

Abstract

A novel, isoform-selective inhibitor of histone deacetylase 8 (HDAC8) has been discovered by the repurposing of a diverse compound collection. Medicinal chemistry optimization led to the identification of a highly potent (0.8 nM) and selective inhibitor of HDAC8.
Keywords: Histone deacetylase, HDAC, histone deacetylase 8, HDAC8, triazole, epigenetic
Histone/protein deacetylase (HDAC) enzymes constitute a family of proteins involved in epigenetic regulation of gene expression by deacetylation of histones.1 Many HDACs are also responsible for the deacetylation of non-histone cellular proteins, a process that is critical for regulation function and localization. There are 11 known “classical” zinc-dependent HDAC isoforms, which are divided into three classes according to their sequence homology and organization. In recent years, HDAC enzymes have emerged as attractive targets for disease biology2 including cancer,3 neurodegenerative diseases,4 autoimmunity, and transplant rejection.5
It is well established that metal-chelating motifs bind tightly to the Zn(II) ion in the active site of HDAC enzymes and can lead to reversible inhibition of their biological function.6 HDAC inhibitors (HDACi) have great potential as therapeutic agents, with several advancing into clinical trials; and multiple pan-active HDACi are FDA approved for T-cell lymphoma and multiple myeloma.2 There is clear interest in continued development of isoform-specific inhibitors as potential therapeutic agents or as tools to further understand selectivity and to discern the function of HDAC isoforms.
In an effort to discover new small molecules with HDAC subtype selectivity,7 we looked to take advantage of an existing small molecule library available at the Boston University Center for Molecular Discovery (BU-CMD). We designed a study to repurpose complex libraries and advanced synthetic intermediates to focus them toward HDAC inhibition activity by addition of strong Zn (II)-chelating moieties.8,9 Utilizing established methodologies, we carried out direct, mild conversion of esters to hydroxamic acids and methyl hydroxymates (Scheme 1, A). In the presence of catalytic cyanide and excess hydroxylamine, a set of methyl esters (1), were converted to hydroxamic acids 2,10 and a similar transformation was carried out with trimethyl aluminum and methanolamine to afford methyl hydroxamates 3.11
Scheme 1. General Reactions for Preparation of Hydroxamic Acids/Esters (A) and Representative Methyl Esters Utilized in the Derivatization (B).

A structurally diverse set of 134 esters (Scheme 1B) was reacted in parallel on a one milligram scale. Products were purified by mass-directed HPLC affording a total of 120 hydroxamic acids and methyl hydroxamates. Purified compounds were assayed against HDAC isoforms 1–9 and a single compound, triazole hydroxamic acid 4, showed good activity (Figure 1). This compound was subsequently validated as a potent inhibitor of HDAC812−16 (IC50 = 10 nM) with modest inhibition of HDAC617 (3600 nM).
Figure 1.
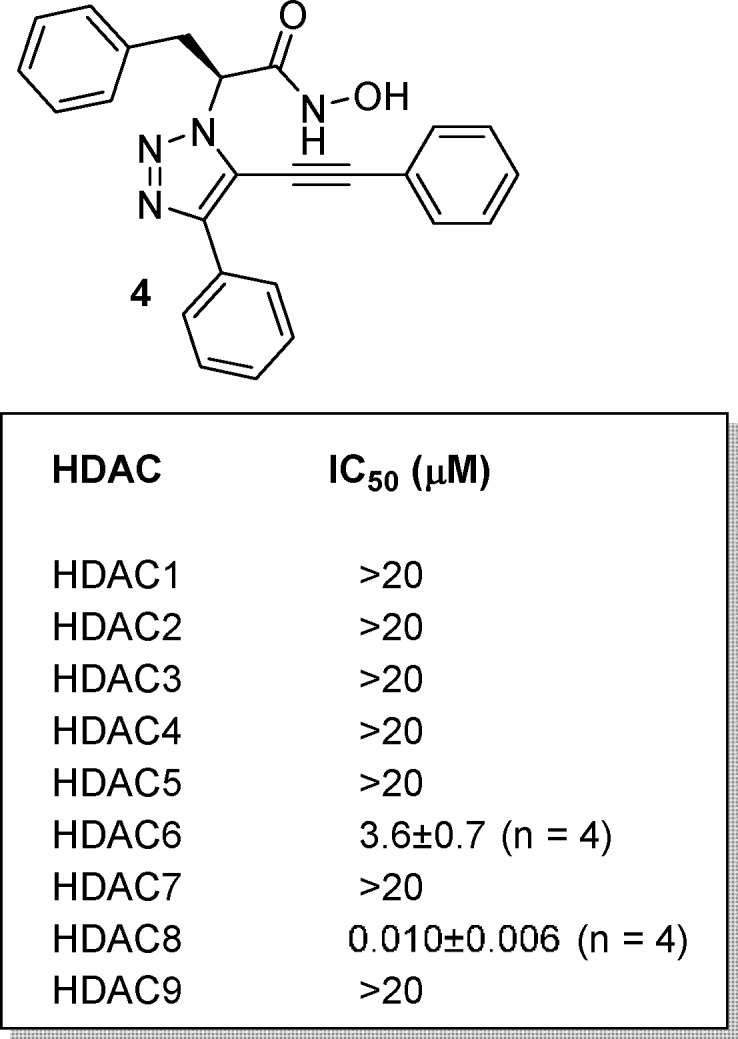
HDAC activity profile for triazole 4.
HDAC8 is classified as a class I HDAC but is unusual in many respects. Unlike other isoforms, little is known about the functions of HDAC8. Classical pan-active HDACi, such as SAHA (vorinostat), bind to HDAC8 with substantially diminished activity (IC50 = 2 μM), reflecting a unique binding site of this isoform.18−21
Cellular functions of HDAC8 have only recently begun to be identified.22 Deardorff and co-workers showed the correlation of HDAC8 mutations to specific phenotypes in patients with Cornelia de Lange syndrome and the apparent role of HDAC8 in deacetylation of SMC3, a critical protein in the cohesin complex.23−25 Recently, Cristea and co-workers reported interaction of HDAC8 with multiple cohesin proteins, SMC3, SMC1a, STAG2, as well as an additional mitosis related protein CROCC.26 These studies have begun to highlight the potential role of HDAC8 in maintaining proper function of cohesin. There is also evidence that HDAC8 modulates acetylation of other proteins such as Oct3/4, Nanog, Cdh1, Rex1, p53, ERRα, and CREB.27,28 A recent study by Lin and co-workers provides evidence that HDAC8 may also have enzymatic function for hydrolysis of larger fatty acids.29
The trisubstituted triazole scaffold was originally obtained via an unusual copper-catalyzed tandem, [3 + 2]-cycloaddition-coupling reaction between azide 5 and phenylacetylene.30 As this approach was limited in yield and scope, a simple, high-yielding synthetic route was developed to facilitate construction of orthogonally substituted analogues (Scheme 2). Copper-mediated Huisgen cycloaddition with phenylacetylene iodide 6 afforded iodotriazole 7.31 Sonogashira coupling with terminal acetylenes afforded alkynyl triazoles 9, which were quantitatively converted to the desired hydroxamic acids 10.
Scheme 2. General Synthetic Scheme for the Synthesis of Triazole Analogues.

In order to determine critical structure–activity relationships (SAR), we first synthesized the corresponding carboxylic acid 11, methyl ester 12, and methyl hydroxamate 13 derivatives, which were all found to be inactive against the entire profile of HDACs, thereby establishing that the hydroxamic acid moiety was necessary for HDAC8 inhibition. The corresponding (R)-enantiomer 14 and disubstituted triazole 15 were synthesized and also found to be inactive (Figure 2).
Figure 2.
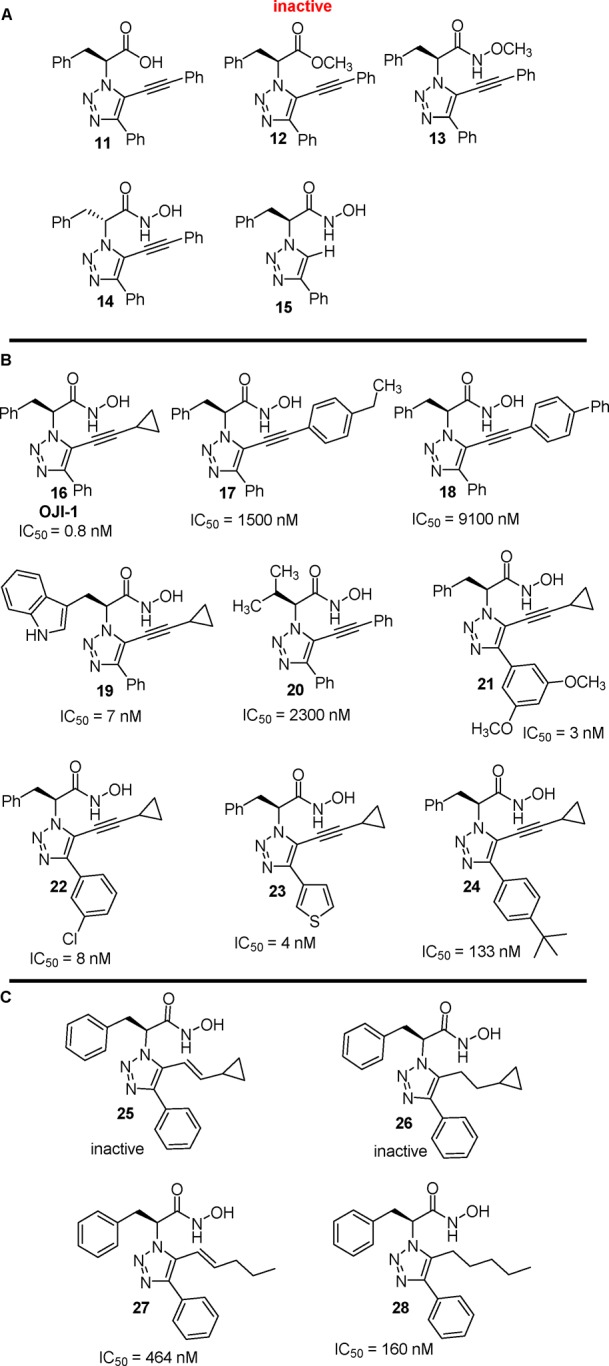
(A) Preliminary inactive analogues. (B) Representative R1–R3 analogues and HDAC8 IC50. (C) Representative reduced analogues.
With this preliminary information in hand, we synthesized an array of analogs to determine more detailed SAR. Position R1 was varied utilizing alternative amino acids, and the R2 and R3 positions were modified using a series of terminal acetylenes. We also synthesized saturated analogues to determine the requirement of the alkyne. In total, 70 analogues were synthesized and assayed for inhibition of HDAC8.
Across the total set of analogues, there was little effect on isoform selectivity, but we observed significant impact on HDAC8 inhibition potency.14 Small hydrophobic groups at the R3 position, such as cyclopropane, were found to be beneficial and afforded greater inhibition, highlighted by the 0.8 nM activity of cyclopropyl congener 16 (OJI-1). Larger substituents were not tolerated as exemplified by the dramatic loss of activity in p-ethylphenyl derivative 17 and biphenyl 18. Introduction of alternative amino acids resulted in a loss of activity compared to phenylalanine. For instance, indole derivative 19 showed a 10-fold loss of potency and the valine derived 20 was essentially inactive. Substitution at the 4-position (R2) of the triazole was the most tolerant. Both electron-donating (21) and electron-withdrawing (22) substituents were tolerated. Furthermore, heteroaromatic groups such as thiophene (23) were well tolerated. However, there was some loss of activity when larger hydrophobic substituents were added such as t-butyl phenyl 24.
We also synthesized a number of analogues that evaluated the oxidation level of the alkyne. Interestingly, reduction of the alkyne resulted in loss of activity for all substrates except for the n-pentyl and pentenyl derivatives 27 and 28, which were found to be significantly less active.
Based on the available SAR, there appears to be five crucial structural features necessary for selective HDAC8 inhibition (Figure 3): (1) S-stereochemistry; (2) an aromatic moiety adjacent to the hydroxamic acid; (3) small, aromatic ring fused directly to the 4-position; (4) a small, hydrophobic acetylenic functionality at the 5-position of the triazole; and (5) an alkyne linker between the triazole and R3.
Figure 3.

SAR for HDAC8 Inhibition.
We also carried out evaluation of the most active analogue (OJI-1) against a panel of HDACs (1–9) to verify selectivity (Figure 4). As expected, there was very little activity against other HDACs. Exceptions were HDAC6 and HDAC1, which were mildly inhibited with IC50s of 1.2 and 4.3 μM, respectively. OJI-1 is more potent and selective than what is reported for PCI-34051 (HDAC8 IC50 = 10 nM, HDAC6 IC50 = 2.9 μM, HDAC1 IC50 = 4 μM),15 which is a tool molecule most often utilized to study the effects of HDAC8 inhibition.
Figure 4.
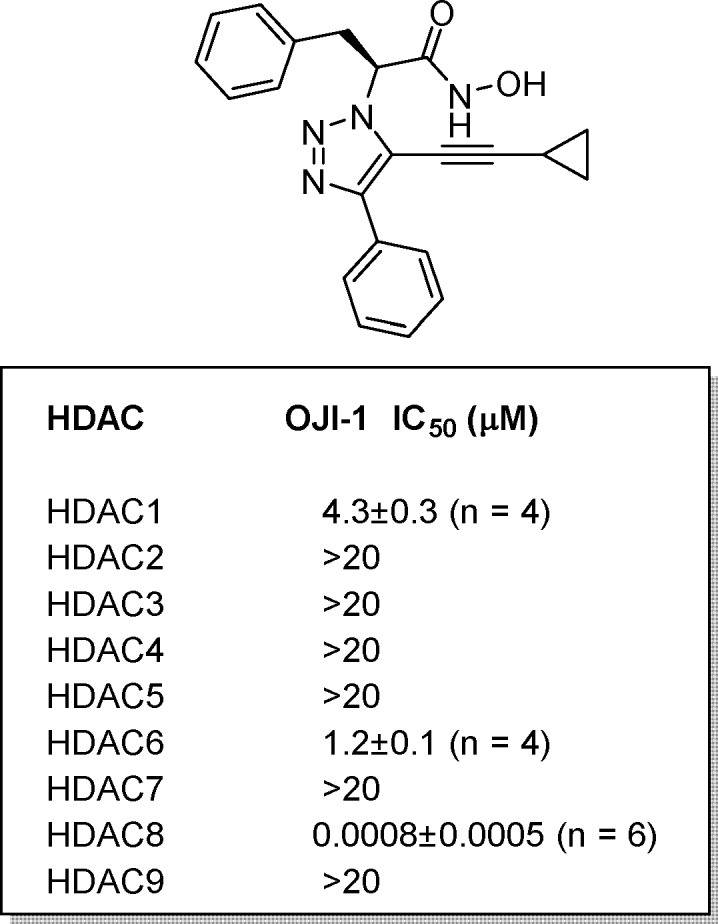
HDAC activity profile for triazole 16.
In conclusion, by repurposing an existing collection of structurally diverse molecules, we have discovered a selective HDAC8 inhibitor. Medicinal chemistry efforts afforded robust SAR resulting in one of the most potent and selective HDAC8 inhibitors reported. Ongoing efforts are focused on refining a binding model through unambiguous crystallization studies and utilizing OJI-1 to better understand the role of HDAC8 in cellular function and its potential as a therapeutic target.
Acknowledgments
We thank Dr. Norman Lee (Boston University) for high-resolution mass spectrometry data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00239.
Experimental procedures, characterization data, and HDAC inhibition data for all analogues (PDF)
Financial support from Boston University and the Center for Chemical Methodology and Library Development at Boston University (GM-067041) are gratefully acknowledged. Work at the BU-CMD is supported by R24-GM111625. NMR (CHE-0619339) and MS (CHE- 0443618) facilities at Boston University are supported by the NSF.
The authors declare no competing financial interest.
Supplementary Material
References
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg K. J.; Johnstone R. W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discovery 2014, 13, 673–691. 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Minucci S.; Pelicci P. G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Fischer A.; Sananbenesi F.; Wang X.; Dobbin M.; Tsai L.-H. Recovery of learning and memory is associated with chromatin remodeling. Nature 2007, 447, 178–182. 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Wang L.; de Zoeten E. F.; Greene M. I.; Hancock W. W. Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nat. Rev. Drug Discovery 2009, 8, 969–981. 10.1038/nrd3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith D. M.; Szőcs B.; Keogh T.; Suponitsky K. Y.; Farkas E.; Buglyó P.; Marmion C. J. Suberoylanilide hydroxamic acid, a potent histone deacetylase inhibitor; its X-ray crystal structure and solid state and solution studies of its Zn (II), Ni (II), Cu (II) and Fe (III) complexes. J. Inorg. Biochem. 2011, 105, 763–769. 10.1016/j.jinorgbio.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Riester D.; Hildmann C.; Grünewald S.; Beckers T.; Schwienhorst A. Factors affecting the substrate specificity of histone deacetylases. Biochem. Biophys. Res. Commun. 2007, 357, 439–445. 10.1016/j.bbrc.2007.03.158. [DOI] [PubMed] [Google Scholar]
- Sternson S. M.; Wong J. C.; Grozinger C. M.; Schreiber S. L. Synthesis of 7200 Small Molecules Based on a Substructural Analysis of the Histone Deacetylase Inhibitors Trichostatin and Trapoxin. Org. Lett. 2001, 3, 4239–4242. 10.1021/ol016915f. [DOI] [PubMed] [Google Scholar]
- Tang W.; Luo T.; Greenberg E. F.; Bradner J. E.; Schreiber S. L. Discovery of histone deacetylase 8 selective inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2601–2605. 10.1016/j.bmcl.2011.01.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; Lee C.-J.; Zhao J.; Toone E. J.; Zhou P. Synthesis, structure, and antibiotic activity of aryl-substituted LpxC inhibitors. J. Med. Chem. 2013, 56, 6954–66. 10.1021/jm4007774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrung M. C.; Chau J. H-L. A convenient procedure for the preparation of amino acid hydroxamates from esters. J. Org. Chem. 1995, 60, 8084–8085. 10.1021/jo00129a059. [DOI] [Google Scholar]
- Suzuki T.; Muto N.; Bando M.; Itoh Y.; Masaki A.; Ri M.; Ota Y.; Nakagawa H.; Lida S.; Shirahige K.; Miyata N. Design, synthesis, and biological activity of NCC149 derivatives as histone deacetylase 8-selective inhibitors. ChemMedChem 2014, 9, 657–664. 10.1002/cmdc.201300414. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Ota Y.; Ri M.; Bando M.; Gotoh A.; Itoh Y.; Tsumoto H.; Tatum P. R.; Mizukami T.; Nakagawa H.; Iida S.; Ueda R.; Shirahige K.; Miyata N. Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. J. Med. Chem. 2012, 55, 9562–9575. 10.1021/jm300837y. [DOI] [PubMed] [Google Scholar]
- Galletti P.; Quintavalla A.; Ventrici C.; Giannini G.; Cabri W.; Penco S.; Gallo G.; Vincenti S.; Giacomini D. Azetidinones as Zinc-Binding groups to design selective HDAC8 inhibitors. ChemMedChem 2009, 4, 1991–2001. 10.1002/cmdc.200900309. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S.; Ramos J.; Luo W.; Sirisawad M.; Verner E.; Buggy J. J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- KrennHrubec K.; Marshall B. L.; Hedglin M.; Verdin E.; Ulrich S. M. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2874–2878. 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
- Olson D. E.; Wagner F. F.; Kaya T.; Gale J. P.; Aidoud N.; Davoine E. L.; Lazzaro F.; Weïwer M.; Zhang Y.-L.; Holson E. B. Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem. 2013, 56, 4816–4820. 10.1021/jm400390r. [DOI] [PubMed] [Google Scholar]
- Decroos C.; Clausen D. J.; Haines B. E.; Wiest O.; Williams R. M.; Christianson D. W. Variable active site loop conformations accommodate the binding of macrocyclic largazole analogues to HDAC8. Biochemistry 2015, 54, 2126–2135. 10.1021/acs.biochem.5b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.; Zhang X.; Wu Y.-D.; Wiest O. Inhibition and mechanism of HDAC8 revisited. J. Am. Chem. Soc. 2014, 136, 11636–11643. 10.1021/ja501548p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estiu G.; Greenberg E.; Harrison C. B.; Kwiatkowski N. P.; Mazitschek R.; Bradner J. E.; Wiest O. Structural origin of selectivity in class II-selective histone deacetylase inhibitors. J. Med. Chem. 2008, 51, 2898–2906. 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- Gallinari P.; Di Marco S.; Jones P.; Pallaoro M.; Steinkühler C. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Cell Research. 2007, 17, 195–211.17325692 [Google Scholar]
- Chakrabarti A.; Oehme I.; Witt O.; Oliveira G.; Sippl W.; Romier C.; Pierce R.; Jung M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. 10.1016/j.tips.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Deardorff M. A.; Bando M.; Nakato R.; Watrin E.; Itoh T.; Minamino M.; Saitoh K.; Komata M.; Katou Y.; Clark D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroos C.; Bowman C. M.; Moser J.-A. S.; Christianson K. E.; Deardorff M. A.; Christianson D. W. Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chem. Biol. 2014, 9, 2157–2164. 10.1021/cb5003762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser F. J.; Ansari M.; Braunholz D.; Gil-Rodríguez M. C.; Decroos C.; Wilde J. J.; Fincher C. T.; Kaur M.; Bando M.; Amor D.; et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet. 2014, 23, 2888–2900. 10.1093/hmg/ddu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P.; Greco T. M.; Guise A. J.; Luo Y.; Yu F.; Nesvizhskii A. I.; Cristea I. M. The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 2013, 9, 1–21. 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson N. A.; Pitcairn C. A.; Fierke C. A. HDAC8 substrates: Histones and beyond. Biopolymers 2013, 99, 112–126. 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson D. E.; Udeshi N. D.; Wolfson N. A.; Pitcairn C. A.; Sullivan E. D.; Jaffe J. D.; Svinkina T.; Natoli T.; Lu X.; Paulk J.; McCarren P.; Wagner F. F.; Barker D.; Howe E.; Lazzaro F.; Gale J. P.; Zhang Y.-L.; Subramanian A.; Fierke C. A.; Carr S. A.; Holson E. B. An Unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem. Biol. 2014, 9, 2210–2216. 10.1021/cb500492r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramsangtienchai P.; He B.; Miller S. P.; Dai L.; Zhao Y.; Lin H.; Spiegelman N. A. HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine. ACS Chem. Biol. 2016, 10.1021/acschembio.6b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard B.; Ryan J.; Beeler A. B.; Porco J. A. Jr. Synthesis of 1,4,5-trisubstituted-1,2,3-triazoles by copper-catalyzed cycloaddition-coupling of azides and terminal alkynes. Tetrahedron 2006, 62, 6405–6411. 10.1016/j.tet.2006.04.025. [DOI] [Google Scholar]
- Hein J. E.; Tripp J. C.; Krasnova L. B.; Sharpless K. B.; Fokin V. V. C. Copper (I)-catalyzed cycloaddition of organic azides and 1-iodoalkynes. Angew. Chem., Int. Ed. 2009, 48, 8018–8021. 10.1002/anie.200903558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.