

Abstract
Numerous RNA binding proteins are deposited onto an mRNA transcript to modulate posttranscriptional processing events ensuring proper mRNA maturation. Defining the interplay between RNA binding proteins that couple mRNA biogenesis events is crucial for understanding how gene expression is regulated. To explore how RNA binding proteins control mRNA processing, we investigated a role for the evolutionarily conserved polyadenosine RNA binding protein, Nab2, in mRNA maturation within the nucleus. This study reveals that nab2 mutant cells accumulate intron-containing pre-mRNA in vivo. We extend this analysis to identify genetic interactions between mutant alleles of nab2 and genes encoding a splicing factor, MUD2, and RNA exosome, RRP6, with in vivo consequences of altered pre-mRNA splicing and poly(A) tail length control. As further evidence linking Nab2 proteins to splicing, an unbiased proteomic analysis of vertebrate Nab2, ZC3H14, identifies physical interactions with numerous components of the spliceosome. We validated the interaction between ZC3H14 and U2AF2/U2AF65. Taking all the findings into consideration, we present a model where Nab2/ZC3H14 interacts with spliceosome components to allow proper coupling of splicing with subsequent mRNA processing steps contributing to a kinetic proofreading step that allows properly processed mRNA to exit the nucleus and escape Rrp6-dependent degradation.
INTRODUCTION
Gene expression is temporally and spatially regulated to produce a precise protein expression profile that dictates the function of each cell. Although much of this control occurs at the level of transcription, both co- and posttranscriptional events also play key regulatory roles. Newly synthesized mRNAs undergo numerous processing events, including 5′ capping, splicing, 3′-end processing, and export to the cytoplasm (1, 2). Ensuring the synchrony of mRNA biogenesis requires RNA binding proteins that not only perform the processing tasks but also couple the events to ensure that only properly processed mRNAs are available for translation in the cytoplasm (3).
Key processing events that must be coordinated include splicing and 3′-end processing. Although steps in mRNA processing are often depicted and studied as separate events, there is a growing body of evidence that these processing events are intimately coupled to one another (2). For example, splicing and 3′-end processing are coupled in humans as mutations in splice site and polyadenylation consensus sequences mutually disrupt both splicing and polyadenylation (4–6). In addition, a number of splicing factors copurify with the 3′-end processing complex (7–10). For example, there is evidence that the splicing factor U2AF2/U2AF65 functions as a bridge between the U2 snRNP and the 3′-end processing machinery (11, 12). Given the extensive coupling between RNA processing steps, it is important to consider the consequences when one step of the process is disrupted in vivo. Cells have developed numerous overlapping mechanisms to ensure that faulty mRNA transcripts are not translated (13–15), suggesting that there must be constant mRNA surveillance during mRNA biogenesis.
One facet of mRNA surveillance is the nuclear retention of pre-mRNA transcripts. The pre-mRNA retention and splicing (RES) complex is composed of splicing factors (Snu17, Bud13, and Pml1) that are critical for both recognizing intron-containing transcripts and retaining them within the nucleus (16, 17). The RES complex relies on intact splice site sequences, as well as the correct composition of RNA binding proteins to form an mRNP (16, 17). Accordingly, mutations in genes encoding the Saccharomyces cerevisiae early splicing factors Mud2 and Msl5 result in a stalled commitment complex (16–20). Once the pre-mRNA is recognized as misprocessed, nuclear pore components such as the Mlp proteins are required for nuclear retention (21–23). By retaining pre-mRNA transcripts, a choice can be made to continue with mRNA maturation or to proceed with mRNA decay (24). In addition, some pre-mRNAs may be exported to the cytoplasm where they can be degraded by cytoplasmic exonucleases or translation-dependent nonsense-mediated decay (25, 26). Both of these mechanisms prevent the translation of unspliced RNAs into potentially defective proteins.
The evolutionarily conserved and essential RNA exosome complex mediates both RNA processing and degradation (27, 28). The nuclear and cytoplasmic core exosome consists of 10 subunits that form a ring-like structure with a 3′-5′ exoribonuclease subunit, Dis3/Rrp44, at the base (24, 29, 30). The nuclear exosome also contains an additional exoribonuclease subunit, Rrp6, which is located at the top of the complex (24, 29, 30). The exosome processes key noncoding RNAs, including rRNA, small nuclear RNA (snRNA), and small nucleolar RNA (snoRNA) (31, 32), but also degrades improperly processed RNAs in both the nucleus and the cytoplasm (27). The nuclear exosome subunit, Rrp6, contributes to the degradation of transcripts with aberrant 3′ polyadenosine tails (33), pre-mRNA transcripts containing introns (34), and transcripts with improper messenger ribonucleoprotein (mRNP) composition (35, 36). A variety of nuclear and cytoplasmic cofactors that are often composed of one or more RNA binding proteins modulate the function of the exosome in vivo (37). The best characterized exosome cofactor is the Trf4/5-Air1/2-Mtr4 polyadenylation (TRAMP) complex that adds a short poly(A) tail to promote exosome-mediated degradation within the nucleus (38). Understanding how exosome cofactors and other RNA binding proteins regulate the exosome is critical to understanding how cells ensure the production of only correctly processed RNAs.
RNA binding proteins that play roles in multiple mRNA processing events are candidates for performing surveillance functions to ensure that only properly processed mRNAs are exported to the cytoplasm. Two key examples are the nuclear polyadenosine RNA binding proteins, Saccharomyces cerevisiae Nab2 and Schizosaccharomyces pombe Pab2. Nab2 is an evolutionarily conserved zinc finger polyadenosine RNA binding protein that functions in poly(A) tail length control and mRNA export (39–42). Nab2 has been implicated in either targeting bound RNAs to Rrp6 for decay (43) or in protecting RNAs from widespread exosome-mediated decay (44). Pab2 is an RNA recognition motif (RRM)-containing polyadenosine RNA binding protein that functions in poly(A) tail metabolism (45). Pab2 recruits Rrp6 to degrade polyadenylated pre-mRNA transcripts (46). Interestingly, Nab2 can impede Pab2/Rrp6-mediated degradation of unspliced rpl30-2 mRNA in S. pombe (47); however, whether this function of Nab2 is conserved in other organisms is not yet clear. These studies suggest a complicated interplay between RNA binding proteins in protecting or targeting RNAs for destruction by the exosome.
The human orthologue of Nab2 is the zinc finger polyadenosine RNA binding protein, ZC3H14 (48, 49). Mutations in the ZC3H14 gene cause autosomal-recessive intellectual disability, indicating that ZC3H14 plays a critical role in the brain (50, 51). Like Nab2, ZC3H14 regulates poly(A) tail length (49). Notably, ZC3H14 colocalizes with nuclear speckles that contain the splicing factor SC-35 (48, 52, 53), suggesting a potential role for ZC3H14 in processing events such as splicing. In support of a link between Nab2/ZC3H14 and splicing, a recent proteomic study found that Nab2 associates with several splicing factors in an RNA-dependent manner (54).
Here, we use molecular, genetic, and biochemical approaches to demonstrate that the polyadenosine RNA binding protein, Nab2/ZC3H14, can link splicing to 3′-end processing. We show that Nab2 associates with the early branch point binding factors Mud2 and Msl5. The requirement for the association between Nab2 and early splicing factors is alleviated when nuclear exosome activity is impaired by removing Rrp6. We extended the work from budding yeast to identify proteins that interact with the human Nab2 orthologue, ZC3H14. This analysis identified the spliceosome as a major interacting complex for ZC3H14. Our data support a model where Nab2 and Mud2 serve as RNA surveillance factors to coordinate splicing, 3′-end processing, and mRNA decay. Furthermore, the proteomic analysis of ZC3H14 suggests that this function could be evolutionarily conserved.
MATERIALS AND METHODS
Plasmids, strains, and chemicals.
All media were prepared by standard procedures, and all DNA manipulations were performed according to standard methods (55). Chemicals were obtained from Sigma-Aldrich, U.S. Biological (Swampscott, MA), or Fisher unless otherwise noted. The S. cerevisiae plasmids and strains used are described in Tables S1 and S2, respectively, in the supplemental material. All deletion mutants were generated by using PCR-based gene disruption as described previously (56). The MUD2 gene on a MUD2 plasmid (generous gift from S. Shuman [57]) was subcloned into pRS316 to create a URA3 CEN MUD2 plasmid. Mud2 amino acid substitutions were generated by site-directed mutagenesis using a QuikChange site-directed mutagenesis kit (Stratagene). All plasmids were sequenced to ensure the presence of desired changes and the absence of additional changes.
Total RNA isolation.
To prepare total RNA, 10-ml cultures were grown in appropriate media to an A600 of 0.4 to 0.6 at 30°C. Total RNA was isolated using the TRIzol method (58). Glass beads (200 μl) were added to the cell pellets, and bead beating was performed for 2 min. For each sample, 100 μl of 1-bromo-3-chloropropane was added, and each sample was vortexed for 15 s and incubated at room temperature for 2 min. The samples were centrifuged for 8 min at 16,300 × g at 4°C, and the upper layer was transferred to a fresh tube. RNA was precipitated with 500 μl of isopropanol, vortexed, and centrifuged for 8 min at 16,300 × g at 4°C. The supernatant was decanted, and the RNA pellet was washed with 75% ethanol. Samples were centrifuged for 5 min at 16,300 × g at 4°C, the supernatant was decanted, and the pellet was air dried. The RNA pellet was resuspended in 30 μl of 10 mM Tris-HCl (pH 7.5).
Quantitative RT-PCR.
For quantitative reverse transcription-PCR (qRT-PCR), 1 μg of total RNA was transcribed to cDNA using a QuantiTect reverse transcription kit (Qiagen) according to the manufacturer's protocol. Quantitative RT-PCR analysis was performed in triplicate using an iCycler iQ real-time machine (Bio-Rad). The real-time PCR mixture contained 10 ng cDNA, 0.1 μg of primers, and QuantiTect SYBR green master mix. Pre-mRNA levels were detected by using primers spanning the intron-exon junction, and total RNA levels were detected using primers in the exon. The mRNA level was normalized to the PDA1 housekeeping gene. Relative quantification of RNA levels was analyzed using the ΔΔCT method (59). Statistical significance was determined using one-way analysis of variance (ANOVA).
Splicing microarray.
All microarray analyses were carried out as previously described (60) with the following modification: yeast cultures were grown to log phase in 50 ml of yeast extract-peptone-dextrose (YEPD) at 30°C and then shifted to the nonpermissive temperature at 16°C for 1 h. Each microarray profile shown is an average of two biological replicates. The gene axis was ordered by hierarchical clustering using the C Clustering Library version 1.32 (61). Data were clustered using an average linkage and the Pearson correlation as the distance measure.
In vitro splicing assay.
Splicing extracts were prepared using the liquid nitrogen method as described earlier (62), except that frozen cells were disrupted in a ball mill (MM301; Retsch) for 3 min at 10 Hz for five cycles. Splicing reaction mixtures containing splicing buffer (3% [wt/vol] polyethylene glycol [PEG] 8000, 2.5 mM MgCl2, 60 mM potassium phosphate [pH 7.0]), 40% (vol/vol) budding yeast extract, 2 mM ATP, and 0.4 nM radiolabeled ACT1 pre-mRNA substrate were incubated for 10 to 30 min at 20°C (62). Splicing gels were exposed to a phosphor screen and imaged with a Storm PhosphorImager (GE Healthcare). Extracts were prepared at least three independent times, and the most active nab2-C437S lysate is shown and quantified in Fig. 4.
FIG 4.

In vitro splicing assay shows lysate prepared from nab2 mutant cells is competent for splicing. (A) Radiolabeled ACT1 pre-mRNA was incubated with lysates isolated from wild-type or nab2-C437S cells. Radiolabeled splicing products were analyzed at 10, 15, and 30 min of incubation by separation on an acrylamide gel. The positions of the pre-mRNA, lariat intermediate, 5′ exon, excised intron, and mature mRNA are indicated. (B and C) The branching efficiency (B) and the efficiency of exon ligation (C) were quantitated from five independent experiments and graphed with error bars that represent the SEM. The branching efficiency was calculated as (LI + mRNA)/(LI + mRNA + pre), and the efficiency of exon ligation was calculated as mRNA/(LI + mRNA), where “pre” represents pre-mRNA, and “LI” represents lariat intermediates.
Cell growth assays.
Cultures were grown to saturation, serially diluted, and spotted onto selective medium plates at 16, 25, 30, and 37°C for 3 to 5 days. For growth curve analysis, the cells were grown overnight to saturation, diluted 100-fold in selective media, and added to the wells of a MicroWell F96 microtiter plate (Nunc) in triplicate. Samples were grown at 25°C for 20 h while measuring the A600 every 30 min.
To assay for synthetic growth defects, various nab2 mutants were combined with the deletion of candidate genes using a plasmid shuffle assay. The shuffle Δnab2 strain (ACY427) was targeted for deletion of MUD2 and CUS2. Cells with nab2 deleted (ACY427) were maintained by a wild-type NAB2 URA3 plasmid (pAC636) and were transformed with mutant LEU2 nab2 plasmids. Transformants were grown on 5-fluoroorotic acid to select against the wild-type NAB2 URA3 plasmid (63). Cells were grown to saturation, serially diluted, and spotted onto plates lacking leucine at 16, 25, 30, and 37°C for 3 to 5 days. Mud2 rescue experiments were performed by transforming the nab2 Δmud2 (ACY2273) strain with mud2 variant plasmids, and the resulting cells were grown to saturation, serially diluted, and spotted onto Ura− Leu− glucose plates at 25°C for 3 days.
Protein binding assays.
To assess binding between Nab2 and either Mud2 or Msl5, tandem affinity purification (TAP)-tagged strains (Srp1-TAP, Pub1-TAP, Mud2-TAP, and Msl5-TAP) were transformed with plasmids expressing C-terminally Myc-tagged variants of Nab2. Cells were grown in 50 ml of minimal media to an A600 of 0.4 to 0.6. Cell pellets were resuspended in lysis buffer (10 mM Tris-HCl [pH 8.0], 100 mM NaCl, 0.1% NP-40, 1 mM phenylmethanesulfonyl fluoride, and 10 mM pepstatin leupeptin aprotinin chymostatin [PLAC]) and lysed by bead beating 4 times for 2 min each. A total of 3 mg of protein lysate was incubated with IgG-Sepharose beads for 3 h. The beads were then washed three times for 1 min to purify the TAP-tagged protein and any associated proteins. Bound and unbound samples were analyzed by SDS-PAGE/immunoblotting using a 1:2,500 dilution of anti-Myc mouse monoclonal antibody (Cell Signaling) to detect Myc-tagged Nab2 copurified with TAP-tagged proteins.
Poly(A) tail length assay.
The bulk poly(A) tail length assay was performed as described previously (64, 65). Briefly, the total RNA was end labeled with [32P]pCp and T4 RNA ligase. RNA was digested using RNase A/T1 to remove nonpoly(A) RNA, purified with phenol chloroform, and ethanol precipitated. Poly(A) tails were resolved on an 8 M urea–Tris-borate-EDTA–10% polyacrylamide gel. Poly(A) tails were quantified using ImageJ to measure the pixel densitometry along the length of the tail.
FISH.
Poly(A) RNA localization was assayed by a fluorescence in situ hybridization (FISH) as described previously (66). Yeast cells were grown in YEPD to log phase at 30°C. The cells were fixed with formaldehyde, digested with zymolyase, and permeabilized. A digoxigenin-labeled oligo(dT)50 probe coupled with a fluorescein isothiocyanate-conjugated anti-digoxigenin antibody (Roche Molecular Biochemicals) was used to detect poly(A) RNA. DAPI (4′,6′-diamidino-2-phenylindole) was used to stain chromatin and indicate the position of the nucleus. Differential interference contrast (DIC) images were obtained to visualize whole cells.
ZC3H14 IP and MS.
Brain tissue was collected from wild-type Black 6 mice and homogenized in immunoprecipitation (IP) buffer (50 mM Tris-HCl [pH 7.4], 100 mM NaCl, 32 mM NaF, and 0.5% NP-40 in diethyl pyrocarbonate [DEPC]-treated water) supplemented with 1 cOmplete mini protease inhibitor tablet (Roche; 1 tablet/10 ml of buffer). Homogenate was sonicated on ice five times at 0.5% output for 10 s and then passed five times through a 27-gauge syringe. Lysates were spun at 13,000 rpm for 10 min at 4°C, and protein concentrations were determined using a standard bicinchoninic acid (BCA) assay (Pierce). Protein G-magnetic beads (Dynabeads; Invitrogen) were suspended in IP buffer and incubated with preimmune rabbit serum or an equal volume of ZC3H14 antibody for 1 h at room temperature. Bead/antibody and bead/preimmune samples were added to clarified cell lysates, followed by incubation at 4°C overnight while tumbling end over end (10% removed prior to overnight incubation for input samples). After incubation, for mass spectrometric (MS) analysis, the beads were magnetized, and unbound samples were collected (10% of input) and washed five times with ice-cold IP buffer. For immunoblotting, ZC3H14 protein complexes were eluted with reducing sample buffer (250 mM Tris-HCl, 500 mM dithiothreitol [DTT], 10% SDS, 0.5% bromophenol blue, and 50% glycerol).
To identify ZC3H14 interacting proteins, bead mass spectrometry was used. After several washing steps, Sepharose beads from control and ZC3H14 immunoprecipitates were resuspended in 8 M urea–100 mM NaHPO4 (pH 8.5; 50 μl, final volume) and treated with 1 mM DTT at 25°C for 30 min, followed by 5 mM iodoacetamide at 25°C for 30 min in the dark. The samples were then diluted to 1 M urea with 50 mM ammonium bicarbonate (final volume, 400 μl) and digested with lysyl endopeptidase (Wako; 1.25 ng/μl, final concentration) at 25°C for 4 h and further digested overnight with trypsin (Promega; 1.25 ng/μl, final concentration) at 25°C. The resulting peptides were desalted with a Sep-Pak C18 column (Waters) and dried under vacuum. Each sample was resuspended in loading buffer (0.1% formic acid, 0.03% trifluoroacetic acid, 1% acetonitrile) and analyzed independently by reverses-phase liquid chromatography (LC) coupled with tandem mass spectrometry (MS/MS) as essentially previously described with slight modifications (67, 68). Briefly, peptide mixtures were loaded onto a C18 nanoLC column (75 μm [inner diameter], 15-cm long, 1.9-μm resin [Maisch GmbH]) and eluted over a 5 to 0% gradient (buffer A: 0.1% formic acid; buffer B: 0.1% formic acid in 100% acetonitrile). Eluates were monitored in an MS survey scan, followed by 10 data-dependent MS/MS scans on a Q-Exactive plus Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA). The acquired MS/MS spectra were searched against a concatenated target decoy mouse reference database (v.62) of the National Center for Biotechnology Information (downloaded 14 November 2013 with 30,267 target entries) using the SEQUEST Sorcerer algorithm (version 4.3.0; SAGE-N). The search parameters included the following: fully tryptic restriction, parent ion mass tolerance of ±50 ppm, up to two missed trypsin cleavages, and dynamic modifications for oxidized Met (+15.9949 Da). The peptides were classified by charge state and first filtered by mass accuracy (10 ppm for high-resolution MS) and then dynamically by increasing XCorr and ΔCn values to reduce the protein false discovery rate to <1%. If peptides were shared by multiple members of a protein family, the matched members were clustered into a single group in which each protein identified by a unique peptide represented a subgroup. Each sample was analyzed in biological replicate, and the protein groups, the total peptide counts, and the peptide spectral counts are provided in Table S6 in the supplemental material.
To validate ZC3H14 interactions identified in the mass spectrometry data set, cultured neuronal cells (N2A) were homogenized in IP buffer (50 mM Tris-HCl [pH 7.4], 100 mM NaCl, 32 mM NaF, and 0.5% NP-40 in DEPC-treated water) supplemented with 1 cOmplete mini protease inhibitor tablet (Roche; 1 tablet/10 ml of buffer). Homogenate was sonicated on ice five times at 0.5% output for 10 s and then passed through a 27-gauge syringe five times. Lysates were spun at 13,000 rpm for 10 min at 4°C, and the protein concentrations were determined with a standard BCA assay (Pierce). Protein G-magnetic beads (Dynabeads; Invitrogen) were suspended in IP buffer and incubated with preimmune rabbit serum or an equal volume of ZC3H14 antibody for 1 h at room temperature. Bead/antibody and bead/preimmune samples were added to clarified cell lysates, followed by incubation at 4°C overnight while tumbling end over end (10% removed prior to overnight incubation for input samples). After incubation, the beads were magnetized, and unbound samples were collected (10% of input) and washed five times with ice-cold IP buffer. The ZC3H14 protein complexes were eluted with reducing sample buffer (250 mM Tris-HCl, 500 mM DTT, 10% SDS, 0.5% bromophenol blue, 50% glycerol). The U2AF2 protein was detected with anti-U2AF2 (Bethyl Labs, Inc.).
Gene ontology (GO) enrichment and network.
Functional enrichment of the modules was determined using the GO-Elite (v1.2.5) package (69). The set of total proteins (1,341) identified was used as the background. We analyzed the 153 proteins enriched in IP (peptide spectrum match [PSM] IP/bead fold change of >2). The Z-score determines the overrepresentation of ontologies in a module, and a permutation P value was used to assess the significance of a Z-score cutoff of 5.5 and a P value cutoff of 0.00001, with a minimum of five proteins per category used as filters in pruning the ontologies. A horizontal bar graph was plotted in R. The networks were constructed using the Circlize package in R.
RESULTS
Unspliced pre-mRNA accumulates in nab2 mutants.
To investigate a possible role for Nab2 in splicing, we used two conditional alleles of NAB2: nab2-ΔN (nab2-1) and nab2-C›A5-7 (Fig. 1A). The nab2-ΔN (nab2-1) allele deletes the N-terminal proline-tryptophan-isoleucine (PWI)-like domain of Nab2, which is critical for proper poly(A) RNA export (70). The nab2-C›A5-7 mutant alters conserved cysteine residues within the fifth, sixth, and seventh zinc fingers and severely compromises RNA binding (71). These nab2 alleles both confer cold-sensitive growth although growth of nab2-ΔN is more severely impaired at all temperatures (Fig. 1B).
FIG 1.
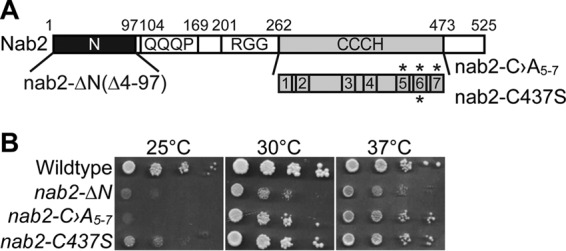
nab2 mutant cells show growth defects. (A) The Nab2 protein consists of four domains: a proline-tryptophan-isoleucine (PWI)-like N-terminal domain, a glutamine-rich domain (QQQP), an arginine-glycine-glycine (RGG) domain required for nuclear localization, and a tandem cysteine-cysteine-cysteine-histidine (CCCH) zinc finger domain comprised of seven zinc fingers (70). Shown below the CCCH zinc finger domain are zinc fingers 1 to 7 with zinc fingers 5 to 7 critical for high-affinity binding to polyadenosine RNA (71). The asterisks above zinc fingers 5, 6, and 7 denote the first cysteine residues (C415, C437, and C458) altered to alanine to generate nab2-C›A5-7. The asterisk below zinc finger 6 denotes the first cysteine residue (C437) altered to serine to generate nab2-C437S. The residues removed (i.e., 4 to 97) to generate the N-terminal nab2 mutant, nab2-ΔN, are also indicated. (B) Wild-type cells and the integrated nab2 mutants nab2-ΔN, nab2-C›A5-7, and nab2-C437S were grown at 30°C, serially diluted, spotted onto YEPD plates, and incubated at the indicated temperatures.
To assay splicing in wild-type and nab2 mutant cells, total RNA was analyzed by quantitative reverse transcription-PCR (qRT-PCR) using primers to detect unspliced and total transcripts. The levels of several well-characterized and abundant intron-containing S. cerevisiae transcripts were measured by qRT-PCR using primers flanking the intron-exon boundary to detect the unspliced pre-mRNA and primers within the exon to detect total mRNA (Fig. 2A). To obtain a representative sample across different classes of mRNA, the analysis was performed with the nonribosomal protein transcripts, ACT1 and TUB1, and the ribosomal protein transcript, RPL28. Relative to wild-type cells, the nab2 mutant cells exhibited a 2- to 3-fold increase in unspliced pre-mRNA (Fig. 2B to D). In contrast, nab2 mutants did not show significant change in total mRNA levels compared to the wild type (Fig. 2B to D). As a control, a splicing factor mutant, prp2-1, which impairs spliceosome activation (72, 73), showed a significant increase in unspliced pre-mRNA and significant decrease in total mRNA levels. These data provide the first evidence that nab2 mutant cells could have defects that lead to the accumulation of improperly spliced transcripts.
FIG 2.
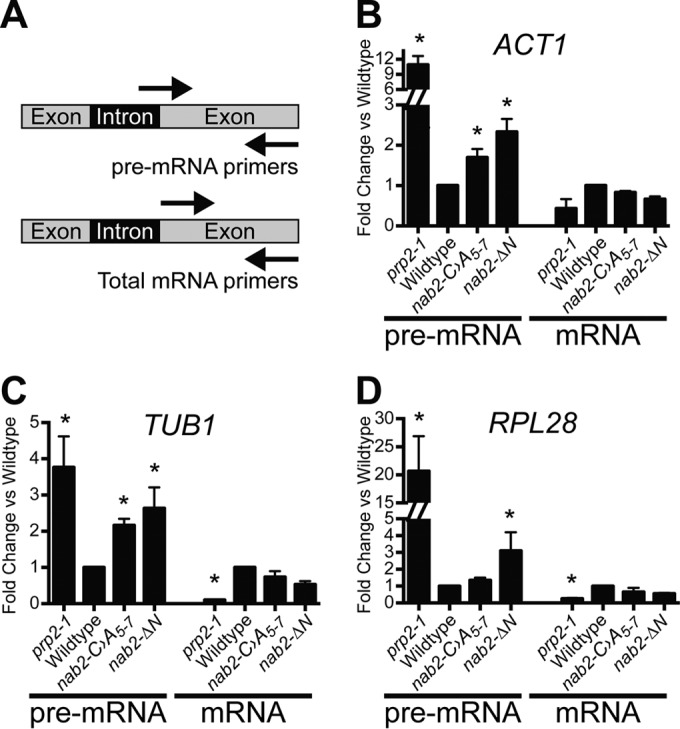
Unspliced transcripts accumulate in nab2 mutant cells. (A) Schematic of primer locations used to amplify unspliced pre-mRNA and total mRNA. For each transcript, unspliced pre-mRNA was detected with primers that span intron-exon boundaries, and total mRNA was detected with primers within the exon. (B to D) Total RNA was prepared from wild-type, nab2-C›A5-7, and nab2-ΔN cells, as well as a control splicing mutant (prp2-1) (72, 73). Quantitative RT-PCR was performed to measure both the pre-mRNA and the total mRNA of the ACT1 (B), TUB1 (C), and RPL28A (D) transcripts. The results were normalized to the PDA1 transcript, and values were plotted relative to wild-type cells, where the value for each transcript analyzed was set to 1.0. Experiments were performed in triplicate, error bars indicate the standard errors of the mean (SEM), and statistical significance (indicated by the asterisks) was calculated for both pre-mRNA and mRNA using one-way ANOVA.
Nab2 is required for proper splicing of a subset of pre-mRNAs.
To assess the requirement for Nab2 in splicing within the transcriptome of S. cerevisiae, we used a splicing microarray (60, 74). This microarray platform contains three probes that target each of 249 intron-containing transcripts in S. cerevisiae. The probes are located (i) within the intron to detect unspliced pre-mRNA (intron), (ii) across the exon-exon junction to detect mature, spliced mRNA (junction), and (iii) within the exon to detect total RNA (Exon) (Fig. 3A). As shown in the heat map depicting the full splicing array in Fig. 3B, both nab2 mutants display a mild increase in intron-containing, unspliced pre-mRNA relative to the wild type. The nab2-ΔN mutant shows elevated intron-containing pre-mRNA at both 16 and 30°C with some transcript-dependent variability, whereas the nab2-C›A5-7 mutant predominantly shows increased intron-containing pre-mRNA at 16°C. More than half of the transcripts analyzed in both nab2 alleles show intron accumulation (Fig. 3B; also see Table S3 in supplemental material); however, only a small number of transcripts exceed a log2 ratio of >1.
FIG 3.

A splicing microarray reveals a modest accumulation of unspliced transcripts in nab2 mutants. (A) The splicing microarray contains probes located within the intron (I) that detect pre-mRNA, located at the exon-exon junction (J) that detect mature mRNA, and located within an exon (E) that detect total mRNA for each intron-containing gene. (B) Splicing profile of nab2-C›A5-7 (C>A) and nab2-ΔN mutants compared to wild-type cells grown at 16 and 30°C for all 249 different intron-containing genes present on the microarray (see Table S3 in the supplemental material). Intron-containing pre-mRNA levels are elevated in nab2-C›A5-7 mutant at 16°C and in nab2-ΔN mutant at both temperatures. The cDNA from total RNA of nab2 mutants grown at 16 and 30°C was hybridized to the splicing array as described in Materials and Methods. Each horizontal line shows the behavior of a single transcript. (C) A closeup view of the splicing profile in nab2 mutants reveals that many ribosomal protein (RPL/RPS) intron-containing genes (e.g., RPL28) and some two-intron-containing genes (e.g., RPL7A) show elevated pre-mRNA levels. Two ribosomal protein two-intron-containing genes, RPL7B and RPS22B, show decreased total and mature mRNA levels. For comparison, the splicing profile of an SR protein mutant (Δnpl3) at 37°C is depicted, which was generated at the same time as the nab2 mutants and has previously been shown to cause pre-mRNA accumulation (112). (D) A Venn diagram illustrates the number of shared pre-mRNA transcripts that accumulate in nab2-C›A5-7 and nab2-ΔN cells. (E) The relative levels of expression of 20 transcripts measured by qRT-PCR and the splicing microarray show a positive correlation (r2 = 0.628). The relative levels of expression of the transcripts assayed by qRT-PCR and microarray were paired, plotted, and analyzed by linear regression.
Interestingly, transcripts that exhibit the greatest intron accumulation in the nab2 mutants (Fig. 3C) include ribosomal protein gene (RPG) transcripts (e.g., RPL28, RPL30, RPL32, RPL21B, RPL22B, and RPS14B), transcripts with large introns (e.g., RPGs [∼400 nucleotides] [nt], DBP2 [1,002 nt], and YRA1 [766 nt]), and transcripts containing snoRNAs in their introns (e.g., ASC1 [snR24] and TEF4 [snR38]) (Fig. 3C). In addition, three ribosomal protein transcripts containing two introns (e.g., RPL7A, RPL7B, and RPS22B) show elevated intron accumulation in the nab2 mutants. RPL7A and RPL7B contain noncanonical branch-point sequences (TGCTAAC) in their first introns, and RPL7A, RPL7B, and RPS22B contain snoRNAs in their second introns (RPL7A [snR39], RPL7B [snR59], and RPS22B [snR44]). Strikingly, RPL7B and RPS22B total RNA (exon) levels and spliced RNA (junction) levels are also greatly reduced in the nab2 mutants (Fig. 3C). These results indicate that Nab2 affects the splicing of transcripts containing introns with embedded snoRNAs and suggest that impaired splicing of snoRNA introns may be linked to impaired processing of snoRNAs in nab2 mutants.
More than 60% of transcripts analyzed in both nab2 alleles show decreased exon expression; however, the transcripts with decreased total RNA do not correlate with an increase in intron accumulation (r2 = 0.032). A Venn diagram shows a shared population of unspliced pre-mRNA transcripts that are affected in each of the nab2 mutants (Fig. 3D; also see Table S4 in supplemental material). However, the shared RNA expression levels varied between the two nab2 mutants as indicated by an average Pearson correlation value of 0.42. These data suggest that each nab2 mutant differentially affects the same population of intron-containing transcripts and could reflect the fate of RNA during the different mRNA processing events.
To confirm the results obtained from the microarray analysis, qRT-PCR was utilized to validate the results. Eight different intron-containing and intronless transcripts were chosen for validation. For the intron-containing transcripts, the unspliced pre-mRNA, mature spliced mRNA, and total RNA levels were measured for each specific transcript. Specific mRNA transcripts were assessed by qRT-PCR, and the fold changes observed were compared to the log2 ratio obtained from the splicing microarray. A positive correlation was observed between the microarray and qRT-PCR (Fig. 3E; also see Table S5 in supplemental material). These observations indicate that nab2 mutant cells have a mild splicing defect.
Nab2 function is not required for efficient splicing in vitro.
The results of the splicing array show the accumulation of intron-containing RNAs in nab2 mutant cells, but we did not assess whether the Nab2 protein is directly required for splicing in vitro. To determine whether Nab2 is required for splicing in vitro, we utilized a well-characterized in vitro splicing assay using total yeast extract and the ACT1 pre-mRNA (62). We analyzed the in vitro splicing activity of cell lysates prepared from mutant cells with an allele of NAB2, nab2-C437S (Fig. 1). The C437S amino acid substitution is a conservative cysteine to serine change in the sixth zinc finger of Nab2 that confers a cold sensitive growth defect (Fig. 1). As shown in Fig. 4A, extracts from nab2-C437S mutant cells carry out splicing of ACT1 pre-mRNA in a manner comparable to the control NAB2 wild-type cell extract. An in vitro splicing assay was quantified to assess whether distinct steps in splicing are impaired in the nab2 mutant cell extract (Fig. 4B and C). Consistent with the representative data shown in Fig. 4A, there was no significant change in the branching efficiency when nab2 mutant extract was compared to a wild-type control extract (Fig. 4B). We detected a modest decrease in the efficiency of exon ligation mediated by the nab2 mutant extract compared to the control extract (Fig. 4C). Thus, although we detected a slight decrease in exon ligation, we did not detect a defect in branching, so we cannot explain the accumulation of pre-mRNA in vivo by a direct effect of Nab2 on splicing.
Genetic interactions between NAB2 and genes encoding splicing factors.
To further explore how reduced Nab2 function could lead to an increase in unspliced transcripts, we assessed the genetic interactions between NAB2 and genes encoding splicing factors. We analyzed growth in double mutants consisting of nab2 alleles combined with deletion of nonessential splicing factors involved in multiple steps in splicing (Table 1). The early spliceosome assembly factors involved in complex E and A formation include the U1 snRNP (Mud2 and Nam8) and U2 snRNP (Lea1, Msl1, Mud1, and Nam8). The catalytic transesterification reaction factors involved in complex B and C formation include the tri-snRNP U5·U4/6 snRNP (Snu66), as well as Prp18, Snt309, and Isy1. Other factors, such as Cus2, modulate the conformation of the U2 snRNA (75). For these studies, we used a series of nab2 alleles: nab2-C347S, nab2-C›A5-7, nab2-ΔN, nab2-ΔRGG, and nab2-ΔQQQP (70). Genetic interactions for the double mutants were analyzed by serial dilution and spotting growth assays. The complete results of this analysis are summarized in Table 1. Mutants of NAB2 genetically interact with a subset of splicing factors. A profound synthetic lethal phenotype is observed when the nab2-ΔN mutant is combined with the Δlea1 or Δmsl1 mutant. A synthetic growth phenotype is also observed when C-terminal NAB2 mutants are combined with Δbud13, Δmud2, Δmsl1, Δprp18, Δsnt309, and Δsnu66 mutants. Figure 5A shows a serial dilution growth assay for two representative splicing factors, Mud2 and Cus2 (30, 75). A negative genetic interaction was identified between NAB2 and MUD2, where the double-mutant Δmud2 nab2-C437S cells show severely impaired growth compared to either single mutant (Fig. 5). The doubling times for both wild-type and Δmud2 cells is 1.6 h, whereas the doubling time for nab2-C437S cells is 2.4 h, and the doubling time for Δmud2 nab2-C437S cells is 3.9 h. Mud2 is an RRM-containing RNA binding protein that associates with branch point binding protein (BBP/Msl5/SF1) to assemble spliceosomes and commit to splicing (16, 18, 30). Loss of Mud2 results in reduced splicing efficiency (30) and pre-mRNA leakage (76, 77). In contrast, no genetic interaction was detected with genes encoding a number of factors involved in splicing (Table 2), including CUS2. As shown in Fig. 5A, even when the Δcus2 mutant is combined with the very severe nab2-C›A5-7 mutant, no change in cell growth was observed. Cus2 is an assembly factor important for U2 snRNP folding involved later in the splicing process than Mud2 (75). CUS2 shows a number of genetic interactions with other splicing factor genes, such as PRP5 (78, 79) and BRE1 (80). Thus, the genetic interactions between NAB2 and genes encoding splicing factors are specific for a subset of splicing factors.
TABLE 1.
Genetic interaction between nab2 mutant alleles and alleles of splicing factors
| Genotype | Spliceosomal complexd | Approximate colony diama |
|||||
|---|---|---|---|---|---|---|---|
| NAB2 | nab2-C437S | nab2-C›A5-7 | nab2-ΔN | nab2-ΔQQQP | nab2-ΔRGG | ||
| Wild type | +++ | +++ | ++ | ++ | +++ | +++ | |
| Δmud2 | E | +++ | ++ | − | + | +++ | +++ |
| Δnam8 | E and A | +++ | +++ | ++ | ++ | +++ | +++ |
| Δcus2 | A | +++ | +++ | ++ | ++ | +++ | +++ |
| Δmud1 | A and B | +++ | +++ | + | ++ | +++ | +++ |
| Δmsl1 | A and B | +++ | + | − | − | +++ | +++ |
| Δlea1 | A and B | +++ | +++ | ND | − | ND | ND |
| Δsnu66c | B and C | +++ | + | + | ++ | +++ | +++ |
| Δsnt309 | B and C | +++ | + | + | + | +++ | +++ |
| Δprp18b | C | +++ | + | + | ++ | +++ | +++ |
| Δisy1 | B, C, and Post | +++ | +++ | ++ | ++ | +++ | +++ |
| Δbud13 | B, C, and Post | +++ | ++ | ++ | ++ | +++ | +++ |
Approximate colony diameters at 30°C: −, no growth; +, 0.5 mm; ++, 1.0 mm; +++, 2.0 mm. ND, not determined.
+++ growth for nab2-ΔN at 16°C.
Growth at 16°C and 37°C.
E, early spliceosomal complex; A, prespliceosome; B, pre-catalytic spliceosome; C, catalytic spliceosome; Post, postspliceosomal complex, formed after the second catalytic step of splicing.
FIG 5.
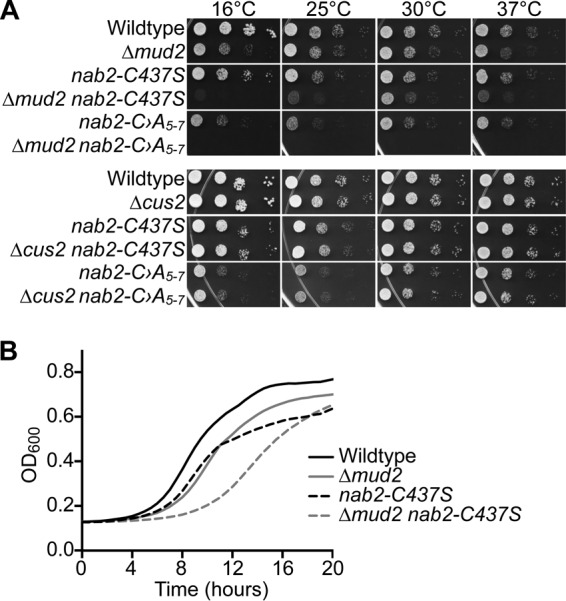
Growth assays to examine genetic interactions between nab2 mutants and alleles of splicing factors. (A) As described in Materials and Methods, a plasmid shuffle assay was used to analyze the growth of the splicing factor mutants in combination with nab2 mutants. Δnab2, Δnab2 Δmud2, and Δnab2 Δcus2 cells maintained by a URA3 plasmid encoding wild-type NAB2 were transformed with LEU2 plasmids encoding wild-type NAB2, nab2-C›A5-7, or nab2-C437S. Cells were selected on 5-fluoroorotic acid (5-FOA) to eliminate the NAB2 maintenance plasmid and grown, serially diluted, and spotted onto plates lacking leucine to assess growth at 16, 25, 30, and 37°C. (B) Growth curves were generated for Δnab2 or Δnab2 Δmud2 cells carrying wild-type NAB2, nab2-C›A5-7, or nab2-C437S mutant plasmids in liquid media. The cells were grown to saturation and diluted, and their optical density at 600 nm (OD600) was measured at A600 for 20 h at 25°C. The growth curves are representative of three independent experiments.
TABLE 2.
Genetic interaction between nab2 mutant alleles and alleles of retention factors
| Genotypeb | Spliceosomal complex | Approximate colony diama |
||
|---|---|---|---|---|
| NAB2 | nab2-C437S | nab2-ΔN | ||
| Wild type | +++ | +++ | ++ | |
| Δmud2 | E | +++ | ++ | + |
| Δmsl1 | A and B | +++ | + | − |
| Δprp18* | C | +++ | + | ++ |
| Δbud13* | B, C, and Post | +++ | ++ | ++ |
| Δpml1 | B, C, and Post | +++ | ++ | ++ |
| Δpml39 | +++ | +++ | +++ | |
| Δmlp1 | +++ | ++ | +++ | |
| Δmlp2 | +++ | ++ | +++ | |
Approximate colony diameters at 30°C: −, no growth; +, 0.5 mm; ++, 1.0 mm; +++, 2.0 mm.
*, +++ growth for nab2-ΔN at 16°C.
A number of splicing factors, including Mud2, Prp18, Bud13, and Pml1, contribute to nuclear retention of pre-mRNA to prevent pre-mRNA export/leakage to the cytoplasm (22, 77, 81). The branch point binding protein BBP/Msl5/SF1 also contributes to the nuclear retention of pre-mRNA by interacting with the nuclear pore-associated protein, Mlp1 (22). As nab2 mutants genetically interact with the splicing factor genes MUD2, PRP18, and BUD13 that are implicated in splicing and retention (22, 77, 81), we hypothesized that Nab2 may be involved in ensuring that improperly spliced pre-mRNA transcripts are retained in the nucleus. To test this hypothesis, we assessed the genetic interactions between nab2 alleles and alleles of genes encoding retention factors. Consistent with previous studies that identified a genetic interaction between nab2-ΔN and genes encoding the retention machinery (21), a synthetic growth phenotype was observed when nab2-C437S was combined with Δmud2, Δprp18, Δbud13, Δpml1, Δmsl1, or Δmlp1/2 mutations (Table 2). Taken together, these results confirm genetic interactions between NAB2 and the splicing and retention machinery in the nucleus.
MUD2 RRM3 is functionally important for NAB2.
To further examine the strong negative genetic interaction identified between NAB2 and MUD2, an integrated nab2 mutant containing a mutation in the sixth zinc finger, nab2-C437S, was combined with a deletion mutant of MUD2. As initially observed in the plasmid shuffle assay (Fig. 5A), the loss of MUD2 in combination with nab2-C437S exacerbates the growth phenotype, resulting in severe growth defects at all of the temperatures analyzed (Fig. 6A). These data confirm that NAB2 genetically interacts with the early splicing and retention factor gene MUD2. To refine the functional basis for the genetic interaction between NAB2 and MUD2, we altered specific residues in Mud2 implicated in distinct functions and performed a rescue experiment in the Δmud2 nab2-C437S double mutant.
FIG 6.
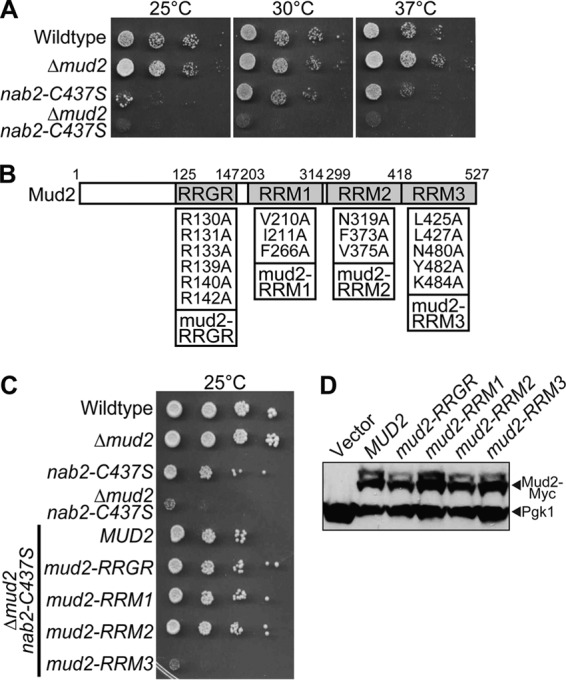
Interaction between Mud2 and Msl5 is required when Nab2 function is impaired. (A) Isogenic integrated strains were generated by deleting MUD2 in wild-type and nab2-C437S cells to produce Δmud2 and Δmud2 nab2-C437S cells. Wild-type, Δmud2, nab2-C437S, and Δmud2 nab2-C437S cells were grown, serially diluted, and spotted onto YEPD plates at 25, 30, and 37°C. (B) Schematic of Mud2 protein showing the position of the tandem RRGR motif domain and three RNA recognition motifs (RRMs): RRM1, RRM2, and RRM3. Amino acid substitutions designed to impair the function of each domain are illustrated. The amino acid substitutions were as follows: the tandem RRGR domain (57) (R130A, R131A, R133A, R139A, R140A, and R142A), RRM1 (R208A, V210A, I211A, and F266A), RRM2 (N310A, I311A, and F373A), and RRM3 (L425A, L427A, N480A, Y482A, and Y484A). (C) Wild-type cells and integrated mutant Δmud2, nab2-C437S, and Δmud2 nab2-C437S cells were transformed with a vector plasmid and are shown as controls. Δmud2 nab2-C437S cells were transformed with plasmids expressing wild-type MUD2 or mud2 mutants in the RRGR, RRM1, RRM2, and RRM3 domains illustrated in panel B. Cells were grown, serially diluted, and spotted onto plates lacking uracil to assess growth at 25°C. (D) The Mud2 variants were all tagged with a Myc epitope to assess the relative expression levels by immunoblotting yeast cell lysates expressing the indicated Mud2 variants with anti-Myc antibody. Control cells that do not express Mud2-Myc (Vector) are shown as a negative control. Pgk1 was used as a loading control.
S. cerevisiae Mud2 is the presumptive orthologue of human U2AF2/U2AF65 (30, 82). Both Mud2 and U2AF2 contain three RRMs (Fig. 6B), which are distantly related (30), with approximately 11% sequence identity in RRM1 and RRM2 and 31% identity in RRM3 (see Fig. S1 in the supplemental material). While U2AF2 contains a conventional arginine-serine (RS)-rich domain, this domain is absent in Mud2 (30, 82). Mud2 does, however, contain tandem RRGR motifs within the N-terminal domain (Fig. 6B) that are critical for Mud2 function (57). For human U2AF2, the RS domain is proposed to interact with the pre-mRNA branch point (83), RRM1 and RRM2 bind to polypyrimidine tract RNA (84), and RRM3 mediates binding to the branch-point binding protein/SF1 (85). Importantly, RRM3 within Mud2 is required for interaction with the yeast orthologue of branch-point binding protein, Msl5 (82). Based on previous analysis of Mud2 (57), as well as sequence alignment of Mud2 with both human U2AF2 and the fission yeast orthologue, Prp2 (see Fig. S1 in the supplemental material) (84–86), we generated a series of mud2 mutants to test for genetic interactions with nab2: a mud2-RRM1 mutant (V210A, I211A, and F266A), a mud2-RRM2 mutant (N319A, F373A, and V375A), and a mud2-RRM3 (57) mutant (L425A, L427A, N480A, Y482A, and K484A). In addition, we created the previously characterized (57) mud2-RRGR mutant (R130A, R131A, R133A, R139A, R140A, and R142A). Of these mud2 mutants, only mud2-RRM3 could not rescue the growth of Δmud2 nab2-C437S double-mutant cells (Fig. 6C), although all of the mud2 proteins were expressed at relatively equal levels (Fig. 6D). These data suggest that interaction of Mud2 RRM3 with Msl5 is critical when Nab2 function is impaired.
Nab2 physically associates with the commitment complex.
Given that NAB2 genetically interacts with the early splicing factor gene MUD2, we hypothesized that these factors might physically interact either directly or via binding to shared mRNA targets. Mud2 physically interacts with the essential branch point binding protein, Msl5, which is crucial for commitment complex formation (17, 77). As shown in Fig. 6C, our results suggest that Mud2 interaction with Msl5 is critical when Nab2 is impaired. We therefore examined whether Nab2 physically associates with Mud2 and/or Msl5. To examine these interactions, we analyzed copurification of Myc-tagged Nab2 with TAP-tagged Mud2 or Msl5 expressed in S. cerevisiae cells. As a control, we included TAP-tagged Pub1, a factor that interacts with Nab2, and Srp1, a nuclear import protein that does not interact with Nab2 (87). Each of the TAP-tagged proteins, which are all approximately the same molecular mass (51 to 60 kDa), is readily detected by immunoblotting (Fig. 7A). As shown in Fig. 7B, Myc-tagged Nab2 copurifies with Pub1, Mud2, and Msl5, whereas no copurification is observed with the negative-control protein, Srp1. The addition of RNase (+RNase) reduces the interaction between Nab2 and members of the commitment complex, Mud2 and Msl5, a finding largely indicative of RNA-dependent binding. To determine which domain of Nab2 is required for the physical association with the commitment complex proteins, we examined the binding of Myc-tagged Nab2 mutants, Nab2-ΔN-Myc or Nab2-ΔZnF5-7-Myc, in each of the TAP-tagged strains (Fig. 7C). The zinc finger deletion mutant, ΔZnF5-7, removes the last zinc fingers, 5 to 7, critical for polyadenosine RNA binding (71, 88). Although Nab2-ΔN-Myc still bound to Mud2 and Msl5, no binding of Nab2-ΔZnF5-7-Myc to Mud2 or Msl5 was detected (Fig. 7C). These data suggest that Nab2 interacts with the same RNAs as Mud2 and Msl5 to physically associate with these commitment complex proteins.
FIG 7.
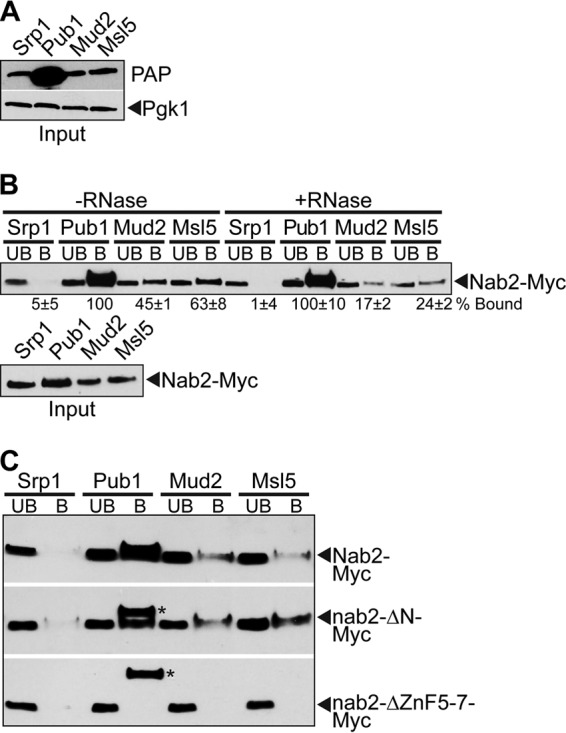
Nab2 physically interacts with the commitment complex in an RNA-dependent manner. (A) An immunoblot shows the expression of the TAP-tagged proteins Srp1, Pub1, Mud2, and Msl5 (all of which are approximately the same molecular weight). Immunoblotting was performed with a peroxidase-antiperoxidase (PAP) antibody to detect TAP-tagged protein expression and with an anti-Pgk1 antibody to detect 3-phosphoglycerate kinase as a loading control. (B) Cells expressing the C-terminal TAP-tagged proteins Srp1, Pub1, Mud2, or Msl5 were transformed with a Nab2-Myc plasmid. The TAP-tagged proteins were purified, and Nab2-Myc was detected by immunoblotting in the input, unbound (UB), and bound (B) fractions. Samples were treated with (+RNase) or without RNase (−RNase) to assess the RNA dependence of the interaction. The percentage of bound Nab2 protein relative to the amount of input protein and Nab2 bound to Pub1 (%Bound) is indicated below the bound fractions. Experiments were performed in triplicate, and the standard deviations were calculated. (C) To map the domain of Nab2 required for interaction with Mud2/Msl5, cells expressing TAP-tagged Srp1, Pub1, Mud2, or Msl5 were transformed with full-length Nab2-Myc, nab2-ΔN-Myc, or nab2-ΔZnF5-7-Myc plasmids. The TAP-tagged proteins were precipitated from yeast lysates, and immunoblotting was performed with anti-Myc antibody to detect Nab2 in the unbound (UB) and bound (B) lanes. The additional bands observed in the Pub1 bound lane in the nab2-ΔN-Myc and nab2-ΔZnF5-7-Myc immunoblot panels (indicated by asterisks) are cross-reacting bands similar in size to the Nab2 protein.
Δmud2 nab2-C437S mutant cells show accumulation of pre-mRNA and extended poly(A) tails.
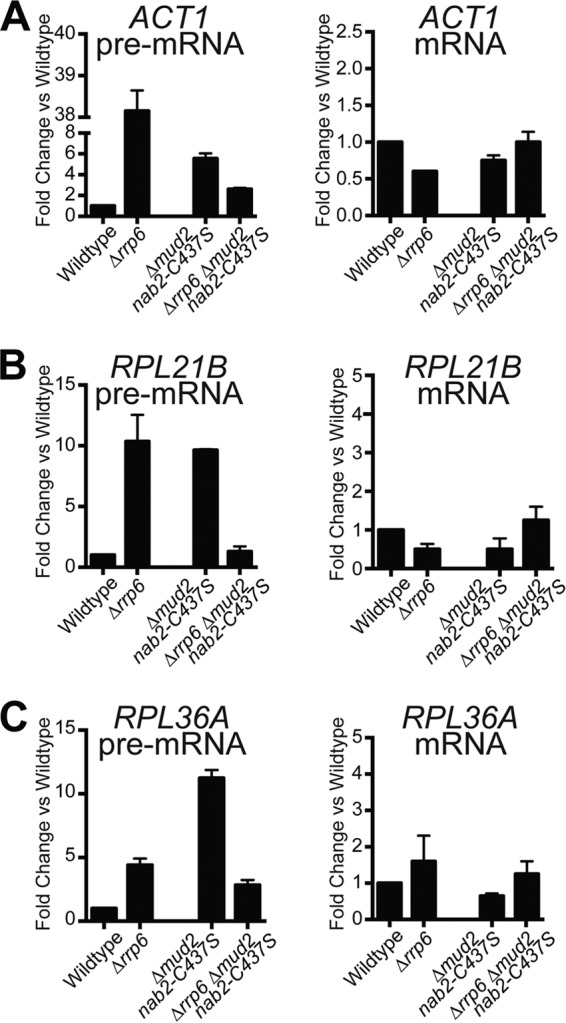
To begin to understand the functional relationship between Nab2 and Mud2, we utilized the Δmud2 nab2-C437S double mutant and assessed several aspects of mRNA processing. Nab2 has been implicated in limiting poly(A) tail length, as well as promoting the export of properly processed mRNAs (39). Mud2 is implicated in splicing (30) and nuclear retention of pre-mRNA (76, 77). Since Mud2 and Nab2 are important for proper splicing, one possibility is that splicing defects are exacerbated in a double mutant. To determine the amount of unspliced pre-mRNA, qRT-PCR was performed for ACT1, RPL21B, and RPL36A transcripts. For all transcripts examined, there was a significant increase in pre-mRNA detected in the double mutant compared to either single mutant (Fig. 8A to C). The increase in pre-mRNA levels for the double mutant compared to either mutant alone suggests that Nab2 and Mud2 likely function in parallel mRNA processing pathways to influence splicing. Interestingly, the amount of total mRNA detected for RPL21B and RPL36A decreased in Δmud2 nab2-C437S cells, a finding consistent with the inability to efficiently complete nuclear processing steps.
FIG 8.

nab2 and mud2 mutants show defects in poly(A) tail length control and mRNA splicing. (A to C) Total RNA was isolated from wild-type and integrated mutant Δmud2, nab2-C437S, and Δmud2 nab2-C437S cells grown at 30°C. Quantitative real-time PCR analysis was performed to amplify ACT1 (A), RPL21B (B), and RPL36A (C) intron-containing transcripts. For each transcript, unspliced pre-mRNA was detected with primers that span intron-exon boundaries, and total mRNA was detected with primers within the exon. The mRNA levels were normalized to the PDA1 transcript and plotted relative to wild-type cells, which were set to 1.0. Experiments were performed in triplicate, error bars represent the SEM, and statistical significance (indicated by the asterisks) was calculated for both pre-mRNA and mRNA using one-way ANOVA. (D) Total RNA isolated from wild-type, Δmud2, nab2-C437S, and Δmud2 nab2-C437S cells grown at 30°C was analyzed for bulk poly(A) tail length as described in Materials and Methods. (E) Poly(A) tails were quantified using ImageJ by calculating pixel intensity along the length of the poly(A) tail and plotted relative to the number of A's, as described in Materials and Methods. (F) To examine poly(A) RNA export, wild-type, nab2-ΔN, nab2-C437S, Δmud2, and Δmud2 nab2-C437S cells grown at 30°C were examined by FISH with an oligo(dT) probe to detect poly(A) RNA and with DAPI to detect DNA. DIC images are also shown.
Nab2 is implicated in the control of poly(A) tail length (39), and we reasoned that the growth defect in Δmud2 nab2-C437S cells could reflect defects in poly(A) tail length control. We assessed poly(A) RNA tail length by utilizing a bulk poly(A) RNA tail length assay. As previously described, nab2-C437S mutant cells accumulate extended poly(A) tails compared to wild-type cells (Fig. 8D and E). In contrast, deletion of MUD2 results in shortening of poly(A) tails. Interestingly, Δmud2 nab2-C437S double-mutant cells show long, heterogeneous poly(A) tails compared to the Δmud2 mutation or even nab2-C437S alone.
Nab2 physically interacts with quality control factors, Mlp1 and Mlp2, present at the nuclear pore (89, 90), and Mud2 genetically interacts with pore-associated quality control factors, Nup60 and Pml39 (91, 92), leading to the idea that Nab2 and Mud2 could contribute to the nuclear retention of improperly processed RNAs. Therefore, Δmud2 nab2-C437S mutant cells could show export defects due to nuclear accumulation of 3′-end extended, intron-containing pre-mRNA transcripts. To examine poly(A) RNA export, we performed fluorescence in situ hybridization. As previously described, poly(A) RNA accumulates in the nucleus in nab2-ΔN cells, but no nuclear accumulation was observed in the nab2-C437S mutant (71). We detected no changes in bulk poly(A) RNA localization when nab2 mutants were combined with Δmud2 (Fig. 8F). Although we cannot rule out the possibility that a specific subset of RNAs accumulates within the nucleus in this bulk poly(A) RNA export assay, this result suggests that the Δmud2 nab2-C437S double-mutant cells do not have a global defect in poly(A) RNA export.
Loss of Rrp6 rescues the growth, pre-mRNA accumulation, and poly(A) tail length defect in Δmud2 nab2-C437S mutants.
One prediction from these data is that Nab2 could play a role in ensuring that unspliced RNAs are targeted for decay. In this scenario, such RNAs would likely be turned over by the RNA exosome, which is a multisubunit complex with 3′-5′ exoribonuclease activity that relies on cofactors to assist in RNA processing and degradation (29). One key catalytic exosome subunit is the nuclear specific component, Rrp6 (93, 94), and an important exosome cofactor is the TRAMP complex, which consists of the RNA binding protein, Air1 or Air2, the poly(A) polymerase, Trf4 or Trf5, and the RNA helicase, Mtr4 (38, 95, 96).
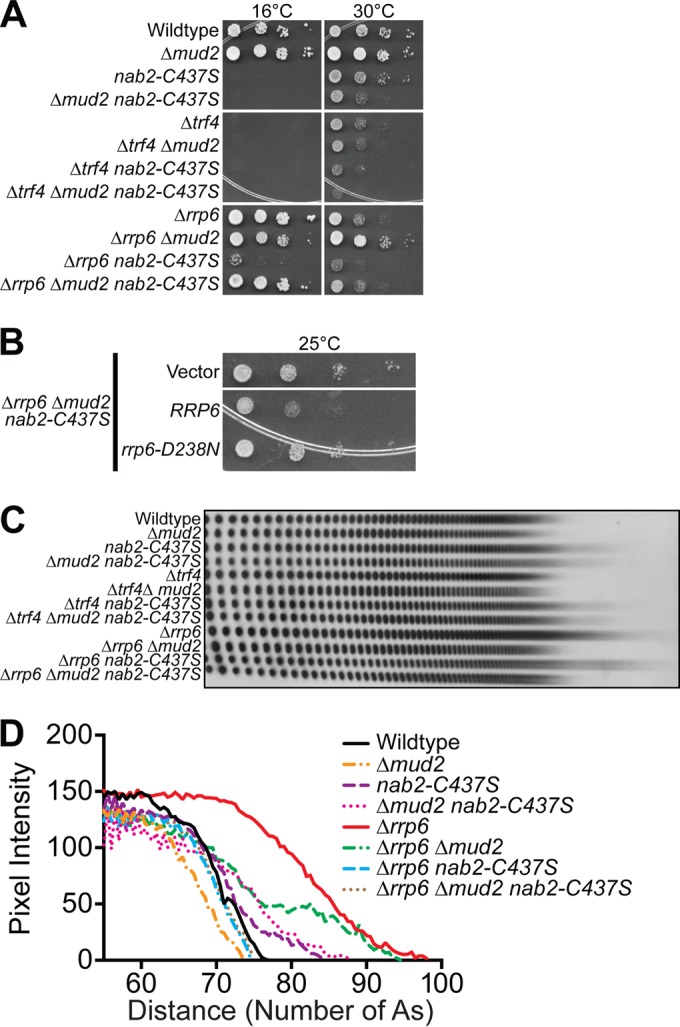
To determine whether Δmud2, nab2-C437S, and Δmud2 nab2-C437S mutants show genetic interactions with the RNA exosome and/or RNA exosome cofactors, we combined Δmud2, nab2-C437S, and Δmud2 nab2-C437S mutants with Δrrp6 or Δtrf4 mutants. The Δtrf4 cells are cold sensitive and grow slowly at 30°C (97, 98), whereas Δrrp6 cells are temperature sensitive and grow slowly at 30°C (93). The Δtrf4 Δmud2 cells do not show any additional changes in growth, and Δtrf4 nab2-C437S cells exhibit only a slight growth defect compared to Δtrf4 cells alone at 30°C (Fig. 9A). However, deletion of RRP6 in combination with nab2-C437S confers some modest growth at 16°C, whereas deletion of RRP6 in Δmud2 nab2-C437S cells confers a significant rescue of the cold-sensitive growth (Fig. 9A). In fact, growth is nearly restored to wild-type levels in the Δrrp6 Δmud2 nab2-C437S triple mutant. These data suggest that the absence of RRP6, presumably a decrease in the activity of the nuclear exosome, can rescue the growth defect of Δmud2 nab2-C437S cells. To test whether the catalytic activity of Rrp6 is required for the improved growth in the Δrrp6 Δmud2 nab2-C437S triple mutant, we transformed cells with a plasmid encoding either wild-type Rrp6 or the Rrp6 catalytic mutant (rrp6-D238N) (99). As shown in Fig. 9B, growth of the rescued Δrrp6 Δmud2 nab2-C437S triple mutant is reduced upon expression of wild-type Rrp6 but not the rrp6-D238N mutant. These results indicate that the loss of Rrp6 catalytic activity rescues the poor growth of nab2-C437S and Δmud2 nab2-C437S mutant cells.
FIG 9.
Genetic interactions between the nuclear exosome subunit, RRP6, and NAB2. (A) Wild-type cells and integrated mutant Δmud2 (ACY2270), nab2-C437S (ACY2202), and Δmud2 nab2-C437S (ACY2273) cells were combined with deletion of the TRAMP poly(A) polymerase, TRF4, or nuclear exosome component, RRP6, gene. The cells were grown, serially diluted, and spotted onto YEPD plates at 25, 30, and 37°C. (B) Δrrp6 Δmud2 nab2-C437S (ACY2313) cells were transformed with vector plasmid and are shown as a control. Δrrp6 Δmud2 nab2-C437S cells were transformed with Rrp6 plasmids expressing wild-type RRP6 or rrp6-D238N catalytic mutant. The cells were grown, serially diluted, and spotted onto plates lacking uracil to assess growth at 16°C. (C) Total yeast RNA was isolated from wild-type, Δmud2, nab2-C437S, and Δmud2 nab2-C437S cells combined with Δtrf4 or Δrrp6 mutant and analyzed for bulk poly(A) tail length as described in Materials and Methods. (D) Poly(A) tails were quantified using ImageJ by calculating pixel intensity along the length of the poly(A) tail and plotted relative to the number of A's, as described in Materials and Methods.
To determine whether the poly(A) tail length is impacted upon the loss of Rrp6, we examined bulk poly(A) tail length. As shown previously (Fig. 8E), extended heterogeneous tails are observed in Δmud2 nab2-C437S cells compared to Δmud2 or nab2-C437S cells alone. Consistent with the growth assay, no additional changes in poly(A) tail length were detected in nab2-C437S or Δmud2 mutants in combination with Δtrf4 (Fig. 9C and D). In contrast, poly(A) tails in Δrrp6 cells are extremely long but are shortened when combined with nab2-C437S or Δmud2 mutants. Interestingly, Δrrp6 Δmud2 nab2-C437S poly(A) tails are as long as poly(A) tails in wild-type cells, a finding consistent with the rescue of cell growth observed in this triple mutant.
To determine whether Rrp6 plays a role in regulating the pre-mRNA that accumulates in Δmud2 nab2-C437S cells, we purified total RNA from wild-type, Δrrp6, Δmud2 nab2-C437S, and Δrrp6 Δmud2 nab2-C437S cells and examined both pre-mRNA and total mRNA levels for ACT1, RPL21B, and RPL36B (Fig. 10). The results indicate that the loss of Rrp6 decreases the accumulation of pre-mRNA in Δmud2 nab2-C437S cells. We detected a corresponding increase in the amount of mRNA when Rrp6 was absent, consistent with a model where cells lacking Rrp6 have more time to allow pre-mRNA processing to occur before mis- or unprocessed transcripts are targeted for decay (see model in Fig. 12).
FIG 10.
Deletion of RRP6 reduces the accumulation of pre-mRNA in Δmud2 nab2-C437S cells. To assess how deletion of RRP6 impacts both pre-mRNA and mRNA levels, total RNA was isolated from wild-type and Δmud2 nab2-C437S cells combined with Δrrp6 mutant. Quantitative real-time PCR analysis was performed to amplify ACT1 (A), RPL21B (B), and RPL36A (C) intron-containing transcripts (pre-mRNA) with primers that span the intron-exon boundary or total mRNA (mRNA) using primers within the exon. The results were normalized to the PDA1 transcript, and values were plotted relative to the wild type, which was set to 1.0.
FIG 12.

Kinetic proofreading model for Nab2 and Mud2 in splicing and mRNA surveillance. (A) In wild-type cells, Mud2 and Nab2 work together to promote efficient splicing, polyadenylation, and export of mRNA (denoted by thick lines). Nab2 interacts with Mlp proteins at the inner face of the nuclear pore to facilitate the efficient export of processed transcripts. (B) In mud2Δ nab2-C437S mutant cells which lack functional MUD2 and NAB2, transcript processing is impaired (thin arrows), triggering degradation by the nuclear exosome subunit, Rrp6 (thick arrow). Although some transcripts may escape nuclear surveillance and exit the nucleus, they may be improperly processed, resulting in extended poly(A) tails and/or missplicing. (C) In Δrrp6 Δmud2 nab2-C437S mutant cells lacking functional MUD2, NAB2, and RRP6, RNA processing is still inefficient, but more time is available in the nucleus to produce mature transcripts (thick arrows) in the absence of Rrp6-mediated degradation (thin arrow). As a result, the transcript is eventually processed properly and exported to the cytoplasm.
The mammalian orthologue of Nab2, ZC3H14, interacts with the spliceosome.
Our data suggest functional interactions between Nab2 and the spliceosome. Previously physical interactions between Nab2 and spliceosomal components have been reported (54). To determine whether the Nab2 orthologue in mammals, ZC3H14, also interacts with components of the spliceosome, we immunoprecipitated endogenous ZC3H14 from mouse brain and used mass spectrometry to identify interacting proteins, as described in Materials and Methods. The full set of interesting proteins identified is provided in Table S6 in the supplemental material. Figure 11A shows a GO analysis where interacting proteins are grouped based on biological process, cellular component, and molecular function and plotted based on the Z-score. Note that the top cellular component identified is spliceosomal complex, while mRNA processing and RNA splicing are the top biological processes identified. Figure 11B provides an eye graph indicating the specific proteins identified in each of the GO categories (see Table S7 in the supplemental material). The colors correspond to the GO categories, while the individual proteins identified are shown in blue. To validate interactions between ZC3H14 and the spliceosome, we immunoprecipitated ZC3H14 from neuronal N2A cells and probed for coimmunoprecipitation of U2AF2/U2AF65, the presumptive orthologue of yeast Mud2 (82). As shown in Fig. 11C, the U2AF2 spliceosomal protein is detected in the bound fraction with ZC3H14 but not with the IgG control. Taken together, these data provide a physical link between ZC3H14 and the spliceosome, suggesting a conserved role for this class of proteins.
FIG 11.

ZC3H14 interacts with spliceosome components. As described in Materials and Methods, endogenous ZC3H14 was precipitated from mouse brain lysate using a polyclonal ZC3H14 antibody (48), and bound proteins were identified by on bead mass spectrometry (see Table S7 in the supplemental material). Three independent biological replicates were performed. (A) The gene ontology (GO) terms for the proteins that are mostly highly enriched with ZC3H14 compared to an IgG control are illustrated. The inclusion criteria for this analysis were a Z-score of ≥5.5, a P value of <0.00001, and ≥5 genes per GO term. (B) The genes that cluster into the listed GO terms in panel A are depicted. The corresponding GO terms are shown by a connecting line in the color corresponding to the GO term. The U2AF2 protein is indicated by an asterisk. (C) We validated the interaction between ZC3H14 and U2AF2 by coimmunoprecipitation from mouse brain lysate. The input and bound samples for anti-ZC3H14 or control IgG immunoprecipitation are shown. Samples were blotted to detect ZC3H14, which is alternatively spliced to produce protein isoforms of ∼100 and ∼75 kDa (48), and U2AF2. The results shown are typical of three independent experiments.
DISCUSSION
This study provides evidence that the polyadenosine RNA binding protein, Nab2, cooperates with splicing factors to ensure proper nuclear mRNA processing. We identify a family of splicing factors, including the commitment complex protein, Mud2, which shows interactions with Nab2. Consistent with a functional overlap, Δmud2 nab2-C437S double-mutant cells show severe growth defects, significant pre-mRNA accumulation, and extended poly(A) tails. Interestingly, the accumulation of aberrant pre-mRNA in the Δmud2 nab2-C437S double-mutant cells is eliminated upon deletion of the nuclear exosome subunit gene, RRP6. These findings provide evidence that Nab2 associates with pre-mRNA to promote proper RNA processing and prevent accumulation of aberrant pre-mRNA via interaction with the nuclear exosome. Consistent with an evolutionarily conserved role for the Nab2 protein in coordinating mRNA processing events through interactions with the splicing factors, the Nab2 orthologue, ZC3H14, is associated with a number of splicing proteins.
Although nab2 mutant cells show an accumulation of pre-mRNA, our results do not support a direct role for Nab2 in splicing. The accumulation of unspliced transcripts that we observed in vivo is modest, and we did not uncover a requirement for Nab2 function in in vitro splicing assays. Although Nab2 is not a component of purified spliceosomal complexes (100), Nab2 does physically interact with a number of splicing factors in an RNA-dependent manner (54), and here we show that both Nab2 and ZC3H14 physically interact with splicing factors. Our data therefore support a model where Nab2 performs an RNA quality control function that monitors splicing to ensure that only correctly and efficiently spliced mRNAs undergo further processing and export. Nab2 may monitor and/or modulate the efficiency of pre-mRNA splicing via interaction with branch point-associated splicing factors, Mud2 and Msl5. In fact, our genetic data suggest that the interaction between Mud2 and Msl5 is critical when Nab2 function is impaired. In addition, Nab2 may modulate splicing efficiency through interactions with intronic noncoding RNAs, such as snoRNAs, or proteins associated with subsequent mRNA processing steps, such as polyadenylation and/or export factors.
The core exosome and Rrp6 play a key role in the turnover of pre-mRNA in the nucleus (34, 101, 102). More recently, depletion of Nab2 has been shown to increase the steady-state levels of pre-mRNAs that are also increased by deletion of RRP6, suggesting that Nab2 is required for efficient Rrp6-dependent turnover of pre-mRNA (43). Nab2 and Rrp6 may thus interface with the same pre-mRNA transcripts to ensure that only properly processed mRNA is produced and exported to the cytoplasm. As shown in Fig. 10 and illustrated in the model for mRNA production in Fig. 12, in wild-type cells, mRNA processing is efficient, and only a fraction of pre-mRNA is targeted by Rrp6 for decay (Fig. 12A). When both Nab2 and Mud2 function is impaired, mRNA processing is less efficient and more unprocessed mRNA accumulates and is targeted by Rrp6 for decay (Fig. 12B). When mRNA processing is impaired, the removal of Rrp6 provides more time for mRNA processing to be completed and thus, although the processing remains inefficient, enough mRNA can be produced and exported to fulfill the needs of the cell (Fig. 12C). This model is consistent with the concept of “kinetic proofreading,” where there is a finite amount of time for processing to occur. mRNAs that are processed in a timely manner are exported to the cytoplasm and have a productive lifetime, whereas RNAs that are not processed sufficiently rapidly are targeted for decay.
One implication of this mRNA production model is that there is extensive competition between mRNA polyadenylation and decay. In support, Nab2 binds to a greater extent to synthetic CYC1 poly(A) transcripts made in whole-cell extracts that lack Rrp6 than to poly(A) transcripts made in wild-type cell extracts (43). Nab2 also precipitates more efficiently with nuclear mRNPs from Δrrp6 cells than wild-type cells (43). These data suggest Nab2 and Rrp6 compete for the same transcript poly(A) tails (43). As shown here, the deletion of RRP6 suppresses the slow growth of nab2-C437S cells at a cold temperature, and previous results have shown that the deletion of RRP6 which rescues the lethality of Δnab2 cells that would otherwise die as NAB2 is essential (103). Deletion or catalytic inactivation of RRP6 also rescues the slow growth and partially restores the poly(A)+ mRNA of a poly(A) polymerase mutant (pap1-1) (99). It therefore seems likely that removal of Rrp6 provides more time and less competition for the nab2-C437S RNA binding mutant to access mRNA transcripts to facilitate proper mRNA processing and mRNP assembly.
This mRNA production model also implies that there is significant competition between mRNA splicing and decay. A previous transcriptome-wide study of rrp6 and dis3 catalytic yeast mutants revealed that most intron-containing pre-mRNAs are targeted for decay by Dis3 rather than the Rrp6 subunit of the exosome (102). This study found many pre-mRNA transcripts targeted by Dis3 are functional and can reenter the splicing pathway to become mature mRNA when Dis3-dependent degradation is impaired (102). However, this study also showed that several intron-containing transcripts that harbored snoRNAs (e.g., RPL7B) are targeted for decay to a similar extent by Rrp6 and Dis3. Moreover, an in vivo RNA cross-linking and analysis of cDNAs (CRAC) study with Rrp6 and Dis3 also showed that Rrp6 binds across introns and that Dis3 is enriched at the 5′ ends of introns (101). These data support the notion that a subset of intron-containing transcripts are targets of Rrp6. Since Nab2 does not directly affect splicing, the accumulation of pre-mRNA in nab2 zinc finger mutants is therefore consistent with impaired pre-mRNA processing and/or decay. Nab2 thus most likely recruits Rrp6 to specific intron-containing transcripts in a novel pre-mRNA processing/decay pathway. Consistent with this idea, Nab2 physically interacts with Rrp6 (43, 54). The reduction in pre-mRNA and increase in mature mRNA upon deletion of RRP6 in Δmud2 nab2-C437S cells (Fig. 10) could therefore reflect pre-mRNA reentry into splicing upon loss of Rrp6.
Although the polyadenosine RNA binding protein, Nab2, should bind all mRNA transcripts containing poly(A) tails (104), results presented here show that Nab2 impairment only causes the accumulation of a subset of intron-containing pre-mRNAs. This raises the following question: what are the features of Nab2 and the pre-mRNAs affected by Nab2 that provide such specificity? With regard to Nab2, the Nab2 zinc finger domain that binds polyadenosine RNA could recognize other RNA sequences in transcripts in addition to or separately from the poly(A) tail. In fact, a recent structural analysis of C. thermophilum Nab2 zinc fingers 3 to 5, which are analogous to S. cerevisiae Nab2 zinc fingers 5 to 7 that bind polyadenosine RNA with high affinity (71), reveals that the zinc fingers recognize specific adenosines within a sequence but that those adenosines may be interspersed with other nucleotides (105). A previous genome-wide analysis of the transcripts bound by Nab2 also identified the Nab2 binding motif as (A)11G, suggesting that Nab2 could recognize other nucleotides in addition to adenosine (106). In addition to Nab2 zinc fingers 5 to 7, Nab2 zinc fingers 1 to 4, which bind polyadenosine RNA with low affinity (107), could also help to recognize novel RNA sequences. The Nab2 zinc finger domain or other domains, such as the N-terminal domain or RGG domain, could further interact with other RNA binding proteins to recognize specific intron-containing transcripts. Notably, here, the Nab2 zinc finger domain is also required for the interaction with RNA binding proteins, Mud2 and Msl5, which recognize the branch point sequence in introns. In addition, the Nab2 zinc finger domain has previously been shown to interact with the RNA binding protein, Pub1 (87).
Among the 249 intron-containing genes on the splicing array, the greatest accumulation of pre-mRNA in nab2 mutants is observed with ribosomal protein genes (RPGs). Notably, intron-containing RPGs represent nearly 40% (102) of all intron-containing genes in S. cerevisiae, are highly regulated by environmental stress, and often contain larger introns (median RPG intron length, ∼400 nt) compared to non-RPGs containing introns (median RPG intron length, ∼130 nt) (60, 74). One feature of pre-mRNA targets of Nab2 could therefore be that they contain large introns that may require more time for proper splicing than smaller introns. Interestingly, the DBP2 gene, which contains the largest intron in S. cerevisiae (1,002 nt), shows increased pre-mRNA in nab2 mutants. Three of the eight genes that contain two introns (RPGs RPL7A, RPL7B, and RPS22B) are prominently affected in nab2 mutants. RPL7A shows pre-mRNA accumulation, whereas RPL7B and RPL22B show a mature and total mRNA decrease. A second feature of some Nab2 pre-mRNA targets could thus be that they contain two introns. Most of the eight genes containing introns that harbor embedded small nucleolar RNAs (snoRNAs) are also affected in nab2 mutants. Affected RPGs with snoRNAs in their second intron include RPL7A (snR39; box C/D), RPL7B (snR59; box C/D), and RPS22B (snR44; box H/ACA). Other affected genes with intronic snoRNAs include TEF4 (snR38; box C/D), ASC1 (snR24; box C/D), IMD4 (snR54; box C/D), and NOG2 (snR191; box H/ACA). A third feature of certain Nab2 pre-mRNA targets could therefore be that their introns contain snoRNAs. Additional features of Nab2 pre-mRNA targets could include adenosine-rich sequences or stem-loop structures within the introns. In support of this idea, RPL22B, which shows elevated pre-mRNA in nab2 mutants, has recently been shown to contain an intronic stem-loop that regulates its expression (108). Novel intronic stem-loop structures in S. cerevisiae have also been identified and validated (109). Future studies will be required to determine why a subset of transcripts seem to depend on Nab2 for proper maturation.
An in vivo RNA cross-linking study showed that Nab2 binds within the coding and intronic regions of mRNA transcripts in addition to the 3′ ends (110), supporting the notion that Nab2 can bind introns to influence splicing. How could Nab2 indirectly affect splicing? One possibility, given the presence of snoRNAs in introns, is that Nab2 plays a role in the processing and biogenesis of snoRNAs. Impairments in snoRNA processing in nab2 mutants could lead to reduced splicing efficiency. Since snoRNAs are often located in introns in mammals, understanding whether Nab2 functions directly in snoRNA processing or the splicing events that liberate introns containing snoRNAs could provide insights into the coupling of splicing and snoRNA processing. Recent work also uncovers a potential function for Nab2 in the surveillance of tRNAs (111), suggesting that Nab2 could play a general role in the regulation of noncoding RNAs.
Previous large-scale studies have revealed physical interactions between Nab2 and numerous RNA processing factors, including components of mRNA splicing (54). Here, we provide evidence for a functional link between Nab2 and splicing. Although Nab2 has been studied fairly extensively, little is known about the molecular functions of ZC3H14 (48), which is lost in a form of inherited intellectual disability (50). Posttranscriptional regulation is particularly critical in neurons, which rely on sophisticated spatial and temporal control of gene expression. We present evidence that physically links ZC3H14 to the spliceosome, suggesting that interplay with the splicing machinery could be an evolutionarily conserved function of Nab2/ZC3H14. Previous work has shown that ZC3H14 is localized to nuclear speckles (48, 52, 53); however, the mass spectrometry data reveal that the spliceosomal complex is the top cellular component identified for ZC3H14 using GO analysis. Given the increased complexity of splicing in higher eukaryotes, ZC3H14 could play a role in modulating splicing or even alternative splicing that is critical for brain function.
Like other RNA binding proteins, this work reveals the complex roles of Nab2 in coupling mRNA splicing, 3′-end processing, and decay. Given the recent finding that Nab2 is required for the protection and stabilization of mRNA from degradation by the exosome (44) but also that Nab2 plays a role in the processing/decay and destabilization of pre-mRNA via the exosome (43), it seems clear that Nab2 possesses at least two different functions in S. cerevisiae. Further work will be required to gain insight into the mechanisms by which Nab2 regulates splicing, polyadenylation, and the nuclear exosome. This study lays the groundwork for studying the function of ZC3H14 in the regulation of mRNA splicing and understanding the complex interplay between polyadenosine RNA binding proteins and mRNA decay factors in neuronal cells.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to members of the Corbett laboratory for helpful discussions and comments. We thank Stewart Shuman (Sloan-Kettering Institute) and Beate Schwer (Weill Cornell Medical College) for generously providing the Mud2 and Mud2-RRM3 mutant plasmids. We thank Christopher Scharer (Emory University) for help with generating the splicing array heat maps.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00402-16.
REFERENCES
- 1.Moore MJ. 2005. From birth to death: the complex lives of eukaryotic mRNAs. Science 309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 2.Singh G, Pratt G, Yeo GW, Moore MJ. 2015. The clothes make the mRNA: past and present trends in mRNP fashion. Annu Rev Biochem 84:325–354. doi: 10.1146/annurev-biochem-080111-092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dreyfuss G, Kim VN, Kataoka N. 2002. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 4.Niwa M, Berget SM. 1991. Mutation of the AAUAAA polyadenylation signal depresses in vitro splicing of proximal but not distal introns. Genes Dev 5:2086–2095. doi: 10.1101/gad.5.11.2086. [DOI] [PubMed] [Google Scholar]
- 5.Nesic D, Cheng J, Maquat LE. 1993. Sequences within the last intron function in RNA 3′-end formation in cultured cells. Mol Cell Biol 13:3359–3369. doi: 10.1128/MCB.13.6.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooke C, Hans H, Alwine JC. 1999. Utilization of splicing elements and polyadenylation signal elements in the coupling of polyadenylation and last-intron removal. Mol Cell Biol 19:4971–4979. doi: 10.1128/MCB.19.7.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lutz CS, Alwine JC. 1994. Direct interaction of the U1 snRNP-A protein with the upstream efficiency element of the SV40 late polyadenylation signal. Genes Dev 8:576–586. doi: 10.1101/gad.8.5.576. [DOI] [PubMed] [Google Scholar]
- 8.Gunderson SI, Beyer K, Martin G, Keller W, Boelens WC, Mattaj LW. 1994. The human U1A snRNP protein regulates polyadenylation via a direct interaction with poly(A) polymerase. Cell 76:531–541. doi: 10.1016/0092-8674(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 9.Gunderson SI, Polycarpou-Schwarz M, Mattaj IW. 1998. U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol Cell 1:255–264. doi: 10.1016/S1097-2765(00)80026-X. [DOI] [PubMed] [Google Scholar]
- 10.Kyburz A, Friedlein A, Langen H, Keller W. 2006. Direct interactions between subunits of CPSF and the U2 snRNP contribute to the coupling of pre-mRNA 3′ end processing and splicing. Mol Cell 23:195–205. doi: 10.1016/j.molcel.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 11.Vagner S, Vagner C, Mattaj IW. 2000. The carboxyl terminus of vertebrate poly(A) polymerase interacts with U2AF 65 to couple 3′-end processing and splicing. Genes Dev 14:403–413. [PMC free article] [PubMed] [Google Scholar]
- 12.Millevoi S, Loulergue C, Dettwiler S, Karaa SZ, Keller W, Antoniou M, Vagner S. 2006. An interaction between U2AF 65 and CF I(m) links the splicing and 3′ end processing machineries. EMBO J 25:4854–4864. doi: 10.1038/sj.emboj.7601331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lykke-Andersen S, Jensen TH. 2015. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- 14.Siwaszek A, Ukleja M, Dziembowski A. 2014. Proteins involved in the degradation of cytoplasmic mRNA in the major eukaryotic model systems. RNA Biol 11:1122–1136. doi: 10.4161/rna.34406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Hoof A, Wagner EJ. 2011. A brief survey of mRNA surveillance. Trends Biochem Sci 36:585–592. doi: 10.1016/j.tibs.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legrain P, Seraphin B, Rosbash M. 1988. Early commitment of yeast pre-mRNA to the spliceosome pathway. Mol Cell Biol 8:3755–3760. doi: 10.1128/MCB.8.9.3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rutz B, Seraphin B. 1999. Transient interaction of BBP/ScSF1 and Mud2 with the splicing machinery affects the kinetics of spliceosome assembly. RNA 5:819–831. doi: 10.1017/S1355838299982286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abovich N, Rosbash M. 1997. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89:403–412. doi: 10.1016/S0092-8674(00)80221-4. [DOI] [PubMed] [Google Scholar]
- 19.Kistler AL, Guthrie C. 2001. Deletion of MUD2, the yeast homolog of U2AF65, can bypass the requirement for sub2, an essential spliceosomal ATPase. Genes Dev 15:42–49. doi: 10.1101/gad.851301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rymond BC. 2010. The branchpoint binding protein: in and out of the spliceosome cycle. Adv Exp Med Biol 693:123–141. doi: 10.1007/978-1-4419-7005-3_9. [DOI] [PubMed] [Google Scholar]
- 21.Palancade B, Zuccolo M, Loeillet S, Nicolas A, Doye V. 2005. Pml39, a novel protein of the nuclear periphery required for nuclear retention of improper messenger ribonucleoparticles. Mol Biol Cell 16:5258–5268. doi: 10.1091/mbc.E05-06-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galy V, Gadal O, Fromont-Racine M, Romano A, Jacquier A, Nehrbass U. 2004. Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell 116:63–73. doi: 10.1016/S0092-8674(03)01026-2. [DOI] [PubMed] [Google Scholar]
- 23.Saroufim MA, Bensidoun P, Raymond P, Rahman S, Krause MR, Oeffinger M, Zenklusen D. 2015. The nuclear basket mediates perinuclear mRNA scanning in budding yeast. J Cell Biol 211:1131–1140. doi: 10.1083/jcb.201503070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fasken MB, Corbett AH. 2005. Process or perish: quality control in mRNA biogenesis. Nat Struct Mol Biol 12:482–488. doi: 10.1038/nsmb945. [DOI] [PubMed] [Google Scholar]
- 25.Hilleren PJ, Parker R. 2003. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol Cell 12:1453–1465. doi: 10.1016/S1097-2765(03)00488-X. [DOI] [PubMed] [Google Scholar]
- 26.Semlow DR, Staley JP. 2012. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends Biochem Sci 37:263–273. doi: 10.1016/j.tibs.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chlebowski A, Lubas M, Jensen TH, Dziembowski A. 2013. RNA decay machines: the exosome. Biochim Biophys Acta 1829:552–560. doi: 10.1016/j.bbagrm.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. 1997. The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′→5′ exoribonucleases. Cell 91:457–466. doi: 10.1016/S0092-8674(00)80432-8. [DOI] [PubMed] [Google Scholar]
- 29.Garneau NL, Wilusz J, Wilusz CJ. 2007. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 30.Abovich N, Liao XC, Rosbash M. 1994. The yeast MUD2 protein: an interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev 8:843–854. doi: 10.1101/gad.8.7.843. [DOI] [PubMed] [Google Scholar]
- 31.Allmang C, Kufel J, Chanfreau G, Mitchell P, Petfalski E, Tollervey D. 1999. Functions of the exosome in rRNA, snoRNA, and snRNA synthesis. EMBO J 18:5399–5410. doi: 10.1093/emboj/18.19.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allmang C, Mitchell P, Petfalski E, Tollervey D. 2000. Degradation of ribosomal RNA precursors by the exosome. Nucleic Acids Res 28:1684–1691. doi: 10.1093/nar/28.8.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milligan L, Torchet C, Allmang C, Shipman T, Tollervey D. 2005. A nuclear surveillance pathway for mRNAs with defective polyadenylation. Mol Cell Biol 25:9996–10004. doi: 10.1128/MCB.25.22.9996-10004.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bousquet-Antonelli C, Presutti C, Tollervey D. 2000. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell 102:765–775. doi: 10.1016/S0092-8674(00)00065-9. [DOI] [PubMed] [Google Scholar]
- 35.Jensen TH, Dower K, Libri D, Rosbash M. 2003. Early formation of mRNP: license for export or quality control? Mol Cell 11:1129–1138. doi: 10.1016/S1097-2765(03)00191-6. [DOI] [PubMed] [Google Scholar]
- 36.Vinciguerra P, Stutz F. 2004. mRNA export: an assembly line from genes to nuclear pores. Curr Opin Cell Biol 16:285–292. doi: 10.1016/j.ceb.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 37.Lykke-Andersen S, Tomecki R, Jensen TH, Dziembowski A. 2011. The eukaryotic RNA exosome: same scaffold but variable catalytic subunits. RNA Biol 8:61–66. doi: 10.4161/rna.8.1.14237. [DOI] [PubMed] [Google Scholar]
- 38.LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D. 2005. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121:713–724. doi: 10.1016/j.cell.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 39.Hector RE, Nykamp KR, Dheur S, Anderson JT, Non PJ, Urbinati CR, Wilson SM, Minvielle-Sebastia L, Swanson MS. 2002. Dual requirement for yeast hnRNP Nab2p in mRNA poly(A) tail length control and nuclear export. EMBO J 21:1800–1810. doi: 10.1093/emboj/21.7.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green DM, Marfatia KA, Crafton EB, Zhang X, Cheng X, Corbett AH. 2002. Nab2p is required for poly(A) RNA export in Saccharomyces cerevisiae and is regulated by arginine methylation via Hmt1p. J Biol Chem 277:7752–7760. doi: 10.1074/jbc.M110053200. [DOI] [PubMed] [Google Scholar]
- 41.Kelly SM, Pabit SA, Kitchen CM, Guo P, Marfatia KA, Murphy TJ, Corbett AH, Berland KM. 2007. Recognition of polyadenosine RNA by zinc finger proteins. Proc Natl Acad Sci U S A 104:12306–12311. doi: 10.1073/pnas.0701244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson JT, Wilson SM, Datar KV, Swanson MS. 1993. NAB2: a yeast nuclear polyadenylated RNA-binding protein essential for cell viability. Mol Cell Biol 13:2730–2741. doi: 10.1128/MCB.13.5.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmid M, Poulsen MB, Olszewski P, Pelechano V, Saguez C, Gupta I, Steinmetz LM, Moore C, Jensen TH. 2012. Rrp6p controls mRNA poly(A) tail length and its decoration with poly(A) binding proteins. Mol Cell 47:267–280. doi: 10.1016/j.molcel.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmid M, Olszewski P, Pelechano V, Gupta I, Steinmetz LM, Jensen TH. 2015. The nuclear polyA-binding protein Nab2p is essential for mRNA production. Cell Rep 12:128–139. doi: 10.1016/j.celrep.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 45.Perreault A, Lemieux C, Bachand F. 2007. Regulation of the nuclear poly(A)-binding protein by arginine methylation in fission yeast. J Biol Chem 282:7552–7562. [DOI] [PubMed] [Google Scholar]
- 46.Lemieux C, Marguerat S, Lafontaine J, Barbezier N, Bahler J, Bachand F. 2011. A pre-mRNA degradation pathway that selectively targets intron-containing genes requires the nuclear poly(A)-binding protein. Mol Cell 44:108–119. doi: 10.1016/j.molcel.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 47.St-Sauveur VG, Soucek S, Corbett AH, Bachand F. 2013. Poly(A) tail-mediated gene regulation by opposing roles of Nab2 and Pab2 nuclear poly(A)-binding proteins in pre-mRNA decay. Mol Cell Biol 33:4718–4731. doi: 10.1128/MCB.00887-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leung S, Apponi L, Cornejo O, Kitchen C, Valentini S, Pavlath G, Dunham C, Corbett A. 2009. Splice variants of the human ZC3H14 gene generate multiple isoforms of a zinc finger polyadenosine RNA binding protein. Gene 439:71–78. doi: 10.1016/j.gene.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kelly SM, Leung SW, Pak C, Banerjee A, Moberg KH, Corbett AH. 2014. A conserved role for the zinc finger polyadenosine RNA binding protein, ZC3H14, in control of poly(A) tail length. RNA 20:681–688. doi: 10.1261/rna.043984.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pak C, Garshasbi M, Kahrizi K, Gross C, Apponi LH, Noto JJ, Kelly SM, Leung SW, Tzschach A, Behjati F, Abedini SS, Mohseni M, Jensen LR, Hu H, Huang B, Stahley SN, Liu G, Williams KR, Burdick S, Feng Y, Sanyal S, Bassell GJ, Ropers HH, Najmabadi H, Corbett AH, Moberg KH, Kuss AW. 2011. Mutation of the conserved polyadenosine RNA binding protein, ZC3H14/dNab2, impairs neural function in Drosophila and humans. Proc Natl Acad Sci U S A 108:12390–12395. doi: 10.1073/pnas.1107103108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kelly S, Pak C, Garshasbi M, Kuss A, Corbett AH, Moberg K. 2012. New kid on the ID block: neural functions of the Nab2/ZC3H14 class of Cys(3)His tandem zinc-finger polyadenosine RNA binding proteins. RNA Biol 9:555–562. doi: 10.4161/rna.20187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guthrie CR, Greenup L, Leverenz JB, Kraemer BC. 2011. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum Mol Genet 20:1989–1999. doi: 10.1093/hmg/ddr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyakura S, Hara M. 2015. Molecular characterization of UKp83/68, a widespread nuclear proteins that bind poly(A) and colocalize with a nuclear Speckle's component. J Med Dent Sci 62:43–56. doi: 10.11480/620203. [DOI] [PubMed] [Google Scholar]
- 54.Klass DM, Scheibe M, Butter F, Hogan GJ, Mann M, Brown PO. 2013. Quantitative proteomic analysis reveals concurrent RNA-protein interactions and identifies new RNA-binding proteins in Saccharomyces cerevisiae. Genome Res 23:1028–1038. doi: 10.1101/gr.153031.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ausubel MF, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 2001. Current protocols in molecular biology. John Wiley & Sons, New York, NY. [Google Scholar]
- 56.Wach A, Brachat A, Pohlmann R, Philippsen P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 57.Chang J, Schwer B, Shuman S. 2010. Mutational analyses of trimethylguanosine synthase (Tgs1) and Mud2: proteins implicated in pre-mRNA splicing. RNA 16:1018–1031. doi: 10.1261/rna.2082610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chomczynski P. 1993. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 15:532–537. [PubMed] [Google Scholar]
- 59.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 60.Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. 2007. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol Cell 27:928–937. doi: 10.1016/j.molcel.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Hoon MJ, Imoto S, Nolan J, Miyano S. 2004. Open source clustering software. Bioinformatics 20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 62.Umen JG, Guthrie C. 1995. Prp16p, Slu7p, and Prp8p interact with the 3′ splice site in two distinct stages during the second catalytic step of pre-mRNA splicing. RNA 1:584–597. [PMC free article] [PubMed] [Google Scholar]
- 63.Boeke JD, Trueheart J, Natsoulis G, Fink GR. 1987. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol 154:164–175. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- 64.Chekanova JA, Belostotsky DA. 2003. Evidence that poly(A) binding protein has an evolutionarily conserved function in facilitating mRNA biogenesis and export. RNA 9:1476–1490. doi: 10.1261/rna.5128903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minvielle-Sebastia L, Winsor B, Bonneaud N, Lacroute F. 1991. Mutations in the yeast RNA14 and RNA15 genes result in an abnormal mRNA decay rate: sequence analysis reveals an RNA-binding domain in the RNA15 protein. Mol Cell Biol 11:3075–3087. doi: 10.1128/MCB.11.6.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wong DH, Corbett AH, Kent HM, Stewart M, Silver PA. 1997. Interaction between the small GTPase Ran/Gsp1p and Ntf2p is required for nuclear transport. Mol Cell Biol 17:3755–3767. doi: 10.1128/MCB.17.7.3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dammer EB, Duong DM, Diner I, Gearing M, Feng Y, Lah JJ, Levey AI, Seyfried NT. 2013. Neuron enriched nuclear proteome isolated from human brain. J Proteome Res 12:3193–3206. doi: 10.1021/pr400246t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herskowitz JH, Seyfried NT, Gearing M, Kahn RA, Peng J, Levey AI, Lah JJ. 2011. Rho kinase II phosphorylation of the lipoprotein receptor LR11/SORLA alters amyloid-beta production. J Biol Chem 286:6117–6127. doi: 10.1074/jbc.M110.167239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zambon AC, Gaj S, Ho I, Hanspers K, Vranizan K, Evelo CT, Conklin BR, Pico AR, Salomonis N. 2012. GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics 28:2209–2210. doi: 10.1093/bioinformatics/bts366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marfatia KA, Crafton EB, Green DM, Corbett AH. 2003. Domain analysis of the Saccharomyces cerevisiae heterogeneous nuclear ribonucleoprotein, Nab2p: dissecting the requirements for Nab2p-facilitated poly(A) RNA export. J Biol Chem 278:6731–6740. [DOI] [PubMed] [Google Scholar]
- 71.Kelly SM, Leung SW, Apponi LH, Bramley AM, Tran EJ, Chekanova JA, Wente SR, Corbett AH. 2010. Recognition of polyadenosine RNA by the zinc finger domain of nuclear poly(A) RNA-binding protein 2 (Nab2) is required for correct mRNA 3′-end formation. J Biol Chem 285:26022–26032. doi: 10.1074/jbc.M110.141127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim SH, Lin RJ. 1996. Spliceosome activation by PRP2 ATPase prior to the first transesterification reaction of pre-mRNA splicing. Mol Cell Biol 16:6810–6819. doi: 10.1128/MCB.16.12.6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.King DS, Beggs JD. 1990. Interactions of PRP2 protein with pre-mRNA splicing complexes in Saccharomyces cerevisiae. Nucleic Acids Res 18:6559–6564. doi: 10.1093/nar/18.22.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. 2007. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol 5:e90. doi: 10.1371/journal.pbio.0050090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yan D, Perriman R, Igel H, Howe KJ, Neville M, Ares M Jr. 1998. CUS2, a yeast homolog of human Tat-SF1, rescues function of misfolded U2 through an unusual RNA recognition motif. Mol Cell Biol 18:5000–5009. doi: 10.1128/MCB.18.9.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Q, Zhang L, Lynn B, Rymond BC. 2008. A BBP-Mud2p heterodimer mediates branchpoint recognition and influences splicing substrate abundance in budding yeast. Nucleic Acids Res 36:2787–2798. doi: 10.1093/nar/gkn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rain JC, Legrain P. 1997. In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J 16:1759–1771. doi: 10.1093/emboj/16.7.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu YZ, Query CC. 2007. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell 28:838–849. doi: 10.1016/j.molcel.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perriman R, Ares M Jr. 2010. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol Cell 38:416–427. doi: 10.1016/j.molcel.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moehle EA, Ryan CJ, Krogan NJ, Kress TL, Guthrie C. 2012. The yeast SR-like protein Npl3 links chromatin modification to mRNA processing. PLoS Genet 8:e1003101. doi: 10.1371/journal.pgen.1003101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dziembowski A, Ventura AP, Rutz B, Caspary F, Faux C, Halgand F, Laprevote O, Seraphin B. 2004. Proteomic analysis identifies a new complex required for nuclear pre-mRNA retention and splicing. EMBO J 23:4847–4856. doi: 10.1038/sj.emboj.7600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rain JC, Rafi Z, Rhani Z, Legrain P, Kramer A. 1998. Conservation of functional domains involved in RNA binding and protein-protein interactions in human and Saccharomyces cerevisiae pre-mRNA splicing factor SF1. RNA 4:551–565. doi: 10.1017/S1355838298980335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Valcarcel J, Gaur RK, Singh R, Green MR. 1996. Interaction of U2AF65 RS region with pre-mRNA branch point and promotion of base pairing with U2 snRNA. Science 273:1706–1709. doi: 10.1126/science.273.5282.1706. [DOI] [PubMed] [Google Scholar]
- 84.Sickmier EA, Frato KE, Shen H, Paranawithana SR, Green MR, Kielkopf CL. 2006. Structural basis for polypyrimidine tract recognition by the essential pre-mRNA splicing factor U2AF65. Mol Cell 23:49–59. doi: 10.1016/j.molcel.2006.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Selenko P, Gregorovic G, Sprangers R, Stier G, Rhani Z, Kramer A, Sattler M. 2003. Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol Cell 11:965–976. doi: 10.1016/S1097-2765(03)00115-1. [DOI] [PubMed] [Google Scholar]
- 86.Chang J, Schwer B, Shuman S. 2012. Structure-function analysis and genetic interactions of the yeast branchpoint binding protein Msl5. Nucleic Acids Res 40:4539–4552. doi: 10.1093/nar/gks049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Apponi LH, Kelly SM, Harreman MT, Lehner AN, Corbett AH, Valentini SR. 2007. An interaction between two RNA binding proteins, Nab2 and Pub1, links mRNA processing/export and mRNA stability. Mol Cell Biol 27:6569–6579. doi: 10.1128/MCB.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brockmann C, Soucek S, Kuhlmann SI, Mills-Lujan K, Kelly SM, Yang JC, Iglesias N, Stutz F, Corbett AH, Neuhaus D, Stewart M. 2012. Structural basis for polyadenosine-RNA binding by Nab2 Zn fingers and its function in mRNA nuclear export. Structure 20:1007–1018. doi: 10.1016/j.str.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Green DM, Johnson CP, Hagan H, Corbett AH. 2003. The C-terminal domain of myosin-like protein 1 (Mlp1p) is a docking site for heterogeneous nuclear ribonucleoproteins that are required for mRNA export. Proc Natl Acad Sci U S A 100:1010–1015. doi: 10.1073/pnas.0336594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vinciguerra P, Iglesias N, Camblong J, Zenklusen D, Stutz F. 2005. Perinuclear Mlp proteins downregulate gene expression in response to a defect in mRNA export. EMBO J 24:813–823. doi: 10.1038/sj.emboj.7600527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS, Ideker T, Guthrie C, Krogan NJ. 2008. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell 32:735–746. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M, Deshpande R, Li Z, Lin ZY, Liang W, Marback M, Paw J, San Luis BJ, Shuteriqi E, Tong AH, van Dyk N, Wallace IM, Whitney JA, Weirauch MT, Zhong G, Zhu H, Houry WA, Brudno M, Ragibizadeh S, Papp B, Pal C, Roth FP, Giaever G, Nislow C, Troyanskaya OG, Bussey H, Bader GD, Gingras AC, Morris QD, Kim PM, Kaiser CA, et al. 2010. The genetic landscape of a cell. Science 327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Briggs MW, Burkard KT, Butler JS. 1998. Rrp6p, the yeast homologue of the human PM-Scl 100-kDa autoantigen, is essential for efficient 5.8 S rRNA 3′ end formation. J Biol Chem 273:13255–13263. doi: 10.1074/jbc.273.21.13255. [DOI] [PubMed] [Google Scholar]
- 94.van Hoof A, Lennertz P, Parker R. 2000. Yeast exosome mutants accumulate 3′-extended polyadenylated forms of U4 small nuclear RNA and small nucleolar RNAs. Mol Cell Biol 20:441–452. doi: 10.1128/MCB.20.2.441-452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. 2005. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol 3:e189. doi: 10.1371/journal.pbio.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Regnault B, Devaux F, Namane A, Seraphin B, Libri D, Jacquier A. 2005. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121:725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 97.Sadoff BU, Heath-Pagliuso S, Castano IB, Zhu Y, Kieff FS, Christman MF. 1995. Isolation of mutants of Saccharomyces cerevisiae requiring DNA topoisomerase I. Genetics 141:465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Iwanejko L, Smith KN, Loeillet S, Nicolas A, Fabre F. 1999. Disruption and functional analysis of six ORFs on chromosome XV: YOL117w, YOL115w (TRF4), YOL114c, YOL112w (MSB4), YOL111c, and YOL072w. Yeast 15:1529–1539. doi:. [DOI] [PubMed] [Google Scholar]
- 99.Burkard KT, Butler JS. 2000. A nuclear 3′-5′ exonuclease involved in mRNA degradation interacts with poly(A) polymerase and the hnRNA protein Npl3p. Mol Cell Biol 20:604–616. doi: 10.1128/MCB.20.2.604-616.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Warkocki Z, Odenwalder P, Schmitzova J, Platzmann F, Stark H, Urlaub H, Ficner R, Fabrizio P, Luhrmann R. 2009. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol 16:1237–1243. doi: 10.1038/nsmb.1729. [DOI] [PubMed] [Google Scholar]
- 101.Schneider C, Kudla G, Wlotzka W, Tuck A, Tollervey D. 2012. Transcriptome-wide analysis of exosome targets. Mol Cell 48:422–433. doi: 10.1016/j.molcel.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gudipati RK, Xu Z, Lebreton A, Seraphin B, Steinmetz LM, Jacquier A, Libri D. 2012. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell 48:409–421. doi: 10.1016/j.molcel.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gonzalez-Aguilera C, Tous C, Babiano R, de la Cruz J, Luna R, Aguilera A. 2011. Nab2 functions in the metabolism of RNA driven by polymerases II and III. Mol Biol Cell 22:2729–2740. doi: 10.1091/mbc.E11-01-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Batisse J, Batisse C, Budd A, Bottcher B, Hurt E. 2009. Purification of nuclear poly(A)-binding protein Nab2 reveals association with the yeast transcriptome and a messenger ribonucleoprotein core structure. J Biol Chem 284:34911–34917. doi: 10.1074/jbc.M109.062034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kuhlmann SI, Valkov E, Stewart M. 2014. Structural basis for the molecular recognition of polyadenosine RNA by Nab2 Zn fingers. Nucleic Acids Res 42:672–680. doi: 10.1093/nar/gkt876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim Guisbert K, Duncan K, Li H, Guthrie C. 2005. Functional specificity of shuttling hnRNPs revealed by genome-wide analysis of their RNA binding profiles. RNA 11:383–393. doi: 10.1261/rna.7234205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Martinez-Lumbreras S, Santiveri CM, Mirassou Y, Zorrilla S, Perez-Canadillas JM. 2013. Two singular types of CCCH tandem zinc finger in Nab2p contribute to polyadenosine RNA recognition. Structure 21:1800–1811. doi: 10.1016/j.str.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 108.Gabunilas J, Chanfreau G. 2016. Splicing-Mediated Autoregulation modulates Rpl22p expression in Saccharomyces cerevisiae. PLoS Genet 12:e1005999. doi: 10.1371/journal.pgen.1005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hooks KB, Naseeb S, Parker S, Griffiths-Jones S, Delneri D. 2016. Novel intronic RNA structures contribute to maintenance of phenotype in Saccharomyces cerevisiae. Genetics 203:1469–1481. doi: 10.1534/genetics.115.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tuck AC, Tollervey D. 2013. A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell 154:996–1009. doi: 10.1016/j.cell.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Turowski TW, Lesniewska E, Delan-Forino C, Sayou C, Boguta M, Tollervey D. 2016. Global analysis of transcriptionally engaged yeast RNA polymerase III reveals extended tRNA transcripts. Genome Res 26:933–944. doi: 10.1101/gr.205492.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kress TL, Krogan NJ, Guthrie C. 2008. A single SR-like protein, Npl3, promotes pre-mRNA splicing in budding yeast. Mol Cell 32:727–734. doi: 10.1016/j.molcel.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.