

Liu et al. show that disruption of paracrine Hedgehog signaling via genetic ablation of Smoothened (Smo) in stromal fibroblasts in a KrasG12D mouse model increased acinar-to-ductal metaplasia (ADM). Smo-deleted fibroblasts had higher expression of transforming growth factor-α (Tgfα) mRNA and secreted higher levels of TGFα, leading to activation of EGFR signaling in acinar cells and increased ADM.
Keywords: acinar-to-ductal metaplasia, hedgehog signaling, EGFR signaling, stromal fibroblast, AKT signaling
Abstract
The contribution of the microenvironment to pancreatic acinar-to-ductal metaplasia (ADM), a preneoplastic transition in oncogenic Kras-driven pancreatic cancer progression, is currently unclear. Here we show that disruption of paracrine Hedgehog signaling via genetic ablation of Smoothened (Smo) in stromal fibroblasts in a KrasG12D mouse model increased ADM. Smo-deleted fibroblasts had higher expression of transforming growth factor-α (Tgfa) mRNA and secreted higher levels of TGFα, leading to activation of EGFR signaling in acinar cells and increased ADM. The mechanism involved activation of AKT and noncanonical activation of the GLI family transcription factor GLI2. GLI2 was phosphorylated at Ser230 in an AKT-dependent fashion and directly regulated Tgfa expression in fibroblasts lacking Smo. Additionally, Smo-deleted fibroblasts stimulated the growth of KrasG12D/Tp53R172H pancreatic tumor cells in vivo and in vitro. These results define a non-cell-autonomous mechanism modulating KrasG12D-driven ADM that is balanced by cross-talk between Hedgehog/SMO and AKT/GLI2 pathways in stromal fibroblasts.
The pancreas undergoes tightly regulated developmental processes during organogenesis that result in the formation of discrete endocrine and exocrine components in the mature organ (Gittes et al. 1996). This process relies on a classic model of epithelio–mesenchymal interactions to drive the undifferentiated duodenal anlage into a fully developed pancreas with well-defined acinar, ductal, and centroacinar compartments (Fell and Grobstein 1968). Seminal studies by Golosow and Grobstein (1962) showed that in vitro culture of the dorsal pancreatic rudiment occurs only in the presence of mesenchyme. In the years following, many groups unsuccessfully attempted to identify the “mesenchymal factor” in the embryonic pancreas responsible for inducing pancreatic epithelial development (Rutter et al. 1964; Ronzio and Rutter 1973; Filosa et al. 1975; Karasaki 1975). More recent studies that aimed to define epithelial lineage-specific responses to mesenchymal signals demonstrated that removal of the pancreatic mesenchyme disrupted acinar cell morphogenesis, thus inhibiting epithelial branching and outgrowth (Gittes et al. 1996; Miralles et al. 1998). These results clearly defined the dependence of pancreatic epithelial cell fate decisions on mesenchymal cues, and subsequent work has demonstrated that FGF (Bhushan et al. 2001), EGF (Tulachan et al. 2006), retinoic acid (Stafford et al. 2006), WNT (Jonckheere et al. 2008), and bone morphogenetic protein (Ahnfelt-Rønne et al. 2010) are important mediators of this process. Similar to pancreatic epithelial cell development during organogenesis, the paracrine signaling networks between the pancreatic epithelium and its adjacent stroma in cancer development have been the subject of intense scrutiny.
Oncogenic mutations in Kras are present in >90% of pancreatic ductal adenocarcinoma (PDAC) patients (Almoguera et al. 1988; Smit et al. 1988; Morris et al. 2010) and are thought to be the initiating mutation in pancreatic cancer (Eser et al. 2014). Genetically engineered mice expressing mutant Kras have metaplastic acini that transdifferentiate into the ductal lineage (Guerra et al. 2003; Hingorani et al. 2003), a process referred to as acinar-to-ductal metaplasia (ADM). Thus, the pancreatic epithelium maintains a degree of cellular plasticity that mimics pancreatic organogenesis (Puri and Hebrok 2010; Reichert and Rustgi 2011). These ADM events have been characterized as premalignant precursors to PDAC primarily through mouse modeling studies. For example, genetically engineered mouse models (GEMMs) that place mutant Kras under the control of acinus-specific promoters generate ADM lesions that progress to pancreatic intraepithelial neoplasias (PanINs) with ductal morphology (Grippo et al. 2003; Tuveson et al. 2006; Habbe et al. 2008). Initial analysis of this acinar-to-ductal switch defined the importance of Kras in ADM formation (Habbe et al. 2008) and has since expanded to additional molecular components within the epithelium that drive ADM, including transforming growth factor-α (TGF-α) (Song et al. 1999), EGFR (Ardito et al. 2012), MIST1 (Shi et al. 2013), SOX-9 (Kopp et al. 2012), KLF4 (Wei et al. 2016), and phosphoinositide-3-kinase (PI3K) (Hill et al. 2010) signaling. Additionally, multiple groups have demonstrated in human studies that ADM occurs during pancreatic cancer progression, justifying the potential significance of defining the underlying pathways that drive acinar cell transdifferentiation (Parsa et al. 1985; Zhu et al. 2007; Remmers et al. 2013). However, the contribution of the mesenchymal stroma to ADM has not garnered the same attention, and non-cell-autonomous signaling networks that drive ADM remain poorly defined. This is surprising given the observations that the desmoplastic stromal reaction increases in the metaplasic pancreas and the previously discussed role of the mesenchyme during epithelial cell fate determination in pancreatic organogenesis.
Hedgehog signaling is one of the most widely studied pancreatic paracrine networks, where Kras transformed epithelial cells secrete Hedgehog ligands that bind to and activate stromal fibroblasts (Hebrok 2003). In canonical Hedgehog signaling, ligands (Sonic, Indian, and Desert hedgehog) bind to the receptor Patched1 (PTCH1), releasing its repression of Smoothened (SMO), the key component of the signaling cascade. Activated SMO relocalizes to the cilia membrane and initiates an intracellular downstream signaling cascade, resulting in activation of GLI transcription factors and expression of downstream targets such as Gli1, Ptch1, and C-myc (Onishi and Katano 2014). Other groups have genetically deleted SMO from the pancreatic epithelium in a Kras pancreatic tumor model and observed no significant difference in malignant transformation or progression, establishing that epithelial Smo is dispensable for Kras-mediated transformation (Nolan-Stevaux et al. 2009).
We used a Kras GEMM of ADM to address the function of fibroblast hedgehog signaling on acinar cell fate. To achieve this, we deleted the key signaling component Smo specifically in fibroblasts in the context of acinar cell-specific expression of oncogenic KrasG12D. The results presented here demonstrate that deletion of SMO in fibroblasts caused an increase in ADM formation. The loss of SMO triggered activation of AKT, leading to increased GLI2-mediated secretion of TGF-α in fibroblasts, EGFR activation in epithelial cells, and enhanced ADM formation. These results define a SMO/AKT/TGFα pathway that, when disrupted, promotes a microenvironment favoring ADM formation in the context of mutant Kras.
Results
Fibroblast-specific deletion of Smo increases ADM in a KrasG12D GEMM
In order to study the role of fibroblast SMO on pancreatic ADM progression, we used the Mist1KrasG12D/+ murine model in combination with fibroblast-specific cre (FspCre)-mediated knockout of Smoloxp (Supplemental Fig. 1A; Long et al. 2001; Tuveson et al. 2006; Trimboli et al. 2008, 2009). The use of this model is critical to the approach because it allows for both Cre-loxP deletion of SmoloxP in stromal fibroblasts and expression of KRASG12D in the acinar cell epithelium. Of note, the Mist1KrasG12D/+ model develops ADM, which progresses to acinar cell carcinoma with ductal features (Tuveson et al. 2006). To use the Mist1KrasG12D/+ mice as a model of de novo ADM formation, all mice were harvested at 6 mo of age. Thus, in this study, we specifically used this animal model to focus on the effect that stromal SMO has on pancreatic ADM development. This was achieved by generating mice from two genetic groups: control mice (Mist1-KrasG12D;SmoloxP/− [KS]) and experimental mice (Mist1-KrasG12D;FspCre;SmoloxP/− [KCS]). Dual-color immunohistochemistry (IHC) for SMA and SMO was performed to precisely examine the extent of SMO deletion in the fibroblast compartment (Fig. 1A). The Vectra multispectral platform was used to differentiate and quantify the percentage of SMO-positive cells within the SMA-positive and SMA-negative cellular compartments (see the Materials and Methods) (Bronisz et al. 2011). This analysis revealed that SMO deletion was confined to SMA-positive stromal fibroblasts with no collateral deletion in epithelial ducts or acini (Fig. 1A, top and bottom graphs, respectively). Single-color IHC for SMO also demonstrated efficient ablation of expression in stromal fibroblast (Supplemental Fig. 1B). To additionally confirm SMO deletion in fibroblasts, SMA-positive pancreatic fibroblasts were isolated (Supplemental Fig. 1C) and shown to exhibit markedly decreased SMO protein and RNA expression when isolated from KCS animals (Supplemental Fig. 1D,E). Purity of fibroblast cultures was assessed by quantitative real-time PCR for fibroblast-specific genes Acta2 and S100a4 along with epithelial-specific genes Cdh1, Krt18, Krt7, and Krt8 in KS and KCS fibroblasts and KPC-derived epithelial cell cultures (Supplemental Fig. 1F). SMO expression levels in other stromal cell compartments, including F4/80-positive macrophages, were unaffected, consistent with our previous characterization of the FspCre transgene (Supplemental Fig. 1G; Trimboli et al. 2008, 2009).
Figure 1.

Stromal SMO ablation accelerates pancreatic ADM and cellular proliferation. (A) Dual-color IHC for SMO (brown) and SMA (red) and quantification of SMO levels in fibroblast and epithelial cell compartments. Unmixed composite and component images of dual-color IHC and colocalization map showing SMA and SMO overlap in yellow. Bars, 50 μm. n = 3. Error bars represent means ± standard deviation (SD). (B) Dual-color IHC for β-amylase (red) and cytokeratin 19 (CK19; brown) and quantification of ADM lesions. KS control mice were compared with KCS experimental mice, and each dot represents one individual mouse. The red bar represents the median of each genetic group. Bars, 25 μm. (C) Dual-color IHC and quantitation for SMA (red) and Ki67 (brown) for fibroblasts (left graph) and ductal-like cells (right graph). Bars, 25 μm. (***) P < 0.001; (ns) not significant.
Histological examination showed that loss of SMO in stromal fibroblasts significantly increased the incidence of ADM, characterized by staining for the ductal cell marker cytokeratin 19 (CK19) and the acinar cell marker β-amylase (Fig. 1B). Sox9 staining of the same tissue confirmed that CK19/β-amylase dual-positive cells were committed to the ductal lineage (Supplemental Fig. 2A; Kopp et al. 2011; Shroff et al. 2014). Ki67 staining of pancreatic sections from both genetic groups demonstrated an increase in proliferative SMA-positive fibroblasts (Fig. 1C, quantified in the left graph) and proliferative ductal-like cells in the KCS group versus KS controls (Fig. 1C, quantified in the right graph). Immunofluorescent costaining of the same tissue with CK19 and Ki67 confirmed the increased proliferation of ductal cells in stromal Smo-deleted mice (Supplemental Fig. 2B).
Additionally, vascular density, measured by MECA32 staining, was significantly increased upon disruption of stromal Smo (Supplemental Fig. 3A). Infiltration of immune cells was assessed. The total T-cell infiltrate, as measured by CD3 staining, was significantly decreased in KCS mice relative to KS mice (Supplemental Fig. 3B). However, infiltration of FOXP3-positive T-regulatory cells increased in KCS mice relative to KS mice (Supplemental Fig. 3C). In contrast, infiltration of GR-1-positive myeloid cells was not significantly altered (Supplemental Fig. 3D).
Smo-deleted fibroblasts promote ADM through activation of EGFR in epithelial cells
In order to elucidate the mechanisms by which Smo-deleted fibroblasts influence acinar cell transformation, an established three-dimensional (3D) acinar cell culture system was used to examine whether fibroblast-secreted factors could promote acinar-to-ductal transformation (Navas et al. 2012; Eser et al. 2013). Wild-type acinar cells treated with conditioned medium (CM) from KCS fibroblasts formed dramatically greater numbers of ductal-like structures compared with CM from control KS cells (Fig. 2A). These structures were confirmed to represent ADM by costaining with CK19 and β-amylase, revealing dual staining of cells similar to ADM observed in vivo (Fig. 2B).
Figure 2.
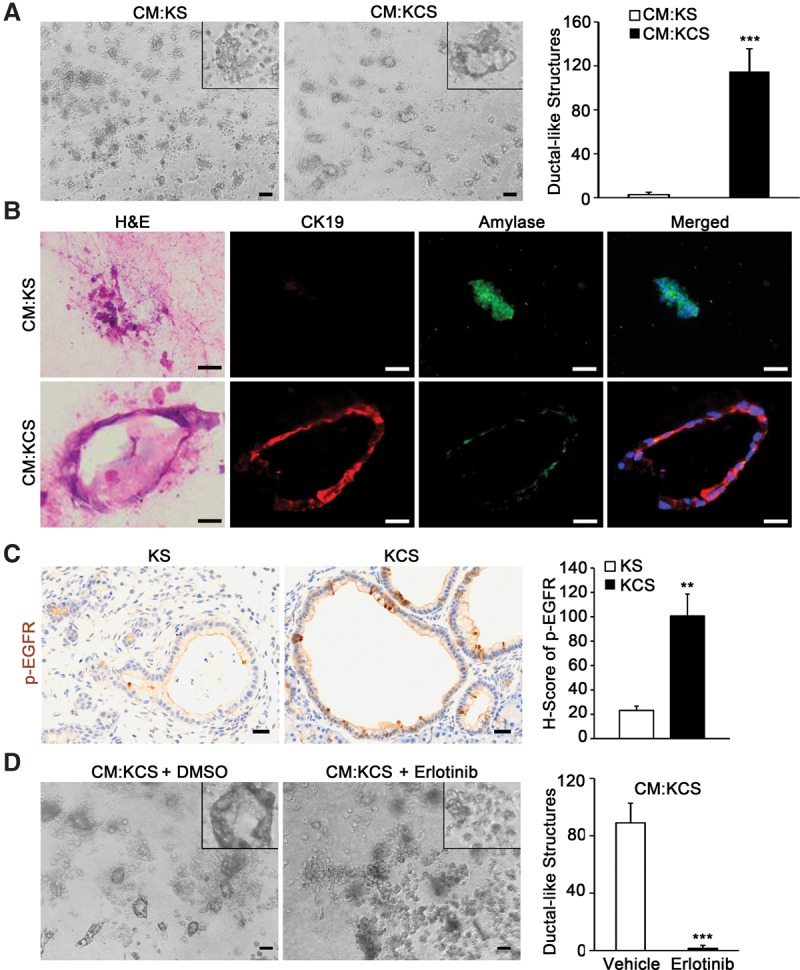
Loss of SMO in pancreatic fibroblasts induces ADM in vitro. (A) Images and quantitation of ductal-like structures in 3D ADM formation assay treated with the indicated CM. Insets show a higher magnification view of representative cellular structures. The graph represents quantitation of ductal-like structures. Bar, 40 μm. n = 3. Error bars represent means ± SD. (B) Matched H&E and coimmunofluorescence images of representative cellular structures from 3D ADM formation assay stained for CK19 (red) and β-amylase (green) after treatment with the indicated CM. (C) IHC and quantitation of phosphorylated EGFR (Tyr1068). The graph represents quantification of the H score for each genotype. Bar, 25 μm. n = 3. Error bars represent means ± SD. (D) Quantitation of ductal-like structures in 3D ADM formation assay treated with KCS CM and DMSO or erlotinib. Bar, 40 μm. n = 3. Error bars represent means ± SD. (**) P < 0.01; (***) P < 0.001.
EGFR is a key regulator of ADM and pancreatic carcinogenesis (Miyamoto et al. 2003; Moore et al. 2007; Ardito et al. 2012), and IHC with antibodies specific for phosphorylated EGFR at Tyr1068 revealed robust activation of the receptor in KCS mice relative to controls (Fig. 2C). In parallel experiments, the EGFR antagonist erlotinib blocked KCS fibroblast CM-stimulated transformation of acinar cells in vitro (Fig. 2D). To further analyze the downstream pathways activated as a result of EGFR phosphorylation, staining was performed for p-ERK, p-STAT3, and HES1. ERK phosphorylation was unchanged in the epithelial compartment between KS and KCS (Supplemental Fig. 4A), as predicted for cells expressing KrasG12D. STAT3 phosphorylation at Tyr705 was markedly increased in the epithelium of KCS mice relative to KS mice (Supplemental Fig. 4B). The Notch pathway target HES1 was also up-regulated, as expected in EGFR-activated pancreatic epithelium (Supplemental Fig. 4C; Miyamoto et al. 2003).
Smo-deleted fibroblasts increase growth of KrasG12D;p53R172H/+ pancreatic tumor cells in vivo and in vitro
To study the effect of deletion of Smo in fibroblasts on pancreatic tumor cell growth and proliferation, KPC cells (a KrasG12D;p53R172H/+ mouse PDAC tumor cell line) (see the Materials and Methods) were admixed with either KS or KCS fibroblasts and coinjected into nude mice. KPC tumor cells injected alone or mixed with wild-type fibroblasts produced tumors of the same mass after 5 wk (Fig. 3A). In contrast, KPC cells injected with fibroblasts lacking SMO were significantly larger than the SMO-intact control (Fig. 3A).
Figure 3.

Deletion of SMO in fibroblasts enhances tumor cell growth and proliferation through EGFR. (A) Quantitation of tumor mass 5 wk after implantation (tumors images are shown below the graph). Bar, 5 mm. n = 5. Each dot represents one tumor, and the red dot corresponds to the image below the graph. (B) Images and quantitation of the percentage of p-Histone H3+ over total CK19+ cells treated with the indicated CM. n = 3. Error bars represent means ± SD. (C) Images and quantitation of the percentage of p-Histone H3+ over total CK19+ cells cotreated with the indicated CM and DMSO or erlotinib. Bar, 40 μm. n = 3. Error bars represent means ± SD. (*) P < 0.05; (***) P < 0.001; (ns) not significant.
Parallel in vitro experiments demonstrated increased staining for proliferation marker phospho-Histone H3Ser10 in KPC cells treated with CM from KCS fibroblasts compared with CM from control KS cells (Fig. 3B). Erlotinib treatment blocked the enhanced proliferation of KPC treated with CM from Smo-deleted fibroblasts (Fig. 3C). Erlotinib efficacy was confirmed by Western blot analysis of phosphorylated EGFR at Tyr1068 (Supplemental Fig. 4D).
TGF-α is up-regulated in Smo-deleted fibroblasts and promotes ADM
To identify the EGFR-activating ligand produced by KCS fibroblasts, mRNA expression levels of several known EGFR-activating ligands were analyzed. A striking increase in Tgfa mRNA was detected in KCS fibroblasts (Fig. 4A). Analysis of KCS CM revealed that TGF-α protein was significantly increased relative to control CM (Fig. 4B, SPARC expression served as an internal control). Consistent with this data, TGF-α, measured by IHC staining, was increased in the stroma of KCS animals (Fig. 4C). Pretreatment of CM from KCS fibroblasts with TGF-α-neutralizing antibody significantly decreased formation of ductal-like structures, thus providing evidence that TGF-α was necessary for ADM induction by KCS fibroblasts (Fig. 4D).
Figure 4.
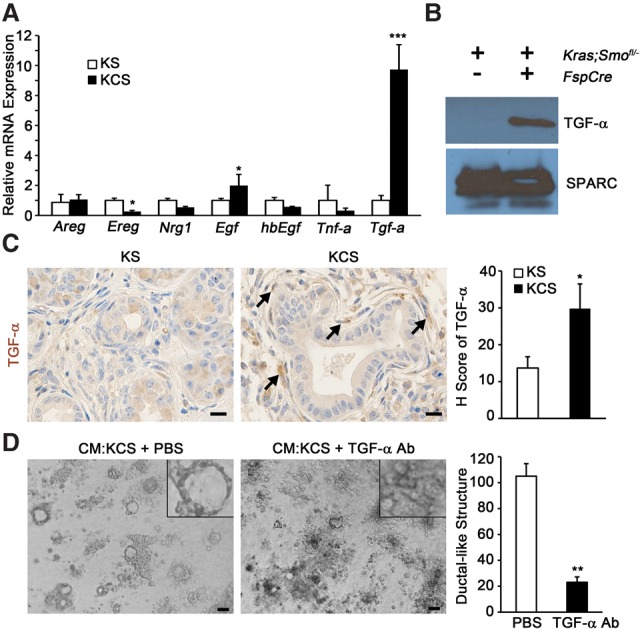
Deletion of SMO in fibroblasts promotes ADM via TGF-α. (A) Quantitative real-time PCR analysis of known EGFR-activating ligands. n = 3. Error bars represent means ± SD. (B) Western blot of TGF-α in CM; SPARC was used for a loading control. (C) IHC and quantitation for TGF-α; the graph represents the quantification of the H score for each genotype. Bar, 25 μm. n = 3. Error bars represent means ± SD. (D) Images and quantitation of ductal-like structures in 3D ADM formation assay with pretreatment of KCS CM with TGF-α-neutralizing antibody. The insets show a higher magnification view of representative cellular structures. The graph represents the quantitation of ductal-like structures. Bar, 40 μm. n = 3. Error bars represent means ± SD. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Smo deletion in fibroblasts selectively activates GLI2-mediated TGF-α production
To address the mechanism by which loss of SMO signaling up-regulates TGF-α production in fibroblasts, we performed global gene expression profiling on SMO-intact and SMO-deleted fibroblasts. Analysis of the data revealed that 516 genes were significantly differentially expressed (greater than twofold, P < 0.05) (the top 100 differentially expressed genes are in Supplemental Table 1). Interestingly, Gli2, a canonical hedgehog signaling transcription factor, was one of the top 25 up-regulated genes in KCS cells (Supplemental Table 1). This result was surprising given the observation that KCS cells had decreased overall hedgehog signaling by gene set enrichment analysis (GSEA) (Fig. 5A). Furthermore, quantitative real-time PCR of SMO signaling components Gli1/2/3 and Ptch1 showed that only Gli2 levels were highly enriched in KCS cells relative to KS cells (Fig. 5B). Gli1, Gli2, Gli3, and Ptch1 all responded as expected to Sonic hedgehog (SHH) treatment in SMO-intact KS cells but not in SMO knockout KCS cells (Fig. 5C; Supplemental Fig. 5A).
Figure 5.

Loss of SMO leads to Gli2 up-regulation and enhanced Tgfa synthesis. (A) GSEA enrichment plot for Hedgehog signaling upon deletion of Smo. (FDR) False discovery rate. (B) Quantitative real-time PCR analysis of Gli1, Gli2, Gli3, and Ptch1 in pancreatic fibroblasts. n = 3. Data are represented as fold change relative to KS. Error bars represent means ± SD. (C) Quantitative real-time PCR analysis of Gli1, Gli2, and Gli3 upon treatment of pancreatic fibroblasts with recombinant SHH. n = 3. Data are represented as fold change relative to untreated KS. Error bars represent means ± SD. (D,E) Western blot analysis and quantification of GLI2 in KS and KCS fibroblasts transfected with nonsilencing control (NC) siRNA or siRNA targeting Gli2. (F) Quantitative real-time PCR analysis of Tgfa in KS and KCS mouse pancreatic fibroblasts transfected with nonsilencing control or siRNA targeting Gli2. (G) Schematic map of the GLI-binding site in the promoter of Tgfa. (H) Relative GLI2 enrichment at the Tgfa promoter by chromatin immunoprecipitation (ChIP) analysis in both KS and KCS mouse pancreatic fibroblasts. n = 3. Error bars represent means ± SD. (I). Relative luciferase activities measured in KCS cells transfected with mouse Tgfa luciferase reporter (pGL3 vector) and Gli2 expression vector (FUW vector). (**) P < 0.01; (***) P < 0.001.
Knockdown of Gli2 by siRNA in KCS cells (Fig. 5D,E; Supplemental Fig. 5B) resulted in decreased levels of Tgfa expression (Fig. 5F). Analysis of the Tgfa promoter region identified a putative GLI consensus binding region that was conserved between humans and mice 0.8 kb upstream of the transcriptional start site, suggesting that GLI2 might directly regulate Tgfa (Fig. 5G). Chromatin immunoprecipitation (ChIP) analysis of the Tgfa promoter region demonstrated enrichment of GLI2 at the putative binding site (Fig. 5H). Furthermore, luciferase reporter assays using the proximal Tgfa promoter region demonstrated that Gli2 overexpression in Smo-null fibroblasts increased Tgfa promoter activation (Fig. 5I).
Smo deletion activates AKT–GLI2 signaling upstream of TGF-α transcription
GSEA also revealed that KCS cells had altered PI3K signaling compared with KS cells (Fig. 6A). Since previous work indicated that AKT activation is one mediator of the oncogenic functions of cancer-associated fibroblasts, we focused attention on this pathway in our model (Okawa et al. 2007). Western blot analysis demonstrated that AKT-activating phosphorylation events (at both Ser473 and Thr308) increased in SMO-deleted cells (Fig. 6B; Supplemental Fig. 6A). Importantly, treatment of KCS fibroblasts with the AKT inhibitor MK-2206 (Fig. 6C; Supplemental Fig. 6B; Hirai et al. 2010) reduced both Tgfa and Gli2 levels (Fig. 6D).
Figure 6.

AKT phosphorylation is induced upon deletion of Smo in pancreatic fibroblasts. (A) GSEA enrichment plots for PI3K signaling upon deletion of Smo. (B) Western blots of three pancreatic fibroblast cultures for total AKT and phosphorylated AKT (Ser473 and Thr308). Quantification is in Supplemental Figure 6. (C) Western blots for p-AKTS473 in DMSO-treated or MK-2206-treated KCS fibroblasts. Quantification is in Supplemental Figure 6. (D) Quantitative real-time PCR analysis for Tgfa and Gli2 in DMSO-treated or MK-2206-treated fibroblasts. n = 3. Error bars represent means ± SD. (E) Coimmunofluorescence of SMA and phosphorylated AKTT308 in KPC-BRCA1CKO mice treated with GDC-0449 or DMSO. n = 3. Error bars represent means ± SD. (F) Images and quantitation of the percentage of p-Histone H3+ over total CK19+ cells cotreated with the indicated CM and DMSO or MK-2206. Bar, 40 μm. n = 3. Error bars represent means ± SD.
To determine whether AKT signaling in SMO-depleted fibroblasts was relevant at later stages of PDAC progression, we performed two types of experiments. First, previous work demonstrated that a small molecule inhibitor of SMO resulted in increased growth of pancreatic tumors in the KPC model (Rhim et al. 2014). We examined the in vivo effect of the SMO inhibitor GDC-0449 (see the Materials and Methods; Von Hoff et al. 2009) on stromal AKT activation. The KPC-BRCA1CKO mice, representing a rapidly progressing PDAC mouse model (Shakya et al. 2011), were used for this approach. Inhibition of Hedgehog signaling was confirmed by decreased expression of Gli1 mRNA in pancreatic tissue from treated mice relative to the vehicle controls (Supplemental Fig. 6C). The inhibition of SMO in this model resulted in robust activation of p-AKT in SMA-positive fibroblasts (Fig. 6E). Second, CM isolated from KCS fibroblasts treated with AKT inhibitor (MK-2206) decreased KPC hyperproliferation relative to vehicle-treated fibroblasts in vitro (Fig. 6F). Activation of EGFR in KPC cells by CM from MK-2206-treated fibroblasts was also significantly diminished (Supplemental Fig. 6D).
AKT–GLI2 signaling is activated in Smo-deleted fibroblasts
GLI2 is stabilized through phosphorylation of residue Ser230 by AKT, resulting in increased expression of downstream target genes (Shi et al. 2015). To test whether the observed increase in GLI2 protein in KCS cells could be due to AKT-dependent phosphorylation, GLI2 was immunoprecipitated from KS and KCS fibroblasts, and its phosphorylation status was determined by Western blot analysis using an antibody specific for phospho-(Ser/Thr) AKT substrates (Zhang et al. 2002). Mouse GLI2 Ser230 and the corresponding human GLI2 Ser234 are within a perfect AKT recognition motif (Supplemental Fig. 7A). Importantly, this is the only perfectly matched AKT recognition site in GLI2. The results showed that endogenous GLI2 was phosphorylated at AKT substrate sites in KCS cells (Fig. 7A; Supplemental Fig. 7A). Comparison of the relative ratio of p-GLI2 to total GLI2 protein in KS cells versus KCS cells indicated that GLI2 phosphorylation levels were increased ∼2.5-fold in KCS cells (Supplemental Fig. 7B). To confirm that GLI2 phosphorylation occurs at Ser230, Flag-tagged GLI2 wild type and GLI2-S230A (lacking the AKT phosphorylation site) were expressed in KS and KCS cells. Robust phosphorylation of Flag-tagged GLI2 wild type could be detected in KCS but not KS cells. In contrast, phosphorylation of Flag-GLI2-S230A was not detected in KCS cells or KS cells (Fig. 7B).
Figure 7.
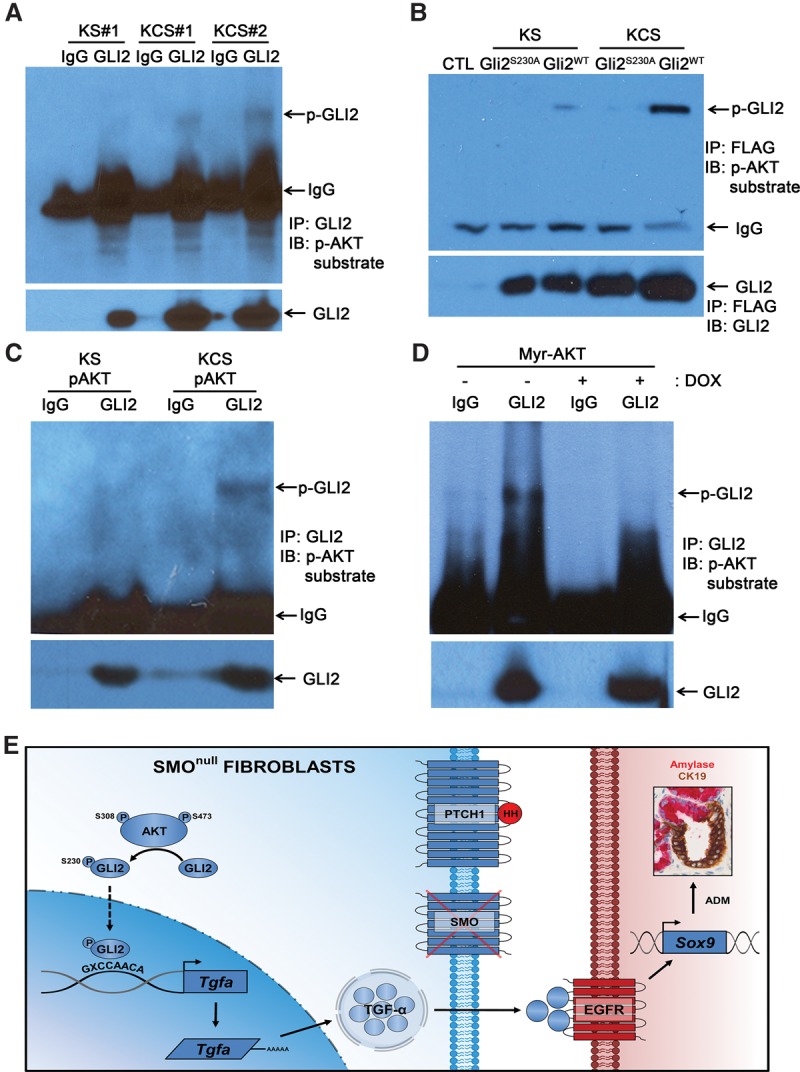
p-AKTS473 directly modulates GLI2 protein. (A) Western blots for phospho-(Ser/Thr) AKT substrate after coimmunoprecipitation with IgG or GLI2 antibody in both KS and KCS mouse pancreatic fibroblasts. Quantification is in Supplemental Figure 7. (B) Western blots for phospho-(Ser/Thr) AKT substrate (top panel) or GLI2 (bottom panel) in KS and KCS pancreatic fibroblasts transfected with control vector, Gli2S320A mutant, or Gli2WT vectors after coimmunoprecipitation with Flag antibody. (C, top panel) p-AKTS473 isolated from KS or KCS was incubated with fresh KS lysate followed by immunoprecipitation with GLI2 antibody and immunoblotting for phospho-(Ser/Thr) AKT substrate. The bottom panel shows total GLI2 levels. (D) Western blots for phospho-(Ser/Thr) AKT substrate after immunoprecipitation with GLI2 antibodies in Tet-off myristoylated AKT1 (myr-AKT)-expressing fibroblasts with the indicated doxycycline treatment. (E) Smo deletion leads to activation of the Akt pathway, which phosphorylates and stabilizes GLI2. GLI2 directly binds to the Tgfa promoter and induces its transcription. Secreted TGF-α then triggers ADM events.
To confirm that GLI2 Ser230 is directly phosphorylated by AKT in KCS fibroblasts, in vitro kinase assays were performed (see the flow diagram in Supplemental Fig. 7C for the experimental design). p-AKTS473 antibody was used to immunoprecipitate activated AKT from either KS or KCS fibroblasts, and the isolated p-AKT was incubated with whole-cell lysates prepared from Smo-positive KS fibroblasts. After incubation, endogenous GLI2 was immunoprecipitated, and the pellet was analyzed by Western blot with AKT substrate antibody. The results showed robust GLI2 phosphorylation by p-AKT isolated from KCS fibroblasts but not KS fibroblasts (Fig. 7C).
To further demonstrate that GLI2 phosphorylation was AKT-dependent and required for Tgfa expression, we isolated pancreatic fibroblasts from Tet-off-inducible myristoylated AKT1 (myr-AKT) transgenic mice (see the Materials and Methods; Belteki et al. 2005; Sun et al. 2005; ). The phosphorylation of GLI2 was compared between vehicle-treated cells and doxycycline-treated cells (Fig. 7D). Doxycycline treatment decreased myr-AKT activity as expected (Supplemental Fig. 7D) and resulted in decreased GLI2 Ser230 phosphorylation (Fig. 7D). Doxycycline treatment also resulted in decreased expression of Tgfa and Gli2 mRNA (Supplemental Fig. 7E). These biochemical assays establish that Smo deletion leads to increased GLI2 phosphorylation at position Ser230 in an AKT-dependent manner.
Discussion
The prevailing view is that the Hedgehog pathway functions in paracrine signaling between epithelial cells with activating Kras mutations and stromal fibroblasts during pancreatic adenocarcinoma progression (Hebrok 2003). In this study, we focused on the function of the Hedgehog pathway in fibroblasts at the initial stage of Kras-dependent acinar transformation. Our rationale for focusing on this earliest event in pancreatic cancer progression was that the influence of the stroma can be more easily dissected at very early stages of the malignant cascade before heterogeneity in tumor cells and consequently, in the stroma, make it difficult to discern cell-autonomous from non-autonomous gene function. Unexpectedly, these studies elucidated a connection between loss of Hedgehog signaling, activation of AKT signaling, and secretion of TGF-α by fibroblasts that synergized with KrasG12D in acinar cells to increase ADM (Fig. 7E). Ablation of Smo in fibroblasts instigated the noncanonical activation of GLI2 expression, and GLI2 directly regulated Tgfa expression. The mechanism involved AKT-dependent phosphorylation of Ser230 in GLI2, which, previous work demonstrated, leads to enhanced stability and transactivation activity (Shi et al. 2015).
The stroma undergoes a dramatic expansion in concert with the stepwise development of human PDAC, suggesting that the stroma is an active partner in PDAC initiation and progression (Feig et al. 2012). However, recent studies have challenged this view, demonstrating that deletion of SHH in epithelial cells increases pancreatic cancer progression (Rhim et al. 2014). Similarly, ablation of stromal SMA-positive cells increased growth of pancreatic tumors (Ozdemir et al. 2014). Our present study supports the suppressive function of the Hedgehog–SMO pathway even at very early stages of preneoplastic transformation. Importantly, our study provides mechanistic insight into how this occurs, revealing the significance of cross-talk between Hedgehog/Smo signaling and AKT signaling. We propose that activation of the AKT/GLI2 axis in stromal fibroblasts and subsequent activation of the EGFR pathway in acinar cells via TGF-α are critical for the increased formation of Kras-dependent pancreatic ADM lesions when Hedgehog signaling is disrupted.
EGFR activation is a potent oncogenic mediator of Kras-driven pancreatic cancer that has been shown to be essential for PDAC formation and epithelial cell proliferation from the earliest stages of detectable disease (Ardito et al. 2012; Navas et al. 2012). Additionally, treatment with the EGFR inhibitor erlotinib prolongs survival in PDAC mouse models (Singh et al. 2010; Miyabayashi et al. 2013). Analysis of downstream EGFR signaling components revealed that activation of STAT3 signaling is seen in acinar cells undergoing transformation (Navas et al. 2012). Consistent with these studies, we show that disruption of fibroblast Hedgehog signaling leads to increased production of TGF-α and subsequent activation of EGFR and STAT3 in the preneoplastic epithelium. Thus, stromal fibroblasts as well as epithelial cells (Smith et al. 1987; Navas et al. 2012) are also a source of TGF-α production during ADM transformation. Enhanced Tgfa expression by Smo-deleted fibroblasts is driven by noncanonical activation of the GLI2 transcription factor. In canonical Hedgehog signaling, GLI1, GLI2, and GLI3 are known to have discrete but overlapping functions in controlling expression of pathway target genes. The noncanonical activation of GLI2, but not GLI1 or GLI3, supports that individual members of this transcription factor family can selectively respond to upstream signaling events in an independent fashion (Varjosalo and Taipale 2008).
Cross-talk between the Hedgehog and PI3K signaling pathways has been identified previously in medulloblastoma (Buonamici et al. 2010). These cancers harbor mutations in multiple Hedgehog pathway components, and treatment of patients with SMO inhibitors leads to an initial positive clinical response, but resistance to this therapy subsequently develops (Yauch et al. 2009). One mechanism of resistance in a subset of patients treated with SMO inhibitors is activation of PI3K signaling, and PI3K inhibitors are able to overcome the resistance to SMO antagonists in preclinical studies (Buonamici et al. 2010). A second potential mechanism of resistance to SMO inhibitors is genomic amplification of GLI2 (Dijkgraaf et al. 2011; Kool et al. 2014). Similarly, in our study, Smo deletion in pancreatic stromal fibroblasts led to activation of PI3K/AKT signaling, as evidenced by AKT phosphorylation at Ser470 and Thr308 and activation of GLI2. We also observed a striking increase of Gli2 mRNA levels when Smo was deleted, and in silico analysis predicts GLI-binding sites in the Gli2 promoter region. Previous reports have suggested that GLI transcription factors can positively autoregulate their own promoters to amplify Hedgehog signaling (Regl et al. 2002; Weiner et al. 2002; Lai et al. 2004; Hu et al. 2006). Future work will test the hypothesis that AKT activation stabilizes GLI2 and induces an autocrine feed-forward loop that reinforces Gli2 expression.
In conclusion, we identified cross-talk between Hedgehog/SMO and AKT/GLI2 pathways involved in cross-talk between epithelial and mesenchymal cell compartments in the pancreas that suppresses ADM. Future studies using recently developed mouse models that combine a FLP recombinase system to drive oncogenic Kras expression and pancreatic cancer (Schönhuber et al. 2014) can be combined with stromal Cre drivers to test whether this pathway cross-talk is also involved during later stages of malignant transformation.
Materials and methods
Animal strains, husbandry, and maintenance
The use of animals was in compliance with federal and university laboratory animal resource regulations. Mist1KrasG12D/+; FspCre; SmoloxP/− animals were generated by crossing the previously described Mist1KrasG12D/+ (Tuveson et al. 2006), SmoloxP (Long et al. 2001), Smo− (Long et al. 2001), and FspCre (Trimboli et al. 2008) strains. FspCre; LSL-tTA; TetO-myrAkt1 mice were used in the myr-AKT experiments (Belteki et al. 2005; Sun et al. 2005). The experiments were performed using littermate mice from a mixed C57BL/6; 129/Sv and FVBN genetic background. KPC epitheial cell lines were isolated from Elas-CreER; LSL-KrasG12D/+; TP53R172H/+ mice and were a generous gift from Dr. Stephen F. Konieczny.
Athymic nude mice used for xenograft experiments were provided by the Ohio State University Target Validation Shared Resource Core. For subcutaneous injection, 0.5 × 106 KPC tumor cells admixed in a 1:1 ratio with fibroblasts were injected into the flanks of nude mice; tumor volumes were measured once per week by caliper.
Multispectral analysis of dual-color IHC
Dual-stained samples were imaged using the PerkinElmer Vectra multispectral slide analysis system. For mouse samples, at least three multispectral images per animal for at least three mice per genotype (unless otherwise noted) were taken manually. The image acquisition work flow consisted of the following: (1) monochrome imaging of the entire slide, (2) RGB low-power imaging of the tumor tissue using an inForm tissue-finding algorithm, and (3) multispectral high-power imaging of one field containing tumor epithelium and stroma by means of an inForm HPF-finding algorithm.
For visualization of the component images (i.e., SMO/brown and SMA/red), the multispectral images were spectrally unmixed using Nuance software. Nuance colocalization tool was used to create the colocalization maps displaying the SMO-positive cells in the SMA-positive cellular compartment overlap in yellow.
For quantification of the SMO staining, the multispectral images were reviewed and analyzed using inForm tissue finder software. A pattern recognition algorithm was used for processing as follows: (1) trainable tissue segmentation to segment the SMA-positive regions, (2) cell segmentation of the SMA-positive compartment, and (3) scoring to bin the spectrally unmixed DAB signal into four categories depending on the staining intensity (0+, 1+, 2+, and 3+). The H score, which ranges from 0 to 300, was calculated using following formula: 1 × (% cells 1+) + 2 × (% cells 2+) + 3 × (% cells 3+). Thus, H score measures staining intensity as well as the percentage of positive cells in a given cellular compartment. Of note, the overlap in IHC staining was a direct quantitative measure of staining for one antigen (SMO) within a distinct cell population defined by a second antigen (SMA) and was not simply the overlap in signal between the two stains.
Statistics
Pearson's correlation, Wilcoxon rank sum test, and Student's t-test were calculated using R 3.0.1 unless otherwise noted. The P-values from Student's t-tests are listed unless otherwise specified. In all graphs, median, means (bar), and standard deviations (lines) are denoted. Microarray data were processed by the robust multiarray (RMA) method and analyzed using the moderated t-test approach (Yu et al. 2011). Comparison-wise, P-value of 0.05 was considered significant.
Histology analysis, IHC, and immunofluorescent staining
Dissected mouse pancreas tissues were fixed, processed, embedded in paraffin, sectioned, and mounted. For IHC, all sections were stained using a Bond Rx autostainer (Leica) unless otherwise noted. Antibodies for the following markers were diluted in antibody diluent (Leica): rabbit antibodies αSMA (1:1500; Abcam), Ki67 (1:100; Abcam), amylase (1:400; Cell Signaling Technology), EGFR Y1068 (1:50; Cell Signaling Technology), SMO (1:200; Bioss), HES1 (Santa Cruz Biotechnology), p-STAT3 (1:100; Cell Signaling Technology), p-AKT (1:50; Cell Signaling Technology), and p-ERK (1:400; Cell Signaling Technology) and rat antibody CK19 (TROMA-III) (1:150; Developmental Studies Hybridoma Bank [DSHB], University of Iowa).
For TGF-α and Sox9, single-antibody IHC staining was performed by conventional methods. The following primary antibodies were used: rabbit anti-SMO (1:100; Abcam) and TGF-α (1:100; Abcam). Staining was visualized by using an immunoperoxidase technique (VectaStain Elite ABC kit, Vector Laboratories) and 3,3′-diaminobenzadine followed by counterstaining with Meyer's hematoxylin.
Fluorescent immunostaining required antigen retrieval, which was performed using a 1× target retrieval solution (pH 6.0) (Dako). Antibodies for the following markers were diluted in antibody diluent (Dako) and applied overnight at 4°C: rabbit antibodies Ki67 (1:100; Abcam), phospho-histone H3 (1:100; Cell Signaling Technology), and amylase (1:400; Cell Signaling Technology); rat antibodies CK19 (TROMA-III) (1:150; DSHB, University of Iowa), CK8 (TROMA-I) (1:300; DSHB, University of Iowa), and Meca32 (1:200; BD Pharm); and mouse antibody α-SMA (1:400; Sigma).
Image acquisition and analysis of ADM and proliferation
Dual-IHC-stained slides were imaged using the PerkinElmer Vectra multispectral slide analysis system. The automatic work flow consisted of the following: (1) monochrome imaging of the entire slide, (2) RGB low-power imaging, and (3) high-power color imaging of all of the fields containing at least a 90% of pancreatic tissue using inForm tissue-finding and HPF-finding algorithms. All HPF images (0.4-mm2 area each) were reviewed to manually assess the density of ADM lesions, which was expressed as number per square millimeter. Twenty HPF images were randomly selected per mouse. Ten HPF images were randomly selected to manually assess the stromal fibroblast Ki67-labeling index (Ki67-positive fibroblasts/total fibroblasts per square millimeter). All histology was validated by pancreatic pathologists.
Gli2 knockdown assay
Gli2 siRNA (On-Target plus mouse Gli2 siRNA SMART pool) and negative control siRNA (On-Target plus mouse nontargeting pool) were purchased from Dharmacon. In brief, transfection was performed at 70% confluency with Lipofectamine 2000 in Opti-MEM medium (Invitrogen). After transfection, the cell culture medium was replaced with fresh medium, and the cells were incubated for an additional 48 h for RNA or protein extraction.
Luciferase assay
Luciferase assay were performed as described previously (Acharyya et al. 2010). After 24 h of transfection, Gli2 expression was induced by doxycycline. Luciferin reagent (Sigma) was added to lysates, and luciferase activity was measured. FUW-Gli2 and control FUW vector were generous gifts from Fanxin Long. Mouse Tgfa promoter (2.0 kb) was inserted into a pGL3 luciferase reporter to generate the pTGFa vector.
ChIP assays
ChIP assays were performed as described previously (Bronisz et al. 2011). Primary KS and KCS cells (2 × 106 cells) were used for each experiment. In brief, cells were cross-linked for 10 min with 1% formaldehyde and lysed. Chromatin was collected and sonicated. Anti-GLI2 ChIP-grade antibody (ab26056) and protein G agarose slurry (Millipore, 16-266) were used to pull down chromatin. After reverse-cross-linking, the DNA was recovered, purified, and analyzed by real-time PCR. Gli2 forward primer 5′-CAATAAACTGGAATTTTCAACTCTGA-3′ and reverse primer 5′-AATCAGACTGACCCCTCTCCT-3′ were used.
Pancreatic fibroblast and isolation of CM
Primary pancreatic fibroblasts were purified based on the following protocol. Briefly, pancreata were dissected from 6-mo-old mice, minced, and digested with collagenase (0.5% collagenase II, 120 U mL−1 DNase I in 1× PBS) with shaking at 225 rpm for 1 h at 37 °C. Digested tissue was resuspended in medium and gravity-purified for 10 min. Supernatants were aspirated, and pellets were washed three times, subjected to two additional gravity sedimentations, and then seeded on tissue culture dishes.
3D acinar culture
3D acinar cell culture was established using previously published protocols (Shi et al. 2015). In brief, mouse pancreata were minced, and acini were released by collagenase P digestion, washed three times in cold 5% fetal bovine serum (FBS), and then filtered through a 105-μm nylon mesh. Cell suspensions were layered on top of FBS, and acini were collected by centrifugation and then resuspended in 3D culture base medium. Twenty-four-well tissue culture plates were coated with a 250-μL per well collagen layer before acini isolation. The cell suspension was then mixed with collagen and plated in a 1:1 ratio. Erlotinib (LC libraries) was dissolved in DMSO and stocked in −20°C. The final concentration in treatment was 10 µM. TGF-α antibody (Abcam) was preincubated with CM from KCS cells as suggested by the manufacturer.
Tumor cell culture
Tumor cells isolated from Elas-CreER; LSL-KrasG12D/+; TP53R172H/+ mice were plated and starved with 0% FBS medium overnight. After starvation, cells were treated for 48 h with CM as indicated in the figure legends. Erlotinib (10 µM) treatment was coadministered at the same time. For AKT inhibition, fibroblasts were treated with 5 µM MK-2206 prior to harvesting CM.
AKT inhibitor (MK-2206) treatment assays
Primary pancreatic fibroblasts were cultured to 80% confluence and washed with PBS followed by fresh 10% FBS-DMEM. AKT inhibitor MK-2206 (Selleckchem, S1078) was used at final concentration of 5 µM, and cellular lysates were collected with RIPA buffer after different time points for Western blot.
GDC-0449 drug treatment
Smo inhibitor GDC-0049 was custom-synthesized by the Medicinal Chemistry Shared Resource at Ohio State University and used for in vitro studies dissolved in DMSO and at the concentrations specified in the figure legends. For in vivo experiments, GDC-0449 was orally administrated to KPC-BRCA1CKO mice. The dosage was 100 mg/kg once per day for 4 d. Four hours after the last dose, mice were dissected, and pancreata were harvested for histology as well as staining as described above.
Coimmunoprecipitation and Western blotting
Primary pancreatic fibroblasts were harvested by trypsinization and lysed in RIPA lysis buffer for Western blot or in nondenaturing extraction buffer (Cell Signaling, 9803). Immunoprecipitation was performed with 8 µg of Gli2 (ab26056) or IgG antibody. After incubation, PRO-A magnetic beads (Millipore, LSKMAGA02) were added. Samples were then washed, and Laemmli buffer was added for Western blot analysis. Primary antibody concentrations were as follows: rabbit polyclonal antibody against SMO (1:500); p-Akt, total Akt, and mouse monoclonal anti-β-actin (1:5000; Sigma-Aldrich) from Cell Signaling Technology; GAPDH (sc-25778) from Santa Cruz Biotechnology; Gli2 from Abcam; and horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies (both at 1:10,000). Signal was developed with the ECL detection reagent (Amersham). Flag-tagged Gli2 mutant (S230A) vector was a generous gift from Fanxin Long.
In vitro AKT kinase assay
p-AKTS473 antibody was added to KS and KCS lysates. After incubation, protein A beads were added into the lysates to immunoprecipitate the p-AKT protein. The precipitated p-AKT beads were added into fresh KS lysate together with ATP and incubated for 2 h at 30°C. Beads were removed, and the remaining lysate was immunoprecipitated with Gli2 antibody and analyzed by Western blot with p-AKT substrate antibody as mentioned above.
RNA, microarray, and real-time PCR
RNA was harvested with Trizol according to the manufacturer's instructions (Invitrogen). RNA samples were hybridized to Affymetrix GeneChip mouse genome 430 2.0 platform at the Ohio State University Comprehensive Cancer Center Microarray Shared Resource Facility. The microarray data were deposited with Gene Expression Omnibus (GEO) and can be viewed at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=afqbqaeoxbgltor&acc=GSE61707.
Supplementary Material
Acknowledgments
We acknowledge Jason Bice, Daphne Bryant, Nicole Drummond, and Lisa Rawahneh from the Solid Tumor Biology Histology Core for their technical support. We also thank Chelsea K. Martin for assisting in initial analysis of pancreatic tumor histology. The Flag-tagged wild-type and S230A mutant vectors were kind gifts from Dr. Longxing Fan's laboratory. This study was supported by National Institutes of Health grant PO1 CA097189 (to M.C.O. and G.L.), National Research Service Award F31 CA189757 (to J.R.P.), and R01 CA124586 (to S.F.K.). This work was also supported by the Department of Defense (W81XWH-14-1-0040 to G.M.S.) and the Pelotonia Fellowship Program (J.R.P. and G.M.S).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.283499.116.
References
- Acharyya S, Sharma SM, Cheng AS, Ladner KJ, He W, Kline W, Wang H, Ostrowski MC, Huang TH, Guttridge DC. 2010. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS One 5: e12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnfelt-Ronne J, Ravassard P, Pardanaud-Glavieux C, Scharfmann R, Serup P. 2010. Mesenchymal bone morphogenetic protein signaling is required for normal pancreas development. Diabetes 59: 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. 1988. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53: 549–554. [DOI] [PubMed] [Google Scholar]
- Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al. 2012. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 22: 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, Nagy A. 2005. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res 33: e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan A, Itoh N, Kato S, Thiery JP, Czernichow P, Bellusci S, Scharfmann R. 2001. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development 128: 5109–5117. [DOI] [PubMed] [Google Scholar]
- Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ, Martin CK, Li F, et al. 2011. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol 14: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, et al. 2010. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med 2: 51ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, et al. 2011. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res 71: 435–444. [DOI] [PubMed] [Google Scholar]
- Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et al. 2013. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 23: 406–420. [DOI] [PubMed] [Google Scholar]
- Eser S, Schnieke A, Schneider G, Saur D. 2014. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 111: 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. 2012. The pancreas cancer microenvironment. Clin Cancer Res 18: 4266–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell PE, Grobstein C. 1968. The influence of extra-epithelial factors on the growth of embryonic mouse pancreatic epithelium. Exp Cell Res 53: 301–304. [DOI] [PubMed] [Google Scholar]
- Filosa S, Pictet R, Rutter W. 1975. Positive control of cyclic AMP on mesenchymal factor controlled DNA synthesis in embryonic pancreas. Nature 257: 702–705. [DOI] [PubMed] [Google Scholar]
- Gittes GK, Galante PE, Hanahan D, Rutter WJ, Debase HT. 1996. Lineage-specific morphogenesis in the developing pancreas: role of mesenchymal factors. Development 122: 439–447. [DOI] [PubMed] [Google Scholar]
- Golosow N, Grobstein C. 1962. Epitheliomesenchymal interaction in pancreatic morphogenesis. Dev Biol 4: 242–255. [DOI] [PubMed] [Google Scholar]
- Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. 2003. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res 63: 2016–2019. [PubMed] [Google Scholar]
- Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, Campuzano V, Barbacid M. 2003. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 4: 111–120. [DOI] [PubMed] [Google Scholar]
- Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, et al. 2008. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci 105: 18913–18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebrok M. 2003. Hedgehog signaling in pancreas development. Mech Dev 120: 45–57. [DOI] [PubMed] [Google Scholar]
- Hill R, Calvopina JH, Kim C, Wang Y, Dawson DW, Donahue TR, Dry S, Wu H. 2010. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res 70: 7114–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450. [DOI] [PubMed] [Google Scholar]
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al. 2010. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9: 1956–1967. [DOI] [PubMed] [Google Scholar]
- Hu MC, Mo R, Bhella S, Wilson CW, Chuang PT, Hui CC, Rosenblum ND. 2006. GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development 133: 569–578. [DOI] [PubMed] [Google Scholar]
- Jonckheere N, Mayes E, Shih HP, Li B, Lioubinski O, Dai X, Sander M. 2008. Analysis of mPygo2 mutant mice suggests a requirement for mesenchymal Wnt signaling in pancreatic growth and differentiation. Dev Biol 318: 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasaki S. 1975. Cell proliferation and subcellular localization of alkaline phosphatase activity in rat liver parenchyma during azo dye carcinogenesis. Cancer Res 35: 482–491. [PubMed] [Google Scholar]
- Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, et al. 2014. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25: 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M. 2011. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138: 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP IV, Pan FC, Akiyama H, Wright CV, Jensen K, et al. 2012. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22: 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Robertson MJ, Schaffer DV. 2004. The Sonic hedgehog signaling system as a bistable genetic switch. Biophys J 86: 2748–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon AP. 2001. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128: 5099–5108. [DOI] [PubMed] [Google Scholar]
- Miralles F, Czernichow P, Scharfmann R. 1998. Follistatin regulates the relative proportions of endocrine versus exocrine tissue during pancreatic development. Development 125: 1017–1024. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM, Wolfe MS, et al. 2003. Notch mediates TGFα-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3: 565–576. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al. 2007. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25: 1960–1966. [DOI] [PubMed] [Google Scholar]
- Morris JP IV, Wang SC, Hebrok M. 2010. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer 10: 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi K, Ijichi H, Mohri D, Tada M, Yamamoto K, Asaoka Y, Ikenoue T, Tateishi K, Nakai Y, Isayama H, et al. 2013. Erlotinib prolongs survival in pancreatic cancer by blocking gemcitabine-induced MAPK signals. Cancer Res 73: 2221–2234. [DOI] [PubMed] [Google Scholar]
- Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. 2012. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 22: 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, Hanahan D. 2009. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev 23: 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa T, Michaylira CZ, Kalabis J, Stairs DB, Nakagawa H, Andl CD, Johnstone CN, Klein-Szanto AJ, El-Deiry WS, Cukierman E, et al. 2007. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev 21: 2788–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi H, Katano M. 2014. Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J Gastroenterol 20: 2335–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. 2014. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25: 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa I, Longnecker DS, Scarpelli DG, Pour P, Reddy JK, Lefkowitz M. 1985. Ductal metaplasia of human exocrine pancreas and its association with carcinoma. Cancer Res 45: 1285–1290. [PubMed] [Google Scholar]
- Puri S, Hebrok M. 2010. Cellular plasticity within the pancreas—lessons learned from development. Dev Cell 18: 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regl G, Neill GW, Eichberger T, Kasper M, Ikram MS, Koller J, Hintner H, Quinn AG, Frischauf AM, Aberger F. 2002. Human GLI2 and GLI1 are part of a positive feedback mechanism in basal cell carcinoma. Oncogene 21: 5529–5539. [DOI] [PubMed] [Google Scholar]
- Reichert M, Rustgi AK. 2011. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest 121: 4572–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmers N, Anderson JM, Linde EM, DiMaio DJ, Lazenby AJ, Wandall HH, Mandel U, Clausen H, Yu F, Hollingsworth MA. 2013. Aberrant expression of mucin core proteins and o-linked glycans associated with progression of pancreatic cancer. Clin Cancer Res 19: 1981–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. 2014. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25: 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronzio RA, Rutter WJ. 1973. Effects of a partially purified factor from chick embryos on macromolecular synthesis of embryonic pancreatic epithelia. Dev Biol 30: 307–320. [DOI] [PubMed] [Google Scholar]
- Rutter WJ, Wessells NK, Grobstein C. 1964. Control of specific synthesis in the developing pancreas. Natl Cancer Inst Monogr 13: 51–65. [PubMed] [Google Scholar]
- Schonhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, Eser S, Feyerabend TB, Paul MC, Eser P, et al. 2014. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat Med 20: 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, deRooij DG, Hirsch S, Ravi K, Hicks JB, Szabolcs M, Jasin M, Baer R, Ludwig T. 2011. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334: 525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, DiRenzo D, Qu C, Barney D, Miley D, Konieczny SF. 2013. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 32: 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chen J, Karner CM, Long F. 2015. Hedgehog signaling activates a positive feedback mechanism involving insulin-like growth factors to induce osteoblast differentiation. Proc Natl Acad Sci 112: 4678–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff S, Rashid A, Wang H, Katz MH, Abbruzzese JL, Fleming JB, Wang H. 2014. SOX9: a useful marker for pancreatic ductal lineage of pancreatic neoplasms. Hum Pathol 45: 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, Thompson JD, Cheng JH, Bou Reslan H, Ho CC, et al. 2010. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models Nat Biotechnol 28: 585–593. [DOI] [PubMed] [Google Scholar]
- Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. 1988. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 16: 7773–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Derynck R, Korc M. 1987. Production of transforming growth factor α in human pancreatic cancer cells: evidence for a superagonist autocrine cycle. Proc Natl Acad Sci 84: 7567–7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SY, Gannon M, Washington MK, Scoggins CR, Meszoely IM, Goldenring JR, Marino CR, Sandgren EP, Coffey RJ Jr, Wright CV, et al. 1999. Expansion of Pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor α. Gastroenterology 117: 1416–1426. [DOI] [PubMed] [Google Scholar]
- Stafford D, White RJ, Kinkel MD, Linville A, Schilling TF, Prince VE. 2006. Retinoids signal directly to zebrafish endoderm to specify insulin-expressing β-cells. Development 133: 949–956. [DOI] [PubMed] [Google Scholar]
- Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, et al. 2005. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci 102: 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, Creasap N, Rosol TJ, Robinson ML, Eng C, et al. 2008. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res 68: 937–945. [DOI] [PubMed] [Google Scholar]
- Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, et al. 2009. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 461: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulachan SS, Doi R, Hirai Y, Kawaguchi Y, Koizumi M, Hembree M, Tei E, Crowley A, Yew H, McFall C, et al. 2006. Mesenchymal epimorphin is important for pancreatic duct morphogenesis. Dev Growth Differ 48: 65–72. [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Zhu L, Gopinathan A, Willis NA, Kachatrian L, Grochow R, Pin CL, Mitin NY, Taparowsky EJ, Gimotty PA, et al. 2006. Mist1–KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res 66: 242–247. [DOI] [PubMed] [Google Scholar]
- Varjosalo M, Taipale J. 2008. Hedgehog: functions and mechanisms. Genes Dev 22: 2454–2472. [DOI] [PubMed] [Google Scholar]
- Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et al. 2009. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 361: 1164–1172. [DOI] [PubMed] [Google Scholar]
- Wei D, Wang L, Yan Y, Jia Z, Gagea M, Li Z, Zuo X, Kong X, Huang S, Xie K. 2016. KLF4 is essential for induction of cellular identity change and acinar-to-ductal reprogramming during early pancreatic carcinogenesis. Cancer Cell 29: 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL, Bakst R, Hurlbert MS, Ruggiero J, Ahn E, Lee WS, Stephen D, Zagzag D, Joyner AL, Turnbull DH. 2002. Induction of medulloblastomas in mice by Sonic hedgehog, independent of Gli1. Cancer Res 62: 6385–6389. [PubMed] [Google Scholar]
- Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, et al. 2009. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326: 572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Gulati P, Fernandez S, Pennell M, Kirschner L, Jarjoura D. 2011. Fully moderated T-statistic for small sample size gene expression arrays. Stat Appl Genet Mol Biol 10.2202/1544-6115.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zha X, Tan Y, Hornbeck PV, Mastrangelo AJ, Alessi DR, Polakiewicz RD, Comb MJ. 2002. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J Biol Chem 277: 39379–39387. [DOI] [PubMed] [Google Scholar]
- Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. 2007. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol 171: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.