

Abstract
Allergic asthma and obesity are the leading health problems in the world. Many studies have shown that obesity is a risk factor of development of asthma. However, the underlying mechanism has not been well established. In this study, we demonstrate that leptin, an adipokine elevated in obese individuals, promoted proliferation and survival of pro-allergic type 2 helper T cells and group 2 innate lymphoid cells and production of type 2 cytokines, which together contribute to allergic responses. Leptin activates mTORC1, MAPK, and STAT3 pathways in TH2 cells. The effects of leptin on TH2 cell proliferation, survival, and cytokine production are dependent on the mTORC1 and MAPK pathways as revealed by specific inhibitors. In vivo, leptin-deficiency led to attenuated experimental allergic airway inflammation. Our results thus support that obesity-associated elevation of leptin contributes to the increased susceptibility of asthma via modulation of pro-allergic lymphocyte responses.
Keywords: asthma, inflammation, innate immunity, leptin, T helper cells
Introduction
Allergic asthma, one of the leading health problems, affects 300 million people worldwide and leads to ∼250,000 deaths per year (as of 2009) (1). Allergic asthma is characterized by increased numbers of airway inflammatory cells, including CD4+ T helper (TH)5 lymphocytes, eosinophils, and activated mast cells, leading to airway remodeling, which involves epithelial cell mucus metaplasia, smooth muscle hypertrophy/hyperplasia, sub-epithelial fibrosis, and increased angiogenesis (2, 3). TH cells, especially TH2 cells, have been shown to play an essential role in allergic airway inflammation. TH2 cells release IL-4, IL-5, and IL-13. Among these cytokines, IL-5 recruits and activates eosinophils that (together with IgE-triggered mast cells) produce IL-13 and TGF-β (3). IL-4 from TH2 cells stimulates TH2 cells themselves and also promotes TH9 differentiation. TH9 cells secrete IL-9 (4–6), a cytokine that is involved in allergic disease likely through induction of IL-13 expression in epithelial cells and activation of mast cells and eosinophils (3, 7, 8). Besides, regulatory T cells produce TGF-β and IL-10 and inhibit conventional lymphocyte proliferation and differentiation, therefore attenuating allergic responses (9). In addition to TH cells, group 2 innate lymphocytes (ILC2s), a recently defined subset of innate lymphocytes, were shown to play a key role in airway inflammation through secretion of the type 2 cytokines, IL-5, IL-13, and IL-9 (10–14). IL-13, produced by ILC2s, enhances dendritic cell migration and promotes TH2 cell differentiation (15). IL-9 secreted by TH9 cells and ILC2s is critical for ILC2 survival and proliferation (16–18). Finally, IL-13 and TGF-β, released by inflammatory cells, directly act on pulmonary epithelial and muscle cells and promote their proliferation, therefore resulting in airway tissue remodeling and dysfunction (3). Thus, the induction of airway inflammation is a complex process that involves multiple cell types and cytokines. How deregulation of pro-allergic mediators causes allergic airway disease is not well understood.
Recently, obesity was identified as a major risk factor for the development of asthma by meta-analysis (19–21). Adipose tissue is an active endocrine organ secreting adipokines (such as leptin and adiponectin) and cytokines (such as IL-6, IL-8, TNF-α). These mediators regulate systemic metabolism and is implicated in the regulation of immune responses. In obese individuals, hypertrophic adipose tissue recruits more proinflammatory infiltrates including macrophages, eosinophils, and NK, T, and B cells (22). After activation, the fat-resident macrophages and adipocytes produce elevated proinflammatory cytokines and adipokines (including leptin), and decreased adiponectin (21). A number of human studies have examined the relationship between leptin and/or adiponectin and asthma. Most of studies show a positive relationship between serum leptin but a negative association between serum adiponectin and the risk of asthma (reviewed in Ref. 21). However, how these two adipokines impact allergic responses remains unclear.
Leptin (encoded by ob gene) is mainly secreted by the adipose tissue and is also present in lymphoid organs (reviewed in Refs. 23, 24). Leptin receptor (ObR), a member of the class I cytokine receptor superfamily, has at least six isoforms resulting from alternative splicing. The functional leptin receptor (ObRb) is expressed in the hypothalamus, where it regulates energy homeostasis and neuroendocrine function, and is expressed broadly on immune cells. Binding of leptin to its functional receptor activates JAK2-STAT3, MAPK, and PI3K-AKT pathways (23, 24). Besides its well-known function in energy homeostasis, leptin also plays an important role in regulating immunity. In humans, leptin deficiency leads to higher incidence of infection-related death during childhood (25). Leptin is involved in the activation, differentiation, proliferation and function of immune cells, such as macrophages and NK cells (reviewed in Refs. 23, 24). In adaptive immunity, leptin is shown to promote TH1 response in both humans and mice, but its role in pro-allergic TH2 response remains controversial (see more details in “Discussion”) (26–28). In this study, we examined the effect of leptin in pro-allergic TH2 and ILC2 responses in a murine allergic asthma model. We found that leptin (ob)-deficient mice had attenuated asthma symptoms with decreased eosinophilia, TH2 and ILC2 proliferation and type 2 cytokine production. Consistent with this, leptin treatment promotes TH2 cell proliferation and survival in vitro in an mTOC1 and MAPK pathway-dependent manner. Our results thus demonstrate a pathogenic role of leptin in asthmatic reactions.
Results
Leptin-deficiency Impairs Type 2 Immune Responses and Attenuates Allergic Airway Inflammation
To define the role of leptin in allergic responses, we utilized an experimental allergic asthma model induced by papain, a well-characterized protease-based allergen involved in human occupational allergic airway disease (29, 30), which is through induction of cytokines IL-33 (an alarmin), TSLP, and IL-25 by the airway epithelium (31, 32). Ob−/− mice and their WT littermates were subjected to induction of allergic asthma by papain and Ova as described (15) (Fig. 1A). After induction of asthma, both Ob−/− and WT mice developed allergic airway inflammation displaying increased immune infiltrates and eosinophilia in the airway compared with unchallenged controls. Ob−/− mice had significantly decreased infiltrates of eosinophils and lymphocytes in the bronchoalveolar lavage fluids (BALFs) compared with WT mice (Fig. 1B), whereas unchallenged Ob−/− mice had only a few eosinophils and lymphocytes in the BALFs that are comparable with unchallenged WT mice (supplemental Fig. S1A). To evaluate lung inflammation, we prepared lung sections from the WT and Ob−/− mice and stained the sections with hematoxylin and eosin. We observed that Ob−/− lungs exhibited decreased mononuclear cell infiltration in the peribronchovascular spaces that contained decreased numbers of eosinophils (Fig. 1C). In addition, papain and Ova challenge elicited IgE responses in both WT and Ob−/− mice; sera and BLAFs of Ob−/− mice contained decreased amounts of Ova-specific IgE (Fig. 1D). IgE expression in unchallenged WT and Ob−/− mice was only at the basal levels (supplemental Fig. S1B). Together, these data revealed a critical role of leptin in allergic airway disease.
FIGURE 1.

Leptin-deficiency attenuates allergic airway inflammation. A, induction of allergic asthma. B, cellular profile of BALFs. C, hematoxylin-and-eosin stain of lung sections from WT and Ob−/− mice after induction of experimental asthma. Arrowheads indicate immune infiltrates. Scale bar, 100 μm. Eosinophil counts were average numbers per field per section (100×). D, ELISA of Ova-specific IgE in sera and BALFs. B, C, right panel, and D, values are means and S.D. Student's t test, *, p ≤ 0.05 and **, p ≤ 0.005. Data shown are a representative of two experiments (n = 5–6 per group).
Type 2 immune responses manifest one of the hallmarks of allergic asthma. To understand the impact of leptin in type 2 immune responses, we assessed the frequencies of TH2 cells and ILC2s in the LLNs collected from asthmatic WT and Ob−/− mice. TH2 cells were defined as LIN+CD4+IL-13+ cells and ILC2s were defined as LIN−CD4−IL-13+ cells by flow cytometry (LIN refers to lineage surface markers for T and B cells, macrophages, eosinophils, neutrophils, basophils, mast cells, and erythrocytes, including CD3, CD5, B220, CD11b, CD11c, Gr-1, Ter119, and IgE (IgE stains FcϵRI+ basophils and mast cells)). We found that there were no differences in the frequencies of TH2 cells, ILC2s, and TH1 cells between Ob−/− and WT mice (Fig. 2A, supplemental Fig. S2A). However, the LLNs of Ob−/− mice contained decreased numbers of TH2 cells and ILC2s but comparable numbers of TH1 cells compared with WT mice (Fig. 2B, supplemental Fig. S2B). Compared with challenged animals, unchallenged Ob−/− and WT mice had many fewer TH2 cells, ILC2s and TH1 cells in LLNs (supplemental Fig. S1C). Upon ex vivo recall with Ova, Ob−/− LLN cells expressed less amounts of TH2 cytokines, IL-4, IL-5, and IL-13 and slightly decreased amounts of TH1 cytokine IFN-γ (which did not reach significance) compared with WT cells (Fig. 2C, supplemental Fig. S2C), whereas LLN cells from unchallenged Ob−/− and WT mice only expressed basal amounts of these cytokines (supplemental Fig. S1D). These results suggest that leptin-deficiency render TH2 cells but not TH1 cells less responsive to antigen stimulation after a type 2 challenge.
FIGURE 2.

Leptin-deficiency impairs type 2 immune responses in vivo. A, intracellular stain of TH2 cells (LIN+CD4+IL-13+) and ILC2s (LIN−CD4−IL-13+) in LLN cells from WT and Ob−/− mice after induction of experimental asthma. B, quantification of TH2 cells and ILC2s in LLNs. C, ELISA of type 2 cytokine expression by LLN cells after ex vivo recall with Ova at indicated concentrations. B and C, values are means and S.D. Student's t test; *, p ≤ 0.05 and **, p ≤ 0.005. Data represent two experiments (n = 5–6 per group).
Leptin Promotes TH2 Responses in Vitro
Because leptin-deficiency did not alter TH2 cell frequencies in vivo (Fig. 2A), we asked whether leptin affects TH2 cell differentiation. We skewed naïve CD4+ T cells under the TH2 condition in the presence or absence of leptin. We found that leptin treatment had little effect on the percentages of IL-13+ cells (Fig. 3A). We next examined the impacts of leptin treatment on TH2 cytokine expression. Interestingly, addition of leptin enhanced the expression levels of IL-4, IL-5, and IL-13 (Fig. 3B), in agreement with our in vivo observations (Fig. 2). These results further suggest that leptin promotes type 2 immune responses by regulating activity of TH2 cells rather than the development of TH2 cells. Although under type 2 immunization, leptin did not impact TH1 cell responses (supplemental Fig. S2), it had the same effects on TH1 cells as on TH2 cells in vitro (supplemental Fig. S3). In summary, our results demonstrate that leptin promotes type 2 immune responses both in vivo and in vitro that in turn, exacerbate allergic reactions.
FIGURE 3.
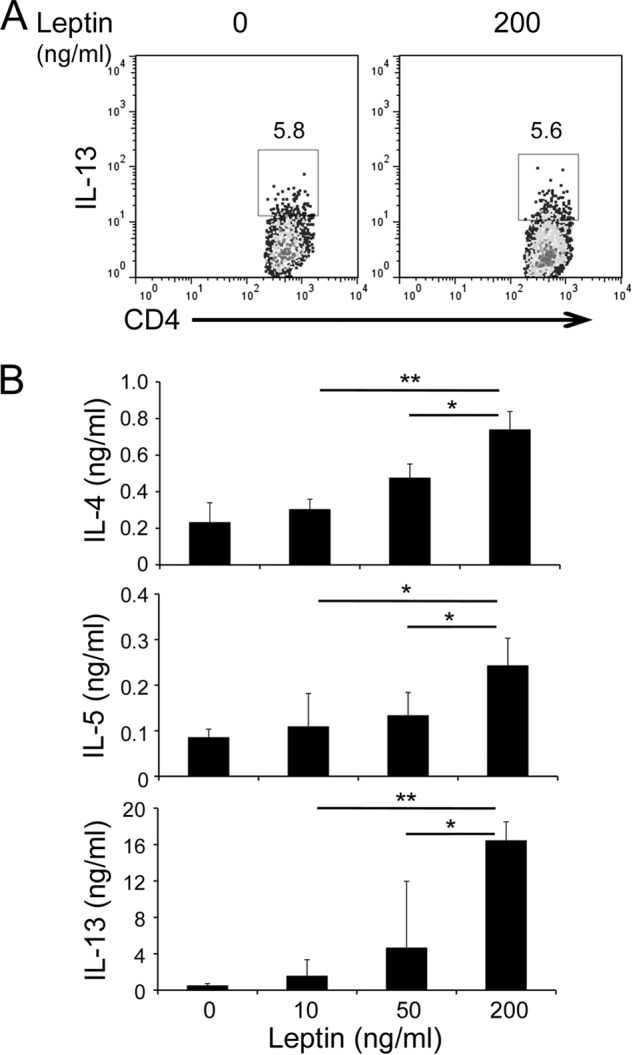
Leptin promotes TH2 cell responses in vitro. A, intracellular stain of TH2 cells differentiated in vitro with or without leptin treatment. B, ELISA of type 2 cytokines expressed by TH2 cells re-stimulated overnight with plate-bound anti-CD3 in the presence of leptin at indicated concentrations. Values are means and S.D. (n = 4 in each group). Student's t test; *, p ≤ 0.05 and **, p ≤ 0.005. Data represent three experiments.
Leptin Promotes TH2 and ILC2 Expansion
Our above data show that leptin promotes TH2 cytokine production but not TH2 cell (and ILC2) differentiation in vivo and in vitro (Figs. 2 and 3). To investigate if leptin regulates TH2 cell (and also ILC2) proliferation, we first examined expression of Ki67, a cell proliferation-associated nucleic protein that marks cells at active phases (G1, S, G2, and M) but not the resting phase (G0), in TH2 and ILC2 cells generated after induction of allergic asthma. We observed that the frequencies and numbers of Ki67+ cells were greater in WT TH2 cells and ILC2s than the corresponding leptin-deficient cells (Fig. 4A). Interestingly, leptin-deficiency did not significantly change Ki67+ cell frequencies inTH1 cells (supplemental Fig. S2D), which was due to the type 2 immune environment eliciting by papain. We next assessed the effect of leptin on the proliferation of in vitro generated TH1 and TH2 cells by CFSE dilution. Interestingly, leptin treatment did not affect TH2 nor TH1 cell proliferation during the primary differentiation (Fig. 4B, supplemental Fig. S4 A). However, during 1-day restimulation of rested TH2 cells, leptin treatment boosted both TH1 and TH2 cell proliferation in a dose-dependent manner (Fig. 4C, supplemental Fig. S4B). These data suggest that leptin affects proliferation of effector TH2 as well as TH1 cells but not primarily differentiating TH2 and TH1 cells.
FIGURE 4.

Leptin promotes proliferation of TH2 cells and ILC2s. A, intracellular stain of Ki67 in LLN TH2 cells and ILC2s from WT and Ob−/− mice after induction of experimental asthma. Bar graphs depict means and S.D. in each group mice (n = 5–6 per group); Student's t test; *, p ≤ 0.05 and **, p ≤ 0.005. B, flow cytometry of CFSE dilution in TH2 cells during in vitro differentiation with leptin at indicated concentrations. C, flow cytometry of CFSE dilution in TH2 cells during 1-day restimulation with plate-bound anti-CD3 in the presence of leptin at indicated concentrations. Data shown are representative of two (A) or three (B, C) experiments.
Leptin Promotes TH2 Cell Survival
In addition to proliferation, survival of TH2 cells also affects their cytokine secretion. We measured activation induced cell death in in vitro differentiated TH2 cells and found that leptin treatment protected TH2 cells from cell death induced by plate-bound anti-CD3 restimulation in a dose-dependent manner (Fig. 5A). Consistently, leptin treatment induced expression of an anti-apoptotic protein Bcl-2 that promotes cell survival (33) (Fig. 5, B and C). In addition to TH2 cells, leptin induced expression of Bcl-2 in TH1 cells and promoted TH1 cell survival (supplemental Fig. S4, C and D). Thus, leptin promotes not only proliferation but also survival of TH2 and TH1 cells.
FIGURE 5.

Leptin protects TH2 cells from activation induced cell death. A–C, cells were cultured in the absence or presence of leptin at indicated concentrations. A, flow cytometry of activation induced cell death in TH2 cells after restimulation with plate-bound anti-CD3 for 24 h. B, intracellular stain of Bcl-2 in in vitro polarized TH2 cells. C, immunoblot of Bcl-2 expression in polarized TH2 cells. Data shown are a representative of two (A, C) or three (B) experiments.
Leptin Treatment Activates STAT3, mTORC1, and MAPK
In an antigen-presenting cell-free system shown in Fig. 3, we observed that leptin promoted TH2 cytokine production. To further understand how leptin directly acts on TH2 and ILC2 cells, we examined whether leptin receptor is present on both TH2 and ILC2 cells. Flow cytometry analysis of the LLN cells isolated from asthmatic mice showed that both TH2 cells and ILC2s expressed ObR (Fig. 6A). To further determine whether leptin itself regulates ObR expression, we treated TH2 cells with or without leptin, and found that ObR was expressed on TH2 cells but leptin treatment did not alter the expression levels of ObR (Fig. 6A). TH1 cells generated either from the asthmatic response or in vitro differentiation also expressed ObR and the expression levels of ObR was not affected by the concentrations of leptin (supplemental Fig. S5A). Taken together, these results imply that leptin directly targets and regulates the activities of TH2 cells, ILC2s, and also TH1 cells.
FIGURE 6.

Leptin activates STAT3, MAPK, and mTOR signal pathways through targeting leptin receptor (ObR). A, flow cytometry of ObR on LLN TH2 cells and ILC2s generated in the asthmatic responses, and on in vitro differentiated TH2 cells. B, immunoblot of phospho- and total S6K, p38, and ERK1/2 in TH2 cells treated with leptin for indicated times. C, intracellular stain of p-STAT3 in TH2 cells treated with leptin for 20 m. Data shown represent two (B, C) or three (A) experiments.
To gain further insights into the role of leptin in facilitating proliferation and survival of TH2 cells, we investigated several pivotal signal cascades that were reported to regulate proliferation and survival of T cells, including JAK2-STAT3, MAPK, and PI3K-AKT-mTOR pathways (34–36). By using immunoblot, we observed that leptin treatment strongly stimulated phosphorylation of S6K, a kinase downstream of mTOR complex 1 (mTORC1) in in vitro polarized TH2 and TH1 cells (Fig. 6B, supplemental Fig. S5B). Similarly, leptin treatment induced p38 and ERK1/2 MAPK phosphorylation in both TH2 and TH1 cells (Fig. 6B, supplemental Fig. S5B). In addition, we observed STAT3 activation upon treatment with leptin in both TH2 and TH1 cells (Fig. 6C, supplemental Fig. S5C). These findings indicate that leptin activates mTOR, MAPK, and JAK2-STAT3 pathways in TH2 and TH1 cells (maybe also ILC2s).
MEK and mTOR Inhibitors Block the Effects of Leptin on TH2 Cell Proliferation, Survival, and Cytokine Production
To understand the specific signal cascade induced by leptin governs TH2 cell proliferation and survival, two different types of signal inhibitors, PD98059 (an inhibitor of MEK1/2 blocking MAPK pathways) and rapamycin (mTOR inhibitor) were utilized in proliferation and survival assay. We found that either PD98059 or rapamycin inhibited leptin induced proliferation of TH2 cells (Fig. 7A), in which the proliferation are comparable with no leptin-treated TH2 cells. In addition, treatment with either PD98059 or rapamycin blocked the protective effect of leptin on activation induced TH2 cell death (Fig. 7B). Moreover, we examined the effect of both inhibitors on TH2 cytokines expression. As expected, enhanced expression of TH2 type cytokines induced by leptin treatment were reversed by addition of either of these inhibitors (Fig. 7C). These findings suggest that both MAPK and mTOR signal cascades are indispensable for the comprehensive function of leptin, which results in promotion of TH2 cell proliferation, survival, and effector cytokine production (outlined in Fig. 7D). Taken together, our results demonstrate an essential role of leptin in allergic airway disease through activation of JAK2-STAT3, MAPK and PI3K-AKT-mTOR signal cascades and promotion of survival and proliferation of pro-allergic lymphocytes.
FIGURE 7.

Effect of MEK and mTOR inhibitors on leptin-induced TH2 cell proliferation, survival, and cytokine expression. A–C, in vitro differentiated TH2 cells were restimulated with plate-bound anti-CD3 in the presence or absence of leptin (A and B, 200 ng/ml; C, 50 ng/ml) with or without PD98059 (20 μm) or rapamycin (200 nm) for 24 h. A, flow cytometry of CFSE dilution. B, flow cytometry of activation induced cell death in TH2 cells. C, ELISA of cytokine expression. Values are means and S.D. (n = 4 in each group). Student's t test; *, p ≤ 0.05 and **, p ≤ 0.005. Data shown are a representative of two (C) or three (A, B) experiments. D, outline of the effects of leptin on TH2 cells.
Discussion
Meta-analysis revealed a link between obesity and the risk of developing allergic asthma (19–21). However, the underlying mechanism remains unclear. Although several studies demonstrated that leptin promotes TH1 responses both in vitro and in vivo, the role of leptin in pro-allergic TH2 responses has not been thoroughly investigated and the past studies have shown unconvincing results (26–28). In a mixed lymphocyte reaction assay, leptin treatment enhanced IFN-γ but inhibited IL-4 producion; this effect of leptin was only observed in memory but not naïve T cells (26). IFN-γ is known to inhibit TH2 cell differentiation, prolierfation, and cytokine production (37, 38), therefore in the mixed lymphocyte reaction assay, massive production of IFN-γ by polyclonal memory TH1 cells might inhibit the TH2 response. Whether leptin affects an antigen-specific TH2 response need further clarification. In another study, Batra et al. revealed that leptin enhanced TH2-mediated colitis and promoted the development of Gata3+ (TH2) cells (28). Interestingly, the same authors observed controversial results in vitro, in which treatment with extremely high concentration of leptin (1 μg ml−1) reduced IL-4+ TH2 cell frequences in repeated polarization cultures but not the primary skewing culture (28). High concentration of leptin may be toxic and overwhelm its physiological effects, because the serum leptin levels are 7.5 ± 9.3 ng ml−1 in non-obese human (39). In a childhood asthma and obesity study, the obese asthma cohort had higher plasma levels of both IL-4 and IFN-γ, correlated with their leptin levels, compared with the controls; although the non-obese asthma group had even higher levels of IL-4, the IgE levels between the obese and non-obese asthma groups were comparable (27), suggesting different mechanisms underlying the regulation of TH2 responses in these two conditions. Our current studies showed that both in vitro and in vivo, leptin did not alter the differentiation of TH2 cells but promoted their survival and proliferation, which resulted in elevated expression of TH2 cell cytokines, IL-4, IL-5, and IL-13. Interestingly, we observed that leptin had the same effects on TH1 cells as on TH2 cells, consistent with the literatures mentioned above. Therefore, the outcome will likely be determined by the skewing condition. Under a type 2 condition, such as allergic asthma, leptin predominantly affects TH2 cells but not TH1 cells. Whether a coincidence of type 1 immune response (e.g. bacterial or fungal infection), in which leptin also affects TH1 cells, impacts the effects of leptin on TH2 responses needs further study.
Recently, ILC2s were shown to be the early responders in experimental asthma induced by proteinase allergens, papain, and house dust mite (HDM) extract (10–14). In addition to type 2 cytokine production, ILC2s promote TH2 cell polarization and memory responses via IL-13-dependent dendritic cell migration to the mediastinal lymph node (15, 40) and major histocompatibility complex class II (MHCII)-mediated crosstalk (41, 42). In agreement with this, mice deficient in ILC2s fail to mount efficient TH2 responses (15, 41). On the other hand, activated TH2 cells, via secretion of IL-2, also reciprocally promotes ILC2 development (29, 42). However, whether leptin regulates the development of asthma through an ILC2-dependent mechanism remains unknown. We observed reduced total number but comparable frequencies of ILC2s in Ob−/− mice in the asthma model compared with WT control mice. Consistently, leptin-deficiency resulted in the decrease of proliferation of ILC2s. Therefore, leptin enhances ILC2 proliferation and contributes to asthmatic responses.
Leptin acts to modulate immune cell function through multiple signaling pathways including JAK2-STAT3, MAPK, and PI3K-AKT (23, 24). The leptin-STAT3 pathway is known to mediate cell survival in hippocampal neurons (43). In line with this, activation of STAT3 by leptin is required for IL-6-mediated anti-apoptotic function in T cells (36). However, whether the leptin-STAT3 axis also impacts T cell proliferation and survival remains unclear. On the other hand, leptin has been shown to exert anti-apoptotic effect and activate ERK1/2 and AKT-mTOR pathways in TH1/TH17 cells (35). Interestingly, the anti-apoptotic effects of leptin are dependent on MAPK activation, rather than the PI3K pathway in human Jurkat T cells (34). Our present studies showed that leptin activated STAT3, MAPK, and AKT-mTOR pathways in TH2 cells and up-regulated Bcl-2, therefore contributing to leptin-mediated proliferation and survival. Indeed, treatment with either PD98059 or rapamycin blocked the effects of leptin on TH2 cell proliferation, survival, and cytokine production. In summary, our study demonstrate that leptin enhances TH2 and ILC2 responses and promotes allergic asthma via activation of STAT3, MAPK, and PI3K pathways, supporting the meta-analysis results that obesity-associated elevation of leptin increases the risk of development of allergic asthma.
Materials and Methods
Animals
Ob−/− (leptin-deficient) mice on C57BL/6 background were generated by crossing female and male heterozygotes; homozygotes and wild-type (WT) littermates at age of 5–8 weeks were used at the initial of experiments. All mice were housed in the specific pathogen-free animal facility at the University of New Mexico Health Sciences Center. All experiments were performed with protocols approved by the Institutional Animal Care and Use Committee of the University of New Mexico.
Induction of Allergic Asthma
For induction of allergic asthma, age, and sex-matched Ob−/− and WT littermates were immunized intranasally with 25 μg of papain and 50 μg of chicken ovalbumin (Ova) for three times on day 0, day 1, and day 14. On day 15, BALFs were collected for analysis of airway infiltrates as described (44). Lung draining mediastinal lymph nodes (LLNs) were collected for cytokine expression and proliferation assays. To reveal lung inflammation, the left upper lobes of the asthmatic lungs were collected for hematoxylin and eosin stain.
In Vitro TH2 Cells Differentiation
CD4+CD25−CD62L+ naïve T cells were sorted from C57BL/6 WT mice and differentiated in a TH2-polarizing condition (5 μg ml−1 anti-IFN-γ and 10 ng ml−1 IL-4) using plate-bound α-CD3/α-CD28 and low serum (3–5% FBS)-containing RPMI medium with or without addition of leptin as indicated. Afterward, the resulting cells were re-stimulated in serum-free medium (OpTmizerTM CTSTM T-Cell Expansion SFM, Life Technologies) for intracellular cytokine expression, apoptosis, and proliferation assay in the presence or absence of leptin.
Proliferation Assay
In vitro TH2 cell proliferation was assessed by carboxyfluorescein succinimidyl ester (CFSE) dilution. During polarization, CFSE-labeled naïve cells were cultured under the TH2 condition in low serum RPMI medium (3–5% FBS) for 3 days; during re-activation, rested TH2 cells were labeled with CFSE and re-stimulated with plate-bound anti-CD3 in serum-free medium with or without addition of leptin for 20 h. For in vivo proliferation, single-cell suspensions of LLNs from the asthmatic mice were prepared, and Ki67 expression in TH2 and ILC2 cells was measured by intracellular stain on a CD4+LIN+IL-13+ or LIN−CD4−IL-13+ gate, respectively. LIN (lineage marker) includes CD3, CD5, B220, CD11b, CD11c, Gr-1, Ter119, and IgE.
Cell Death Assay
Following restimulation with plate-bound anti-CD3 with or without addition of leptin as indicated for 24 h, activation induced cell death of TH2 cells were assessed by 7-AAD marking DNA contents in damaged cells or LIVE/DEAD Green (LIVE/DEAD® Fixable Dead Cell Stain Kits, Invitrogen) reactive to free amines both in the interior and on the cell surface.
ELISA
LLN cells (4 × 106 cells ml−1) from the asthmatic mice were recalled with various concentrations of Ova for 3 days, and the supernatants were collected for measurement of cytokine expression by ELISA using a standard protocol. For in vitro differentiated TH2 cells, the cells were polarized for 4 days and restimulated for overnight in the presence of various concentrations of leptin as indicated, and the supernatants were used to measure cytokines expression. To measure Ova-specific IgE, plate-bound Ova (100 μg ml−1) was used as capture and anti-mouse IgE (23G3, eBioscience) as detection antibody.
Immunoblot
In vitro differentiated TH2 cells were starved for 24 h in serum-free medium and then treated with leptin (200 ng ml−1) for different time spans. Afterward, whole-cell lysates were prepared and subjected to immunoblot analysis for multiple leptin-associated signaling proteins expression. To evaluate Bcl-2 expression, in vitro differentiated TH2 cells were further cultured in serum-free medium with or without leptin (200 ng ml−1) for 24 h, and whole-cell lysates were used for immunoblot. Antibodies were anti-p70 S6 kinase (S6K) (CG1396, Cell Applications), anti-phospho (p)-S6K (Thr-389) (9205, Cell Signaling), anti-p-p38 MAPK (Thr-180/Tyr-182) (9211, Cell Signaling), anti-p38 MAPK (8690, Cell Signaling), anti-Bcl-2 (BCL/10C4, Biolegend).
Flow Cytometry Antibodies
CD3e (145-2C11), CD4 (GK1.5), CD5 (53-7.3), B220 (RA3-6B2), CD11b (M1/70), CD11c (N418), Gr-1 (RB6-8C5), Ter119 (TER-119), IgE (23G3), IL-13 (eBio13A), and Ki67 (SolA15) were purchased from eBioscience, p-STAT3 (Tyr-705) (4/P-STAT3) from BD Biosciences, ObR (AF497), and anti-goat IgG (NL002) from RnD Systems, and goat IgG isotype control (sc-3887) from Santa Cruz Biotechnology.
Phospho-STAT Stain
Phospho-STAT3 stain was performed using a previously described protocol with minor modifications (45). In brief, in vitro differentiated TH2 cells were starved for 24 h in serum-free medium and then treated with or without leptin (200 ng ml−1) for 20 min. The cells were then fixed by 1.6% paraformaldehyde for 20 min, resuspended in 50% ethanol for 1 min, and permeabilized with methanol for 20 min. The resulting cells were stained with anti-p-STAT3 in cold 0.1% BSA-PBS for 30 min.
Statistical Analysis
The statistical significance of differences between groups was calculated with the unpaired Student's t test. p values of 0.05 or less were considered significant.
Author Contributions
X. O. Y. and M. L. designed the project. H. Z., X. Z., E. F. C., and Y. L. conducted the experiments. H. Z. analyzed the results. H. Z. and X. O. Y. wrote the manuscript. All authors reviewed and approved the manuscript.
Supplementary Material
This work was supported in part by National Institutes of Health Grants R56AI110442 and R56AI116772, American Lung Association Grant RG-268131, and UNM Cancer Center Pilot Award through contract National Institutes of Health Grant P30CA118100 (to X. O. Y.), and American Diabetes Association Junior Faculty Research Award 1-13-JF-37, American Heart Association Grant-in-Aid Award 15GRNT24940018, and CoBRE Pilot Award through National Institutes of Health Grant P30GM103400 and UNM RAC Pilot Awards (to M. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S5.
- TH
- helper T cell
- ILC2
- group 2 innate lymphoid cell
- Ob−/−
- leptin deficient
- ObR
- leptin receptor
- Ova
- chicken ovalbumin
- BALF
- bronchoalveolar lavage fluid
- LLN
- lung draining mediastinal lymph node
- CFSE
- carboxyfluorescein succinimidyl ester
- MHCII
- major histocompatibility complex class II
- S6K
- p70 S6 kinase
- STAT3
- signal transducer and activator of transcription protein 3
- MAPK
- mitogen-activated protein kinase
- mTORC1
- mammalian target of rapamycin complex 1
- JAK2
- Janus kinase 2.
References
- 1. Fanta C. H. (2009) Asthma. Asthma. N. Engl. J. Med. 360, 1002–1014 [DOI] [PubMed] [Google Scholar]
- 2. Pascual R. M., and Peters S. P. (2005) Airway remodeling contributes to the progressive loss of lung function in asthma: an overview. J. Allergy Clin. Immunol. 116, 477–486 [DOI] [PubMed] [Google Scholar]
- 3. Doherty T., and Broide D. (2007) Cytokines and growth factors in airway remodeling in asthma. Curr. Opin. Immunol. 19, 676–680 [DOI] [PubMed] [Google Scholar]
- 4. Dardalhon V., Awasthi A., Kwon H., Galileos G., Gao W., Sobel R. A., Mitsdoerffer M., Strom T. B., Elyaman W., Ho I. C., Khoury S., Oukka M., and Kuchroo V. K. (2008) IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat. Immunol. 9, 1347–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmitt E., Germann T., Goedert S., Hoehn P., Huels C., Koelsch S., Kühn R., Müller W., Palm N., and Rüde E. (1994) IL-9 production of naive CD4+ T cells depends on IL-2, is synergistically enhanced by a combination of TGF-β and IL-4, and is inhibited by IFN-γ. J. Immunol. 153, 3989–3996 [PubMed] [Google Scholar]
- 6. Veldhoen M., Uyttenhove C., van Snick J., Helmby H., Westendorf A., Buer J., Martin B., Wilhelm C., and Stockinger B. (2008) Transforming growth factor-beta 'reprograms' the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 9, 1341–1346 [DOI] [PubMed] [Google Scholar]
- 7. Soroosh P., and Doherty T. A. (2009) Th9 and allergic disease. Immunology 127, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Townsend J. M., Fallon G. P., Matthews J. D., Smith P., Jolin E. H., and McKenzie N. A. (2000) IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity 13, 573–583 [DOI] [PubMed] [Google Scholar]
- 9. Sakaguchi S., Yamaguchi T., Nomura T., and Ono M. (2008) Regulatory T cells and immune tolerance. Cell 133, 775–787 [DOI] [PubMed] [Google Scholar]
- 10. Walker J. A., and McKenzie A. N. (2013) Development and function of group 2 innate lymphoid cells. Curr. Opin. Immunol. 25, 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanos S. L., and Diefenbach A. (2013) Innate lymphoid cells: from border protection to the initiation of inflammatory diseases. Immunol. Cell Biol. 91, 215–224 [DOI] [PubMed] [Google Scholar]
- 12. Spits H., and Cupedo T. (2012) Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol. 30, 647–675 [DOI] [PubMed] [Google Scholar]
- 13. Spits H., and Di Santo J. P. (2011) The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 12, 21–27 [DOI] [PubMed] [Google Scholar]
- 14. Tait Wojno E. D., and Artis D. (2012) Innate lymphoid cells: balancing immunity, inflammation, and tissue repair in the intestine. Cell Host Microbe 12, 445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Halim T. Y., Steer C. A., Mathä L., Gold M. J., Martinez-Gonzalez I., McNagny K. M., McKenzie A. N., and Takei F. (2014) Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilhelm C., Hirota K., Stieglitz B., Van Snick J., Tolaini M., Lahl K., Sparwasser T., Helmby H., and Stockinger B. (2011) An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat. Immunol. 12, 1071–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilhelm C., Turner J. E., Van Snick J., and Stockinger B. (2012) The many lives of IL-9: a question of survival? Nat. Immunol. 13, 637–641 [DOI] [PubMed] [Google Scholar]
- 18. Turner J. E., Morrison P. J., Wilhelm C., Wilson M., Ahlfors H., Renauld J. C., Panzer U., Helmby H., and Stockinger B. (2013) IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 210, 2951–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Camargo C. A. Jr, Weiss S. T., Zhang S., Willett W. C., and Speizer F. E. (1999) Prospective study of body mass index, weight change, and risk of adult-onset asthma in women. Arch. Intern. Med. 159, 2582–2588 [DOI] [PubMed] [Google Scholar]
- 20. Beuther D. A., and Sutherland E. R. (2007) Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am. J. Respir. Crit. Care Med. 175, 661–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dixon A. E., Holguin F., Sood A., Salome C. M., Pratley R. E., Beuther D. A., Celedón J. C., and Shore S. A. (2010) An official American Thoracic Society Workshop report: obesity and asthma. Proc. Am. Thorac. Soc. 7, 325–335 [DOI] [PubMed] [Google Scholar]
- 22. Bapat S. P., Myoung Suh J., Fang S., Liu S., Zhang Y., Cheng A., Zhou C., Liang Y., LeBlanc M., Liddle C., Atkins A. R., Yu R. T., Downes M., Evans R. M., and Zheng Y. (2015) Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528, 137–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matarese G., Moschos S., and Mantzoros C. S. (2005) Leptin in immunology. J. Immunol. 174, 3137–3142 [DOI] [PubMed] [Google Scholar]
- 24. Fantuzzi G., and Faggioni R. (2000) Leptin in the regulation of immunity, inflammation, and hematopoiesis. J. Leukoc. Biol. 68, 437–446 [PubMed] [Google Scholar]
- 25. Ozata M., Ozdemir I. C., and Licinio J. (1999) Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J. Clin. Endocrinol. Metab. 84, 3686–3695 [DOI] [PubMed] [Google Scholar]
- 26. Lord G. M., Matarese G., Howard J. K., Baker R. J., Bloom S. R., and Lechler R. I. (1998) Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 394, 897–901 [DOI] [PubMed] [Google Scholar]
- 27. Youssef D. M., Elbehidy R. M., Shokry D. M., and Elbehidy E. M. (2013) The influence of leptin on Th1/Th2 balance in obese children with asthma. J. Bras. Pneumol. 39, 562–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Batra A., Okur B., Glauben R., Erben U., Ihbe J., Stroh T., Fedke I., Chang H. D., Zeitz M., and Siegmund B. (2010) Leptin: a critical regulator of CD4+ T-cell polarization in vitro and in vivo. Endocrinology 151, 56–62 [DOI] [PubMed] [Google Scholar]
- 29. Milne J., and Brand S. (1975) Occupational asthma after inhalation of dust of the proteolytic enzyme, papain. Br. J. Ind. Med. 32, 302–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Soto-Mera M. T., López-Rico M. R., Filgueira J. F., Villamil E., and Cidrás R. (2000) Occupational allergy to papain. Allergy 55, 983–984 [DOI] [PubMed] [Google Scholar]
- 31. Oboki K., Ohno T., Kajiwara N., Arae K., Morita H., Ishii A., Nambu A., Abe T., Kiyonari H., Matsumoto K., Sudo K., Okumura K., Saito H., and Nakae S. (2010) IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl. Acad. Sci. U.S.A. 107, 18581–18586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu H. S., Angkasekwinai P., Chang S. H., Chung Y., and Dong C. (2010) Protease allergens induce the expression of IL-25 via Erk and p38 MAPK pathway. J. Korean Med. Sci. 25, 829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Czabotar P. E., Lessene G., Strasser A., and Adams J. M. (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63 [DOI] [PubMed] [Google Scholar]
- 34. Fernández-Riejos P., Goberna R., and Sánchez-Margalet V. (2008) Leptin promotes cell survival and activates Jurkat T lymphocytes by stimulation of mitogen-activated protein kinase. Clin. Exp. Immunol. 151, 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mattioli B., Giordani L., Quaranta M. G., and Viora M. (2009) Leptin exerts an anti-apoptotic effect on human dendritic cells via the PI3K-Akt signaling pathway. FEBS Lett. 583, 1102–1106 [DOI] [PubMed] [Google Scholar]
- 36. Takeda K., Kaisho T., Yoshida N., Takeda J., Kishimoto T., and Akira S. (1998) Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 161, 4652–4660 [PubMed] [Google Scholar]
- 37. Oriss T. B., McCarthy S. A., Morel B. F., Campana M. A., and Morel P. A. (1997) Crossregulation between T helper cell (Th)1 and Th2: inhibition of Th2 proliferation by IFN-γ involves interference with IL-1. J. Immunol. 158, 3666–3672 [PubMed] [Google Scholar]
- 38. Szabo S. J., Sullivan B. M., Peng S. L., and Glimcher L. H. (2003) Molecular mechanisms regulating Th1 immune responses. Annu. Rev. Immunol. 21, 713–758 [DOI] [PubMed] [Google Scholar]
- 39. Considine R. V., Sinha M. K., Heiman M. L., Kriauciunas A., Stephens T. W., Nyce M. R., Ohannesian J. P., Marco C. C., McKee L. J., Bauer T. L., et al. (1996) Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 334, 292–295 [DOI] [PubMed] [Google Scholar]
- 40. Halim T. Y., Hwang Y. Y., Scanlon S. T., Zaghouani H., Garbi N., Fallon P. G., and McKenzie A. N. (2016) Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat. Immunol. 17, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oliphant C. J., Hwang Y. Y., Walker J. A., Salimi M., Wong S. H., Brewer J. M., Englezakis A., Barlow J. L., Hams E., Scanlon S. T., Ogg G. S., Fallon P. G., and McKenzie A. N. (2014) MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 41, 283–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mirchandani A. S., Besnard A. G., Yip E., Scott C., Bain C. C., Cerovic V., Salmond R. J., and Liew F. Y. (2014) Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J. Immunol. 192, 2442–2448 [DOI] [PubMed] [Google Scholar]
- 43. Guo Z., Jiang H., Xu X., Duan W., and Mattson M. P. (2008) Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 283, 1754–1763 [DOI] [PubMed] [Google Scholar]
- 44. Yang X. O., Zhang H., Kim B. S., Niu X., Peng J., Chen Y., Kerketta R., Lee Y. H., Chang S. H., Corry D. B., Wang D., Watowich S. S., and Dong C. (2013) The signaling suppressor CIS controls proallergic T cell development and allergic airway inflammation. Nat. Immunol. 14, 732–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krutzik P. O., and Nolan G. P. (2003) Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A 55, 61–70 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.