

ABSTRACT
Mountain ecosystems are characterized by a diverse range of climatic and topographic conditions over short distances and are known to shelter a high biodiversity. Despite important progress, still little is known on bacterial diversity in mountain areas. Here, we investigated soil bacterial biogeography at more than 100 sampling sites randomly stratified across a 700-km2 area with 2,200-m elevation gradient in the western Swiss Alps. Bacterial grassland communities were highly diverse, with 12,741 total operational taxonomic units (OTUs) across 100 sites and an average of 2,918 OTUs per site. Bacterial community structure was correlated with local climatic, topographic, and soil physicochemical parameters with high statistical significance. We found pH (correlated with % CaO and % mineral carbon), hydrogen index (correlated with bulk gravimetric water content), and annual average number of frost days during the growing season to be among the groups of the most important environmental drivers of bacterial community structure. In contrast, bacterial community structure was only weakly stratified as a function of elevation. Contrasting patterns were discovered for individual bacterial taxa. Acidobacteria responded both positively and negatively to pH extremes. Various families within the Bacteroidetes responded to available phosphorus levels. Different verrucomicrobial groups responded to electrical conductivity, total organic carbon, water content, and mineral carbon contents. Alpine grassland bacterial communities are thus highly diverse, which is likely due to the large variety of different environmental conditions. These results shed new light on the biodiversity of mountain ecosystems, which were already identified as potentially fragile to anthropogenic influences and climate change.
IMPORTANCE This article addresses the question of how microbial communities in alpine regions are dependent on local climatic and soil physicochemical variables. We benefit from a unique 700-km2 study region in the western Swiss Alps region, which has been exhaustively studied for macro-organismal and fungal ecology, and for topoclimatic modeling of future ecological trends, but without taking into account soil bacterial diversity. Here, we present an in-depth biogeographical characterization of the bacterial community diversity in this alpine region across 100 randomly stratified sites, using 56 environmental variables. Our exhaustive sampling ensured the detection of ecological trends with high statistical robustness. Our data both confirm previously observed general trends and show many new detailed trends for a wide range of bacterial taxonomic groups and environmental parameters.
INTRODUCTION
The ecology of mountain grassland ecosystems has been extensively studied by macroecologists but less so by microbial ecologists (1). In pioneering studies, King and coworkers (2) analyzed the bacterial habitat distribution along a mountain slope. Most authors have studied soil grassland bacterial diversity on a single or a few mountain transects, notably to emphasize the elevation factor in mountain ecosystems (3–8). However, alpine studies that span an important horizontal surface area, along with the vertical elevation gradient, are much fewer, despite that mountain ecosystems are composed of a large spectrum of different topographic, climatic, soil physical, and chemical conditions, interspersed with a wide range of microclimates occupied by varied vegetation and animal populations (2, 9–12).
In general, soil pH has been identified as the strongest single abiotic parameter influencing soil bacterial diversity among 1,000 soil cores across Great Britain and the American continent (13, 14). This trend was also found in a number of other soil-related mountain bacterial studies in the Colorado Rockies, Tibetan Plateau, the European Alps, and the Gorbeia Natural Park in Spain (2, 5, 8, 10, 15, 16). Other environmental drivers of importance to grassland soil bacterial communities in mountain ecosystems include annual radiation, mean annual temperature, soil ammonium content, litter C:N ratio, snow depth, and plant diversity (2, 5–8, 10, 17).
The effect of elevation in shaping soil bacterial communities has remained controversial. Mostly, elevation in itself does not appear to affect bacteria (18), but other drivers, such as mean annual temperature and mean annual precipitation, that are influenced by elevation have been shown to impact soil bacteria in mountain ecosystems (5, 17). Furthermore, the soil composition can change over an elevation gradient due to the geological and/or land use history of mountain areas, giving each mountain ecosystem a unique geochemical profile that shapes the local endemic bacterial communities. For instance, the soil bacterial communities along the Changbai Mountain in China were driven by the soil total carbon and nitrogen, the C:N ratio, and dissolved organic carbon, which changed across the elevation strata (19), whereas in the Colorado Rockies, soil pH and elevation were strongly negatively correlated with each other (3). Elevational differences may also relate to human impact, as was shown in a study by Marini and coworkers (20) on the effect of farm size in the Italian Alps. In addition, Geremia and coworkers (10) showed that anthropogenically managed alpine grasslands had different soil bacterial communities than those of natural alpine grasslands. These examples show that observed ecological trends may be difficult to systematically generalize for all alpine soil bacterial communities, notably as a function of elevation. It is therefore important to study the ecological dynamics associated with mountain areas within their local environmental conditions and constraints.
Bacteria have been repeatedly shown to be crucial players in ecosystem functioning (21–23). Given the importance yet functional fragility of mountain ecosystems (24, 25), it is important to more comprehensively study and understand the environmental drivers of bacterial diversity across a wider spatial scale in such ecosystems, across both large horizontal and vertical gradients. This may be advantageous for follow-up studies that attempt to predict the effects of changing land usage and climatic conditions. The focus area of our study is a unique 700-km2 region in the western Swiss Alps covering altitudinal differences between 500 and 3,000 m, which has been intensively characterized for plant and fungal diversity patterns during a number of years but with unknown bacterial diversity (1) (http://rechalp.unil.ch).
The goal of our study was to characterize the main bacterial diversity patterns in the alpine grassland soils across the study area using high-throughput sequencing and taxonomic inference of amplified 16S rRNA gene fragments. More than 100 randomly stratified sampling sites were selected, which were further characterized for a wide range of 56 local climatic and soil physicochemical parameters, in order to detect ecological trends with high confidence. Our hypothesis was that mountainous grasslands, because of the large elevational and microclimatic differences, would harbor a wide beta diversity, correlating to a large number of environmental variables instead of only a few major parameters. Although bacterial biogeographic trends across mountain ecosystems have been reported before, the strength of our study is the well-conceived randomly stratified design and high sample density in an otherwise well-characterized alpine area. Indeed, we report many new detailed ecological trends at the bacterial taxon level while confirming previously observed global effects, such as that of pH.
MATERIALS AND METHODS
Sample collection, manipulation, and soil physicochemical analysis. (i) Sample collection, transport, and storage.
The entire 700-km2 study region is located in the western Swiss Alps (Fig. 1). Two sampling campaigns were conducted. The first campaign was conducted during September and October 2011 across an elevation gradient of 500 to 2,000 m in order to validate the overall protocol (including sampling, storage during transport, extraction, and data analysis) on 15 samples. The second campaign took place during summer 2012 across an elevation gradient of 800 to 3,000 m using a balanced random stratification design (26), where the elevation strata were divided into segments of 250 m (Fig. 1). Samples from a total of 105 sites were collected during the months of July to October 2012. For each sampling site, a global positioning system (GPS) was used to find the exact coordinates of the plot, and a premeasured rope was then used to delimit the 2- by 2-m plot that was centered on the GPS coordinates and aligned along the steepest slope. The tools used for collecting soil samples were either presterilized or sterilized on the field prior to each collection using ethanol and a portable butane-based lighter. After removing aboveground plant material, ant nests, and visible insects, 100 g of soil was collected from the 0- to 5-cm-depth layer from each of four corners and the center of the 2- by 2-m plot and deposited into sterile plastic bags (giving a total of 500 g per site). Soil samples were homogenized on site by hand massaging the sterile plastic bag containing the soil, after which the sample was immediately divided in two subsets. One small subset was transferred to a sheet of sterile aluminum foil that had been previously heated at 500°C for 4 h, wrapped tightly, bagged in a ziplock bag, and flash-frozen in liquid nitrogen directly at the field site before being transported to the lab, where the samples were stored at −80°C until further processing for physicochemical analyses. The remainder of the soil sample was stored on ice during the transport and at 4°C at the field lodge. They were processed within 36 h in the laboratory for DNA extraction and further physicochemical analyses. The temperature of the air and the soil at a depth of 5 cm were measured on site. Weather conditions during sampling were reported and further complemented with a list of 15 topographic and climatic recordings across the site (27, 28).
FIG 1.

Map of study area in western Switzerland (base maps reproduced with the permission of swisstopo [BA16075]). The sampling sites were chosen according to a balanced random stratified design along a wide elevation range. The location of the sites are color-coded according to the elevation stratum in which they belong. Site numbers correspond to the numbering system referred to in other studies done within the study area (e.g., Ndiribe et al. [70] and Pellissier et al. [69]).
(ii) Fresh sample processing.
In the laboratory, a subset of each fresh soil sample was sieved through an ethanol and flame-sterilized 2-mm sieve, and 0.25-g aliquots of soil were transferred to sterile microcentrifuge tubes and stored on ice until DNA extraction. The remaining bulk soil was conserved in the cold room at 4°C. Within a week, more soil was sieved through the 2-mm mesh sieve and was weighed to obtain the initial weight of the soil. The samples were then dried at 105°C for 48 h and reweighed to obtain the gravimetric sieved soil water content, calculated as (weight of the wet soil − weight of the dried soil)/weight of the dried soil. Another subset of each soil was dried without prior manipulation at 40°C until constant weight for determination of the bulk soil water content. pH and electrical conductivity were measured on the samples that were dried at 40°C, which were sandwiched between two clean sheets of paper, disaggregated using a kitchen roller, and sieved through a 2-mm sieve according to the U.S. Department of Agriculture (USDA)-National Resources Conservation Service (NRCS) protocols (29). The resulting fine soil was mixed with Milli-Q water at a ratio of 1:2.5 (wt/vol) and mixed every 10 min for 1 h with a glass rod before the pH was measured in the suspension, similarly to Dubuis et al. (28). More Milli-Q water was then added to the soil slurry and mixed to obtain a 1:5 (wt/vol) ratio, and the electrical conductivity was measured.
(iii) Flash-frozen sample processing.
Flash-frozen soil samples were stored at −80°C and were used for the determination of 41 soil physicochemical properties (A. Buri, C. Cianfrani, T. Adatte, E. Pinto-Figueroa, J. Spangenberg, E. Yashiro, E. Verrecchia, A. Guisan, and J.-N. Pradervand, submitted for publication) as follows. While the soil samples were still frozen, they were initially freeze-dried on a Lyovac GT2 freeze dryer (SRK Systemtechnik GmbH, Goddelau, Germany). Dried samples were then transferred under sterile conditions into glass jars and stored at −20°C. Before analysis, the soil was sieved through a 2-mm mesh sieve and ground into powder. A subset of each sample was also decarbonized with 10% HCl. Total phosphorus content was determined by colorimetric analysis after mineralization at 550°C with Mg(NO3)2. The characterization of bulk organic matter, including mineral carbon, total organic carbon (TOC), oxygen index, hydrogen index, and temperature of maximal pyrolytic yield, was done using a Rock-Eval 6 machine (Vinci Technologies, Rueil-Malmaison, France) with a standard whole-rock pyrolysis method (31). Nitrogen, carbon, and hydrogen content analyses were performed using a Carlo Erba CNS2500 CHN elemental analyzer coupled with a Fisons Optima mass spectrometer (32). The carbon and nitrogen isotope compositions were determined using a Carlo Erba 1108 elemental analyzer connected to a Thermo Fisher Scientific Delta Plus V isotope ratio mass spectrometer via a Confo III interface (33). The major element composition was defined using an X-ray fluorescence analysis, and X-ray diffraction analysis was done to determine the soil mineralogy. Finally, the soil organic matter content was measured by loss of ignition (LOI) at 1,050°C. For a complete list of environmental parameters, see Table S1 in the supplemental material.
DNA extraction, manipulation, and sequencing.
DNA was extracted from 0.25 g of soil in triplicate for each sample using the PowerSoil DNA isolation kit (Mo Bio Laboratories, Carlsbad, CA, USA), according to the manufacturer's instructions, except that the bead beating was done in a FastPrep cell disrupter model FP120 at the speed setting of 5.0 for 45 s. The extracted DNA quality was assessed on a 1% agarose electrophoresis gel, and extensively sheared samples were discarded. If needed, another 0.25 g of soil was reextracted to have triplicate DNA extractions for each soil sample. However, for those soil samples with extensively sheared sets of DNA extractions, the entire soil sample was discarded from further processing and downstream analyses. Triplicate extractions were then pooled per individual sample.
Bacterial community analysis was adapted for a protocol using HiSeq Illumina sequencing of a PCR-amplified hypervariable region of the 16S rRNA gene, whose amplicon length was short enough to overlap the 100-bp paired-end generated reads. Primers from nine studies covering the short V3, V5, and V6 regions were queried in silico against the Ribosomal Database Project database (version 10.27) to obtain the proportion of reads that matched the primers. The best-matching primer set (784DEG and 880RDEG) (34) (see Table S1 in the supplemental material) covered the V5 hypervariable region of the 16S rRNA gene and is similar to those published by Lazarevic and coworkers (34). Barcrawl (35) was used to generate 3- to 6-base barcodes to append to the forward and reverse primer, resulting in a total of 39 differently barcoded primer pairs. We used staggered barcode lengths, because the HiSeq Illumina platform requires a large variability in bases at the 5′ end of the DNA strands in order to distinguish the DNA clusters on the flow cell during the cluster calling stage (36). Each pooled sample DNA was amplified in quadruplicate in 50-μl reaction volumes containing 1 μl of DNA, 10 μl of 5× PrimerSTAR buffer, 4 μl of dinucleoside triphosphate (dNTP) mixture (200 μM each), 2.5 μl of barcoded primer (10 μM each), 1 μl of bovine serum albumin (0.2 mg · ml−1; Promega, Madison, WI, USA), and 1.25 U of PrimerSTAR high-sensitivity (HS) DNA polymerase (TaKaRa, Shiga, Japan). The DNA amplification conditions in the Applied Biosystems GeneAmp PCR system model 9700 were 98°C for 2 min and then 30 cycles of 98°C for 1 min, 51°C for 5 s, and 72°C for 20 s. Products from replicate reactions were combined, and the DNA quality was assessed by electrophoresis on a 3% agarose gel. Products (∼125 bp) were purified with the MinElute 96 ultrafiltration (UF) PCR purification kit (Qiagen, Hilden, Germany) and quantified using the Quant-iT assay kit (Life Technologies, Carlsbad, CA, USA). These purified and quantified PCR products from individual samples were pooled, and libraries were prepared with the TruSeq DNA sample preparation kit (Illumina, San Diego, CA, USA), which were sequenced on an Illumina HiSeq 2500 platform according to the manufacturer's instructions for 100-bp paired-end reads (Illumina) at the Genomic Technology Facility of the University of Lausanne.
Data analysis.
The paired-end Illumina sequences were demultiplexed and quality filtered according to both the adapter barcodes and the custom primer barcodes. Notably, only reads with the exact sequence (no mismatches) of the 5′-to-3′ index three-part combined barcodes were kept, and obvious chimeras, such as those containing multiple forward or reverse primers, were discarded. For those reads whose amplicons were short enough that the 3′ region overlapped and/or went beyond the reverse primer sequence, the reverse primer region and everything downstream were deleted. Consensus sequences of paired-end reads (of between 70 and 184 bp, with a mean of 110 bp after removing primer barcodes) were then generated with PANDAseq using the following parameters: pandaseq -f forward.fastq -r reverse.fastq -F -N -o 5 -t 0.6 -l 70 -d bfsrk, where a consensus region of >5 bp was chosen as a parameter due to the short Illumina reads and relatively large heterogeneity in the hypervariable region length (37). In QIIME (version 1.4.0 for the 2011 data set and 1.7.0 for the 2012 data set [38]), the sequences were quality filtered using the split_libraries_fastq.py, with default parameters except for the barcode length of 18, and then clustered into operational taxonomic units (OTUs) at the 97% similarity threshold using the gg_13_8 database from Greengenes as a reference using the pick_closed_reference_otus.py (39). A strict reference-based OTU selection approach was used in order to keep only reads that had the highest probability of being valid bacteria, i.e., within 3% similarity to known bacteria in the curated reference database, and in order to filter out anomalous reads (see, e.g., Caporaso et al. [38]). Resulting OTU tables were then used for downstream analyses in QIIME (alpha diversity, adonis statistic, and weighted and unweighted UniFrac matrices) and R (core, vegan, and ade4 packages [http://www.r-project.org/] [40, 41]). Custom scripts were written for further statistical analyses and data manipulations. The alpha-diversity indices were also generated in QIIME, notably the Simpson's diversity index, Pielou's evenness, which is derived from Shannon's diversity, and Good's coverage. The Good's nonparametric coverage estimator calculates the proportion of the population that is represented by the sample, calculated as 1 − (singleton OTUs/number of reads) (42, 43). For linear model analyses, the adjusted R2 is reported. The adonis statistic was applied on the weighted UniFrac distance matrix. The adonis statistic is a nonparametric permutation-based approach for multivariate analysis of variance (44), which essentially allows a calculation of the proportion of the variation in a community matrix (e.g., the weighted UniFrac dissimilarity matrix) that is explained by the environmental variables of interest. The nonmetric multidimensional scaling (NMDS) ordination plot was generated from the weighted UniFrac distance matrix (45), and the effect of the environmental variables was overlaid with the R function envfit() (40). The NMDS ordination of the weighted UniFrac matrix was used in the ade4 package to calculate the between-class analysis (41). The abundance versus occupancy plot was used to identify specialist and generalist members of the bacterial communities (46). For the cooccurrence network analysis, the Spearman's rank correlation coefficients were generated from the environmental parameters and bacterial OTUs, and the P values were corrected for type I error with the false-discovery rate (FDR) statistic (47). Spearman instead of Pearson correlation was used, because some of the environmental parameters did not vary in a linear fashion relative to other parameters and bacteria but displayed other types of monotonic relationships. In order to reduce the complexity of the correlation network and to remove rare and putatively transient phylotypes that could increase the proportion of spurious correlations, the bacterial OTU table was first filtered of OTUs containing <100 sequences. The remaining 3,589 OTUs were then divided into relatively equal-sized subsets: OTUs that were present in <60 sites, and OTUs that were present in at least 60 sites, comprising 1,706 and 1,883 OTUs, respectively. The actual midpoint for obtaining equal-sized subsets of OTUs was 62 sites (with 1,797 and 1,792 OTUs, respectively), but the final cutoff was rounded to the nearest 10th. The final edge list of the networks containing both bacterial and environmental parameters included only those correlation coefficients that were ±0.75 or above and with an FDR-corrected P value of <0.01 for the global networks, and ±0.7 or above and with an FDR-corrected P value of <0.01 for the networks containing only the direct correlations between bacterial OTUs and environmental parameters. The network visualization software Gephi 0.8.2 was used to explore the resulting correlation network (48). The correlations were examined using the YifanHu multilevel algorithm (49). The largest cluster of the global network containing the more commonly found OTUs was further examined using the ForceAtlas 2 algorithm (50). The modularity and other network-related statistics were also generated in Gephi.
Accession number(s).
Sample reads from the 2011 and 2012 sampling campaigns, respectively, were submitted to the NCBI Sequence Read Archive under BioProject numbers PRJNA327018 and PRJNA327017.
RESULTS
Workflow optimization.
In order to determine the method of soil preparation for DNA extraction that least perturbs the soil bacterial communities sampled, two methods were tested during the 2011 field campaign. In the first method, DNA was extracted from fresh soil samples within 36 h from collection in the field, whereas in the second method, DNA was extracted from air-dried samples, as described previously (1). Sequences of the amplified V5 region of the 16S rRNA gene from all of the samples were normalized to the sample containing the lowest number of reads (i.e., 164,585). The overall mean richness, Simpson's diversity, equitability, and Good's coverage across all the samples were 3,063 (standard deviation [SD], 497.1) OTUs, 0.992 (SD, 0.003), 0.733 (SD, 0.03), and 0.995 (SD, 0.001), respectively. Unweighted UniFrac distance analysis showed no difference in community membership between air-dried and fresh samples (see Fig. S1A in the supplemental material). In contrast, the weighted UniFrac distance metric indicated that the community structures were more similar between replicates of DNA extracted from fresh samples from a single site than from the dried soil samples (Fig. S1B). We concluded that extracting the DNA within 36 h would better conserve overall bacterial diversity than extracting it from air-dried soil samples.
Global site analysis.
Samples from 105 sites were obtained during summer 2012 in the 700-km2 study region in the western Swiss Alps, across an elevation gradient of 800 to 3,000 m (Fig. 1). Of these, samples from 100 sites yielded DNA of adequate quality for further manipulations and sequencing. The amount of sequence reads per sample varied between 99,618 and 3,439,625 reads. For downstream comparative analysis, they were normalized to 99,618 (randomly subsampled) reads per sample. Good's coverage values for read-normalized samples were in the order of 0.990 to 0.995, indicating that overall diversity of the communities is adequately represented.
A total of 62 phyla were represented within the study region, with 10 phyla being more abundant than 1%, with soil-typical high proportions of Acidobacteria and Actinobacteria, as well as Proteobacteria and Bacteroidetes (Table 1 and Fig. 2A). The total richness across the whole study area was estimated at 12,741 OTUs (at 97% identity threshold), with a mean community richness of 2,918 OTUs per site (Fig. 2B). Thereby, the total alpine grassland bacterial community was very rich but within the standard reported in soil for multimillion-read data sets (e.g., see references 51 and 52). Simpson's diversity, evenness, and Good's coverage were homogeneously distributed across the study area and were not affected by latitudinal or longitudinal gradients (see Fig. S2 in the supplemental material). Mean diversity indices were similar for both 2011 and 2012 samples, thereby suggesting good consistency in the methodology. Given that Good's coverage calculates the proportion of the total community at a site that is represented by the normalized sample, the very high-average Good's coverage of almost 1.000 (mean coverage of 0.995 and 0.991 for years 2011 and 2012, respectively) also explains why the mean diversity indices between the two data sets were relatively similar despite the different numbers of reads per sample at which they were normalized.
TABLE 1.
Average taxonomic composition of the alpine grassland soil bacterial communities
| Phyluma | Proportion (%) (mean ± SD)b | Minimum (%) | Maximum (%) |
|---|---|---|---|
| Acidobacteria | 21.00 ± 8.5 | 6.8 | 52.4 |
| Actinobacteria | 11.30 ± 6.0 | 2.1 | 37.6 |
| Bacteroidetes | 7.50 ± 4.0 | 0.8 | 22.8 |
| Chloroflexi | 2.10 ± 1.5 | 0.1 | 7.6 |
| Firmicutes | 0.80 ± 0.6 | 0.0 | 3.0 |
| Gemmatimonadetes | 1.10 ± 0.9 | 0.2 | 4.8 |
| Nitrospirae | 2.40 ± 1.7 | 0.0 | 6.9 |
| Planctomycetes | 1.50 ± 0.6 | 0.3 | 3.9 |
| Proteobacteria | 41.00 ± 7.3 | 21.8 | 63.1 |
| Alphaproteobacteria | 15.40 ± 3.5 | 8.2 | 25.2 |
| Betaproteobacteria | 14.60 ± 5.7 | 3.5 | 30.5 |
| Deltaproteobacteria | 5.50 ± 1.7 | 2.5 | 13.9 |
| Gammaproteobacteria | 5.40 ± 2.3 | 1.7 | 11.3 |
| Verrucomicrobia | 7.70 ± 4.0 | 1.7 | 19.0 |
| WS3 | 1.30 ± 0.9 | 0.0 | 3.6 |
Only the phyla that were represented in at least 0.8% of the total community after normalizing all of the samples to 99,618 reads per sample are shown. Phyla were attributed from the OTUs at a similarity threshold of 97%.
Calculated as the arithmetic mean proportion across 100 samples from 100 sites.
FIG 2.

Distribution of the bacterial communities across the study area. (A) Taxonomic profile of the bacterial communities grouped by phylum. The sites are ordered by their respective soil pH, from lowest to highest pH. (B) OTU richness across the study site. The mean richness was 2,918.08 OTUs (SD, 429.08). See Fig. S3 in the supplemental material for plots of Good's coverage, Simpson's diversity, Faith's phylogenetic diversity, and Pielou's equitability across the study area. (Base map reproduced with the permission of swisstopo [BA16075].)
Effect of environmental parameters on the bacterial community structure and diversity.
A total of 56 climatic, topographical, soil chemical, and physical parameters were determined for every site (Table 2; see also Table S2 in the supplemental material). Correlation analysis indicated clustering and interdependence for a large number of those parameters (see Fig. S3 in the supplemental material). Of the correlated groups, pH explained the highest proportion of the variability in the bacterial community structure in the alpine soils (R2 = 34.27%, Table 2), followed by the hydrogen index (HI; R2 = 15.48%) and the annual average number of frost days during the growing season (sfroyy; R2 = 15.23%). Although many environmental variables were cross-correlated, they exhibited different degrees of covariation with the bacterial community structure. For example, within the pH-correlated variable cluster, soil calcium oxide (CaO; R2 = 15.04%), mineral carbon (MiC; R2 = 13.64%), and silicon dioxide (SiO; R2 = 12.68%) contents also explained a relatively large proportion of the bacterial community structure, but organic matter, quartz, and bulk carbon contents explained only 3% of the variability (Table 2). The hydrogen index (HI) cluster was further dominated by soil water content (R2 = 8.4%) and soluble phosphorus (R2 = 6.43%). Within the annual average number of frost days during the growing season (sfroyy) cluster, the number of precipitation days per growing season (pday; R2 = 11.24%) and elevation itself (Alt; R2 = 10.51%) further explained the highest proportion of the variability in the bacterial community structure (Table 2). The global effect of the environmental variables on community diversity across the study area is further illustrated by the NMDS ordination of the data (Fig. 3). The envfit analysis showed that even among cross-correlated variables, not all are pointing in the exact same direction (Fig. 3).
TABLE 2.
Effect of environmental parameters on soil bacterial community structure
| Environmental parametera | Autocorrelation group no.b | R2c | P value | FDR-corrected P valued |
|---|---|---|---|---|
| pH | 1 | 34.27 | 0.001 | 0.003 |
| CaO | 1 | 15.04 | 0.001 | 0.003 |
| Mineral carbon | 1 | 13.64 | 0.001 | 0.003 |
| SiO2 | 1 | 12.68 | 0.001 | 0.003 |
| Annual avg site water balance accounting for soil properties (0.1 mm · yr-−1) | 1 | 11.07 | 0.001 | 0.003 |
| Calcite (%) | 1 | 9.62 | 0.001 | 0.003 |
| Bulk C:N ratio | 1 | 7.41 | 0.001 | 0.003 |
| Nitrogen stable isotope ratio (per ml vs air-N2) | 1 | 6.43 | 0.001 | 0.003 |
| Organic matter content via loss of ignition | 1 | 3.96 | 0.002 | 0.004 |
| Quartz (%) | 1 | 3.28 | 0.013 | 0.021 |
| Ankerite (%) | 1 | 2.20 | 0.052 | 0.073 |
| Bulk carbon content | 1 | 1.52 | 0.156 | 0.182 |
| Hydrogen index (mg hydrocarbons · g−1 TOC) | 2 | 15.48 | 0.001 | 0.003 |
| Bulk soil gravimetric water content (dried at 40°C) (%) | 2 | 8.40 | 0.001 | 0.003 |
| Soluble phosphorus content (mg · kg [DW]−1) | 2 | 6.53 | 0.002 | 0.004 |
| Sieved soil gravimetric water content (dried at 105°C) (%) | 2 | 5.15 | 0.002 | 0.004 |
| Bulk hydrogen content | 2 | 4.37 | 0.006 | 0.012 |
| Bulk nitrogen content | 2 | 3.67 | 0.013 | 0.021 |
| Oxygen index (mg CO2 · g−1 TOC) | 2 | 3.43 | 0.008 | 0.014 |
| Total organic carbon content | 2 | 3.12 | 0.012 | 0.021 |
| Annual avg no. of frost days during the growing season (days × 100) | 3 | 15.23 | 0.001 | 0.003 |
| No. of precipitation days per growing season (day) | 3 | 11.24 | 0.001 | 0.003 |
| Elevation (m) | 3 | 10.51 | 0.001 | 0.003 |
| Monthly avg temp (°C × 100) | 3 | 9.81 | 0.001 | 0.003 |
| Carbon stable isotope ratio (per ml vs VPDB) | 3 | 9.73 | 0.001 | 0.003 |
| Monthly mean precipitation sum (mm) | 3 | 9.60 | 0.001 | 0.003 |
| Monthly moisture index (0.1 mm · mo−1) | 3 | 9.32 | 0.001 | 0.003 |
| Final latitude | 3 | 9.14 | 0.001 | 0.003 |
| Annual degree days (day × °) | 3 | 8.76 | 0.001 | 0.003 |
| Soil temp (−5 cm) (°C) | 3 | 6.14 | 0.001 | 0.003 |
| Topographic position | 3 | 2.90 | 0.022 | 0.034 |
| NiO | 4 | 9.91 | 0.001 | 0.003 |
| MgO | 4 | 6.17 | 0.002 | 0.004 |
| Phyllosilicates (%) | 4 | 5.07 | 0.003 | 0.006 |
| Na2O | 4 | 4.23 | 0.003 | 0.006 |
| TiO2 | 4 | 2.06 | 0.067 | 0.092 |
| Al2O3 | 4 | 1.65 | 0.132 | 0.161 |
| Fe2O3 | 4 | 0.80 | 0.574 | 0.618 |
| K2O | 4 | 0.74 | 0.619 | 0.642 |
| Cr2O3 | 4 | 0.59 | 0.728 | 0.728 |
| Electrical conductivity (1:1 μS · cm−1) | 5 | 6.67 | 0.001 | 0.003 |
| MnO | 6 | 3.51 | 0.007 | 0.013 |
| Plagioclase-Na (%) | 7 | 3.05 | 0.017 | 0.027 |
| Daily avg global potential shortwave radiation per mo (kJ · day−1) | 8 | 2.67 | 0.037 | 0.055 |
| Sine-transformed direction that a slope faces (° · %−1) | 8 | 2.50 | 0.026 | 0.039 |
| Terrain slope (°) | 9 | 2.34 | 0.04 | 0.057 |
| Direction that a slope faces (° · %−1) | 10 | 2.00 | 0.078 | 0.104 |
| Final longitude | 11 | 1.90 | 0.082 | 0.107 |
| Goethite (%) | 12 | 1.85 | 0.105 | 0.134 |
| Total phosphorus content (mg · g−1) | 13 | 1.66 | 0.123 | 0.153 |
| P2O5 | 13 | 1.59 | 0.152 | 0.181 |
| Topographic wetness index | 14 | 1.31 | 0.238 | 0.272 |
| Indoses (%) | 15 | 0.97 | 0.41 | 0.459 |
| Dolomite (%) | 16 | 0.81 | 0.598 | 0.632 |
| Temp of maximal pyrolytic yield (°C) | 17 | 0.77 | 0.548 | 0.602 |
| Feldspath-K (%) | 18 | 0.68 | 0.669 | 0.681 |
Data are presented as the % (wt), unless otherwise indicated. For a full list of environmental parameters and their abbreviations, see Table S2 in the supplemental material. VPDB, Vienna Pee Dee Belemnite.
The numbers represent the different modules (groups) of autocorrelated environmental parameters derived from the Spearman's rank correlation network (coefficients >±0.5) shown in Fig. S4 in the supplemental material. The modules were calculated using the modularity statistics in Gephi 0.8.2.
Proportion of variability in the bacterial community structure explained by each environmental parameter using adonis() and the weighted UniFrac matrix as the input dissimilarity matrix.
Nonsignificant P values after FDR correction are italicized.
FIG 3.
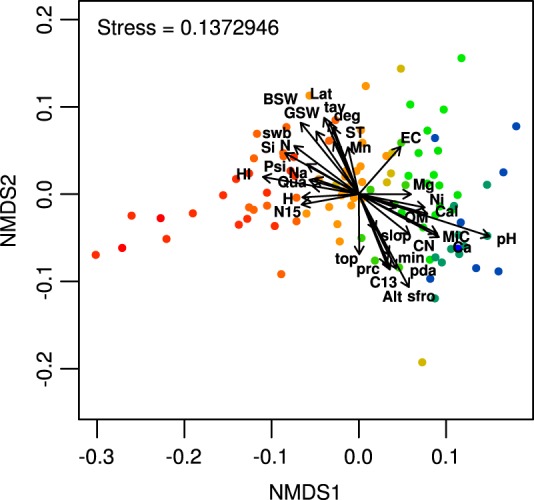
Nonmetric multidimensional scaling ordination from the weighted UniFrac dissimilarity matrix of the alpine soil bacterial communities across the first and second dimensions. The points represent the individual sites according to their pH (red, lowest; blue, highest pH). The effect and direction of environmental parameters are superimposed by arrows (origin = point 0,0). Only parameters with a P value of <0.05 using envfit() are included. BSW, bulk soil gravimetric water content; GSW, bulk soil gravimetric water content; Lat, latitude; tav, monthly average temperature; swb, annual average site water balance accounting for soil properties; ST, soil temperature at depth of −5 cm; N, bulk nitrogen content; Mn, MnO; EC, electrical conductivity; Si, SiO2; Psi, phyllosilicate; Na, Na2; Qua, quartz; Mg, MgO; H, bulk hydrogen content; Ni, NiO; N15, stable isotopic nitrogen ratio; Cal, calcite; OM, organic matter content; slop, terrain slope; MiC, mineral carbon content; CN, C:N ratio; Ca, CaO; top, topographic position; min, monthly moisture index; prc, monthly mean precipitation sum; pda, number of precipitation days per growing season; C13, stable isotopic carbon ratio; sfro, average annual number of frost days during the growing season.
Analyzed solely as a function of pH, all general bacterial diversity indices showed clear hyperbolic trends, with Simpson's diversity, Faith's phylogenetic diversity, and Pielou's equitability indices being highest at neutral pH and lower at the pH extremes (Fig. 4A to D). Good's coverage showed the inverse trend as a function of pH, with the lowest coverage at neutral pH and increasing coverage at acidic and alkaline pHs (Fig. 4D). No clear trends were visible between the hydrogen indices (HIs) and general diversity indices, although Simpson's diversity, Faith's phylogenetic diversity, and Pielou's equitability remained generally high at an HI of <300 mg of hydrocarbons · g−1 TOC, while they decreased at HI values of >300 (see Fig. S4 in the supplemental material). No distinct patterns could be discerned when the diversity indices were analyzed as a function of sfroyy (see Fig. S5 in the supplemental material). Pairs of environmental parameters themselves often displayed clear but nonlinear relationships (see Fig. S6 in the supplemental material). Finally, the bacterial alpha-diversity indices showed no significant trend as a function of elevation (Fig. 4E and F; see also Fig. S7 in the supplemental material).
FIG 4.
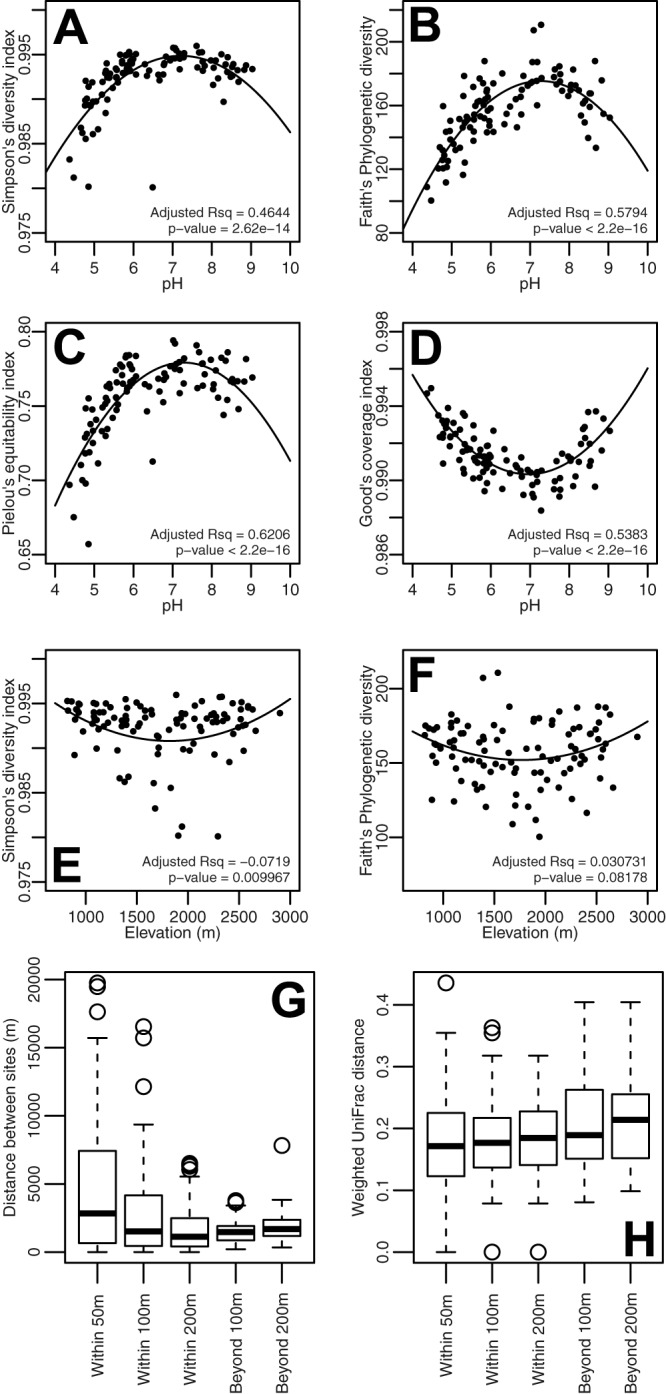
Relationship between soil bacterial diversity and soil pH (A to D) and elevation (E to H). (A and E) Simpson's diversity index. (B and F) Faith's phylogenetic diversity index. (C) Pielou's equitability index, which measures the level of evenness across the OTUs in the communities. (D) Good's coverage index. (G and H) Geographic distance (G) and weighted UniFrac community dissimilarity (H) between each query site and its closest neighboring site within the respective elevation constraints. (A to F) Points correspond to individual sites. Quadratic trend lines are shown, which were calculated using the lm() function in R. The adjusted R2 values indicate the goodness of fit of the bacterial diversity to the respective trend lines. P values indicate the significance of the overall model. (G and H) The term “within” signifies that the neighboring site is within the respective elevation difference from the query site, and the term “beyond” signifies that the neighboring site is further above or further below the indicated elevation difference from the query site. The bottom, middle, and top bands of the box and whisker plots represent the first, median, and third quartiles and the whiskers delimit the 1.5 interquartile range below and above the first and third quartiles, respectively.
Since elevation is a highly prominent component of an alpine study area, its effect on the soil bacterial communities was further investigated by multivariate approaches. Between-class analysis showed clear segregation of bacterial communities across the area as a function of pH, as expected (see Fig. S8A in the supplemental material), but no segregation as a function of elevation (250-m elevation strata; see Fig. S8B). An exception was a single cluster containing the elevation strata above 2,250 m (see Fig. S8B). This, however, is likely due to the higher soil pH at higher-located sites. Next, we determined whether beta diversity would be more dissimilar to sites outside than within elevation strata. By taking advantage of the homogeneous distribution of the sampling sites across the study area (Fig. 2B; see also Fig. S2 in the supplemental material), specific elevation strata were centered around each site while recording its distance to the closest neighboring site. With increasing vertical “width” of each elevation stratum, the median distance to the nearest site within the respective strata decreased (Fig. 4G). In contrast, the median weighted UniFrac beta dissimilarity increased with increasing elevation strata width (Wilcoxon test of medians, P < 2.2 × 10−16; Fig. 4H), indicating the highest median beta dissimilarity with sites outside the examined strata. Further testing showed that the distances between nearest neighboring sites were not significantly different when they were within 100-m elevation and beyond 200-m elevation from each other (Tukey's test adjusted, P = 0.091). Pairwise comparison of means further indicated that significantly more similar communities were found within 100 m elevation of each other than at >200-m elevation differences (adjusted P = 0.014). These tests indicated a weak but statistically significant effect of elevation on bacterial community diversity.
Effect of environmental parameters on bacterial phyla and classes.
Whereas the overall taxonomic profiles of the soil bacterial communities clearly shifted as a function of pH (Fig. 2A; see also Fig. S9 in the supplemental material), both relative abundance and richness of individual taxonomic groups correlated in different ways as a function of pH (see Fig. S10A to N in the supplemental material). For instance, acidobacterial abundance was highest at low pH and decreased at higher pH, whereas richness slightly increased as a function of increasing pH (see Fig. S10A). In contrast, Actinobacteria, Bacteroidetes, and Gemmatimonadetes relative abundance and richness increased with increasing pH (see Fig. S10B, C, and F). Chloroflexi relative abundance but not richness peaked at both pH extremes (see Fig. S10D), whereas both the relative abundance and OTU richness of Firmicutes showed opposite trends, being highest at low pH (see Fig. S10E). In contrast, among the Nitrospirae, Planctomycetes, candidate phylum WS3, Verrucomicrobia, and Proteobacteria, the OTU richness peaked at neutral pH (see Fig. S10G to N), and for Verrucomicrobia, Alphaproteobacteria, and Gammaproteobacteria, their relative abundance decreased with increasing pH (see Fig. S10I, K, and N).
Soil mineral and total organic carbon content were strong drivers for the oligotrophic Verrucomicrobia groups (see Fig. S11 in the supplemental material). Among the different verrucomicrobial groups, the Spartobacteria and Pedosphaerae were the most abundant and diverse in the alpine grassland soils (see Fig. S12 in the supplemental material), and their preferential site occupation differed according to environmental parameters (Fig. 5; see Fig. S13 in the supplemental material). Notably, the methylacidiphilae occurred preferentially in more acid soils (Fig. 5A and B), whereas Pedosphaerae abundance was driven more by soil water content (see Fig. S13B). In contrast, the spartobacterial abundance was highest at neutral pH, and its OTU richness was more evenly distributed (see Fig. S13C). The Verruco 5 and Opitutae groups were further driven by electrical conductivity (Fig. 5C and D; see also Fig. S13G and H).
FIG 5.

Distribution of verrucomicrobial class richness and abundance across ordination space, respectively, of the methylacidiphilae (A and B) and Verruco-5 (C and D). The bubble size depicts the relative richness or abundance of a verrucomicrobial class at each sampling site, while the red contours show the smooth response surface of the respective richness or relative abundance. Only the most relevant environmental variables are shown (envfit vectors).
Cooccurrence patterns of bacterial OTUs.
In order to discern specialist and generalist groups among the alpine soil bacterial communities, we examined OTU abundance versus occupancy, as described in Barberan et al. (46). In essence, this is a plot of the mean relative OTU abundance per site as a function of the number of sites with occurrence. In contrast to previous studies (Barberan et al. [46]), however, no specialist groups were found at a 97% OTU similarity cutoff among the alpine soil bacterial communities using the abundance-versus-occupancy test (see Fig. S14 in the supplemental material). As an alternative method, we built cooccurrence networks between environmental variables and OTU sets. In order to distinguish the ecology of the more and less commonly occurring OTUs, the data set was divided into two relatively equal-sized subsets. This first consisted of a subset of less commonly occurring OTUs, which were present in fewer than 60 sites (Fig. 6A), and the second was a subset of more commonly occurring OTUs, which were present in at least 60 sites (Fig. 6B). The actual midpoint number of sites for having equal numbers of OTUs in each subset was 62 sites, with 1,797 and 1,792 OTUs, respectively, but we rounded off the cutoff to 60 sites. Under the definition of a strong modular network having modularity of higher than 0.4 (53), the network with the less commonly found OTUs was weaker than that containing the more commonly found OTUs (Fig. 6A and B). To illustrate the validity of the correlation networks, one can appreciate the trends of pH dependence of Acidobacteria. Acidobacterial OTUs exclusively negatively correlating with pH in the network (Fig. 6B) belonged to the classes Acidobacteriia and DA052, which decreased dramatically in relative abundance at the class level with increasing pH (see Fig. S15 in the supplemental material). In contrast, the large majority of OTUs within Acidobacteria-6 and Chloracidobacteria (= Acidobacteria group 4 of Ribosomal Database Project) correlated positively with pH (Fig. 6B), and their relative abundances increased at higher pH (see Fig. S15), whereas the Solibacteres OTUs negatively correlated with pH, as confirmed by their class abundance data (see Fig. S15).
FIG 6.

Global cooccurrence networks of detected OTUs (dots) across all sites and environmental parameters (abbreviations), constructed using the Yifan Hu Multilevel algorithm. (A and B) Network based on the less commonly found OTUs (present in fewer than 60 sites, covering 1,706 OTUs) (A) and on the more commonly found bacterial OTUs (present in at least 60 sites, 1,883 OTUs) (B). Distinct cooccurring groups with four or more nodes are encircled and commented (e.g., pH-dependent) or given neutral indications (e.g., module A). Edge colors (lines) indicate positive (red) or negative (blue) correlations. Nodes within the same modules are given arbitrarily the same color. Shown are only those correlations with a Spearman correlation coefficient threshold of ±0.75 and FDR-corrected P value of <0.01. The average degree, i.e., the node connectivity, was 32.173 and 47.512, the average path length, i.e., the distance between all pairs of nodes, was 3.037 and 3.691, the average clustering coefficient was 0.634 and 0.630, and the modularity using default parameters in Gephi was 0.379 and 2.201, respectively.
Both networks showed a major cluster, which included as many as 566 (82.75%, Fig. 6A) and 860 (88.8%, Fig. 6B) OTU nodes, many of which correlated with pH and CaO. Other clusters with at least four nodes included OTUs correlating with the water-saturated anoxic environment (Fig. 6A), soluble phosphorus, putative nitrogen fixers, Firmicutes cluster, or elevation (Fig. 6B). A number of smaller clusters (named modules A, B, etc.) were detected as well but without clear signature or consisting solely of environmental variables. The water-saturated anoxic environment module (Fig. 6A) consisted of OTUs that were highly prevalent in mainly three sites. Field logs indicated that one site was composed of peat soil, while the two other samples came from typical but highly water-saturated prairie grasslands, where the bulk soil gravimetric water content was 56.55% for the peat site and 146.48% and 217.0% for the two prairie sites. This module included OTUs classified as strict anaerobes of the orders Syntrophobacterales, Ignavibacteriales, Chromatiales, and Desulfobacterales and members of the Methylococcales.
The soluble-phosphorus-dependent module (Fig. 6B) correlated strongly with OTUs belonging to Bacteroidetes. Within this module, a single OTU in the family Chitinophagaceae was strongly correlated with soluble phosphorus (ρ = 0.755), while the other OTUs were more moderately correlated with soluble phosphorus (ρ = 0.378 to 0.631) and highly correlated with each other (ρ > 0.75) (see Table S3 in the supplemental material). The four classes representing the Bacteroidetes in the module, the Cytophagia, Flavobacteriia, Saprospirae, and Sphingobacteriia, also had the highest richness and relative abundance among the Bacteroidetes classes found in the alpine grassland soil (see Fig. S16 in the supplemental material). However, despite a positive association of the phylum Bacteroidetes with soluble phosphorus, this trend was lost at the class level, possibly owing to the metabolic diversity of the individual subgroups (see Table S3). Moderately strong correlations reappeared at the family level and included those families associated with the soluble-phosphorus cooccurrence module (see Table S3). The putative nitrogen fixer module (Fig. 6B) was composed of one member of the order Clostridiales and eight proteobacterial OTUs, of which six OTUs belonged to the genus Geobacter, one to the family Desulfobulbaceae, and one to the genus Propionivibrio. The Firmicutes module (Fig. 6B) was made up exclusively of members of the Firmicutes, consisting of the families Bacillaceae, Planococcaceae, Exiguobacteriaceae, and an unidentified family. Finally, 15 environmental parameters did not cluster specifically with bacterial OTUs but mainly among themselves (Fig. 6A, modules B to D). The smaller bacterial modules (data not shown) were predominantly composed of single phyla, which suggests a certain level of preference for similar environmental conditions by closely related bacteria (46). One further big cluster appearing among the network of the more commonly found OTUs (module B, Fig. 6B) showed inconsistent connections, linking with OTUs that both positively and negatively correlated with pH, thereby suggesting that this is a spurious cluster.
When removing connections between OTUs and focusing only on direct correlations between OTUs and environmental parameters, pH and CaO remained the two most important variables, followed by soil bulk water content (Fig. 7). Other environmental variables that had a significant influence on bacterial OTUs included the δ13C, mineral carbon content, monthly moisture index, hydrogen index, and soluble phosphorus. The network in Fig. 7 also displays OTU nodes by their phylogenetic membership, illustrating how most bacterial phyla can be found under all conditions. Apart from the major pH-CaO-dependent cluster, five bacterial OTUs correlated with climatic parameters (annual degree days [deg], monthly moisture index [min], etc.; Fig. 7A and B), and six OTUs were significantly driven by soil mineral carbon content (MiC; Fig. 7A and B). Among those OTUs that were driven by soil mineral carbon content, there was an overrepresentation of Verrucomicrobia relative to its normal proportion within the alpine soil communities (50% of the MiC-associated OTUs, compared with a mean of 7.7% per site).
FIG 7.

Cooccurrence networks displaying only the direct correlations between or among environmental parameters (abbreviations) and bacterial OTUs (colored nodes), constructed by using the Yifan Hu multilevel algorithm, and further manually reorganized. (A and B) Network based on the less commonly found bacterial OTUs (present in fewer than 60 sites, covering 1,706 OTUs) (A), and on the more commonly found bacterial OTUs (present in at least 60 sites, 1,883 OTUs) (B). OTU (dot) colors represent phylogenetic groups, according to the color legend. OTUs negatively correlating with pH and CaO are placed in the left circle inner hemisphere, and those with positive correlations are in the right inner hemisphere. OTUs correlating exclusively with CaO are placed in the upper hemisphere outer rim, and those only correlating with pH are in the lower hemisphere outer rim. Red lines (edges) indicate positive correlations, and blue lines indicate negative correlations. Only correlations with a Spearman correlation coefficient above ±0.7 and with an FDR-corrected P value of <0.01 are shown.
DISCUSSION
Here, we studied bacterial diversity patterns among 100 sites distributed across a 700-km2 area in the western Swiss Alps covering altitudes between 800 and 3,000 m, and further teased apart the most important drivers for the observed diversity differences in terms of as many as 56 different climatic, topographic, and physicochemical parameters. The choice of such a large number of environmental parameters was justified by the large elevational, spatial, and microclimatic differences across the sites within this mountainous study area. Given this greater diversity in environmental conditions, we had hypothesized that a large number of environmental variables would be required to explain the observed bacterial diversity, at the community level, but also notably among individual subsets of the bacterial communities, which is in essence what we observed (Table 2). Our data give unprecedented insight into mountainous bacterial diversity patterns and the major driving environmental factors. Because of the large diversity of environmental conditions across the alpine landscape, several previously observed general correlations could be reinforced, such as between pH and OTU relative abundance, whereas other trends, such as number of frost days, soil mineral carbon content, soil stable isotope carbon ratio, and monthly moisture index, have not been detected in this form before. In contrast to our expectations, the effect of elevation itself on bacterial community structure was weak, albeit statistically significant (Fig. 4E to H).
The finding that pH appeared as the strongest driver of the alpine soil bacterial community structure agrees with previous research in soil bacterial biogeography, where, for instance, researchers have used terminal restriction fragment length polymorphism (TRFLP), pyrosequencing, and Illumina sequencing data to examine pH trends across Great Britain (14) and across the American continent (13, 54), as well as along the elevation gradient in China and Spain (15, 55). In contrast, it should be acknowledged that trends are more complex than pH only, as this is correlating with a group of 11 other parameters that each individually also explain significant parts of the observed diversity variation (notably, e.g., % CaO, % mineral carbon, % SiO2, and annual average site water balance accounting for soil properties; Table 2).
Bacterial communities in the alpine grassland sites showed a noticeable level of decrease in the phylogenetic diversity, especially at the higher pH extreme due to environmental filtering and phylogenetic diversification at neutral pH (Fig. 4B; see also Fig. S10 in the supplemental material). At the bacterial phylum level (and class level for Proteobacteria), many taxonomic groups displayed distinct, and often nonlinear, trends as a function of pH (see Fig. S10). This seems to be in contrast to previously observed linear relationships with pH of, notably, members of the Acidobacteria and the Gammaproteobacteria (56). Acidobacterial classes whose relative abundances drastically changed at pH extremes (e.g., Acidobacteriia and DA052 subclasses; see Fig. S15C in the supplemental material) correlated exclusively with pH in the cooccurrence networks (see Fig. S15A and B), whereas the other classes with lesser dependence on pH (e.g., Acidobacteria-6, Solibacteres, and Chloracidobacteria; see Fig. S15C) consisted of groups of OTUs that scattered both positively and negatively in relation to pH in the network (see Fig. S15A and B).
A surprising influence on community diversity was found for the hydrogen index (HI) and its correlated variables (Table 2). The decrease in overall bacterial diversity and evenness at higher values of HI (see Fig. S4 in the supplemental material) suggests that the bacterial diversity is greatest when the fresh organic matter content is medium to low in proportion to other subsets of the soil matter. HI was also positively correlated with the bulk soil water content (Spearman's ρ = 0.563; see Fig. S4). This suggests that more water-saturated soils tend to be more acidic as well as anoxic, which would in turn slow down the degradation ability of fresh organic matter by both bacterial and fungal communities, as happens in typical high-latitude wetland systems, such as fens and peat bogs (57). In contrast, the small amounts of fresh organic matter at high pH extremes are likely due to the scarcity of plant and animal diversity at the highest elevations. Notably, the western Swiss Alps is composed in large part of calcareous rock that originated from the ancient shallow seas that covered the region prior to the rise of the mountain range. As a result, the soil pH in the region is largely dependent upon the degree of presence of the bedrock near the surface. Hence, at higher elevations, the pH tended to be more alkaline because the bedrock was more exposed near or on the surface from which the soil samples were collected (see Fig. S6 in the supplemental material). The influence of these two different environmental factors on the soil fresh organic matter content likely explains the sinusoidal pattern of HI across the pH gradient.
The reason why HI explains as much as 15.48% of the variation in the bacterial community structure compared to organic matter (OM; R2 = 3.96%) might be because it measures the preferential loss of the more easily transformable fraction of the organic matter that is rich in polysaccharides, lignin, and other compounds (58, 59), as opposed to the total organic matter content (8). Despite the significant proportion of variation explained by the HI group of variables, their contribution was less clear in the network analyses (Fig. 6 and 7). Only a few OTUs belonging to the Actinobacteria, Bacteriodetes, and Deltaproteobacteria negatively correlated with the HI cluster (Fig. 7A). A single OTU belonging to the candidate phylum WS3 was negatively correlated with HI. This OTU was abundant mainly at the highest elevations where the soil pH was among the most alkaline, suggesting also the higher prevalence of the parental mountain components and scarcer topsoil. This association agrees with observations by Will and coworkers (60), who found higher abundances of WS3 members in the deeper B soil horizons compared to the organic matter-rich A horizons.
Among the HI-correlated variables, bulk soil gravimetric water (BSW) content explained a greater proportion of the variation in the bacterial community structure than the sieved soil gravimetric water content (GSW; Table 2). Most of the bacterial OTUs displayed negative correlations with soil water content. The reason for this may be that higher water content also makes a soil more anoxic. In that respect, these OTUs are likely to be aerobic organisms that would be negatively impacted under increasing anoxic conditions. Interestingly, a cooccurrence module was found among the less commonly present OTUs (Fig. 6A), containing mostly known anaerobes or putative methanotrophs and predominating at sites consisting of peat and/or with relatively high soil water contents, even though there was no specific correlation in the network to either BSW or GSW. The significant association of putative methanotrophs with anaerobic conditions is strongly indicative of active methane production in certain regions of the alpine grassland ecosystem, the activity of which could be exacerbated in a potential future with a changed climate.
Surprisingly few correlations were detected with soluble-phosphorus content or other nutrients, except for a Bacteroidetes OTU group (Fig. 6B), suggesting that members of the Bacteroidetes may be generally more responsive to phosphorus availability in the alpine soil ecosystem. Indeed, both richness and abundance of Bacteroidetes increased in response to soil soluble-phosphorus levels (see Table S3 in the supplemental material). Also, at the family level within Bacteroidetes, significant and positive correlations with soluble-phosphorus contents were detected, suggesting that the sensitivity of Bacteroidetes to phosphorus levels extends through a relatively large group of populations within the phylum. This is in agreement with other studies, which, using metabolic and metagenomic approaches, observed positive correlations in Bacteroidetes activity with available phosphorus in marine environments (61, 62). A surprisingly separate cluster of OTUs appeared in the network analysis (Fig. 6B), which contained members being related to known nitrogen-fixing bacteria (63). This suggests that bacteria within this module are independent of the types of soil nitrogen contents that were measured for the current study. Indeed, the total soil nitrogen content, the C:N ratio, and the isotopic nitrogen ratio in the soil had no significant correlations to this group of OTUs nor to any of the other bacterial OTUs (Fig. 6 and 7). The likely explanation for this phenomenon is that bacterial taxa are driven by specific nitrogen compounds, such as ammonia and nitrates, rather than the overall nitrogen contents of soil.
Verrucomicrobia is a relatively poorly known phylum that is commonly reported in soil (64). The abundance of the Verrucomicrobia within the alpine soil bacterial communities was on average 7.7% (Table 1), and different classes of Verrucomicrobia occupied very different soil niches (Fig. 5; see also Fig. S13 in the supplemental material). On average, Verrucomicrobia were overrepresented within the group of OTUs that were negatively correlated with mineral carbon content (Fig. 7B, MiC, blue nodes), and their relative abundance decreased with higher soil mineral carbon content and increased with higher soil total organic carbon content (see Fig. S11 in the supplemental material). In fact, Verrucomicrobia members were particularly rare at higher-elevation sites where mineral carbon contents are high. This and previous observations lend evidence that although a subset of the verrucomicrobial communities may favor oligotrophic lifestyles (65, 66) and Verrucomicrobia may prefer less-nutrient-amended uncultivated soils (67), they generally adopt a life history strategy requiring a nonnegligible amount of organic compounds found more commonly in lower-elevation grasslands in the alpine soil environment.
Neither community-based alpha diversity nor multivariate analyses detected elevation-dependent community turnover in the western Swiss alpine grassland soils, similarly to previously reported accounts (e.g., the study by Fierer et al. [18]). However, a more specific site-by-site examination of the beta dissimilarity revealed weak but statistically significant trends across the elevation gradient. Notably, bacterial communities were more similar to each other when their altitudinal positions were more similar, even if these sites were more distant to each other than neighboring sites at adjacent elevation strata. Furthermore, the neighboring bacterial communities from sites that were within 100 m elevation of each other were significantly more similar to each other than between neighboring sites further than 200 m elevation of each other (Fig. 4H). Thus, although the direction of the alpine soil bacterial community turnover is driven by abiotic environmental factors, such as soil pH, the bacterial community structure tends to be increasingly dissimilar with increasing elevational difference, regardless of the horizontal geographic distance between sites.
Since altitude in mountains results in different elevation-dependent topoclimatic and physicochemical parameters, it was not surprising to find a variety of correlations among bacterial OTUs and elevation-dependent parameters. However, due to their high degree of cross-correlation, it was generally impossible to determine the exact parameters driving these bacterial populations. Exceptionally, certain bacterial OTUs were exclusively correlated with the stable carbon isotope ratio of total soil organic carbon and the monthly moisture index, suggesting their importance for those groups. The soil stable isotope carbon ratio is associated with the plant's physiological state, which therefore may impact the local soil microbiome and may be interesting to follow up in future correlative studies of plant and microbial diversity (68).
In conclusion, we find that the western Swiss alpine grasslands contain highly diverse communities (average 2,918 OTUs per site, with 12,741 total different OTUs across 100 sites), which are shaped as a result of highly variable local environmental conditions. In addition to confirming previously observed general trends of pH, our study indeed shows the many new and detailed contributions of local environmental variables to soil bacterial community diversity. Future studies should focus on correlating bacterial community data to plant and fungal diversity in order to better understand their influence on plants and fungi across the alpine landscape. The bacterial community diversity data can also possibly be integrated into mathematical models that describe landscape distributions similar to those for fungi (69), and which may help develop predictive models to forecast community changes in response to anthropogenic and climate changes.
Supplementary Material
ACKNOWLEDGMENTS
Computations were performed at the Vital-IT Center (http://www.vital-it.ch) for high-performance computing of the SIB Swiss Institute of Bioinformatics. We thank colleagues at the Genomic Technologies Facility and the Vital-IT for their extensive technical help, notably Keith Harshman, Johann Weber, Heinz Stockinger, and Samuel Neuenschwander, as well as Stéphane Dray, the ade4 package expert, and the field and lab assistants who participated in the alpine soil project over the years.
We declare that we have no known conflicts of interest and that all of the above-mentioned coauthors have approved the entire content of the manuscript.
Funding Statement
This work was funded by the FP7-PEOPLE-2010-IIF MP-Alps (grant agreement 273965), Swiss National Fund ProDoc grants MR 135129 and 31003A-1528661, the Agassiz Foundation, and the Pro-Femmes grant from the Faculty of Biology and Medicine of the University of Lausanne.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01170-16.
REFERENCES
- 1.Pellissier L, Niculita-Hirzel H, Dubuis A, Pagni M, Guex N, Ndiribe C, Salamin N, Xenarios I, Goudet J, Sanders IR, Guisan A. 2014. Soil fungal communities of grasslands are environmentally structured at a regional scale in the Alps. Mol Ecol 23:4274–4290. doi: 10.1111/mec.12854. [DOI] [PubMed] [Google Scholar]
- 2.King AJ, Freeman KR, McCormick KF, Lynch RC, Lozupone C, Knight R, Schmidt SK. 2010. Biogeography and habitat modelling of high-alpine bacteria. Nat Commun 1:53. doi: 10.1038/ncomms1055. [DOI] [PubMed] [Google Scholar]
- 3.Bryant JA, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL. 2008. Colloquium paper: microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc Natl Acad Sci U S A 105:11505–11511. doi: 10.1073/pnas.0801920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo G, Kong W, Liu J, Zhao J, Du H, Zhang X, Xia P. 2015. Diversity and distribution of autotrophic microbial community along environmental gradients in grassland soils on the Tibetan Plateau. Appl Microbiol Biotechnol 99:8765–8776. doi: 10.1007/s00253-015-6723-x. [DOI] [PubMed] [Google Scholar]
- 5.Xu M, Li X, Cai X, Gai J, Li X, Christie P, Zhang J. 2014. Soil microbial community structure and activity along a montane elevational gradient on the Tibetan Plateau. Eur J Soil Biol 64:6–14. doi: 10.1016/j.ejsobi.2014.06.002. [DOI] [Google Scholar]
- 6.Yang Y, Gao Y, Wang S, Xu D, Yu H, Wu L, Lin Q, Hu Y, Li X, He Z, Deng Y, Zhou J. 2014. The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J 8:430–440. doi: 10.1038/ismej.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan Y, Si G, Wang J, Luo T, Zhang G. 2014. Bacterial community in alpine grasslands along an altitudinal gradient on the Tibetan Plateau. FEMS Microbiol Ecol 87:121–132. doi: 10.1111/1574-6941.12197. [DOI] [PubMed] [Google Scholar]
- 8.Zinger L, Lejon DPH, Baptist F, Bouasria A, Aubert S, Geremia RA, Choler P. 2011. Contrasting diversity patterns of crenarchaeal, bacterial and fungal soil communities in an alpine landscape. PLoS One 6:e19950. doi: 10.1371/journal.pone.0019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delarze R, Gonseth Y, Eggenberg S, Vust M. 2015. Guide des milieux naturels de Suisse, 3rd ed Editions Rossolis, Bussigny, Switzerland. [Google Scholar]
- 10.Geremia RA, Puşcaş Zinger ML, Bonneville J-M, Choler P. 2016. Contrasting microbial biogeographical patterns between anthropogenic subalpine grasslands and natural alpine grasslands. New Phytol 209:1196–1207. doi: 10.1111/nph.13690. [DOI] [PubMed] [Google Scholar]
- 11.King AJ, Farrer EC, Schmidt SK. 2012. Co-occurrence patterns of plants and soil bacteria in the high-alpine subnival zone track environmental harshness. Front Microbiol 3:347. doi: 10.3389/fmicb.2012.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsiknia M, Paranychianakis NV, Varouchakis EA, Nikolaidis NP. 2015. Environmental drivers of the distribution of nitrogen functional genes at a watershed scale. FEMS Microbiol Ecol 91:fiv052. doi: 10.1093/femsec/fiv052. [DOI] [PubMed] [Google Scholar]
- 13.Fierer N, Jackson R. 2006. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci U S A 103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffiths RI, Thomson BC, James P, Bell T, Bailey M, Whiteley AS. 2011. The bacterial biogeography of British soils. Environ Microbiol 13:1642–1654. doi: 10.1111/j.1462-2920.2011.02480.x. [DOI] [PubMed] [Google Scholar]
- 15.Lanzén A, Epelde L, Garbisu C, Anza M, Martín-Sánchez I, Blanco F, Mijangos I. 2015. The community structures of prokaryotes and fungi in mountain pasture soils are highly correlated and primarily influenced by pH. Front Microbiol 6:1321. doi: 10.3389/fmicb.2015.01321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J-T, Cao P, Hu H-W, Li J, Han L-L, Zhang L-M, Zheng Y-M, He J-Z. 2015. Altitudinal distribution pattern of soil bacterial and archaeal communities along Mt. Shegyla on the Tibetan Plateau. Microb Ecol 69:135–145. doi: 10.1007/s00248-014-0465-7. [DOI] [PubMed] [Google Scholar]
- 17.Singh D, Lee-Cruz L, Kim W-S, Kerfahi D, Chun J-H, Adams JM. 2014. Strong elevational trends in soil bacterial community composition on Mt. Halla, South Korea. Soil Biol Biochem 68:140–149. doi: 10.1016/j.soilbio.2013.09.027. [DOI] [Google Scholar]
- 18.Fierer N, McCain CM, Meir P, Zimmermann M, Rapp JM, Silman MR, Knight R. 2011. Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92:797–804. doi: 10.1890/10-1170.1. [DOI] [PubMed] [Google Scholar]
- 19.Shen C, Ni Y, Liang W, Wang J, Chu H. 2015. Distinct soil bacterial communities along a small-scale elevational gradient in alpine tundra. Front Microbiol 6:582. doi: 10.3389/fmicb.2015.00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marini L, Fontana P, Klimek S, Battisti A, Gaston K. 2009. Impact of farm size and topography on plant and insect diversity of managed grasslands in the Alps. Biol Conserv 142:394–403. doi: 10.1016/j.biocon.2008.10.034. [DOI] [Google Scholar]
- 21.Chapin FS III, Zavaleta ES, Eviner VT, Naylor RL, Vitousek PM, Reynolds HL, Hooper DU, Lavorel S, Sala OE, Hobbie SE, Mack MC, Díaz S. 2000. Consequences of changing biodiversity. Nature 405:234–242. doi: 10.1038/35012241. [DOI] [PubMed] [Google Scholar]
- 22.Knelman JE, Nemergut DR. 2014. Changes in community assembly may shift the relationship between biodiversity and ecosystem function. Front Microbiol 5:424. doi: 10.3389/fmicb.2014.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagg C, Bender SF, Widmer F, van der Heijden MGA. 2014. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc Natl Acad Sci U S A 111:5266–5270. doi: 10.1073/pnas.1320054111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guisan A, Broennimann O, Buri A, Cianfrani C, D'Amen M, Di Cola V, Fernandes R, Gray S, Mateo RG, Pinto E, Pradervand J-N, Scherrer D, von Däniken I, Yashiro E, Vittoz P. 2016. Climate change impact on mountain biodiversity, in press In Lovejoy T, Hannah L (ed), Climate change and biodiversity. Yale University Press, New Haven, CT. [Google Scholar]
- 25.Theurillat J, Guisan A. 2001. Potential impact of climate change on vegetation in the European Alps: a review. Clim Chang 50:77–109. doi: 10.1023/A:1010632015572. [DOI] [Google Scholar]
- 26.Hirzel A, Guisan A. 2002. Which is the optimal sampling strategy for habitat suitability. Ecol Model 157:311–341. [Google Scholar]
- 27.Randin CF, Engler R, Pearman PB, Vittoz P, Guisan A. 2010. Using georeferenced databases to assess the effect of climate change on alpine plant species and diversity, p 149–163. In Spehn EM, Körner C (ed), Data mining for global trends in mountain biodiversity. CRC Press, Boca Raton, FL. [Google Scholar]
- 28.Dubuis A, Giovanettina S, Pellissier L, Pottier J, Vittoz P, Guisan A. 2013. Improving the prediction of plant species distribution and community composition by adding edaphic to topo-climatic variables. J Veg Sci 24:593–606. doi: 10.1111/jvs.12002. [DOI] [Google Scholar]
- 29.National Resources Conservation Service. 2009. Soil survey field and laboratory methods manual. National Resources Conservation Service, US Department of Agriculture, Lincoln, NE. [Google Scholar]
- 30.Reference deleted.
- 31.Lafargue E, Espitalie J, Pillot D. 1998. Rock-Eval 6 applications in hydrocarbon exploration, production, and soil contamination studies. Oil Gas Sci Technol 53:421–437. [Google Scholar]
- 32.Tamburini F, Adatte T, Föllmi K, Bernasconi SM, Steinmann P. 2003. Investigating the history of East Asian monsoon and climate during the last glacial–interglacial period (0–140,000 years): mineralogy and geochemistry of ODP sites 1143 and 1144, South China Sea. Mar Geol 201:147–168. doi: 10.1016/S0025-3227(03)00214-7. [DOI] [Google Scholar]
- 33.Spangenberg JE, Ferrer M, Tschudin P, Volken M, Hafner A. 2010. Microstructural, chemical and isotopic evidence for the origin of late neolithic leather recovered from an ice field in the Swiss Alps. J Archaeol Sci 37:1851–1865. doi: 10.1016/j.jas.2010.02.003. [DOI] [Google Scholar]
- 34.Lazarevic V, Whiteson K, Huse S, Hernandez D, Farinelli L, Osterås M, Schrenzel J, François P. 2009. Metagenomic study of the oral microbiota by Illumina high-throughput sequencing. J Microbiol Methods 79:266–271. doi: 10.1016/j.mimet.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank DN. 2009. BARCRAWL and BARTAB: software tools for the design and implementation of barcoded primers for highly multiplexed DNA sequencing. BMC Bioinformatics 10:1–13. doi: 10.1186/1471-2105-10-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gloor GB, Hummelen R, Macklaim JM, Dickson RJ, Fernandes AD, MacPhee R, Reid G. 2010. Microbiome profiling by Illumina sequencing of combinatorial sequenced-tagged PCR products. PLoS One 5:e15406. doi: 10.1371/journal.pone.0015406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. 2012. PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics 13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dixon P. 2003. VEGAN, a package of R functions for community ecology. J Veg Sci 14:927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- 41.Dray S, Dufour A. 2007. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20. doi: 10.18637/jss.v022.i04. [DOI] [Google Scholar]
- 42.Good IJ. 1953. The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264. [Google Scholar]
- 43.Esty WW. 1986. The efficiency of Good's nonparametric coverage estimator. Ann Stat 14:1257–1260. doi: 10.1214/aos/1176350066. [DOI] [Google Scholar]
- 44.Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- 45.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barberan A, Bates ST, Casamayor EO, Fierer N. 2012. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J 6:343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300. [Google Scholar]
- 48.Bastian M, Heymann S, Jacomy M. 2009. Gephi: an open source software for exploring and manipulating network. Third International AAAI Conference on Weblogs and Social Media, 17 to 20 May 2009, San Jose, CA https://gephi.org/publications/gephi-bastian-feb09.pdf. [Google Scholar]
- 49.Hu Y. 2005. Efficient and high quality force-directed graph drawing. Math J 10:37–71. [Google Scholar]
- 50.Jacomy M, Venturini T, Heymann S, Bastian M. 2014. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS One 9:e98679. doi: 10.1371/journal.pone.0098679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bartram AK, Lynch MDJ, Stearns JC, Moreno-Hagelsieb G, Neufeld JD. 2011. Generation of multimillion sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl Environ Microbiol 77:3846–3852. doi: 10.1128/AEM.02772-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Cong J, Lu H, Li G, Xue Y, Deng Y, Li H, Zhou J, Li D. 2015. Soil bacterial diversity patterns and drivers along an elevational gradient on Shennongjia Mountain, China. Microb Biotechnol 8:739–746. doi: 10.1111/1751-7915.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Newman MEJ. 2006. Modularity and community structure in networks. Proc Natl Acad Sci U S A 103:8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lauber C, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen C, Xiong J, Zhang H, Feng Y, Lin X, Li X, Liang W, Chu H. 2013. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol Biochem 57:204–211. doi: 10.1016/j.soilbio.2012.07.013. [DOI] [Google Scholar]
- 56.Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, Knight R, Fierer N. 2010. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4:1340–1351. doi: 10.1038/ismej.2010.58. [DOI] [PubMed] [Google Scholar]
- 57.Yavitt JB, Yashiro E, Cadillo-Quiroz H, Zinder SH. 2012. Methanogen diversity and community composition in peatlands of the central to northern Appalachian Mountain region, North America. Biogeochemistry 109:117–131. doi: 10.1007/s10533-011-9644-5. [DOI] [Google Scholar]
- 58.Carrie J, Sanei H, Stern G. 2012. Standardisation of Rock–Eval pyrolysis for the analysis of recent sediments and soils. Org Geochem 46:38–53. doi: 10.1016/j.orggeochem.2012.01.011. [DOI] [Google Scholar]
- 59.Disnar JR, Guillet B, Keravis D, Di-Giovanni C, Sebag D. 2003. Soil organic matter (SOM) characterization by Rock-Eval pyrolysis: scope and limitations. Org Geochem 34:327–343. doi: 10.1016/S0146-6380(02)00239-5. [DOI] [Google Scholar]
- 60.Will C, Thürmer A, Wollherr A, Nacke H, Herold N, Schrumpf M, Gutknecht J, Wubet T, Buscot F, Daniel R. 2010. Horizon-specific bacterial community composition of German grassland soils as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol 76:6751–6759. doi: 10.1128/AEM.01063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Larsen PE, Collart FR, Field D, Meyer F, Keegan KP, Henry CS, McGrath J, Quinn J, Gilbert JA. 2011. Predicted Relative Metabolomic Turnover (PRMT): determining metabolic turnover from a coastal marine metagenomic dataset. Microb Inform Exp 1:4. doi: 10.1186/2042-5783-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sebastián M, Gasol JM. 2013. Heterogeneity in the nutrient limitation of different bacterioplankton groups in the Eastern Mediterranean Sea. ISME J 7:1665–1668. doi: 10.1038/ismej.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gaby JC, Buckley DH. 2012. A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One 7:e42149. doi: 10.1371/journal.pone.0042149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA, Knight R, Fierer N. 2011. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 43:1450–1455. doi: 10.1016/j.soilbio.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.da Rocha UN, Andreote FD, de Azevedo JL, van Elsas JD, van Overbeek LS. 2009. Cultivation of hitherto-uncultured bacteria belonging to the Verrucomicrobia subdivision 1 from the potato (Solanum tuberosum L.) rhizosphere. J Soils Sediments 10:326–339. doi: 10.1007/s11368-009-0160-3. [DOI] [Google Scholar]
- 66.Senechkin IV, Speksnijder AGCL, Semenov AM, van Bruggen AHC, van Overbeek LS. 2010. Isolation and partial characterization of bacterial strains on low organic carbon medium from soils fertilized with different organic amendment. Microb Ecol 60:829–839. doi: 10.1007/s00248-010-9670-1. [DOI] [PubMed] [Google Scholar]
- 67.Carbonetto B, Rascovan N, Álvarez R, Mentaberry A, Vázquez MP. 2014. Structure, composition and metagenomic profile of soil microbiomes associated to agricultural land use and tillage systems in Argentine Pampas. PLoS One 9:e99949. doi: 10.1371/journal.pone.0099949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dias ACF, Dini-Andreote F, Hannula SE, Andreote FD, Pereira E Silva MDC, Salles JF, de Boer W, van Veen J, van Elsas JD. 2013. Different selective effects on rhizosphere bacteria exerted by genetically modified versus conventional potato lines. PLoS One 8:e67948. doi: 10.1371/journal.pone.0067948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pellissier L, Pinto E, Niculita-Hirzel H, Moora M, Villard L, Goudet J, Guex N, Pagni M, Xenarios I, Sanders I, Guisan A. 2013. Plant species distributions along environmental gradients: do belowground interactions with fungi matter? Front Plant Sci 4:500. doi: 10.3389/fpls.2013.00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ndiribe C, Pellissier L, Dubuis A, Vittoz P, Salamin N, Guisan A. 2013. Plant functional and phylogenetic turnover correlate with climate and land use in the Western Swiss Alps. J Plant Ecol 7:439–450. doi: 10.1093/jpe/rtt064. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.