer (25 mm Tris, pH 8.0, 2 mm EDTA). 1.5 ml of cold 40% sucrose/25 mm Tris, pH 8.0, were added, and the mixture was incubated on ice for 15 min. After incubation, 15 μl of 1 m MgSO4 were added, and the sample was centrifuged at 16,000 × g at 4 °C for 30 min. The supernatant (periplasmic and outermembrane components) was diluted in 10 ml of buffer 1 with 10 mm imidazole and mixed with 100 μl of resin Ni-NTA resin for 30 min. The bead/lysate mixture was applied to a gravity flow column and washed with 5 CV of buffer 1 with 20 mm followed by 45 mm imidazole. Protein was eluted in 1 CV of buffer 1 with 300 mm imidazole. Samples of each fraction were mixed 1:1 with SDS loading dye, boiled, and analyzed by SDS-PAGE and Western blot. Western blots were probed with mouse monoclonal anti-His antibodies (1:5000 dilution, LifeTein LLC) and rabbit polyclonal anti-N-glycan R1 (1:5000 dilution) (45). Membranes were incubated with IR-dye linked secondary antibodies (Li-COR, IRDye 800CW goat anti-rabbit IgG catalog #926-302211; lot #C51007-08 and IRDye 680LT goat anti-mouse IgG) and visualized with a Li-COR Odyssey IR-Scanner.
For isolation of PEB3 and AcrA from C. jejuni, 250-ml cultures of MH broth supplemented with 10 μg/ml trimethoprim and 15 μg/ml chloramphenicol were inoculated from agar plates and grown for 24 h with shaking at 37 °C under microaerophilic conditions. Cell cultures were centrifuged at 3000 × g at 4 °C for 15 min. Protein from the periplasmic fraction were isolated and purified with the same procedure used for E. coli.
Levels of glycosylated and non-glycosylated protein were quantitated by densitometry with ImageJ and normalized to total protein levels (glycosylated/(glycosylated + non-glycosylated)). Statistical analysis was performed by Student's t test. A p value of <0.02 was considered statistically significant.
Author Contributions
J. M. S. and B. I. conceived the study. J. M. S. designed, performed, and analyzed the experiments. J. M. S. and B. I. wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Prof. Christine Szymanski from the University of Georgia and Dr. Patricia Guerry from the Naval Medical Research Center for C. jejuni strains and plasmids and for expert advice on handling C. jejuni. We also thank Prof. Markus Aebi from ETH Zürich for the pgl expression plasmid and the BioMicroCenter at MIT for assistance with thermofluor assays.
This work was supported by National Institutes of Health Grants GM-039334 (to B. I.) and AI109857 (to J. M. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1 and S2 and Figs. S1 and S2.
- RER
- rough endoplasmic reticulum
- OTase
- oligosaccharyltransferase
- Sec
- general secretion
- Und-PP
- undecaprenol diphosphate
- TAT
- twin arginine translocation
- Ni-NTA
- nickel-nitrilotriacetic acid
- MH
- Mueller Hinton
- RBS
- ribosome-binding site
- CV
- column volume.
References
- 1. Helenius A., and Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 2. Vigerust D. J., and Shepherd V. L. (2007) Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mitra N., Sinha S., Ramya T. N., and Surolia A. (2006) N-linked oligosaccharides as outfitters for glycoprotein folding, form and function. Trends Biochem. Sci. 31, 156–163 [DOI] [PubMed] [Google Scholar]
- 4. Kelleher D. J., and Gilmore R. (2006) An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R [DOI] [PubMed] [Google Scholar]
- 5. Kelleher D. J., Banerjee S., Cura A. J., Samuelson J., and Gilmore R. (2007) Dolichol-linked oligosaccharide selection by the oligosaccharyltransferase in protist and fungal organisms. J. Cell Biol. 177, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kelleher D. J., Karaoglu D., Mandon E. C., and Gilmore R. (2003) Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol. Cell 12, 101–111 [DOI] [PubMed] [Google Scholar]
- 7. Nilsson I., Kelleher D. J., Miao Y., Shao Y., Kreibich G., Gilmore R., von Heijne G., and Johnson A. E. (2003) Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J. Cell Biol. 161, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shibatani T., David L. L., McCormack A. L., Frueh K., and Skach W. R. (2005) Proteomic analysis of mammalian oligosaccharyltransferase reveals multiple subcomplexes that contain Sec61, TRAP, and two potential new subunits. Biochemistry 44, 5982–5992 [DOI] [PubMed] [Google Scholar]
- 9. Pfeffer S., Dudek J., Gogala M., Schorr S., Linxweiler J., Lang S., Becker T., Beckmann R., Zimmermann R., and Förster F. (2014) Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 5, 3072. [DOI] [PubMed] [Google Scholar]
- 10. Ruiz-Canada C., Kelleher D. J., and Gilmore R. (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shrimal S., Trueman S. F., and Gilmore R. (2013) Extreme C-terminal sites are posttranslocationally glycosylated by the STT3B isoform of the OST. J. Cell Biol. 201, 81–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nothaft H., and Szymanski C. M. (2010) Protein glycosylation in bacteria: sweeter than ever. Nat. Rev. Microbiol. 8, 765–778 [DOI] [PubMed] [Google Scholar]
- 13. Szymanski C. M., Yao R., Ewing C. P., Trust T. J., and Guerry P. (1999) Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol. Microbiol. 32, 1022–1030 [DOI] [PubMed] [Google Scholar]
- 14. Glover K. J., Weerapana E., and Imperiali B. (2005) In vitro assembly of the undecaprenylpyrophosphate-linked heptasaccharide for prokaryotic N-linked glycosylation. Proc. Natl. Acad. Sci. U.S.A. 102, 14255–14259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allos B. M. (2001) Campylobacter jejuni Infections: update on emerging issues and trends. Clin. Infect. Dis. 32, 1201–1206 [DOI] [PubMed] [Google Scholar]
- 16. Szymanski C. M., Burr D. H., and Guerry P. (2002) Campylobacter protein glycosylation affects host cell interactions. Infect. Immun. 70, 2242–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hendrixson D. R., and DiRita V. J. (2004) Identification of Campylobacter jejuni genes involved in commensal colonization of the chick gastrointestinal tract. Mol. Microbiol. 52, 471–484 [DOI] [PubMed] [Google Scholar]
- 18. Kowarik M., Young N. M., Numao S., Schulz B. L., Hug I., Callewaert N., Mills D. C., Watson D. C., Hernandez M., Kelly J. F., Wacker M., and Aebi M. (2006) Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 25, 1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wacker M., Linton D., Hitchen P. G., Nita-Lazar M., Haslam S. M., North S. J., Panico M., Morris H. R., Dell A., Wren B. W., and Aebi M. (2002) N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298, 1790–1793 [DOI] [PubMed] [Google Scholar]
- 20. Scott N. E., Parker B. L., Connolly A. M., Paulech J., Edwards A. V., Crossett B., Falconer L., Kolarich D., Djordjevic S. P., Højrup P., Packer N. H., Larsen M. R., and Cordwell S. J. (2011) Simultaneous glycan-peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol. Cell Proteomics 10, M000031–MCP201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kowarik M., Numao S., Feldman M. F., Schulz B. L., Callewaert N., Kiermaier E., Catrein I., and Aebi M. (2006) N-Linked glycosylation of folded proteins by the bacterial oligosaccharyltransferase. Science 314, 1148–1150 [DOI] [PubMed] [Google Scholar]
- 22. Lin J., Michel L. O., and Zhang Q. (2002) CmeABC functions as a multidrug efflux system in Campylobacter jejuni. Antimicrob. Agents Chemother. 46, 2124–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fisher A. C., Haitjema C. H., Guarino C., Çelik E., Endicott C. E., Reading C. A., Merritt J. H., Ptak A. C., Zhang S., and DeLisa M. P. (2011) Production of secretory and extracellular N-linked glycoproteins in Escherichia coli. Appl. Environ. Microbiol. 77, 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rangarajan E. S., Bhatia S., Watson D. C., Munger C., Cygler M., Matte A., and Young N. M. (2007) Structural context for protein N-glycosylation in bacteria: the structure of PEB3, an adhesin from Campylobacter jejuni. Protein Sci. 16, 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Min T., Vedadi M., Watson D. C., Wasney G. A., Munger C., Cygler M., Matte A., and Young N. M. (2009) Specificity of Campylobacter jejuni adhesin PEB3 for phosphates and structural differences among its ligand complexes. Biochemistry 48, 3057–3067 [DOI] [PubMed] [Google Scholar]
- 26. Petrescu A. J., Milac A. L., Petrescu S. M., Dwek R. A., and Wormald M. R. (2004) Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology 14, 103–114 [DOI] [PubMed] [Google Scholar]
- 27. Whitley P., Nilsson I. M., and von Heijne G. (1996) A nascent secretory protein may traverse the ribosome endoplasmic reticulum translocase complex as an extended chain. J. Biol. Chem. 271, 6241–6244 [DOI] [PubMed] [Google Scholar]
- 28. Chen W., and Helenius A. (2000) Role of ribosome and translocon complex during folding of influenza hemagglutinin in the endoplasmic reticulum of living cells. Mol. Biol. Cell 11, 765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kawai F., Paek S., Choi K. J., Prouty M., Kanipes M. I., Guerry P., and Yeo H. J. (2012) Crystal structure of JlpA, a surface-exposed lipoprotein adhesin of Campylobacter jejuni. J. Struct. Biol. 177, 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Müller A., Thomas G. H., Horler R., Brannigan J. A., Blagova E., Levdikov V. M., Fogg M. J., Wilson K. S., and Wilkinson A. J. (2005) An ATP-binding cassette-type cysteine transporter in Campylobacter jejuni inferred from the structure of an extracytoplasmic solute receptor protein. Mol. Microbiol. 57, 143–155 [DOI] [PubMed] [Google Scholar]
- 31. Lizak C., Gerber S., Numao S., Aebi M., and Locher K. P. (2011) X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 [DOI] [PubMed] [Google Scholar]
- 32. Slynko V., Schubert M., Numao S., Kowarik M., Aebi M., and Allain F. H. (2009) NMR structure determination of a segmentally labeled glycoprotein using in vitro glycosylation. J. Am. Chem. Soc. 131, 1274–1281 [DOI] [PubMed] [Google Scholar]
- 33. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pei Z. H., Ellison R. T. 3rd, and Blaser M. J. (1991) Identification, purification, and characterization of major antigenic proteins of Campylobacter jejuni. J. Biol. Chem. 266, 16363–16369 [PubMed] [Google Scholar]
- 35. Linton D., Allan E., Karlyshev A. V., Cronshaw A. D., and Wren B. W. (2002) Identification of N-acetylgalactosamine-containing glycoproteins PEB3 and CgpA in Campylobacter jejuni. Mol. Microbiol. 43, 497–508 [DOI] [PubMed] [Google Scholar]
- 36. Glover K. J., Weerapana E., Numao S., and Imperiali B. (2005) Chemoenzymatic synthesis of glycopeptides with PglB, a bacterial oligosaccharyl transferase from Campylobacter jejuni. Chem. Biol. 12, 1311–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Culyba E. K., Price J. L., Hanson S. R., Dhar A., Wong C. H., Gruebele M., Powers E. T., and Kelly J. W. (2011) Protein native-state stabilization by placing aromatic side chains in N-glycosylated reverse turns. Science 331, 571–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen M. M., Glover K. J., and Imperiali B. (2007) From peptide to protein: comparative analysis of the substrate specificity of N-linked glycosylation in C. jejuni. Biochemistry 46, 5579–5585 [DOI] [PubMed] [Google Scholar]
- 39. Ericsson U. B., Hallberg B. M., Detitta G. T., Dekker N., and Nordlund P. (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357, 289–298 [DOI] [PubMed] [Google Scholar]
- 40. Bagos P. G., Nikolaou E. P., Liakopoulos T. D., and Tsirigos K. D. (2010) Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics 26, 2811–2817 [DOI] [PubMed] [Google Scholar]
- 41. Berks B. C. (2015) The twin-arginine protein translocation pathway. Annu. Rev. Biochem. 84, 843–864 [DOI] [PubMed] [Google Scholar]
- 42. Beckwith J. (2013) The Sec-dependent pathway. Res. Microbiol. 164, 497–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tullman-Ercek D., DeLisa M. P., Kawarasaki Y., Iranpour P., Ribnicky B., Palmer T., and Georgiou G. (2007) Export pathway selectivity of Escherichia coli twin arginine translocation signal peptides. J. Biol. Chem. 282, 8309–8316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lei S. P., Lin H. C., Wang S. S., Callaway J., and Wilcox G. (1987) Characterization of the Erwinia carotovora pelB gene and its product pectate lyase. J. Bacteriol. 169, 4379–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nothaft H., Scott N. E., Vinogradov E., Liu X., Hu R., Beadle B., Fodor C., Miller W. G., Li J., Cordwell S. J., and Szymanski C. M. (2012) Diversity in the protein N-glycosylation pathways within the Campylobacter genus. Mol. Cell Proteomics 11, 1203–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pandhal J., and Wright P. C. (2010) N-Linked glycoengineering for human therapeutic proteins in bacteria. Biotechnol. Lett. 32, 1189–1198 [DOI] [PubMed] [Google Scholar]
- 47. Linton D., Dorrell N., Hitchen P. G., Amber S., Karlyshev A. V., Morris H. R., Dell A., Valvano M. A., Aebi M., and Wren B. W. (2005) Functional analysis of the Campylobacter jejuni N-linked protein glycosylation pathway. Mol. Microbiol. 55, 1695–1703 [DOI] [PubMed] [Google Scholar]
- 48. Feldman M. F., Wacker M., Hernandez M., Hitchen P. G., Marolda C. L., Kowarik M., Morris H. R., Dell A., Valvano M. A., and Aebi M. (2005) Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 102, 3016–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pandhal J., Desai P., Walpole C., Doroudi L., Malyshev D., and Wright P. C. (2012) Systematic metabolic engineering for improvement of glycosylation efficiency in Escherichia coli. Biochem. Biophys. Res. Commun. 419, 472–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pandhal J., Woodruff L. B., Jaffe S., Desai P., Ow S. Y., Noirel J., Gill R. T., and Wright P. C. (2013) Inverse metabolic engineering to improve Escherichia coli as an N-glycosylation host. Biotechnol. Bioeng. 110, 2482–2493 [DOI] [PubMed] [Google Scholar]
- 51. Hitchcock A., Hall S. J., Myers J. D., Mulholland F., Jones M. A., and Kelly D. J. (2010) Roles of the twin-arginine translocase and associated chaperones in the biogenesis of the electron transport chains of the human pathogen Campylobacter jejuni. Microbiology 156, 2994–3010 [DOI] [PubMed] [Google Scholar]
- 52. Chavan M., Yan A., and Lennarz W. J. (2005) Subunits of the translocon interact with components of the oligosaccharyl transferase complex. J. Biol. Chem. 280, 22917–22924 [DOI] [PubMed] [Google Scholar]
- 53. Jaffé S. R., Strutton B., Levarski Z., Pandhal J., and Wright P. C. (2014) Escherichia coli as a glycoprotein production host: recent developments and challenges. Curr. Opin. Biotechnol. 30, 205–210 [DOI] [PubMed] [Google Scholar]
- 54. Schwarz F., Huang W., Li C., Schulz B. L., Lizak C., Palumbo A., Numao S., Neri D., Aebi M., and Wang L. X. (2010) A combined method for producing homogeneous glycoproteins with eukaryotic N-glycosylation. Nat. Chem. Biol. 6, 264–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Valderrama-Rincon J. D., Fisher A. C., Merritt J. H., Fan Y. Y., Reading C. A., Chhiba K., Heiss C., Azadi P., Aebi M., and DeLisa M. P. (2012) An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat. Chem. Biol. 8, 434–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nasab F. P., Schulz B. L., Gamarro F., Parodi A. J., and Aebi M. (2008) All in one: Leishmania major STT3 proteins substitute for the whole oligosaccharyltransferase complex in Saccharomyces cerevisiae. Mol. Biol. Cell 19, 3758–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hese K., Otto C., Routier F. H., and Lehle L. (2009) The yeast oligosaccharyltransferase complex can be replaced by STT3 from Leishmania major. Glycobiology 19, 160–171 [DOI] [PubMed] [Google Scholar]
- 58. Samuelson J., Banerjee S., Magnelli P., Cui J., Kelleher D. J., Gilmore R., and Robbins P. W. (2005) The diversity of dolichol-linked precursors to Asn-linked glycans likely results from secondary loss of sets of glycosyltransferases. Proc. Natl. Acad. Sci. U.S.A. 102, 1548–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yan Q., and Lennarz W. J. (2002) Studies on the function of oligosaccharyl transferase subunits: Stt3p is directly involved in the glycosylation process. J. Biol. Chem. 277, 47692–47700 [DOI] [PubMed] [Google Scholar]
- 60. Mohorko E., Glockshuber R., and Aebi M. (2011) Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 34, 869–878 [DOI] [PubMed] [Google Scholar]
- 61. Cherepanova N. A., Shrimal S., and Gilmore R. (2014) Oxidoreductase activity is necessary for N-glycosylation of cysteine-proximal acceptor sites in glycoproteins. J. Cell Biol. 206, 525–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mohd Yusuf S. N., Bailey U. M., Tan N. Y., Jamaluddin M. F., and Schulz B. L. (2013) Mixed disulfide formation in vitro between a glycoprotein substrate and yeast oligosaccharyltransferase subunits Ost3p and Ost6p. Biochem. Biophys. Res. Commun. 432, 438–443 [DOI] [PubMed] [Google Scholar]
- 63. Mohorko E., Owen R. L., Malojčić G., Brozzo M. S., Aebi M., and Glockshuber R. (2014) Structural basis of substrate specificity of human oligosaccharyl transferase subunit N33/Tusc3 and its role in regulating protein N-glycosylation. Structure 22, 590–601 [DOI] [PubMed] [Google Scholar]
- 64. Kale A., Phansopa C., Suwannachart C., Craven C. J., Rafferty J. B., and Kelly D. J. (2011) The virulence factor PEB4 (Cj0596) and the periplasmic protein Cj1289 are two structurally related SurA-like chaperones in the human pathogen Campylobacter jejuni. J. Biol. Chem. 286, 21254–21265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Matern Y., Barion B., and Behrens-Kneip S. (2010) PpiD is a player in the network of periplasmic chaperones in Escherichia coli. Bmc Microbiol. 10, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Antonoaea R., Fürst M., Nishiyama K., and Müller M. (2008) The periplasmic chaperone PpiD interacts with secretory proteins exiting from the SecYEG translocon. Biochemistry 47, 5649–5656 [DOI] [PubMed] [Google Scholar]
- 67. Davis L., Young K., and DiRita V. (2008) Genetic manipulation of Campylobacter jejuni. Curr. Protoc. Microbiol. 10.1002/9780471729259.mc08a02s10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Figurski D. H., and Helinski D. R. (1979) Replication of an origin-containing derivative of plasmid rk2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U.S.A. 76, 1648–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Larsen J. C., Szymanski C., and Guerry P. (2004) N-Linked protein glycosylation is required for full competence in Campylobacter jejuni 81-176. J. Bacteriol. 186, 6508–6514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yao R., Alm R. A., Trust T. J., and Guerry P. (1993) Construction of new Campylobacter cloning vectors and a new mutational cat cassette. Gene 130, 127–130 [DOI] [PubMed] [Google Scholar]
- 71. Johnson D. E., Ai H. W., Wong P., Young J. D., Campbell R. E., and Casey J. R. (2009) Red fluorescent protein pH biosensor to detect concentrative nucleoside transport. J. Biol. Chem. 284, 20499–20511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jaffee M. B., and Imperiali B. (2013) Optimized protocol for expression and purification of PglB the membrane-bound bacterial oligosaccharyl transferase. Protein Expr. Purif. 89, 241–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.